
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 14 June 2022
Sec. Livestock Genomics
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.894380
 Gao Gong1
Gao Gong1 Yixing Fan2
Yixing Fan2 Xiaochun Yan1
Xiaochun Yan1 Wenze Li1Xiaomin Yan1Hongfu Liu1Ludan Zhang1Yixing Su1Jiaxin Zhang1Wei Jiang1
Wenze Li1Xiaomin Yan1Hongfu Liu1Ludan Zhang1Yixing Su1Jiaxin Zhang1Wei Jiang1 Zhihong Liu1
Zhihong Liu1 Zhiying Wang1Ruijun Wang1Yanjun Zhang1Qi Lv1,3,4,5*Jinquan Li1,3,4,5*Rui Su1,3,4,5*
Zhiying Wang1Ruijun Wang1Yanjun Zhang1Qi Lv1,3,4,5*Jinquan Li1,3,4,5*Rui Su1,3,4,5*Cashmere goat from Inner Mongolia is an excellent local breed in China, and the related cashmere product is a kind of precious textile raw material with high price. Cashmere is generated from secondary hair follicles, which has obvious annual periodicity and includes three different stages: anagen, catagen, and telogen. Therefore, we investigated skin transcriptome data for 12 months using weighted gene co-expression network analysis (WGCNA) to explore essential modules, pathways, and genes responsible for the periodic growth and development of secondary hair follicles. A total of 17 co-expression modules were discovered by WGCNA, and there is a strong correlation between steelblue module and month (0.65, p = 3E−09), anagen (0.52, p = 1E−05), telogen (−0.6, p = 8E−08). Gene expression was generally high during late anagen to catagen (June to December), while expression was downregulated from telogen to early anagen (January–May), which is similar to the growth rule of hair follicle cycle. KEGG pathway enrichment analyses of the genes of steelblue module indicated that genes are mainly enriched in Cell cycle, Wnt signaling pathway, p53 signaling pathway and other important signal pathways. These genes were also significantly enriched in GO functional annotation of the cell cycle, microtubule movement, microtubule binding, tubulin binding, and so on. Ten genes (WIF1, WNT11, BAMBI, FZD10, NKD1, LEF1, CCND3, E2F3, CDC6, and CDC25A) were selected from these modules, and further identified as candidate biomarkers to regulate periodic development of hair follicles using qRT-PCR. The Wnt signaling pathway and Cell cycle play an important role in the periodic development of hair follicles. Ten genes were identified as essential functional molecules related to periodic development of hair follicle. These findings laid a foundation for understanding molecular mechanisms in biological functions such as hair follicle development and hair growth in cashmere goats.
Cashmere goat is a typical economic breed, providing high quality meat and cashmere for human beings. The coat obtained from the cashmere goat is a typical heterogeneous fleece, including both myelinated coarse hairs generated by primary hair follicle (PHF) and unmyelinated cashmere generated by secondary hair follicle (SHF). Cashmere has significant advances in softness, smoothness, slenderness, lightness, elasticity, heat retention, comfort, etc. Therefore, cashmere is known as “fiber gemstone” or “soft gold,” and is the best natural fiber raw material in the textile industry (1). Hair follicles of cashmere goats exhibit an obvious growth cycle, including anagen, catagen, and telogen (Figure 1) (2). Like a tiny organ of hair growth, hair follicles are regulated by various factors, such as heredity, nutrition, season, age, shearing, environment, and so on.
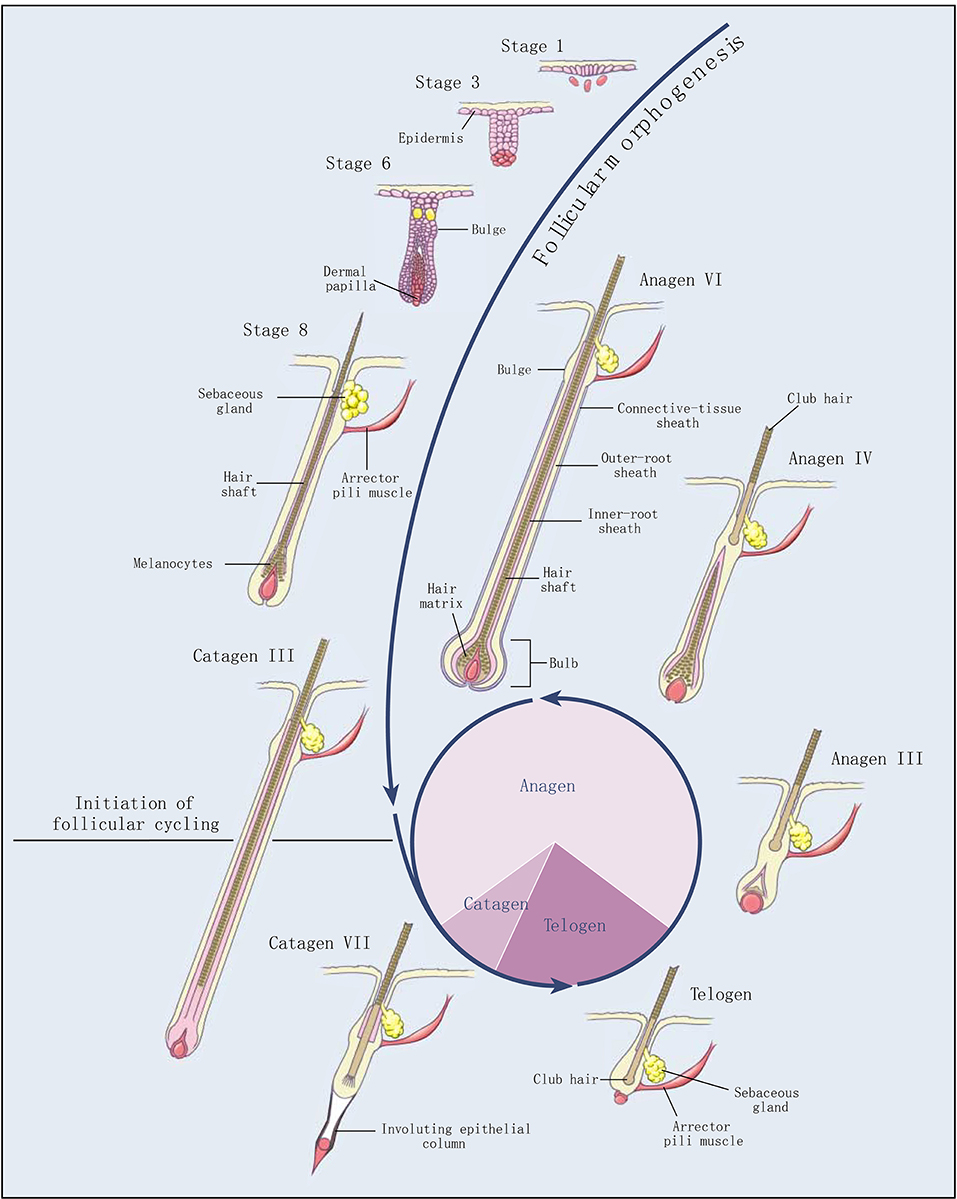
Figure 1. Development and cycling of hair follicles (2). Selected stages of the morphogenesis of hair follicles and the three stages of follicular cycling (anagen, catagen, and telogen) are shown. The roman numerals indicate morphologic substages of anagen and catagen. The pie chart shows the proportion of time the hair follicle spends in each stage.
