
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 30 June 2022
Sec. Livestock Genomics
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.909039
This article is part of the Research TopicOmics Applied to Livestock GeneticsView all 12 articles
 Chunlei Zhang1Jing Zhao1Yanli Guo1
Chunlei Zhang1Jing Zhao1Yanli Guo1 Qinglei Xu1Mingzheng Liu1Meng Cheng1Xiaohuan Chao1
Qinglei Xu1Mingzheng Liu1Meng Cheng1Xiaohuan Chao1 Allan P. Schinckel2
Allan P. Schinckel2 Bo Zhou1*
Bo Zhou1*Copy number variation (CNV) has been considered to be an important source of genetic variation for important phenotypic traits of livestock. In this study, we performed whole-genome CNV detection on Suhuai (SH) (n = 23), Chinese Min Zhu (MZ) (n = 11), and Large White (LW) (n = 12) pigs based on next-generation sequencing data. The copy number variation regions (CNVRs) were annotated and analyzed, and 10,885, 10,836, and 10,917 CNVRs were detected in LW, MZ, and SH pigs, respectively. Some CNVRs have been randomly selected for verification of the variation type by real-time PCR. We found that SH and LW pigs are closely related, while MZ pigs are distantly related to the SH and LW pigs by CNVR-based genetic structure, PCA, VST, and QTL analyses. A total of 14 known genes annotated in CNVRs were unique for LW pigs. Among them, the cyclin T2 (CCNT2) is involved in cell proliferation and the cell cycle. The FA Complementation Group M (FANCM) is involved in defective DNA repair and reproductive cell development. Ten known genes annotated in 47 CNVRs were unique for MZ pigs. The genes included glycerol-3-phosphate acyltransferase 3 (GPAT3) is involved in fat synthesis and is essential to forming the glycerol triphosphate. Glutathione S-transferase mu 4 (GSTM4) gene plays an important role in detoxification. Eleven known genes annotated in 23 CNVRs were unique for SH pigs. Neuroligin 4 X-linked (NLGN4X) and Neuroligin 4 Y-linked (NLGN4Y) are involved with nerve disorders and nerve signal transmission. IgLON family member 5 (IGLON5) is related to autoimmunity and neural activities. The unique characteristics of LW, MZ, and SH pigs are related to these genes with CNV polymorphisms. These findings provide important information for the identification of candidate genes in the molecular breeding of pigs.
Copy number variation (CNV) was discovered in 1936 by Bridges in drosophila (1). The duplication of a segment of the drosophila Bar gene caused failure in the formation of normal compound eyes. The definition of CNV is constantly being refined with the additional research. Redon et al. (2) defined CNV as a DNA fragment whose copy number has changed in contrast to the reference genome, and the size from 1 kb to several Mb. According to its structural characteristics, a CNV can be classified as copy number gain or copy number loss. When both copy number gain and loss occur, it is called both type. The CNV mainly affects gene expression through gene dose-effect and gene interruption (3). When the copy number variation region (CNVR) contains dose-sensitive genes, the gene expression level changes with the copy number or the CNV in the coding region influences the gene function and leads to gene disruption and loss of coding ability.
A previous study detected 3,131 CNVRs in Chinese and European pigs. There were 129 and 147 unique CNVRs in Chinese pigs and European pigs, respectively (4). According to the functional enrichment analysis, the genes containing unique CNVRs in Chinese pig breeds are associated with disease resistance and high fertility, while the genes containing unique CNVRs in European pig breeds are closely related to muscle development (4). These results are consistent with the characteristics of Chinese and European pig breeds. A comprehensive CNV study on 98 Xiang pigs and 22 Kele pigs detected 172 CNVRs in 660 annotated genes, which are enriched in sensory, cognitive, reproductive, and ATP synthesis functions (5). These functions are well-matched with the living environment and breed characteristics of Xiang pigs and Kele pigs. In particular, the genes of propagation-related CNVRs have obvious contact with the number of piglets in the Xiang pigs. In addition, studies on the Italian white pig (6), Taihu pig (7), and Bama pig (8) also found a correlation between the breed characteristics and the functions of genes annotated in CNVRs. These studies indicate that the functions of CNVRs are associated with the phenotypes of pigs.
Large White (LW) pigs are well known for their growth and reproductive performance. Min Zhu (MZ) pigs are distributed in northern China and have the characteristics of substantial fat deposition and excellent stress resistance. Suhuai (SH) pigs are crossbred pigs that contain 75 % LW and 25 % Chinese Huai. The Huai and MZ pig breeds originated in north China. The objective of this study is to explore the characteristics of CNV in European LW, Chinese MZ, and crossbred SH pigs at the whole-genome level.
Twenty-three SH pigs were selected from the Huaiyin pig farm in Huai'an, Jiangsu Province. A standard phenol/chloroform/isoamyl alcohol protocol was used to extract genomic DNA from pig ear tissue samples. The Illumina Hiseq2000 platform was used for whole-genome sequencing. In addition, the whole-genome sequencing data of MZ pigs (n = 11) and LW pigs (n = 12) were downloaded from the public database (https://www.ncbi.nlm.nih.gov/) (Supplementary Table 1). The FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to analyze the quality of the sequencing data and the parameter was as follows: fastqc -o output -t thread seqfile1..seqfileN. Where “-o” indicates the pathway of the out file, “-t” indicates the number of threads running programs, and “seqfile” indicates the input sequencing data. Then the Cutadapt (https://cutadapt.readthedocs.io/en/stable/) was used for quality filtering and reads trimming. The parameter was as follows: cutadapt -q 10,15 –quality-base = 33 -o output.fastq input.fastq. Where “-q” indicates filtering the quality of the reads, 10 and 15 represent the threshold of the 3' and 5', “–quality-base = 33” indicates the phred33 score system, and “-o” indicates the pathway of the out file. The sequencing data were integrated by MultiQC (v1.11) to meet the requirements of CNV detection (Supplementary Figure 1) (9). The sequences were aligned to the reference genome (Sscrofa 11.1) assembly using the Burrows-Wheeler Aligner (BWA) (v 0.7.17) (10). The overall average sequencing depth reaches 12.89 × , up to 16.22 × , at lowest 9.16 × , and 46 samples' average mapping ratio reached 96.47%.
We used the software CNVcaller to detect CNVs and determine the CNVRs (11). All steps were conducted using the default program. First, build a reference genome database. The reference genome was based on the sliding window of the user's specified size, and the GC, repeat, and gap content of each window on the genome were counted on the genome. The command was as follows: Perl CNVReferenceDB.pl reference.fa -w 800. Where “reference.fa” is the reference genome, “-w” indicates the size of the sliding window. According to the author's suggestion, we selected a window size of 800 bp, and a step of 400 bp to generate the reference genome database. Second, the absolute copy number of each window was calculated. The BAM file (BWA comparison generation) of each sample and the number of reads in each window were analyzed. The high similarity reads (≥97%) were merged, and the low-complexity regions were removed. Based on the GC content, the correct the number of reads in each window after merging was used to calculate the absolute copy number. The command was as follows: bash Individual.Process.sh -b sample.bam -h sample -d link. Where “-b” indicates the BAM file, “-h” indicates the label of the BAM file, and “-d” indicates the link files required for correction. The third step was determination of the CNVR. The boundary of each CNVR was preliminarily determined by comprehensively considering the distribution of absolute copy number, the frequency of variation, and the significant correlation between adjacent windows (primaryCNVR). Then, the adjacent CNVRs whose copy number distribution was significantly related to the population were further merged to obtain the final CNV detection results (mergedCNVR). The command was as follows: bash CNV.Discovery.sh -l list -e exclude_list -f 0.1 -h 1 -r 0.5 -p primaryCNVR -m mergeCNVR. Where “-l” indicates the list of results files after the absolute copy number correction; “-e” indicates the samples in this list are not used for the detection of CNVR. “-f” and “-h” represent the difference between the individual's absolute copy number and the reference absolute copy number in frequency and quantity, which greater than the setting value is considered a candidate CNV window; “-r” indicates the correlation coefficient of the absolute copy number of the adjacent candidate CNV window (no overlap), which greater than the setting value will be merged; and “-p” and “-m” indicate the output files primaryCNVR and mergeCNVR. A genome-wide CNVR map was drawn by RIdeogram (12).
