 Yaonan Li
Yaonan Li Yanfang Lan
Yanfang Lan- Wuhan Business University, Wuhan, China
Increasing evidence reveals the importance of gut microbiota in animals for regulating intestinal homeostasis, metabolism, and host health. The gut microbial community has been reported to be closely related to many diseases, but information regarding diarrheic influence on gut microbiota in horses remains scarce. This study investigated and compared gut microbial changes in horses during diarrhea. The results showed that the alpha diversity of gut microbiota in diarrheic horses decreased observably, accompanied by obvious shifts in taxonomic compositions. The dominant bacterial phyla (Firmicutes, Bacteroidetes, Spirochaetes, and Kiritimatiellaeota) and genera (uncultured_bacterium_f_Lachnospiraceae, uncultured_bacterium_f_p-251-o5, Lachnospiraceae_AC2044_group, and Treponema_2) in the healthy and diarrheic horses were same regardless of health status but different in abundances. Compared with the healthy horses, the relative abundances of Planctomycetes, Tenericutes, Firmicutes, Patescibacteria, and Proteobacteria in the diarrheic horses were observably decreased, whereas Bacteroidetes, Verrucomicrobia, and Fibrobacteres were dramatically increased. Moreover, diarrhea also resulted in a significant reduction in the proportions of 31 genera and a significant increase in the proportions of 14 genera. Taken together, this study demonstrated that the gut bacterial diversity and abundance of horses changed significantly during diarrhea. Additionally, these findings also demonstrated that the dysbiosis of gut microbiota may be an important driving factor of diarrhea in horses.
Introduction
Mammal intestines contain more than 1014 microorganisms including bacteria, fungi, viruses, and protozoa, which are approximately 10 times the total quantity of host cells and play crucial roles in intestinal physiological function, metabolism, and host health (1–3). Furthermore, increasing evidence indicated that gut microbiota also functions in epithelial differentiation, intestinal homeostasis, and immunity (4, 5). Early investigations demonstrated that the consistency of the gut microbial community is the precondition for conducting digestive absorption and complicated metabolic functions, whereas gut microbial dysbiosis is closely related to many diseases (6–8). Currently, gut microbial dysbiosis has been shown to be an important driving factor of non-alcoholic fatty liver disease, high blood pressure, and diabetes (9, 10). Recent research on gut microbiota has also provided evidence that obesity, colonitis, and colorectal cancer may be the result of gut microbial dysbiosis (11, 12).
Diarrhea is one of the main reasons for decreased production performance and death in farmed animals and has been regarded as a key factor affecting the development of the livestock industry in many countries. Previous studies indicated that diarrhea was present in nearly all mammals, especially in newborn pigs, chickens, and sheep with susceptible gut microbiota (13–15). Considering the negative impact of diarrhea on animal husbandry, it is important to investigate its etiology and treatment. Numerous studies indicated that gut microbiota played key roles in the prevention, control, and diagnosis of diarrhea (16, 17). Wang et al. revealed that the gut microbial community of diarrheic goats changed dramatically accompanied by high mortality (13). Similarly, Li et al. also reported that the gut microbiota of giraffes changed significantly during diarrhea (18).
Metagenomics is a key tool for investigating shifts in gut microbiota during diseases (19, 20). By systematically exploring and comparing acquired information, the relationship between gut microbiota and diseases could be further understood, and prevention and control measures can be developed to minimize economic losses (21–23). Presently, the complicated composition and structure of gut microbiota in diarrheic pigs, yaks, and giraffes have been successfully analyzed based on the high-throughput sequencing technology (24–26). However, there are few reports on the gut microbiota of horses, and even fewer studies on the composition and structure of gut microbiota in horses in different health statuses. Therefore, the objective of this study was to compare and investigate the composition and discrepancy of gut microbial populations between healthy and diarrheic horses.
Materials and Methods
Animals and Sample Collection
A total of 16 horses (8 healthy horses and 8 diarrheic horses) from Wuhan Business University (Wuhan, China) were used for this experiment. The horses we screened possessed the same immune background. Moreover, the health statuses of the horses were diagnosed and evaluated by a professional veterinarian before sample collection. The rectum was swabbed by a trained technician using sterile swabs in a rotating fashion. The obtained samples including healthy and diarrheic feces were immediately placed into sterile plastic containers and transported to the laboratory and later stored at −80°C for further study.