The secondary hair follicles of Inner Mongolia cashmere goat have a typical cycle, whose shed season is one year, from April to May every year. The growth cycle includes anagen (April–November), catagen (December–January of the following year), and telogen (February–March) (3, 4). During the telogen, the epithelial bud cells at the bottom of the secondary hair follicles begin to extend downward, and the hair papillae are activated to start hair follicle reconstruction. Hair follicles enter the anagen and begin to grow rapidly. After entering the catagen phase, the hair balls begin to atrophy and become thinner, and gradually move upward; the hair follicles entering the telogen are only above the sebaceous glands, and the cells are in a static state (5). The periodic growth of cashmere goat hair follicles is regulated by a series of molecules, including hormone regulation, such as melatonin (6), prolactin (7), thyroxine (8), and so on. Growth factors are also involved in the regulation of hair follicles, including IGFs (9), FGFs (10), VEGF (11), PDGF (12), and so on. In addition, Keratin genes (KRTs) (13), Keratin-associated proteins (KAPs) (14), and bone morphogenetic proteins (BMPs) (15) are proven to regulate hair follicles. Signaling pathways, including the Wnt signaling pathway (16), PI3K-Akt signaling pathway (17), MAPK signaling pathway (18), Ras signaling pathway (19), cell cycle pathway (13), and so on also play important roles in the development of hair follicle (20).
In recent years, with the development of life science, sequencing technology and other high-throughput methods have been well developed. As the cost of high-throughput sequencing is decreasing, a large amount of sequencing data is gradually cumulated. Weighted gene co-expression network analysis (WGCNA) is a useful tool to analyze these high-throughput data (21–23). WGCNA has great advances in understanding the molecular mechanisms of complex traits and diseases such as cell cycle (21), lung cancer (24), pancreatic cancer (25), including genetic diseases in humans, mice, and many other organisms. This method combines high-throughput data with sample phenotypic data for joint analysis to define a small number of modules and identify key markers, which greatly improve the analytical efficiency of high-throughput data (22). The genetic mechanisms of cashmere quality traits can be further analyzed by the WGCNA method. Recently, some researchers applied WGCNA to analyze the hair follicle development and identified the module consistent with the embryonic hair follicle development stage of Inner Mongolia cashmere goat, as well as the key gene (WNT10A) for the mature stage of hair follicle development in Inner Mongolia cashmere goat skin (26). Many researchers have found that ECM-receptor interaction, Focal adhesion, PI3K-Akt signaling pathway, Estrogen signaling pathway, Wnt signaling pathway, Hedgehog signaling pathway, Cell cycle, Arachidonic acid metabolism, cAMP signaling pathway, and other signal pathways are closely related to the periodic growth of hair follicles. The core genes such as COL1A1, C1QTNF6, KRTAP3-1, DLX3, OVOL1, COL1A1, C1QTNF6, KRTAP3-1, WNT5A, LOC102172600, and LOC102191729 were screened (13, 27–30). In this study, we used WGCNA to analyze the gene expression data of 12 months from Inner Mongolia cashmere goats, and identify important modules and genes related to the periodic growth of secondary hair follicles. The genes were further validated by GO, KEGG, gene connectivity network, and real-time fluorescence quantitative technique. This study provides a new insight into the regulation mechanism of periodic growth of hair follicles in cashmere goats.
In this study, we used transcriptome data that our team (SRA: PRJNA832904). The sequenced individuals were six 2-year-old adult ewes of unrelated Inner Mongolia cashmere goats (Alba type). The skin tissues were collected once a month for 12 months. RNA-seq was carried out by using Illumina X Ten sequencing platform system. Sequence alignment was carried out with the goat genome (Capra hircus, ARS1) by HISAT (2.0.4) (31). The gene expression level of each sample was analyzed by HTSeq (v0.6.1) (32), and the gene expression data (FPKM) was calculated. The genes with zero expression in more than 16 samples were filtered, and the differential expression of genes between the two groups was analyzed by DESeq (padj < 0.05) (33), and all the differential genes were merged. The filtered dataset contains 7,320 gene expression data from 72 samples. The skin sample in January was marked as M_1 (_1, _2, _3, _4, _5, _6).
In this study, skin samples were collected in accordance with the International Guiding Principles for Biomedical Research Involving Animals and were approved by the Animal Ethics Committee of the Inner Mongolia Academy of Agriculture and Animal Husbandry Sciences which is responsible for animal care and use in the Inner Mongolia Autonomous Region of China. The experimental animals came from Inner Mongolia Jin Lai Livestock Technology Company (Hohhot, Inner Mongolia, China). All Inner Mongolia cashmere goats are raised according to the standard of cashmere goats. The skin tissues of 3 Albus adult ewes of Inner Mongolia cashmere goat were collected for 12 months. The sampling site was the upper one-third of the left scapula along the mid-dorsal and mid-abdominal lines. About 1% pentobarbital sodium anesthetic was used for muscle anesthesia. After hair shearing and alcohol disinfection, ~1 cm2 of skin tissue was grasped with sterile forceps and quickly cut near the tip using sterile scalpel blades. Each clipping was obtained immediately adjacent to the location of the previous shearing. Yunnan Baiyao powder (Yunnan Baiyao Group Co., Ltd., China) was applied immediately to stop the bleeding. Then the samples were quickly put into the liquid nitrogen and finally stored at −80°C. Skin samples were used for RNA extraction and quantitative real-time PCR.
The hair follicle cycle development of Inner Mongolia cashmere goat was analyzed by weighted gene co-expression network analysis (WGCNA, Version 1.70-3) in R (Version 4.1.0) (21). The specific usage of the R package was referred to the official website of WGCNA (https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/) (22). Firstly, based on the correlations between samples, a clustering tree was drawn to eliminate outliers (h > 16,000). Using the gene expression data, the absolute value of the expression correlation coefficient between the two genes was calculated, and the gene co-expression correlation matrix (Sij = |cor(Xi, Xj)|) was constructed. In order to realize the scale-free network, the adjacency degree β between genes is calculated by pickSoftThreshold function, and the β value of R2 > 0.8 was selected to construct the adjacency matrix (). Finally, in order to evaluate the correlation between genes, a topological overlap matrix [] was constructed. We set the minimum module size to 30, drew the TOM clustering tree by TOM matrix, and the genes with similar expression patterns were clustered into one group through the TOM clustering tree. Merging of modules whose expression profiles were very similar. We choose a Height cutoff set to 0.30, and the corresponding correlation was set to 0.70 (MEDissThres = 0.3). Key gene modules were identified by associating with phenotypic. Different developmental stages of the cashmere goat hair follicle cycle were divided by the month of sample collection. April to November was defined as anagen, December to January was defined as catagen, and February to March was defined as telogen to construct a phenotypic matrix. In order to explore the correlations between modules and traits, module-trait relationships were calculated. A heat map of the expression of the key modules was drawn. Through the correlations between module genes and traits, the key modules related to the hair follicle cycle of cashmere goats were identified.
Kyoto Encyclopedia of Genes and Genomes (KEGG) (34) is a collection of databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances. The Gene Ontology (GO) (35) is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species. The GO covers three domains: cellular component, molecular function, and biological process. KEGG and GO enrichment analysis of genes of module genes was implemented by the clusterProfiler (4.2.2) (36) R package, in which gene length bias was corrected. The enrichment was considered to be significant when the corrected p-values were <0.05. The exportNetworkToCytoscape function in WGCNA is used to derive the network relationship (threshold = 0.10). The output module nodes file and the module edges file were imported into the Cytoscape3.9.0 (37) for network visualization. Combined with the results of weight network and functional enrichment analysis, the candidate was identified, and the gene expression curve was drawn by GraphPad Prism8.3.0 using FPKM data.
The total RNA of the skin was extracted according to the user guide of TRLzol Reagent (Invitrogen). The concentration and quality of total RNA were determined by NanoDrop 2000 (Thermo). The total RNA that passed the test was synthesized cDNA using PrimeScript synthesis RT reagent Kit with gDNA Eraser (RR047A, TAKARA).