The CNVRs detected were used to analyze the genetic structure differences among three pig breeds. We performed principal component analysis (PCA) by PLINK (v 1.90) (13). PLINK was used to convert the CNVRs file into bed format. ADMIXTURE (v 1.3.0) was used to execute population genetic structure analysis (14). We first set the ancestral population number K value between 1 and 5, then compute the Cross-Validation Error for each K values. When the Cross-Validation Error value became the least, the K value was the number of ancestors. MEGAX was used for evolutionary tree analysis to evaluate the genetic distance between the populations. By calculating the VST value (2), we analyze the genetic difference between the two groups.
Where Vtotal is the total variance in copy number between the two groups, V1 and V2 are variances in copy number of population 1 and population 2, respectively. N1 and N2 are the numbers of samples of population 1 and population 2, respectively. Ntotal is the total number of all the samples. We compare the genetic distance between groups by the mean VST values. All diagrams were drawn by ggplot2 (15, 16).
To further study the relationship between CNVRs and the phenotypic characteristics of the population, a Venn diagram was drawn by TBTOOLS (v 1.098661) (17) to observe the differential and common CNVRs. Gene annotation and pathway enrichment were conducted for the population-specific CNVRs using g:Profiler (18) and KOBAS (19), respectively.
QTL data were downloaded from Pig QTLdb (https://www.animalgenome.org/cgi-bin/QTLdb/SS/index). Bedtools (v 2.15.0) (20) was used to overlap the QTLs with the group-specific CNVRs, and the unique corresponding QTL area was obtained after removing the repeat value. According to the description of QTL traits, the group-specific CNVRs that affect the phenotypes of LW, MZ, and SH pigs were analyzed.
We randomly selected 4 CNVRs fragments to detect copy number polymorphisms by qPCR and the 2−ΔCt method, ΔCt value = (Cttarget – Ctreference) (21). Primers used in qPCR were designed by Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast). The highly conserved fragment of the GCG in pigs was selected as an internal reference gene (22). Primer sequences for CNVRs and GCG are shown in Supplementary Table 2. To ensure that the test samples were comparable to the GCG, we first constructed the standard curve of each CNVR after gradient dilution of DNA. Total CNVs were verified on the QuantStudio 5 real-time PCR system (ABI, USA), and PCR amplification conditions were designed according to the manufacturer's description (Vazyme, China). The PCR amplification system was completed in a 20 μL system, including the following ingredients: 10 μL SYBR master Mix, 2 μL DNA (around 5ng), 0.4 μL forward primers, and 0.4 μL reverse primers, and 7.2 μL water. The PCR conditions were as follows: first step 95° C for 30 seconds followed by 40 cycles at 95 ° C for 10 s and 60 ° C for 30 s. The CNV type detected by the above PCR method was the same as those detected by the CNVcaller. Where CNVR-9017 was the gain type in LW pigs, but the normal type in SH pigs. The CNVR-1169, CNVR-9126, and CNVR-1771 were expressed in two pig breeds as the gain type. In addition, we used the Integrative Genomics Viewer (IGV) (23) to visualize the genome of the samples, and its results were the same as qPCR (Supplementary Figure 2). Each CNVR fragment has 4 biological repetitions in both LW and SH pigs, and all samples were performed in triplicate.
A total of 11,173 CNVRs were detected in 46 pigs (Supplementary Table 3). There were 10,917, 10,885, and 10,836 CNVRs detected in SH, LW, and MZ pigs, respectively. The coverage area of these CNVRs in the three populations is more than 43 million bp, which accounts for about 1.8% of the whole genome (Sscrofa 11.1) (Supplementary Table 4). In all samples, there were 3,457, 2390, and 5,326 cases of copy number loss, copy number gain, and both type, respectively (Figure 1). The length of CNVRs ranges from 1.6 to 560 kb, but 61.23% of CNVRs are 1.6 to 3 kb, and only 0.75% CNVRs are more than 30 kb (Supplementary Figure 3). Moreover, a total of 8,247 CNVRs were detected in <5 pigs, and 4,134 CNVRs were found in the unique individual (Supplementary Figure 4).
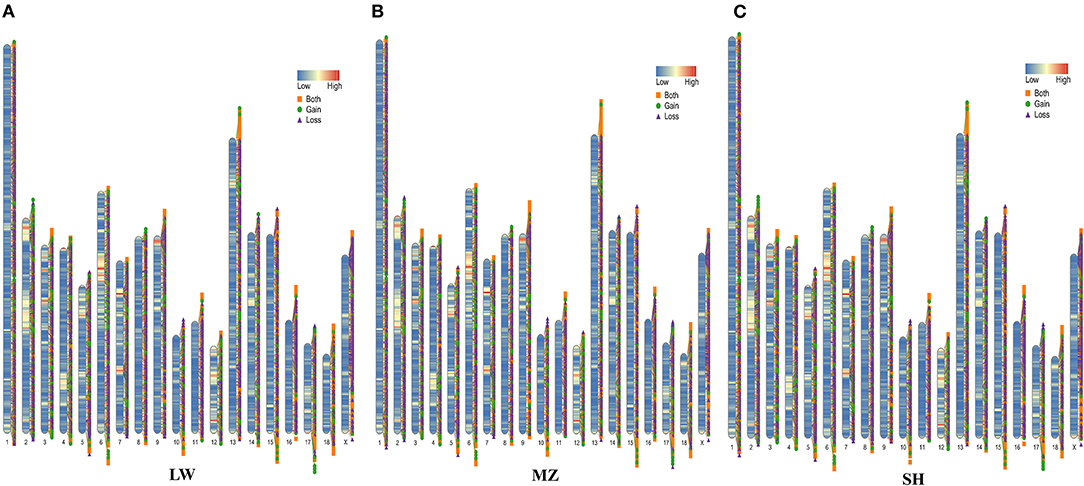
Figure 1. The genome distribution of CNVRs and variation types of LW (A), MZ (B), and SH (C) pigs. The legend of “Low” to “High” indicates the gene density on the pig chromosomes. The yellow square represents the both type, the green circle represents the gain type, and the purple triangle represents the loss type.
A PCA graph was developed with all the samples having been divided into three groups: SH, MZ, and LW pig breeds (Figure 2A). The LW and SH pigs are closer in the PCA diagram, and the individuals are arrayed tight. The MZ pigs are far from them, and the individuals are scattered.