16S rDNA Gene Amplicon Sequencing
Prior to the DNA extraction, 16 fecal samples from control and diarrheic horses were unfrozen and homogenized at room temperature. Afterward, the treated fecal samples were subjected to bacterial DNA extraction based on the manufacturer's protocol. Quantification and electrophoresis of the extracted DNA were performed to ensure that the concentration and integrity of extracts meet analysis demands. To dissect the changes in the gut bacterial community, we amplified the V3/V4 regions utilizing bacterial primers (338F: ACTCCTACGGGAGGCAGCA and 806R: GGACTACHVGGGTWTCTAAT). The PCR amplification procedure was set based on previous studies. PCR products were conducted target fragment recovery and gel electrophoresis detection to acquire purified products. The PCR products were recovered by fluorescence quantification and proportionally mixed following sequencing requirements. The qualified products were used to prepare sequencing libraries by using the PacBio platform (Biomarker Technologies, China). To acquire qualified libraries, the original libraries were required to suitably embellish such as sequence repair, quality evaluation, purification, and fluorescent quantitation. Libraries that passed quality screening were subjected to 2 × 300 bp paired-end sequencing using a MiSeq sequencing machine.
Bioinformatics and Data Analysis
The initial data from Illumina MiSeq sequencing was performed a quality assessment to obtain effective data. Briefly, raw data containing problematical sequences including short, unqualified, and mismatched sequences were subjected to screening and removal of primer sequences to achieve clean reads utilizing the Trimmomatic (v0.33) and Cutadapt software (1.9.1). The Usearch software (v10) was used for splicing clean reads and then the spliced sequences were secondary screened based on sequence length range. Subsequently, identification and elimination of chimera sequences were performed to obtain final effective reads utilizing the UCHIME software (v4.2). Effective reads that passed quality inspection were clustered, and OTUs were partitioned based on 97% similarity. Additionally, Venn maps were also generated to characterize the distribution and richness of bacterial OTUs in each sample. To further investigate the shifts in gut microbial diversity and abundance during diarrhea, we computed multiple alpha diversity indexes based on OTU distribution. Principal component analysis was also conducted to dissect gut bacterial beta diversities between both groups. The sequencing depth and evenness of each sample were evaluated through rank abundance and rarefaction curves. Differential bacterial taxa associated with diarrhea exposure were recognized by Metastats and LEfSe analysis. An SPSS statistical program (v20.0) was used for conducting data analysis, and P-values (means ± SD) <0.05 were determined statistically significant.
Results
Sequences Analyses
In this research, 8 healthy and 8 diarrheic fecal samples were subjected to high-throughput sequencing analysis. After optimizing the original data, a total of 127,8741 high-quality sequences were obtained from the 16 samples (Table 1). In addition, the number of valid sequences in the healthy horses ranged from 794.84 to 803.09, while the number of valid sequences in the diarrheic populations varied from 794.85 to 801.39. The Chao1, Shannon, and Rank abundance curves showed a tendency to saturate, implying eligible depth and evenness (Figures 1A–C). High-quality sequences with 97% nucleotide sequence similarity were identified as one OTU. A total of 1,175 OTUs have been recognized in gut bacterial communities, varying from 1,035 to 1,124 in each sample (Figure 1E). Moreover, there were 1,156 and 1,144 OTUs in the healthy and diarrheic horses, respectively, and 1,125 OTUs in common, accounting for approximately 95.74% of the total OTUs (Figure 1D).
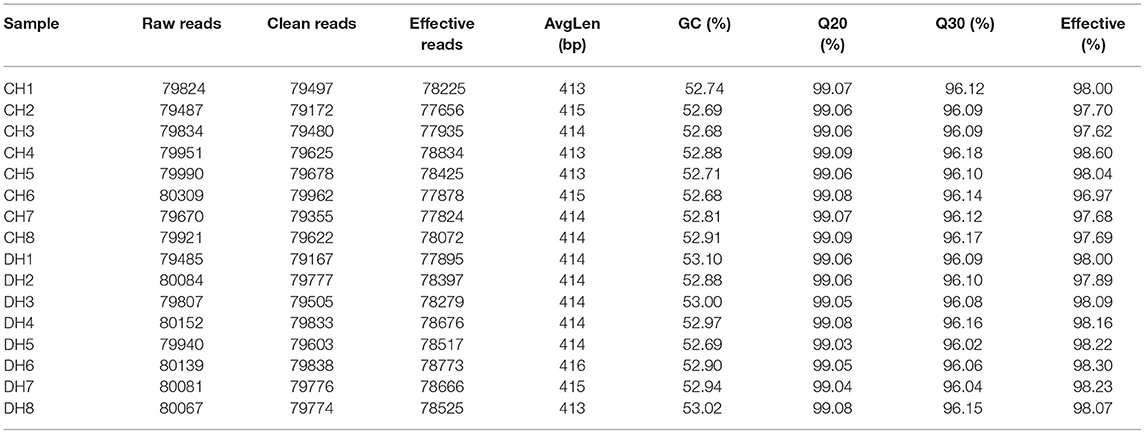
Table 1. Bacterial sequence information of each sample.