The mRNA coding region sequence of the genes was queried by the NCBI database, and the fluorescent quantitative specific primers of the gene were designed by Primer-BLAST (NCBI) (38). The primer parameters were set as follows: PCR product size 90–200 bp, primer size 18–22 bp, primer melting temperatures (Tm) set to 58.0–62.0°C, primer GC content 40–60%, and other parameters as default values. The primer sequence is shown in Supplementary Table 1, and the primer was handed over to Sangon Biotech (Shanghai) Co., Ltd.
In this study, LightCycler ®96 (Roche) instrument and TB Green “Premix Ex Taq” II (RR820A, TAKARA) kit were used for the Quantitative real-time PCR (qRT-PCR) test. The reaction system is TB Green Premix Ex Taq II (Tli RNaseH Plus) (2 ×) 5 μl, cDNA 1 μl, PCR Forward/Reverse Primer (10 μM) 0.5 μl, ddH2O 3 μl. Programs: Preincubation 95°C 30 s, two step amplification (95°C 5 s, Tm 30 s) for a total of 45 cycles, melting, and cooling. Each month tested three samples and made three technical repeats.
Excel 2019 was used to collate and summarize the CQ value. The genes were calibrated by GAPDH and β-actin as double house-keeping genes. The 2−ΔΔCT method (39) was used to calculate the relative gene expression. SAS 9.2 ANOVA was used to analyze the variance of the data. Duncan method was used for multiple comparisons, and the relative gene expression map was drawn by GraphPad Prism 8.3.0. p < 0.05 was considered to be statistically significant.
Based on the gene expression data of skin tissues of Inner Mongolia cashmere goats in different months, we obtained a gene expression matrix including 72 samples and 7,320 genes after filtering. among them. Construct a co-expression network through R packets of WGCNA. The hclust function is used to analyze the samples (Figure 2A). Six outlier samples are found in the hierarchical clustering of the samples and were eliminated in further analyses. In order to build a network with scale-free distribution and retain information as much as possible, a soft threshold method is used to find the best soft-thresholding powers β (Figure 2B). Specific Powervalue information is shown in Supplementary Table 2. We discovered that the connectivity between genes in the network is high (β = 10, R2 = 1.82), indicating a scale-free network (Figure 2C). The TOM clustering tree is constructed to divide modules preliminarily, and then similar modules are combined by a dynamic hybrid cutting method (Figure 2D). Finally, genes are divided into 17 modules (Table 1). The largest module contains 2,152 genes (darkgrey module), while the smallest module contains 35 genes (darkmagenta module). The module gene clustering heat map is drawn by hclust function, which is used to show the connectivity relationship between the two genes (Figure 2E). From the module-trait correlation heat map (Figure 2F), we discovered four modules significant related to months and different stages. Steelblue module is significantly correlated with month (0.65, p = 3E−09), anagen (0.52, p = 1E−05) and telogen (−0.6, p = 8E−08), respectively. Royalblue module have strong correlations with month (−0.62, p = 2E−08), anagen (−0.44, p = 2E−04) and telogen (−0.42, p = 5E−04). Lightcyan module has strong correlations with anagen (0.58, p=4E−07) and telogen (−0.43, p = 3E−04), while orange module is correlated with month (0.58, p = 4E−07). Genes in these modules might be involved in the periodic development of secondary hair follicles.
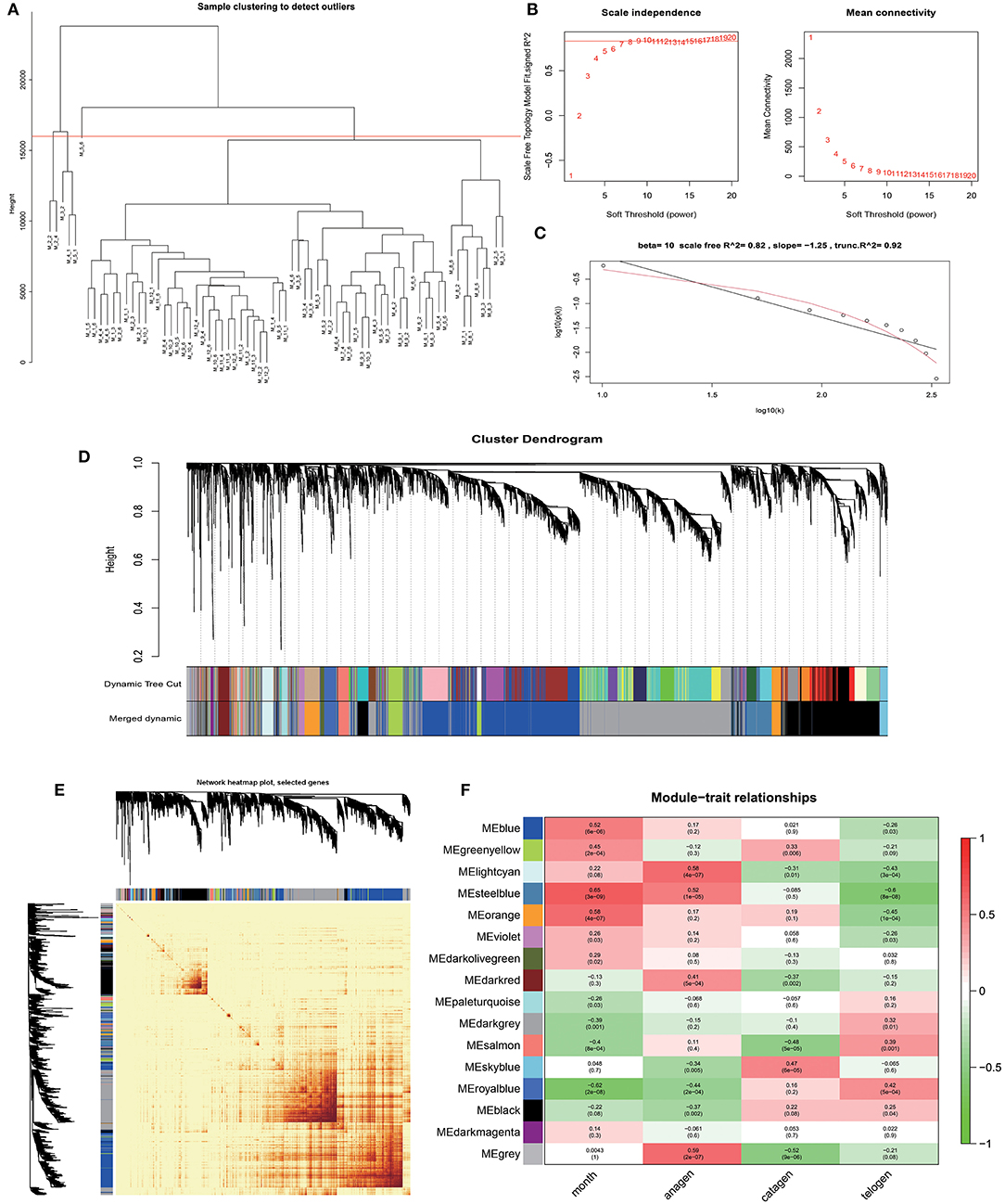
Figure 2. Identification of co-expression modules by WGCNA. (A) Hierarchical clustering information of samples, red line = 16,000. (B) The determination of soft thresholding power. (C) When β = 10, the scale-free network fitting. (D) The gene clustering dendrogram was obtained according to hierarchic clustering of adjacency based dissimilarity. (E) Network heatmap of module-genes, Each tree represents a module, each branch represents a gene, and the darker the color of each dot, the stronger the connectivity between the two genes corresponding to the row and column. (F) Module–trait relationships, Abscissa is the trait, the ordinate is the module, the number of each grid represents the correlation between the module and the trait, and the number in parentheses represents p-value.