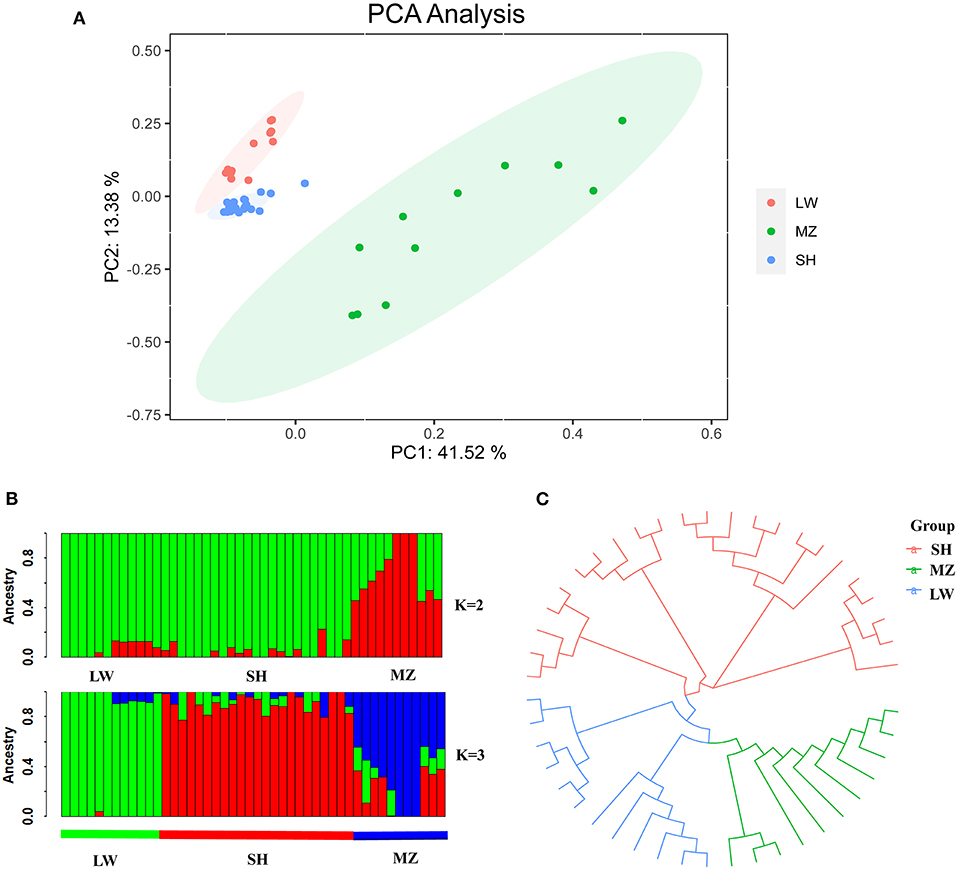
Figure 2. (A) PCA plot of LW, MZ, and SH pigs. Red, green, and blue represent LW, MZ, and SH pigs, respectively. (B) Diagram of genetic structure analysis, the K value represents the number of the hypothetical ancestor. When the ancestral population number K = 2, there are obvious differences between LW and MZ pigs, while the information of SH pigs is covered by LW pigs, green and red represent LW and MZ pigs, respectively. When K = 3, the three pig breeds are well separated. Green, red, and blue represent LW, SH, and MZ pigs, respectively. (C) Evolutionary tree diagram of LW, SH, and MZ pigs. The location of SH pigs is closer to the root, moreover, the genetic distance between SH and LW pigs is less than SH and MZ pigs.
When the ancestral population number K = 2, there are obvious differences between LW and MZ pigs, while the information of SH pigs is covered by that of LW pigs. When K = 3, the Cross-Validation Error is the smallest (Supplementary Table 5), and the three pig breeds are well separated (Figure 2B). The result of phylogenetic tree analysis is similar to that of PCA. Since the genetic background of the SH pig is complicated (containing 75 % Large White and 25 % Chinese Huai), the position of the SH pigs is close to the root of the tree, and the distance to LW pigs is closer than MZ pigs (Figure 2C). The average VST value of SH and LW pigs is just 0.111; but the average VST values are 0.234 and 0.265 in SH and MZ pigs and LW and MZ pigs, respectively (Figure 3). The VST analysis results are the same as the PCA analysis and genetic structural analysis. The genetic distance between SH and LW pigs is smaller than that between LW and MZ pigs.
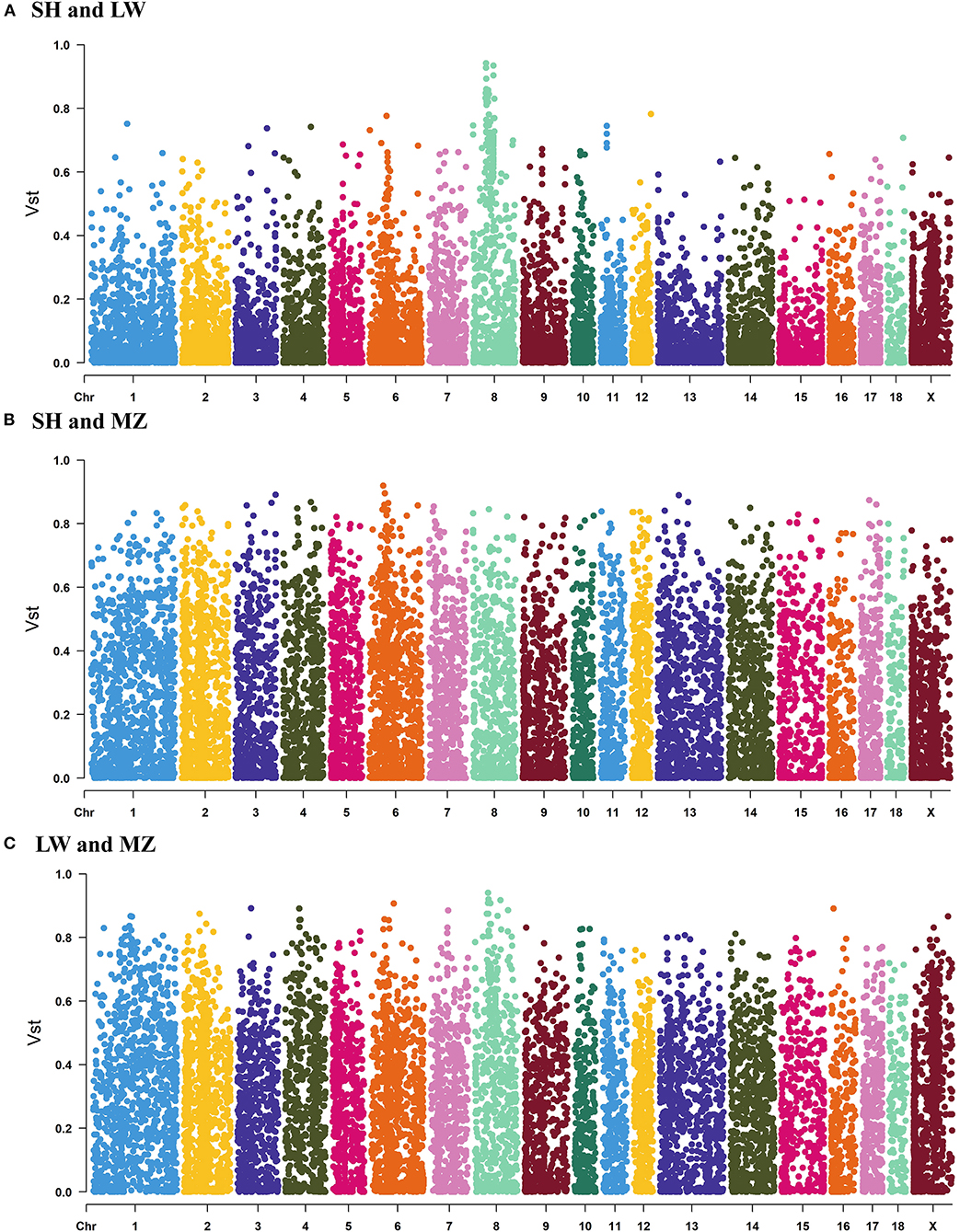
Figure 3. The VST values of all the copy number variation regions (CNVRs) in SH and LW (A) pigs, SH and MZ (B) pigs, LW and MZ (C) pigs. The average VST value of SH and LW pigs is just 0.111; but the average VST values are 0.234 and 0.265 in SH and MZ pigs and LW and MZ pigs, respectively.