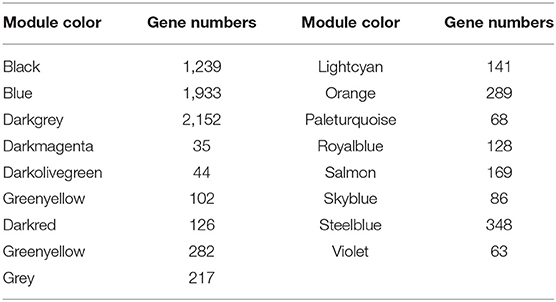
Table 1. Gene number in 17 modules.
Through Module trait relationships analysis, we found that the steelblue module has the strongest correlations with many stages of hair follicle cycle development. Therefore, we will focus on the genes in this module for follow-up analysis. With a scatter plot of steelblue module's module membership and month, anagen, and telogen's gene significance (Figures 3A–C). We identified a high correlation between steelblue module and month (cor=0.7, p = 4.3E−52), indicating that the gene of this module has a strong correlation with monthly changes. In addition, this steelblue module also has moderate correlations with anagen (cor=0.35, p= 2.2E−11) and telogen (cor=0.53, p=2.2E−26). These results indicate that genes in this module are strongly associated with the periodic development of hair follicles. There are 348 genes in the steelblue module, including 322 annotated genes and 26 new transcripts (Supplementary Table 3). Then we analyzed steelblue module gene expression patterns in detail (Figure 3D), and discovered specific expression patterns in different months. During anagen, the gene expressions of this module were generally low from April to May and high from June to November. During catagen, the expressions began to decrease. During telogen, the gene expressions were generally low.
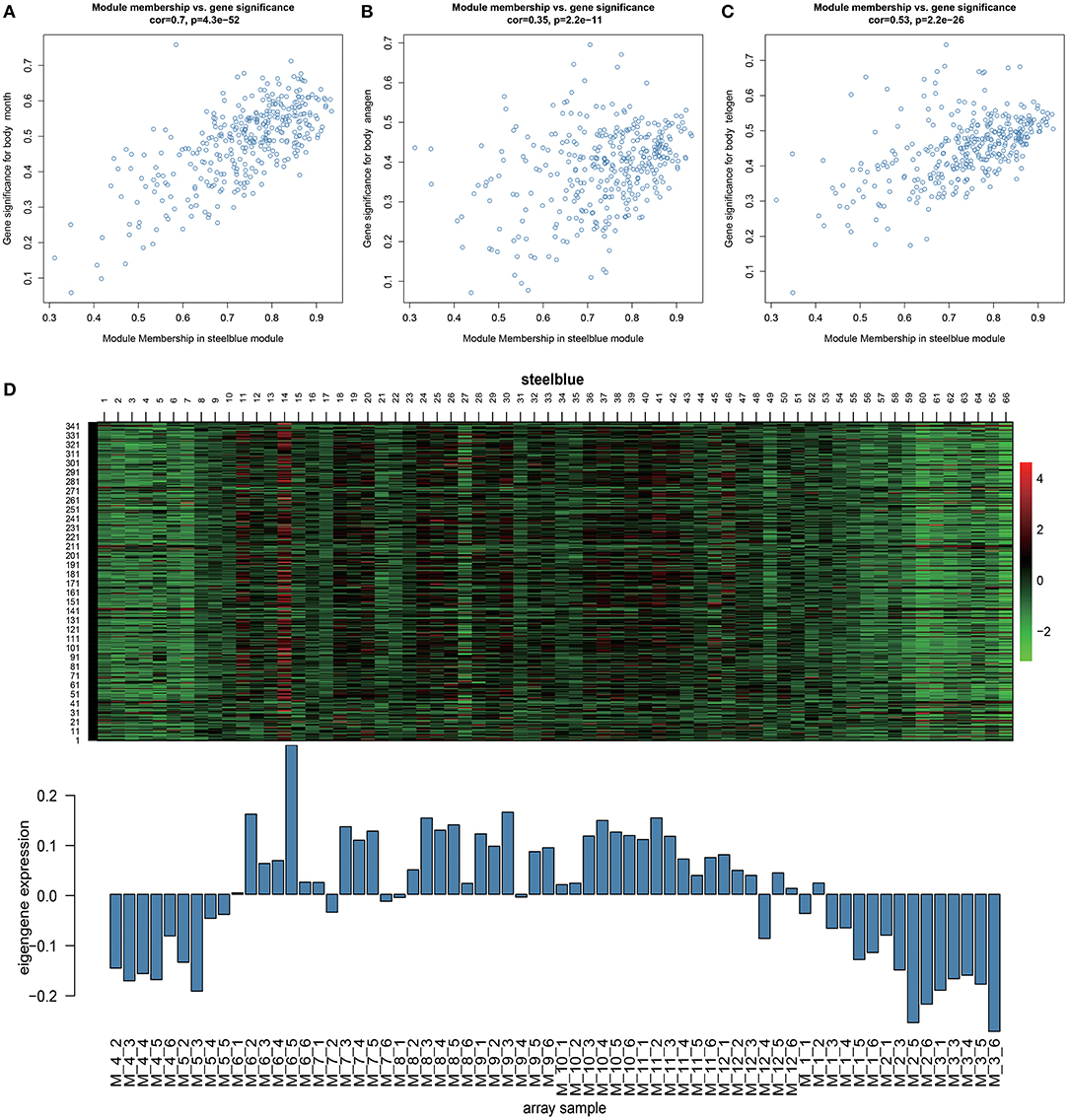
Figure 3. Key module analysis. (A–C) Scatterplot of Gene Significance for Module Membership in steelblue module. (D) Heatmap of steelblue module genes expression pattern. The Abscissa is the sample name, the above picture is the heat map of the expression of the genes in the module in different samples, and the following picture is the expression pattern of the characteristic values of the module in different samples.
In enrichment analysis of KEGG pathways (Figure 4A; Supplementary Table 4), steelblue module genes were significantly enriched in 13 signal pathways, including Cell cycle, Wnt signaling pathway, p53 signaling pathway, pyrimidine metabolism, and drug metabolism-cytochrome P450. Wnt signaling pathway and Cell cycle are typical pathways that regulate the periodic development of hair follicles. Through GO functional enrichment analysis (Figure 4B; Supplementary Table 5). We found that the module genes were significantly enriched in 10 GO annotations, including four biological processes and six molecular functions. The biological processes of enrichment include cell cycle (GO:0007049), microtubule-based movement (GO:0007018), microtubule-based process (GO:0007017), and nuclear division (GO:0000280). The enrichment molecular functions include microtubule binding (GO:0008017), tubulin binding (GO:0015631), microtubule motor activity (GO:0003777), and motor activity (GO:0003774), macromolecular complex binding (GO:0044877) and protein complex binding (GO:0032403).
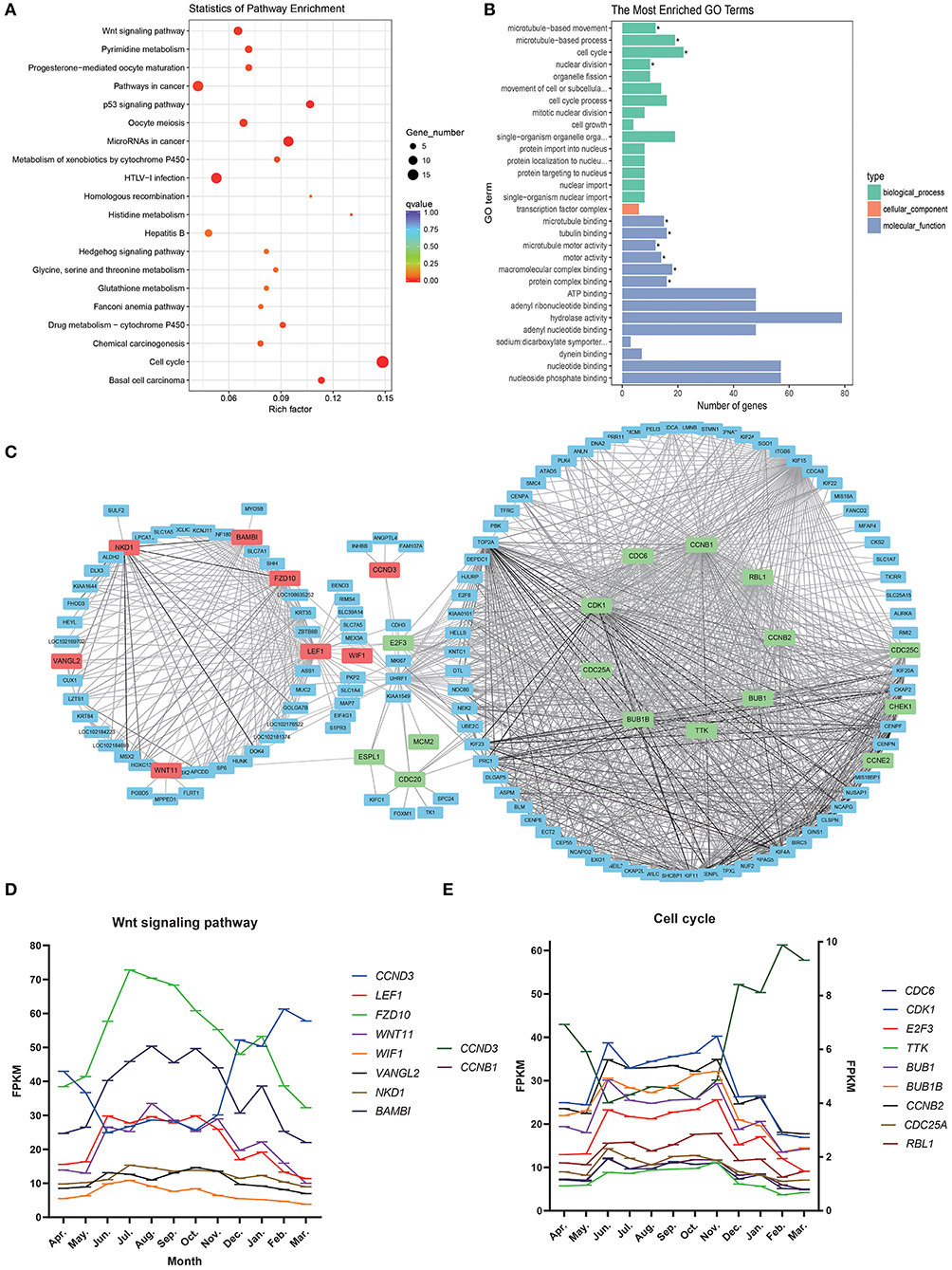
Figure 4. Steelblue module gene analysis map. (A) KEGG enrichment analysis of steelblue module. (B) GO analysis of steelblue module. (C) Gene co-expression network in steelblue modules. (D) Gene expression trend of wnt signaling pathway, Abscissa indicates month, in which the anagen (April–November), catagen (December–January) and telogen (February–March), the ordinate is FPKM. (E) Gene expression trend of cell cycle, CCNB, and CCND3 use the left ordinate axis, other genes use the left coordinate axis.
We constructed a gene network for steelblue module (Figure 4C) and found that genes of the Wnt signaling pathway and Cell cycle pathway were in the center of a network. LEF1, VANGL2, BAMBI, WNT11, FZD10, NKD1, and WIF1 in the Wnt signaling pathway had high connectivity. CCND3 and E2F3 belonged to both the Wnt signaling pathway and cell cycle. CDK1, CCNB1, TTK, BUB1, BUB1B, CCNB2, CDC6, CCND3, CDC25A, RBL1, and other genes in the Cell cycle were also located in the center of the network, and these candidate genes might play an important regulatory role. In the gene expression trend map of these two pathways (Figures 4D,E), we found that the expression trend of CCND3 was opposite to that of other genes in the two pathways. The expression of CCND3 decreased gradually in anagen and began to increase in catagen and telogen. Gene expression trends of other genes in the Wnt signaling pathway and Cell cycle were the same, which increased gradually during anagen, reached the highest level from August to October, then decreased gradually, and continued to decrease in catagen and telogen. Expression levels of most genes in the cell cycle were low.
Total RNA was extracted from 36 skin samples for 12 months. The concentration was above 180 ng/μl, and OD260/280 was between 1.80 and 2.01. A number of candidate genes related to the periodic development of hair follicles were identified by modular gene analysis. 10 genes (WIF1, WNT11, BAMBI, FZD10, NKD1, LEF1, CCND3, E2F3, CDC6, and CDC25A) participating in Wnt signaling pathways and cell cycle were selected and validated by qRT-PCR during 12 months (Figures 5A–J). Through multiple comparisons, it was found that the differences between these candidate genes in different months were significant (p < 0.05), and a statistical table was made (Table 2).
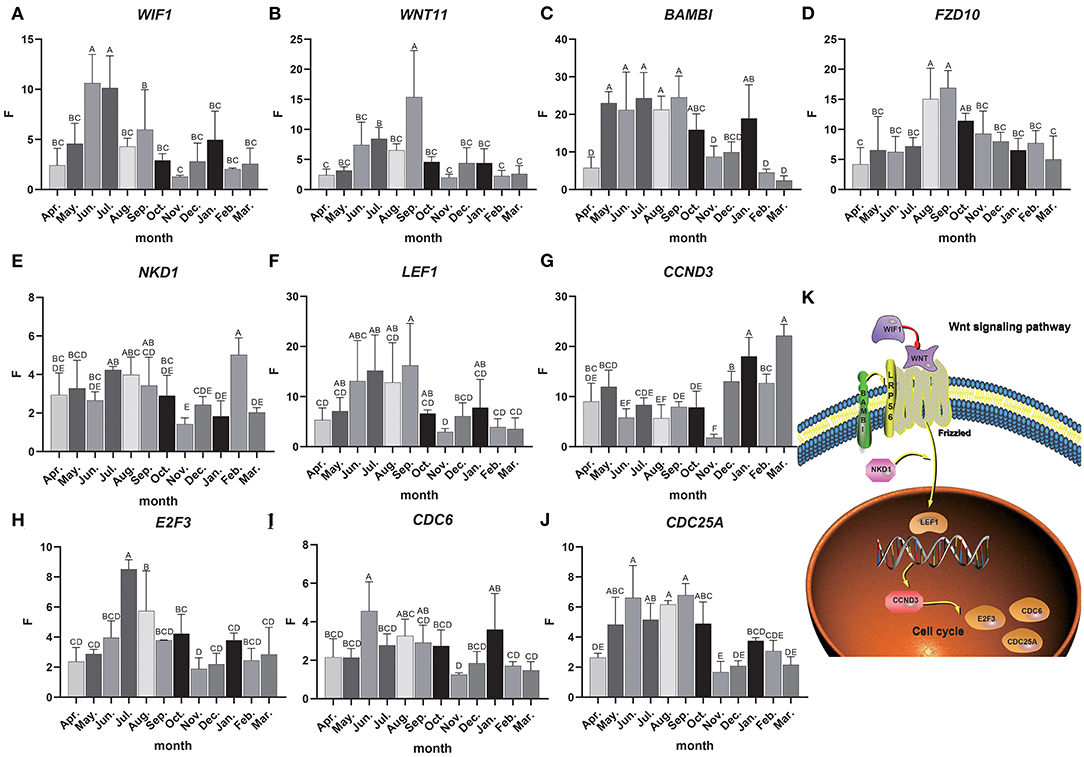
Figure 5. Candidate gene expression analysis. (A–J) The relative expression of candidate genes, the Abscissa indicates the month, the ordinate is the relative expression F, different letters are marked to indicate significant, while the same letters indicate that the differences are not significant, the error bar represents SD. (K) Interactive network control chart.