The differences in CNVRs between pig breeds were compared through the Venn diagram (Figure 4A). A total of 10,671 CNVRs are shared among the three pig breeds. There are 23, 47, and 39 group-specific CNVRs in the SH, MZ, and LW pigs, respectively. A total of 140 CNVRs are common in the SH and LW pigs, while only 83 CNVRs are common in the SH and MZ pigs, and 35 CNVRs are common in the LW and MZ pigs.
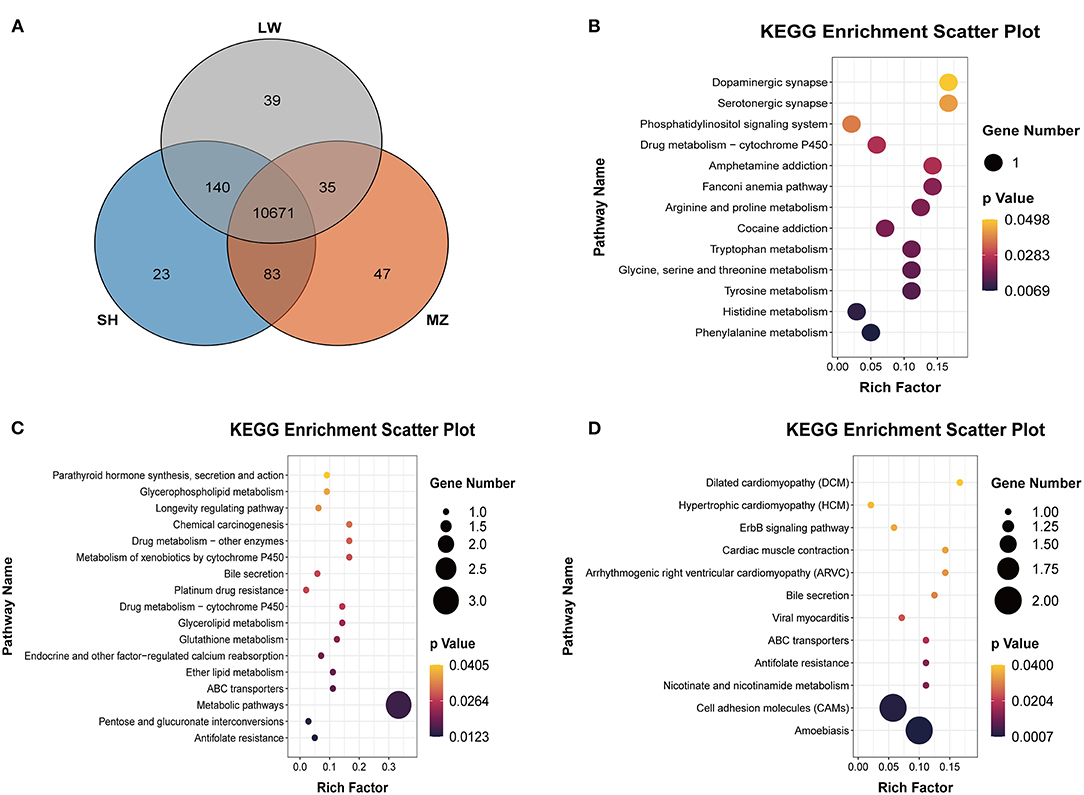
Figure 4. (A) Venn diagram of the CNVRs in LW, SH, and MZ pigs. The known genes of LW (B), MZ (C), and SH (D) pigs in the group-specific CNVRs were analyzed in the KEGG pathway.
We noted the genes associated with group-specific CNVRs and discovered 35 known genes (Table 1) and 25 novel genes (Supplementary Table 6). These known genes were analyzed in the KEGG pathway.
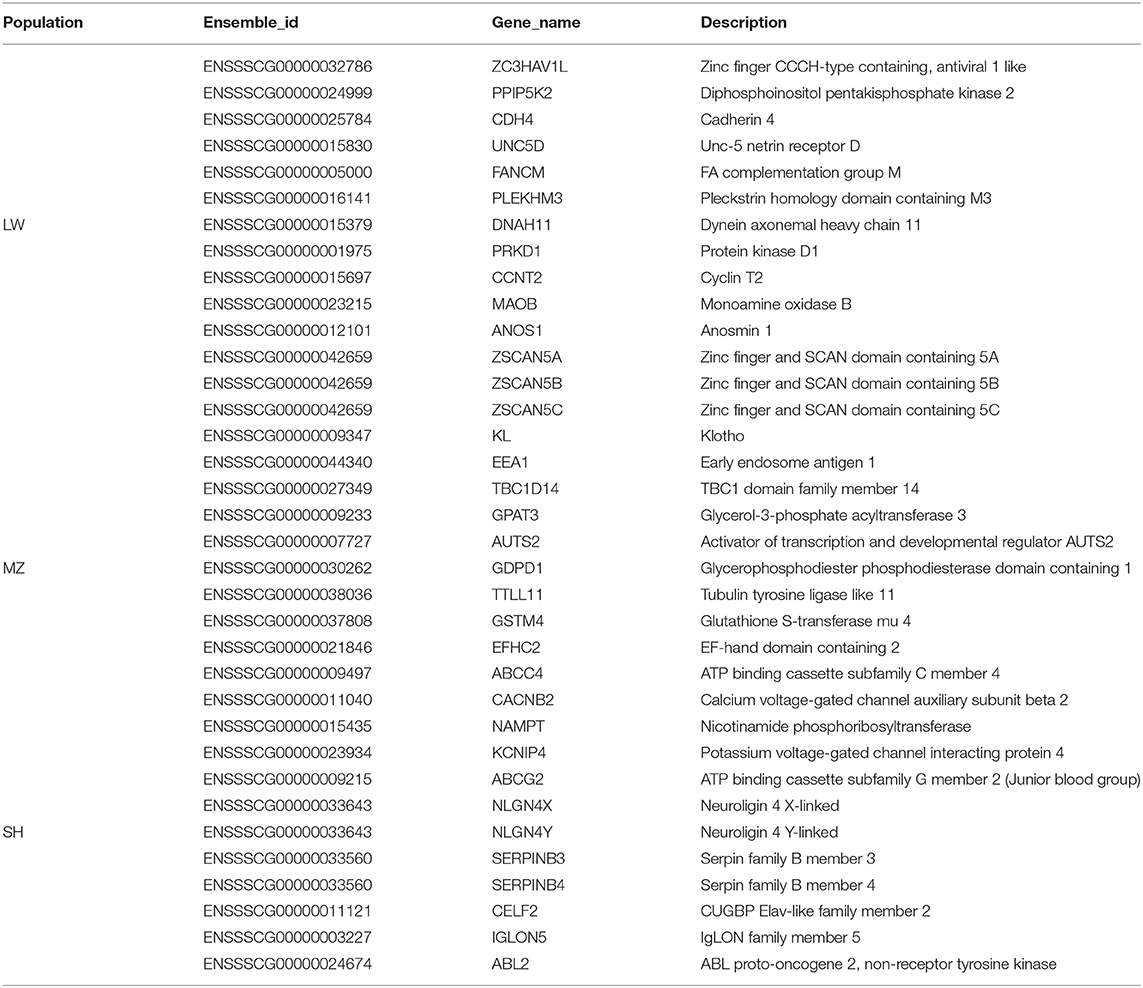
Table 1. Annotated genes in group-Specific CNVRs of LW, MZ, and SH pigs.