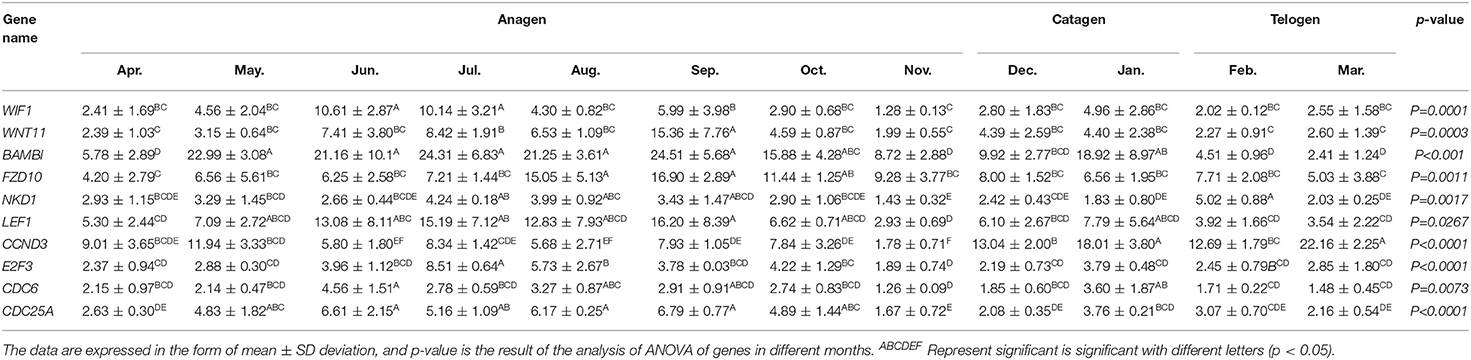
Table 2. Statistical table of relative expression of candidate genes.
We found that the quantitative results were basically consistent with transcriptome data, but were different among genes. For instance, the expression level of WIF1 was higher in anagen, and WIF1 expression in June and July was significantly higher than that in other months (Figure 5A). The expression of WNT11 in September was significantly higher than that in other months, while the expression level was the lowest in the catagen and telogen (Figure 5B). The expression of BAMBI was the highest from May to September and the lowest during the telogen (Figure 5C). Expression of FZD10 was the highest in August and September (Figure 5D). The overall expression of NKD1 was consistent in each month, and the expression level was up-regulated in the anagen (Figure 5E). Expression of LEF1 was the highest from June to September, and the expression level was lower in the catagen and telogen (Figure 5F). The expression level of CCND3 was low in anagen, significantly lower in November than in other months, and gradually increased in catagen and telogen (Figure 5G). Expression of E2F3 was the highest in July and August, and the expression level was lower in the catagen and telogen (Figure 5H). Expression of CDC6 was consistent in each month (Figure 5I). The expression level of CDC25A was higher in the whole anagen, and the expression level from May to October was significantly higher than that in other months (Figure 5J). The expression of these genes changes over time, indicating close correlations to the secondary hair follicle cycle.
Dynamic regulatory network control chart according to the regulatory relationship between Wnt signaling pathway and cell cycle pathways and the expression of candidate genes (Figure 5K). We can find that WIF1 regulates WNT11, WNT11, and BAMBI to activate the receptor Frizzled in the Wnt signaling pathway, and then activates LEF1 under the regulation of the NKD1 gene, and then inversely regulates CCND3. At the same time, it affects E2F3, CDC6, CDC25A, and other genes in the cell cycle pathway. These genes may work together to regulate the periodic growth of the hair follicles.
WGCNA is a systematic method to discover functional modules by phenotype-transcriptome joint analysis, function enrichment analysis, reduction of gene dimension, and biological significance analysis of modules. In this study, based on the gene expression data and phenotypic data of Inner Mongolia cashmere goat for 12 months, 7,320 effective differential genes were divided into modules by WGCNA, and a total of 17 co-expression modules were obtained. In order to mine the modules related to hair follicle cycle development, we identified that the steelblue module had the highest correlation with the hair follicle cycle.
The genes in Steelblue modules were significantly enriched in the Wnt signaling pathway and cell cycle, which are typical cycle-related genes. The Wnt signaling pathway is involved in the regulation of development, wound healing, disease and cancer. The Wnt pathway regulates cell proliferation in the skin, which directly affects skin regeneration of wounds. Hair follicles of cashmere goats are also undergoing continuous reborn. Studies have found that the origin of new hair follicle cells is regulated by the Wnt signal pathway to activate hair follicle stem cells (HFSC) (40). The activation of the Wnt signaling pathway can induce HFSC from the telogen to the anagen, thus accelerating hair regeneration and development (20). In this study, we also found that the expression level of genes in Steelblue module began to increase gradually during anagen, and decreased gradually in catagen and telogen. The expression pattern of these genes was consistent with the cycle of hair follicles, which may be involved in the regeneration of hair follicles. Through Figures 4D,E, we can also find that the genes in these two pathways also have the same expression trend.
WIF1 belongs to the family of Wnt inhibitors, which interferes with Wnt signal transduction by directly binding to WNT proteins (41). The expression of WIF1 increased in the early stage of hair follicle growth (42), which was consistent with the results of this study. It is known that the expression of WNT11 is up-regulated during skin wound healing, indicating that the signal transduction of this gene may be active in the stage of wound healing and play an important role in the regeneration of hair follicles (43). BAMBI is an inhibitor of TGF-β family members and plays a role in hair follicle development (44). BAMBI is different in fetal skin, and the expression from 45 days to 135 days increases gradually in the embryonic stage, which is consistent with the development of hair follicles in the embryonic stage and can positively regulate the growth of hair follicles (45). BAMBI also plays an important role in the periodic growth of hair follicles, and it can be found that the expression of BAMBI in the whole anagen is significantly higher than that in catagen and telogen, indicating that the expression of BAMBI plays a positive role in the rapid maturation of hair follicles during hair follicle growth. The expression of FZD10 is up-regulated during the transition from the resting stage to the growing stage, which contributes to the activation of HFSC and promotes the circulation and regeneration of hair follicles (46). LEF1 is a downstream factor of the Wnt signaling pathway and is expressed in inner and outer root sheath cells, hair stromal cells, and dermis. LEF1 is also highly expressed during the growth period and can promote the growth and development of hair (47). This study has the same results. At the same time, some studies have shown that the expression of LEF1 in the dermis can support the new development of hair follicles in the wound, induce the expression of LEF1 in human dermal cells to enhance the ability of skin repair, and regenerate new hair follicles in the healed wound (48). LEF1 plays an important role in the regulation of hair follicle regeneration. CCND3 plays an important role in cell proliferation and apoptosis affects the normal progress of the cell cycle and is a key molecule in the process of transformation. CCND3 knockdown induces cell cycle arrest and apoptosis (49). This study found that the expression level of CCND3 was higher in the catagen and telogen and the expression of CCND3 may promote the apoptosis of hair follicle cells and enter the next cycle. CDC6 encodes key proteins of DNA replication and is responsible for recruiting MCM helicases to the origin of replication during the G1 phase of the cell division cycle. The overexpression of CDC6 protein in the skin can prolong the retrogression and resting period in the hair growth cycle and promote the preservation of hair (50).
Through this study, it was found that these candidate genes were differentially expressed at different stages of the hair follicle growth cycle. The expression of these genes WIF1, BAMBI, LEF1, CDC25A, WNT11, FZD10, and E2F3 was significantly high in anagen, but low in catagen and telogen, indicating that it plays an important role in the rapid growth and development of hair follicles. The expression of CCND3 was significantly higher in catagen and telogen, indicating that the gene can promote the apoptosis and regeneration of hair follicles. NKD1 and CDC6 showed significant differences only in some months. These findings suggest that these candidate genes may play an important role in the development of secondary hair follicles.
In this study, we used the WGCNA method to study the important regulatory genes of secondary hair follicle development in cashmere goats and obtained 17 co-expression modules. A strong correlation between Steelblue and multiple stages of hair follicle growth and development was discovered, and identified as a key module. Wnt signaling pathway and Cell cycle play an important role in the periodic development of hair follicles. Ten important genes related to the hair follicle cycle were identified and further verified, including WIF1, WNT11, BAMBI, FZD10, NKD1, LEF1, CCND3, E2F3, and CDC6. These findings lay a foundation for further study of molecular mechanisms in biological functions such as hair follicle development and hair growth in cashmere goats.
The transcriptome dataset used in this study can be found in the SRA database and is expected to be made public on June 30, 2024 with the login number PRJNA832904. Other data sets during the current study are available from the corresponding author on reasonable request.Requests to access these datasets should be directed to RS: c3VydWl5dUAxMjYuY29t.
In this study, skins were collected in accordance with the International Guiding Principles for Biomedical Research involving animals and approved by the Special Committee on Scientific Research and Academic Ethics of Inner Mongolia Agricultural University, responsible for the approval of Biomedical Research Ethics of Inner Mongolia Agricultural University [Approval No. (2020) 056].
RS, JL, and QL conceived the study. GG, XiaomY, HL, YS, and LZ collected the samples and extracted RNA. GG, WL, JZ, and XiaocY analyzed the data. RS and GG prepared the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was financially supported by National Natural Science Foundation of China (31860637), China Agriculture Research System of MOF and MARA (CARS-39), Science and Technology Major Project of Inner Mongolia Autonomous Region (2021ZD0012), Scientific Project of Inner Mongolia Agricultural University on High-level Introduced Talented Personnel (NDYB2018-1), Central Government Guides Local Science and Technology Development Fund Projects (2020ZY0007), and Special Funding Project for the Iconic Achievements of the College of Animal Science, Inner Mongolia Agricultural University (No. BZCG202111).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer WB declared a shared affiliation with the author YF to the handling editor at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to the staff of Inner Mongolia Jinlai Livestock Technology Co for providing assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.894380/full#supplementary-material
Supplementary Table 1. Primer sequences of candidate gene and reference genes.
Supplementary Table 2. Power information statistics table.
Supplementary Table 3. Steelblue module gene information statistical table.
Supplementary Table 4. KEGG enrichment analysis table of steelblue module.
Supplementary Table 5. GO analysis table of steelblue module.
1. Ge W, Zhang W, Zhang Y, Zheng Y, Li F, Wang S, et al. A single-cell transcriptome atlas of cashmere goat hair follicle morphogenesis. Genomics Proteomics Bioinformatics. (2021) 19:437–51. doi: 10.1016/j.gpb.2021.07.003
2. Paus R, Cotsarelis G. The biology of hair follicles. New Engl J Med. (1999) 341:491–7. doi: 10.1056/nejm199908123410706
3. Song S, Yang M, Li Y, Rouzi M, Zhao Q, Pu Y, et al. Genome-wide discovery of LincRNAs with spatiotemporal expression patterns in the skin of goat during the cashmere growth cycle. BMC Genomics. (2018) 19:495. doi: 10.1186/s12864-018-4864-x
4. Yuan C, Wang X, Geng R, He X, Qu L, Chen Y. Discovery of cashmere goat (Capra hircus) microRNAs in skin and hair follicles by solexa sequencing. BMC Genomics. (2013) 14:511. doi: 10.1186/1471-2164-14-511
5. He Y, Cheng L, Wang J, Liu X, Luo Y. Identification of the secondary follicle cycle of hexi cashmere goat. Anatom Record. (2012) 295:1520–8. doi: 10.1002/ar.22522
6. Liu J, Mu Q, Liu Z, Wang Y, Liu J, Wu Z, et al. Melatonin regulates the periodic growth of cashmere by upregulating the expression of Wnt10b and B-catenin in Inner Mongolia cashmere goats. Front Genet. (2021) 12:665834. doi: 10.3389/fgene.2021.665834
7. Zhang L, Duan C, Guo Y, Zhang Y, Liu Y. Inhibition of prolactin promotes secondary skin follicle activation in Cashmere goats. J Anim Sci. (2021) 99:skab079. doi: 10.1093/jas/skab079
8. van Beek N, Bodó E, Kromminga A, Gáspár E, Meyer K, Zmijewski MA, et al. Thyroid hormones directly alter human hair follicle functions: anagen prolongation and stimulation of both hair matrix keratinocyte proliferation and hair pigmentation. J Clin Endocrinol Metab. (2008) 93:4381–8. doi: 10.1210/jc.2008-0283
9. Nixon AJ, Ford CA, Oldham JM, Pearson AJ. Localisation of insulin-like growth factor receptors in skin follicles of sheep (Ovis aries) and changes during an induced growth cycle. Comp Biochem Physiol Part A Physiol. (1997) 118:1247–57. doi: 10.1016/s0300-9629(97)00048-0
10. Kawano M, Komi-Kuramochi A, Asada M, Suzuki M, Oki J, Jiang J, et al. Comprehensive analysis of Fgf and Fgfr expression in skin: Fgf18 is highly expressed in hair follicles and capable of inducing anagen from telogen stage hair follicles. J Invest Dermatol. (2005) 124:877–85. doi: 10.1111/j.0022-202X.2005.23693.x
11. Hu X, Hao F, Li X, Xun Z, Gao Y, Ren B, et al. Generation of Vegf knock-in cashmere goat via the Crispr/Cas9 system. Int J Biol Sci. (2021) 17:1026–40. doi: 10.7150/ijbs.55559
12. Pazzaglia I, Mercati F, Antonini M, Capomaccio S, Cappelli K, Dall'Aglio C, et al. PDGFA in Cashmere goat: a motivation for the hair follicle stem cells to activate. Animals. (2019) 9:38. doi: 10.3390/ani9020038
13. Bao Q, Zhang X, Bao P, Liang C, Guo X, Yin M, et al. Genome-wide identification, characterization, and expression analysis of keratin genes (KRTs) family in Yak (Bos grunniens). Gene. (2022) 818:146247. doi: 10.1016/j.gene.2022.146247
14. Gong H, Zhou H, Wang J, Li S, Luo Y, Hickford JGH. Characterisation of an ovine keratin associated protein (KAP) gene, which would produce a protein rich in glycine and tyrosine, but lacking in cysteine. Genes. (2019) 10:848. doi: 10.3390/genes10110848
15. Plikus MV, Baker RE, Chen CC, Fare C, de la Cruz D, Andl T, et al. Self-organizing and stochastic behaviors during the regeneration of hair stem cells. Science. (2011) 332:586–9. doi: 10.1126/science.1201647
16. Bai WL, Zhao SJ, Wang ZY, Zhu YB, Dang YL, Cong YY, et al. LncRNAs in secondary hair follicle of cashmere goat: identification, expression, and their regulatory network in Wnt signaling pathway. Anim Biotechnol. (2018) 29:199–211. doi: 10.1080/10495398.2017.1356731
17. Liu F, Shi J, Zhang Y, Lian A, Han X, Zuo K, et al. Nanog attenuates hair follicle-derived mesenchymal stem cell senescence by upregulating Pbx1 and activating Akt signaling. Oxid Med Cell Longev. (2019) 2019:4286213. doi: 10.1155/2019/4286213