A total of 14 known genes were annotated in 39 unique CNVRs in LW pigs. These genes regulate the metabolism of phenylalanine, histidine, and other amino acids based on the KEGG pathway (Figure 4B). The CCNT2 gene is widely involved in the regulation of cell differentiation and the cell cycle. In fibroblasts of C2C12 cells, the overexpression of CCNT2 strengthened MyoD-dependent transcription and promoted myogenic differentiation (24). A comprehensive study reported that the CCNT2 gene induced the differentiation of muscle cells with the molecular partner Pkn (25), which may play a positive role in the meat production of LW pigs. The FANCM gene is involved in defective DNA repair and reproductive cell development (26). Previous studies found that the FANCM gene was associated with Non-obstructive Azoospermia and ovarian deficiency, which led to male/female infertility (27, 28). It may be related to the reproductive performance of the LW pigs. LW pigs are commonly mated to other maternal lines to produce crossbred commercial sows.
We annotated 10 known genes in 47 unique CNVRs in MZ pigs. These genes are enriched in “Antifolate Resistance,” “Metabolic Pathways,” and “Glycerolipid Metabolism” based on the KEGG pathway (Figure 4C). A previous study reported that the GPAT3 gene plays an important role in lipid metabolism, which causes rapid growth and exquisite meat quality in Yunling cattle (29). The knockout of the GPAT3 gene altered energy balance in diet-induced obesity in mice, indicating that the GPAT3 gene plays a role in regulating energy and lipid homeostasis (30). It may be related to the fat deposition capacity of MZ pigs. The GSTM4 and TBC1D14 genes are considered to participate in detoxification and autophagy (31, 32). These genes are related to “Glutathione Metabolism,” “Platinum Drug Resistance,” and “Metabolism of Xenobiotics by Cytochrome P450” detoxification and resistance gene pathways.
We have annotated 11 known genes in the 23 unique CNVRs in SH pigs. These genes are enriched in resistance and ATP-related pathways (Figure 4D). Interestingly, some genes are associated with neurodevelopment. The NLGN4X and NLGN4Y genes are located on the X and Y chromosomes, respectively. Neurogenesis, neuron differentiation, and muscle development are increasingly disturbed in neuron stem cells with NLGN4X knockdown, including DLG4 and NLGN3 postsynaptic genes also have decreased expression (33). The IGLON5 gene participates in regulating sleep and other neural activities and is also related to autoimmunity (34).
The group-specific CNVRs of LW, SH, and MZ pigs were mapped in the QTLs of the pigs. There are 1,139, 938, and 1,283 QTLs in the SH, LW, and MZ pigs, respectively. A Venn diagram shows that 248 QTLs overlap between the LW and SH pigs, 237 QTLs overlap between the SH and MZ pigs, and 178 QTLs overlap between the MZ and LW pigs. There are 285, 545, and 700 group-specific QTLs in the SH, LW, and MZ pigs, respectively (Supplementary Figure 5). A circus diagram was used to show the location of these unique QTLs (Figure 5A). The effects of QTLs on traits are divided into three levels, “Trait Categories,” “Trait Type,” and “Trait.” The difference in the meat and disease resistance traits of LW, MZ, and SH pigs is more distinct (35) (Figures 5B–D). So QTLs for meat and health trait categories were analyzed.
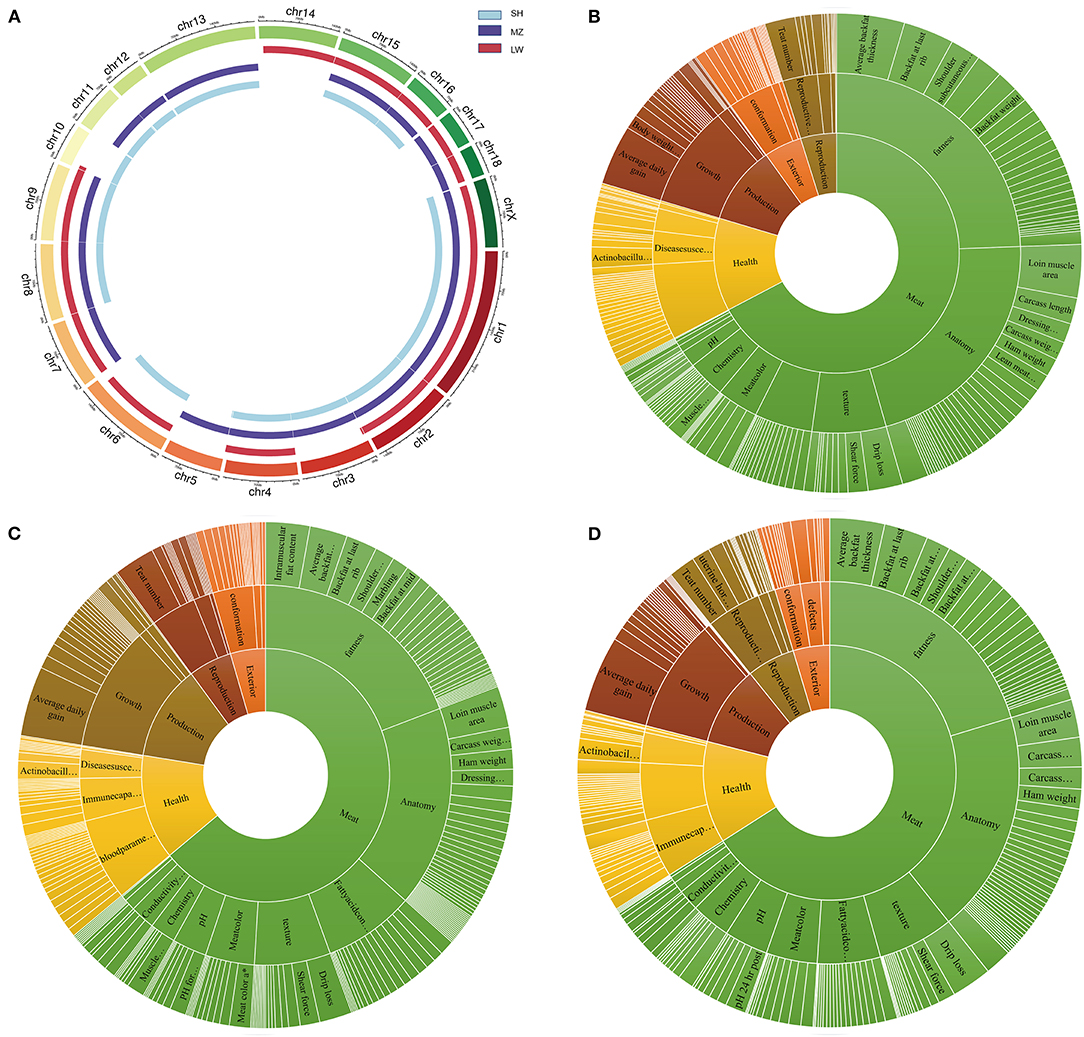
Figure 5. (A) The genome distribution of the group-specific QTLs in SH, MZ, and LW pigs. (B–D) are the group-specific QTLs in SH, LW, and MZ pigs.