18. Su R, Gong G, Zhang L, Yan X, Wang F, Zhang L, et al. Screening the key genes of hair follicle growth cycle in Inner Mongolian Cashmere goat based on RNA sequencing. Arch Anim Breed. (2020) 63:155–64. doi: 10.5194/aab-63-155-2020
19. Kasi RA, Moi CS, Kien YW, Yian KR, Chin NW, Yen NK, et al. Para-phenylenediamine-induces apoptosis via a pathway dependent on Ptk-Ras-Raf-Jnk activation but independent of the Pi3k/Akt pathway in Nrk-52e cells. Mol Med Rep. (2015) 11:2262–8. doi: 10.3892/mmr.2014.2979
20. Hu XM Li ZX, Zhang DY, Yang YC, Fu SA, Zhang ZQ, et al. A systematic summary of survival and death signalling during the life of hair follicle stem cells. Stem Cell Res Ther. (2021) 12:453. doi: 10.1186/s13287-021-02527-y
21. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. (2005) 4:Article17. doi: 10.2202/1544-6115.1128
22. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. (2008) 9:559. doi: 10.1186/1471-2105-9-559
23. Hao D, Wang X, Yang Y, Thomsen B, Holm LE, Qu K, et al. Integrated analysis of MRNA and MicroRNA co-expressed network for the differentiation of bovine skeletal muscle cells after polyphenol resveratrol treatment. Front Vet Sci. (2021) 8:777477. doi: 10.3389/fvets.2021.777477
24. Ding M, Li F, Wang B, Chi G, Liu H. A comprehensive analysis of WGCNA and serum metabolomics manifests the lung cancer-associated disordered glucose metabolism. J Cell Biochem. (2019) 120:10855–63. doi: 10.1002/jcb.28377
25. Mantini G, Vallés AM, Le Large TYS, Capula M, Funel N, Pham TV, et al. Co-expression analysis of pancreatic cancer proteome reveals biology and prognostic biomarkers. Cell Oncol. (2020) 43:1147–59. doi: 10.1007/s13402-020-00548-y
26. Wu Z, Hai E, Di Z, Ma R, Shang F, Wang Y, et al. Using WGCNA (weighted gene co-expression network analysis) to identify the hub genes of skin hair follicle development in fetus stage of Inner Mongolia cashmere goat. PLoS ONE. (2020) 15:e0243507. doi: 10.1371/journal.pone.0243507
27. Wang J, Sui J, Mao C, Li X, Chen X, Liang C, et al. Identification of key pathways and genes related to the development of hair follicle cycle in cashmere goats. Genes. (2021) 12:180. doi: 10.3390/genes12020180
28. Zhang X, Bao P, Ye N, Zhou X, Zhang Y, Liang C, et al. Identification of the key genes associated with the yak hair follicle cycle. Genes. (2021) 13:32. doi: 10.3390/genes13010032
29. Hu S, Li C, Wu D, Huo H, Bai H, Wu J. The dynamic change of gene-regulated networks in cashmere goat skin with seasonal variation. Biochem Genet. (2021) 60:527–42. doi: 10.1007/s10528-021-10114-2
30. Li X, Liu Z, Ye S, Liu Y, Chen Q, Guan W, et al. Integrated analysis of LncRNA and MRNA reveals novel insights into wool bending in Zhongwei goat. Animals. (2021) 11:3326. doi: 10.3390/ani11113326
31. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. (2015) 12:357–60. doi: 10.1038/nmeth.3317
32. Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics. (2015) 31:166–9. doi: 10.1093/bioinformatics/btu638
33. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. (2010) 11:R106. doi: 10.1186/gb-2010-11-10-r106
34. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. (2000) 28:27–30. doi: 10.1093/nar/28.1.27
35. Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. (2005) 21:3674–6. doi: 10.1093/bioinformatics/bti610
36. Yu GC, Wang LG, Han YY, He QY. Clusterprofiler: an R package for comparing biological themes among gene clusters. Omics. (2012) 16:284–7. doi: 10.1089/omi.2011.0118
37. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
38. Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-Blast: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. (2012) 13:134. doi: 10.1186/1471-2105-13-134
39. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
40. Houschyar KS, Momeni A, Pyles MN, Maan ZN, Whittam AJ, Siemers F. Wnt signaling induces epithelial differentiation during cutaneous wound healing. Organogenesis. (2015) 11:95–104. doi: 10.1080/15476278.2015.1086052
41. Kim BK, Baek IC, Lee HY, Kim JK, Song HH, Yoon SK. Gene expression profile of the skin in the 'Hairpoor' (HrHp) mice by microarray analysis. BMC Genomics. (2010) 11:640. doi: 10.1186/1471-2164-11-640
42. Li S, Zheng X, Nie Y, Chen W, Liu Z, Tao Y, et al. Defining key genes regulating morphogenesis of apocrine sweat gland in sheepskin. Front Genet. (2018) 9:739. doi: 10.3389/fgene.2018.00739
43. Fathke C, Wilson L, Shah K, Kim B, Hocking A, Moon R, et al. Wnt signaling induces epithelial differentiation during cutaneous wound healing. BMC Cell Biol. (2006) 7:4. doi: 10.1186/1471-2121-7-4
44. Lv X, Chen L, He S, Liu C, Han B, Liu Z, et al. Effect of nutritional restriction on the hair follicles development and skin transcriptome of Chinese merino sheep. Animals. (2020) 10:1058. doi: 10.3390/ani10061058
45. Han W, Yang F, Wu Z, Guo F, Zhang J, Hai E, et al. Inner Mongolian Cashmere goat secondary follicle development regulation research based on MRNA-MiRNA co-analysis. Sci Rep. (2020) 10:4519. doi: 10.1038/s41598-020-60351-5
46. Kandyba E, Leung Y, Chen YB, Widelitz R, Chuong CM, Kobielak K. Competitive balance of intrabulge BMP/Wnt signaling reveals a robust gene network ruling stem cell homeostasis and cyclic activation. Proc Natl Acad Sci USA. (2013) 110:1351–6. doi: 10.1073/pnas.1121312110
47. Zhao B, Li J, Chen Q, Yang N, Bao Z, Hu S, et al. A treatment combination of IGF and EGF promotes hair growth in the Angora rabbit. Genes. (2020) 12:24. doi: 10.3390/genes12010024
48. Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, et al. Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. Elife. (2020) 9:e60066. doi: 10.7554/eLife.60066
49. Koo KH, Kwon H. MicroRNA miR-4779 suppresses tumor growth by inducing apoptosis and cell cycle arrest through direct targeting of PAK2 and CCND3. Cell Death Dis. (2018) 9:77. doi: 10.1038/s41419-017-0100-x
Keywords: hair follicle cycle, WGCNA, core genes, Inner Mongolia cashmere goats, Wnt signaling pathway
Citation: Gong G, Fan Y, Yan X, Li W, Yan X, Liu H, Zhang L, Su Y, Zhang J, Jiang W, Liu Z, Wang Z, Wang R, Zhang Y, Lv Q, Li J and Su R (2022) Identification of Genes Related to Hair Follicle Cycle Development in Inner Mongolia Cashmere Goat by WGCNA. Front. Vet. Sci. 9:894380. doi: 10.3389/fvets.2022.894380
Received: 11 March 2022; Accepted: 25 April 2022;
Published: 14 June 2022.
Edited by:
Meng-Hua Li, Institute of Zoology (CAS), ChinaReviewed by:
Wenlin Bai, Shenyang Agricultural University, ChinaCopyright © 2022 Gong, Fan, Yan, Li, Yan, Liu, Zhang, Su, Zhang, Jiang, Liu, Wang, Wang, Zhang, Lv, Li and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Lv, bHZxaTEyMDJAaW1hdS5lZHUuY24=; Jinquan Li, bGlqaW5xdWFuX25kQDEyNi5jb20=; Rui Su, c3VydWl5dUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.