In the anatomy type of the meat category, the trait cases of “muscle area and muscle fiber” and “fat to meat ratio and fat-cut percentage” are different in LW, MZ, and SH pigs. The number of muscle-related QTLs is 12.2 times that of fat-related QTLs in LW pigs (61/5). And this ratio is only 3.6 times and 7 times in MZ and SH pigs (55/15, 35/5). Interestingly, the “EnzyMeactivity” QTLs are unique to the MZ pigs. The number of total “NADPH-generation enzyme activity” and “NADP-malate dehydrogenase activity” is 12, which is related to the oxidation reaction in the organism, particularly fatty acids generation (36). In the trait category of health, the number of “Immune capacity” is huge difference among LW, MZ, and SH pigs, with a total of 24 traits, and 64 QTLs related to immune capacity in the MZ pigs, but only 6 traits, 23 QTLs, and 14 traits, 34 QTLs are in LW and SH pigs, respectively.
The role of CNV's is an increasingly discussed academic topic, and previous studies on CNV have been conducted in humans, cattle, sheep, and other species (37–40). CNVs could destroy the normal expression of genes and ultimately cause phenotypic changes mainly through dosage effects, interruption, and position effects of gene deletion and duplication (41–43). As a type of essential variation in the genome, CNV polymorphisms play key roles in species evolution, environmental adaptation, disease resistance, and disease susceptibility (44–46). However, numerous past studies have concentrated on CNV on the chromosomal DNA with little attention given to CNV of non-chromosomal DNA. Mitochondrial DNA (mtDNA) passes through maternal inheritance, which has been confirmed to be related to many traits, including respiratory and cardiovascular disease (47). As a component of ribosomes, rRNA easily becomes a substrate of homologous recombination resulting in CNV due to its repetitive sequence structure (48).
In our present study, we noticed that LW pigs have excellent meat production. Several genes containing the unique CNVRs are involved in the regulation of cell proliferation and cell cycle regulation in LW pigs. These genes have extensive participation in muscle growth and development. We also obtained the same results in the QTL analysis.
Among these genes, the CCNT2 gene is related to cell differentiation and cell cycle, especially regulating the differentiation of muscle cells (24). Many studies have focused on the combined analysis of microRNA (miRNA) and CCNT2. Previous research reported that miR-15a, miR-155-5p, and miR-188-5p inhibit muscle differentiation and skeletal muscle development via target binding CCNT2 (49–51). Due to their great reproductive performance, in the modern pig breeding systems, LW pigs are used to produce crossbred female parents. Among these genes, FANCM is involved in DNA damage repair, and the mutation causes deaths of spermatogenic cells at all levels and stagnation of round spermatids, which causes male reproductive disorders, including sperm deformities, decreased motility, and decreased numbers (27). These results are interesting because these genes may be related to the reproductive performance of LW pigs.
We found GPAT3 related to adipogenesis in unique CNVRs in MZ pigs. The promoter polymorphisms of the GPAT3 were associated with intramuscular fat content in Laiwu pigs, and the knockout of GPAT3 was related to insulin resistance and fatty liver in a mouse model of severe congenital generalized lipodystrophy (30, 52). The GPAT3 accelerated the fat production capacity of MZ pigs. Understandably, the habitat of the MZ pigs is in northern China, where winter temperatures reach minus 40 degrees Celsius. Sufficient fat keeps them resistant to the cold and stores energy. Similarly, MZ pigs have good disease resistance and detoxification capabilities. GSTM4 is a member of the glutathione sulfur transferase family and plays a key role in the detoxification of insecticides and other exogenous substances. In abamectin-resistant tetranychus urticae, the activity of GSTs was significantly increased (53). The QTLs mapped to the group-specific CNVRs in MZ pigs are related to fat and immunity. The genes mentioned above provide favorable conditions for the survival of MZ pigs in cold regions.
The SH pig is crossbred of Chinese and European pigs. The CNV polymorphisms of some genes were unique in SH pigs. SERPINB3 is a homologous substance to chicken ovalbumin protein (OVA) in humans. It takes part in apoptosis and autoimmune diseases and is related to the prognosis (54). The NAMPT is primarily involved in redox reactions, and the signals it transmits act during various stages of cell physiology, including cell cycle and proliferation (55). It is a participant and regulator of many diseases. The results were within our expectations, including genes related to immunity and cell proliferation. What surprised us was that some genes are related to neuroprotection and neurological disorders. NLGN4X and NLGN4Y, as marker molecules of human autism, are considered to play an important role in the etiology of autism, the formation of synapses, and the transmission of information. Autism can lead to stereotypic behavior and communication difficulties in humans and is related to developmental mental disorders (56, 57). In addition, the massive accumulation of IGLON5 antibodies has been proven to damage the cytoskeleton of hippocampal neurons, which can lead to the occurrence of autoimmune diseases and neurodegeneration (34, 58). These findings were interesting as SH pigs are more docile and more easily domesticated than LW pigs. The neurological foundation of these behavioral differences is still unknown.
By analyzing the genetic structure of LW, MZ, and SH pigs, we found that SH and LW pigs are closely related, while MZ pigs are distantly related to pigs of the other two breeds. It indicates that LW and SH pigs have more genetic exchanges than MZ pigs, which have the same trend in PCA, evolutionary tree, VST, and the group-special CNVRs and QTLs analyses. Based on the results of genetic structural analysis, we found that the lineage of SH pigs came from LW pigs, and MZ pigs have a smaller genetic distance from SH pigs than LW pigs. This may be because the MZ pig have genetic exchanges with the LW pig of widespread reproduction, and the habitats of MZ and SH pigs are similar in geographical location, climate, and altitude, which have the same environmental driving forces and adaptability that make them produce the same CNV (59). Understandably, the main source of CNV was inherited from ancestors, followed by adaptation to environmental changes and other reasons that led to random mutations (60, 61).
In summary, we have performed genome-wide CNV detection on LW, MZ, and SH pigs to explore the relationship between CNVs and phenotypic characteristics of pig breeds. The functions of genes containing unique CNVRs are related to the phenotypic traits of pig breeds. From this, we have identified some candidate genes. These CNV polymorphisms provide a theoretical basis for the understanding of the relationship between phenotype and CNVs.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was reviewed and approved by Experimental Animal Welfare and Ethics Committee of Nanjing Agricultural University, Nanjing, China.
BZ came up with the idea and revised the manuscript. CZ wrote the manuscript and performed the experiments. JZ, YG, and QX collected the samples and isolated the genomic DNA. ML, MC, and XC analyzed the data. AS and BZ reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
This work was supported by the National Natural Science Foundation of China (NO. 32172786) and the JBGS Project of Breeding Industry Revitalization in Jiangsu Province [JBGS(2021)101].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.909039/full#supplementary-material
Supplementary Figure 1. All samples are suitable for CNV detection.
Supplementary Figure 2. Genomic visualization of CNVR-9017, CNVR-1169, CNVR-9126, and CNVR-1771 in LW and SH pigs.
Supplementary Figure 3. The length-frequency distribution of CNVRs. The majority of CNVRs are concentrated in 1.6-3 kb, accounting for 61.23% of the total, with only 0.75% exceeding 30 kb.
Supplementary Figure 4. The variable frequency distribution of CNVRs. A total of 8,247 CNVRs were found in <5 individuals, and 4,134 CNVRs were found in a unique individual.
Supplementary Figure 5. A Venn diagram shows 285, 545, and 700 group-specific QTLs in the SH, LW, and MZ pigs, respectively.
Supplementary Table 1. The whole-genome sequencing data of MZ, LW, and SH pigs.
Supplementary Table 2. The standard curve and primers for qPCR, and the verification results of the CNV type.
Supplementary Table 3. A total of CNVRs were detected in 46 pigs and variations of types.
Supplementary Table 4. The distribution of CNVRs on pig chromosomes of the pig.
Supplementary Table 5. The Cross-Validation Error under the ancestral population number K value ranges from 1 to 5.
Supplementary Table 6. Novel genes identified in LW, MZ, and SH pigs.
1. BRIDGES CB. The bar “Gene” a duplication. Science. (1936) 83:210–1. doi: 10.1126/science.83.2148.210
2. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. (2006) 444:444–54. doi: 10.1038/nature05329
3. Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. (2005) 1:e49. doi: 10.1371/journal.pgen.0010049
4. Wang Y, Tang Z, Sun Y, Wang H, Wang C, Yu S, et al. Analysis of genome-wide copy number variations in chinese indigenous and western pig breeds by 60 k snp genotyping arrays. PLoS ONE. (2014) 9:e106780. doi: 10.1371/journal.pone.0106780
5. Xie J, Li R, Li S, Ran X, Wang J, Jiang J, et al. Identification of copy number variations in xiang and kele pigs. PLoS ONE. (2016) 11:e0148565. doi: 10.1371/journal.pone.0148565
6. Schiavo G, Dolezal MA, Scotti E, Bertolini F, Calo DG, Galimberti G, et al. Copy number variants in italian large white pigs detected using high-density single nucleotide polymorphisms and their association with back fat thickness. Anim Genet. (2014) 45:745–9. doi: 10.1111/age.12180
7. Wang Z, Chen Q, Liao R, Zhang Z, Zhang X, Liu X, et al. Genome-wide genetic variation discovery in Chinese Taihu pig breeds using next generation sequencing. Anim Genet. (2017) 48:38–47. doi: 10.1111/age.12465
8. Zhang L, Huang Y, Si J, Wu Y, Wang M, Jiang Q, et al. Comprehensive inbred variation discovery in bama pigs using de novo assemblies. Gene. (2018) 679:81–9. doi: 10.1016/j.gene.2018.08.051
9. Ewels P, Magnusson M, Lundin S, Kaller M. Multiqc: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. (2016) 32:3047–8. doi: 10.1093/bioinformatics/btw354
10. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
11. Wang X, Zheng Z, Cai Y, Chen T, Li C, Fu W, et al. Cnvcaller: highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience. (2017) 6:1–12. doi: 10.1093/gigascience/gix115
12. Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, et al. Rideogram: drawing Svg graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci. (2020) 6:e251. doi: 10.7717/peerj-cs.251
13. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation Plink: rising to the challenge of larger and richer datasets. Gigascience. (2015) 4:7. doi: 10.1186/s13742-015-0047-8
14. Shringarpure SS, Bustamante CD, Lange K, Alexander DH. Efficient analysis of large datasets and sex bias with admixture. BMC Bioinformatics. (2016) 17:218. doi: 10.1186/s12859-016-1082-x
15. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. Mega X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
16. Ito K, Murphy D. Application of Ggplot2 to pharmacometric graphics. CPT Pharmacometrics Syst Pharmacol. (2013) 2:e79. doi: 10.1038/psp.2013.56
17. Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. Tbtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. (2020) 13:1194–202. doi: 10.1016/j.molp.2020.06.009
18. Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. G:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 Update). Nucleic Acids Res. (2019) 47:W191–W8. doi: 10.1093/nar/gkz369
19. Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. Kobas 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. (2011) 39:W316–22. doi: 10.1093/nar/gkr483
20. Quinlan AR, Hall IM. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics. (2010) 26:841–2. doi: 10.1093/bioinformatics/btq033
21. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative Pcr and the 2(-Delta Delta C(T)) method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
22. Ballester M CA, Ibáñez E, Sánchez A, Folch JM. Real-time quantitative pcr-based system for determining transgene copy number in transgenic animals. Biotechniques. (2004) 37:3. doi: 10.2144/04374ST06
23. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (Igv): high-performance genomics data visualization and exploration. Brief Bioinform. (2013) 14:178–92. doi: 10.1093/bib/bbs017
24. Simone C SP, Bagella L, Pucci B, Bellan C, De Falco G, De Luca A, et al. Activation of myod-dependent transcription by cdk9/Cyclin T2. Oncogene. (2002 J) 21:4137–48. doi: 10.1038/sj.onc.1205493
25. Cottone G, Baldi A, Palescandolo E, Manente L, Penta R, Paggi MG, et al. Pkn is a novel partner of cyclin T2a in muscle differentiation. J Cell Physiol. (2006) 207:232–7. doi: 10.1002/jcp.20566
26. Kasak L, Punab M, Nagirnaja L, Grigorova M, Minajeva A, Lopes AM, et al. Bi-allelic recessive loss-of-function variants in fancm cause non-obstructive azoospermia. Am J Hum Genet. (2018) 103:200–12. doi: 10.1016/j.ajhg.2018.07.005
27. Yin H, Ma H, Hussain S, Zhang H, Xie X, Jiang L, et al. A homozygous fancm frameshift pathogenic variant causes male infertility. Genet Med. (2019) 21:62–70. doi: 10.1038/s41436-018-0015-7
28. Jaillard S, Bell K, Akloul L, Walton K, McElreavy K, Stocker WA, et al. New insights into the genetic basis of premature ovarian insufficiency: novel causative variants and candidate genes revealed by genomic sequencing. Maturitas. (2020) 141:9–19. doi: 10.1016/j.maturitas.2020.06.004
29. Zhang F, Hanif Q, Luo X, Jin X, Zhang J, He Z, et al. Muscle transcriptome analysis reveal candidate genes and pathways related to fat and lipid metabolism in yunling cattle. Anim Biotechnol. (2021) 7:1−8. doi: 10.1080/10495398.2021.2009846 [Epub ahead of print].
30. Gao M, Liu L, Wang X, Mak HY, Liu G, Yang H. Gpat3 deficiency alleviates insulin resistance and hepatic steatosis in a mouse model of severe congenital generalized lipodystrophy. Hum Mol Genet. (2020) 29:432–43. doi: 10.1093/hmg/ddz300
31. Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C, et al. Tbc1d14 regulates autophagy via the trapp complex and Atg9 traffic. EMBO J. (2016) 35:281–301. doi: 10.15252/embj.201592695
32. Denson J, Xi Z, Wu Y, Yang W, Neale G, Zhang J. Screening for inter-individual splicing differences in human Gstm4 and the discovery of a single nucleotide substitution related to the tandem skipping of two exons. Gene. (2006) 379:148–55. doi: 10.1016/j.gene.2006.05.012
33. Shi L, Chang X, Zhang P, Coba MP, Lu W, Wang K. The functional genetic link of nlgn4x knockdown and neurodevelopment in neural stem cells. Hum Mol Genet. (2013) 22:3749–60. doi: 10.1093/hmg/ddt226
34. Landa J, Gaig C, Plaguma J, Saiz A, Antonell A, Sanchez-Valle R, et al. Effects of Iglon5 antibodies on neuronal cytoskeleton: a link between autoimmunity and neurodegeneration. Ann Neurol. (2020) 88:1023–7. doi: 10.1002/ana.25857
35. Clapperton M, Bishop SC, Glass EJ. Innate immune traits differ between meishan and large white pigs. Vet Immunol Immunopathol. (2005) 104:131–44. doi: 10.1016/j.vetimm.2004.10.009
36. Belew GD, Silva J, Rito J, Tavares L, Viegas I, Teixeira J, et al. Transfer of glucose hydrogens via acetyl-coa, malonyl-coa, and nadph to fatty acids during de novo lipogenesis. J Lipid Res. (2019) 60:2050–6. doi: 10.1194/jlr.RA119000354
37. Liu M, Li B, Shi T, Huang Y, Liu GE, Lan X, et al. Copy number variation of bovine Shh gene is associated with body conformation traits in Chinese beef cattle. J Appl Genet. (2019) 60:199–207. doi: 10.1007/s13353-019-00496-w
38. Feng Z, Li X, Cheng J, Jiang R, Huang R, Wang D, et al. Copy number variation of the pigy gene in sheep and its association analysis with growth traits. Animals. (2020) 10:6888. doi: 10.3390/ani10040688
39. Locke ME, Milojevic M, Eitutis ST, Patel N, Wishart AE, Daley M, et al. Genomic copy number variation in mus musculus. BMC Genomics. (2015) 16:497. doi: 10.1186/s12864-015-1713-z
40. Khatri B, Kang S, Shouse S, Anthony N, Kuenzel W, Kong BC. Copy number variation study in japanese quail associated with stress related traits using whole genome re-sequencing data. PLoS ONE. (2019) 14:e0214543. doi: 10.1371/journal.pone.0214543
41. Vegesna R, Tomaszkiewicz M, Medvedev P, Makova KD. Dosage regulation, and variation in gene expression and copy number of human Y chromosome ampliconic genes. PLoS Genet. (2019) 15:e1008369. doi: 10.1371/journal.pgen.1008369
42. Iijima-Yamashita Y, Matsuo H, Yamada M, Deguchi T, Kiyokawa N, Shimada A, et al. Multiplex fusion gene testing in pediatric acute myeloid leukemia. Pediatr Int. (2018) 60:47–51. doi: 10.1111/ped.13451
43. Velagaleti GV B-WG, Northup JK, Lockhart LH, Hawkins JC, Jalal SM, Withers M, et al. Position effects due to chromosome breakpoints that map approximately 900 kb upstream and approximately 13 Mb downstream of Sox9 in two patients with campomelic. Am J Hum Genet. (2005) 76:652–62. doi: 10.1086/429252
44. Wang H, Wang C, Yang K, Liu J, Zhang Y, Wang Y, et al. Genome wide distributions and functional characterization of copy number variations between Chinese and western pigs. PLoS ONE. (2015) 10:e0131522. doi: 10.1371/journal.pone.0131522
45. Fernandez AI, Barragan C, Fernandez A, Rodriguez MC, Villanueva B. Copy number variants in a highly inbred Iberian porcine strain. Anim Genet. (2014) 45:357–66. doi: 10.1111/age.12137
46. Revay T, Quach AT, Maignel L, Sullivan B, King WA. Copy number variations in high and low fertility breeding boars. BMC Genomics. (2015) 16:280. doi: 10.1186/s12864-015-1473-9
47. Foote K, Reinhold J, Yu EPK, Figg NL, Finigan A, Murphy MP, et al. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell. (2018) 17:e12773. doi: 10.1111/acel.12773
48. Porokhovnik L. Individual copy number of ribosomal genes as a factor of mental retardation and autism risk and severity. Cells. (2019) 8:1151. doi: 10.3390/cells8101151
49. Teng Y, Wang Y, Fu J, Cheng X, Miao S, Wang L. Cyclin T2: a novel Mir-15a target gene involved in early spermatogenesis. FEBS Lett. (2011) 585:2493–500. doi: 10.1016/j.febslet.2011.06.031
50. Xu S, Chang Y, Wu G, Zhang W, Man C. Potential role of Mir-155-5p in fat deposition and skeletal muscle development of chicken. Biosci Rep. (2020) 40. doi: 10.1042/BSR20193796
51. Wang F ZQ, Liu JZ, Kong DL. Mirna-188-5p alleviates the progression of osteosarcoma via target degrading Ccnt2. Eur Rev Med Pharmacol Sci. (2020) 24:29–35. doi: 10.26355/eurrev_202001_19892
52. Ma C, Sun Y, Wang J, Kang L, Jiang Y. Identification of a promoter polymorphism affecting Gpat3 gene expression that is likely related to intramuscular fat content in pigs. Anim Biotechnol. (2020) 21:1–4. doi: 10.1080/10495398.2020.1858847 [Epub ahead of print].
53. Mounsey KEPC, Arlian LG, Morgan MS, Holt DC, Currie BJ, Walton SF, et al. Increased transcription of glutathione S-Transferases in acaricide exposed scabies mites. Parasit Vectors. (2010) 3:43. doi: 10.1186/1756-3305-3-43
54. Riaz N, Havel JJ, Kendall SM, Makarov V, Walsh LA, Desrichard A, et al. Recurrent Serpinb3 and Serpinb4 mutations in patients who respond to Anti-Ctla4 IMMUNOTHERAPY. Nat Genet. (2016) 48:1327–9. doi: 10.1038/ng.3677
55. Sharif T, Martell E, Dai C, Ghassemi-Rad MS, Kennedy BE, Lee PWK, et al. Regulation of Cancer and Cancer-Related Genes Via Nad(). Antioxid Redox Signal. (2019) 30:906–23. doi: 10.1089/ars.2017.7478
56. Nguyen TA, Wu K, Pandey S, Lehr AW, Li Y, Bemben MA, et al. A cluster of autism-associated variants on X-Linked Nlgn4x functionally resemble Nlgn4y. Neuron. (2020) 106:759–68e7. doi: 10.1016/j.neuron.2020.03.008
57. Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-Linked genes encoding neuroligins Nlgn3 and Nlgn4 are associated with autism. Nat Genet. (2003) 34:27–9. doi: 10.1038/ng1136
58. Ryding M, Gamre M, Nissen MS, Nilsson AC, Okarmus J, Poulsen AAE, et al. Neurodegeneration induced by anti-Iglon5 antibodies studied in induced pluripotent stem cell-derived human neurons. Cells. (2021) 10:837. doi: 10.3390/cells10040837
59. Frantz LA, Schraiber JG, Madsen O, Megens HJ, Cagan A, Bosse M, et al. Evidence of long-term gene flow and selection during domestication from analyses of eurasian wild and domestic pig genomes. Nat Genet. (2015) 47:1141–8. doi: 10.1038/ng.3394
60. Hull RM, Cruz C, Jack CV, Houseley J. Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol. (2017) 15:e2001333. doi: 10.1371/journal.pbio.2001333
Keywords: evolution, genetic structure analysis, economic traits, livestock, crossbreeding
Citation: Zhang C, Zhao J, Guo Y, Xu Q, Liu M, Cheng M, Chao X, Schinckel AP and Zhou B (2022) Genome-Wide Detection of Copy Number Variations and Evaluation of Candidate Copy Number Polymorphism Genes Associated With Complex Traits of Pigs. Front. Vet. Sci. 9:909039. doi: 10.3389/fvets.2022.909039
Received: 31 March 2022; Accepted: 09 June 2022;
Published: 30 June 2022.
Edited by:
Nuno Carolino, Instituto Nacional Investigaciao Agraria e Veterinaria (INIAV), PortugalReviewed by:
Wilson Nandolo, Lilongwe University of Agriculture and Natural Resources, MalawiCopyright © 2022 Zhang, Zhao, Guo, Xu, Liu, Cheng, Chao, Schinckel and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Zhou, emhvdWJvQG5qYXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.