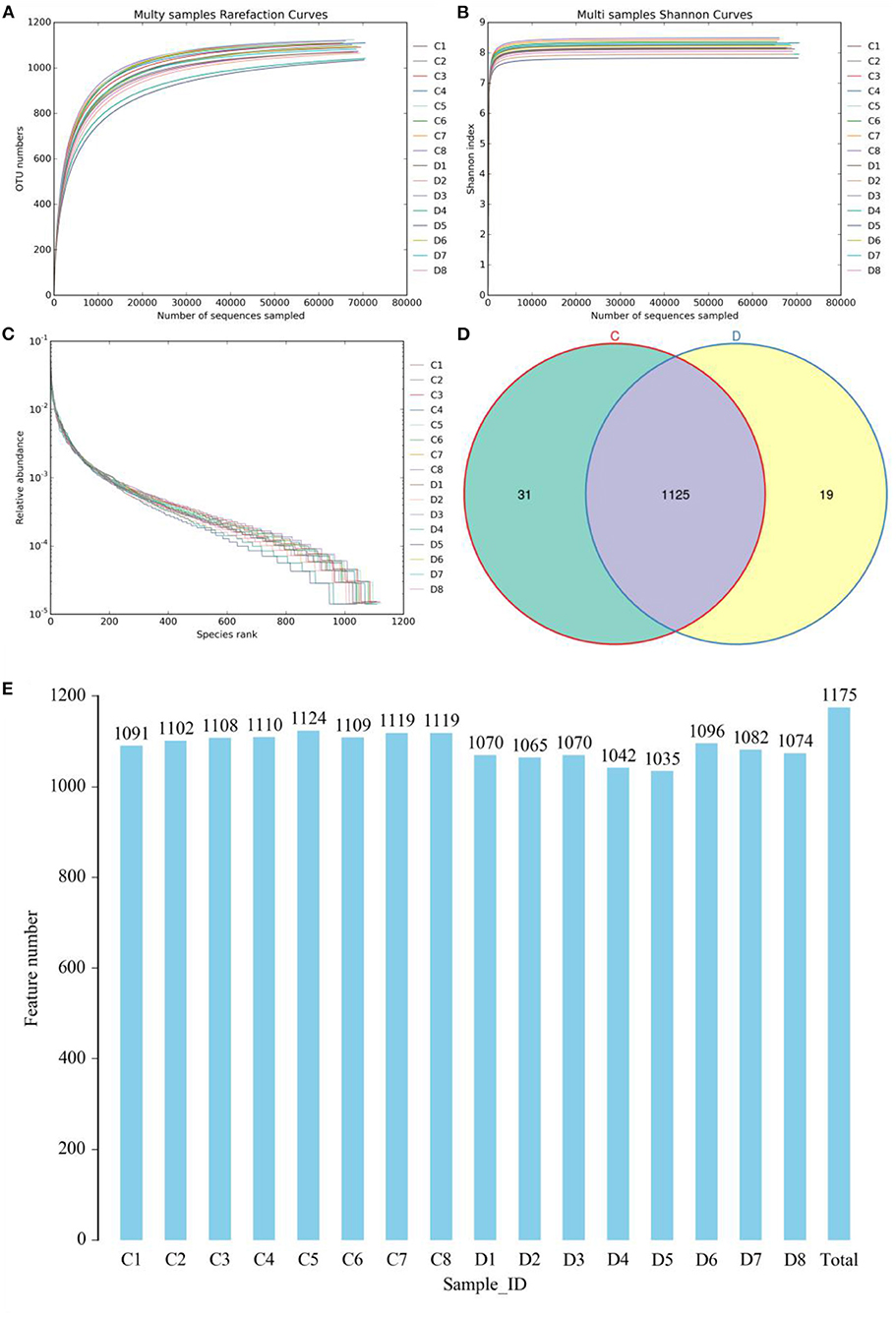
Figure 1. Feasibility analysis of sequencing data. Sequencing depth and evenness of gut microbiota could be assessed with (A,B) rarefaction and (C) rank abundance curves. (D) Venn diagrams for shared and unique operational taxonomic unit (OTU) distribution. (E) Quantity of OTUs in each sample.
Analysis of Microbial Diversity in the Healthy and Diarrheic Horses
The indicates of Chao1, ACE, Shannon, and Good's coverage were calculated to evaluate the alpha diversity of the microbial community. Good's coverage estimates varied from 99.88 to 99.96% for all of the samples, showing excellent coverage. The average Chao1 and ACE indices in the healthy horses were 1,130.99 and 1,123.46, while those in the diarrheic populations were 1,098.34 and 1,089.36 (Figures 2A,B). Furthermore, the average Shannon index was 5.861 and 6.41 in the healthy and diarrheic horses, respectively (Figure 2C). Statistical analysis showed that the diversity indices including Chao1, ACE, and Shannon of the healthy horses were significantly higher than those of the diarrheic populations. The results of Chao1, ACE, and Shannon indices showed that there were significant differences in the richness and diversity of gut microbial population between the healthy and diarrheic horses. The PCoA scatterplot of gut microbiota showed a separation of samples in the healthy and diarrheic horses, which was in line with the UPGMA results, indicating a significant shift in gut microbial principal compositions (Figures 2D–F).
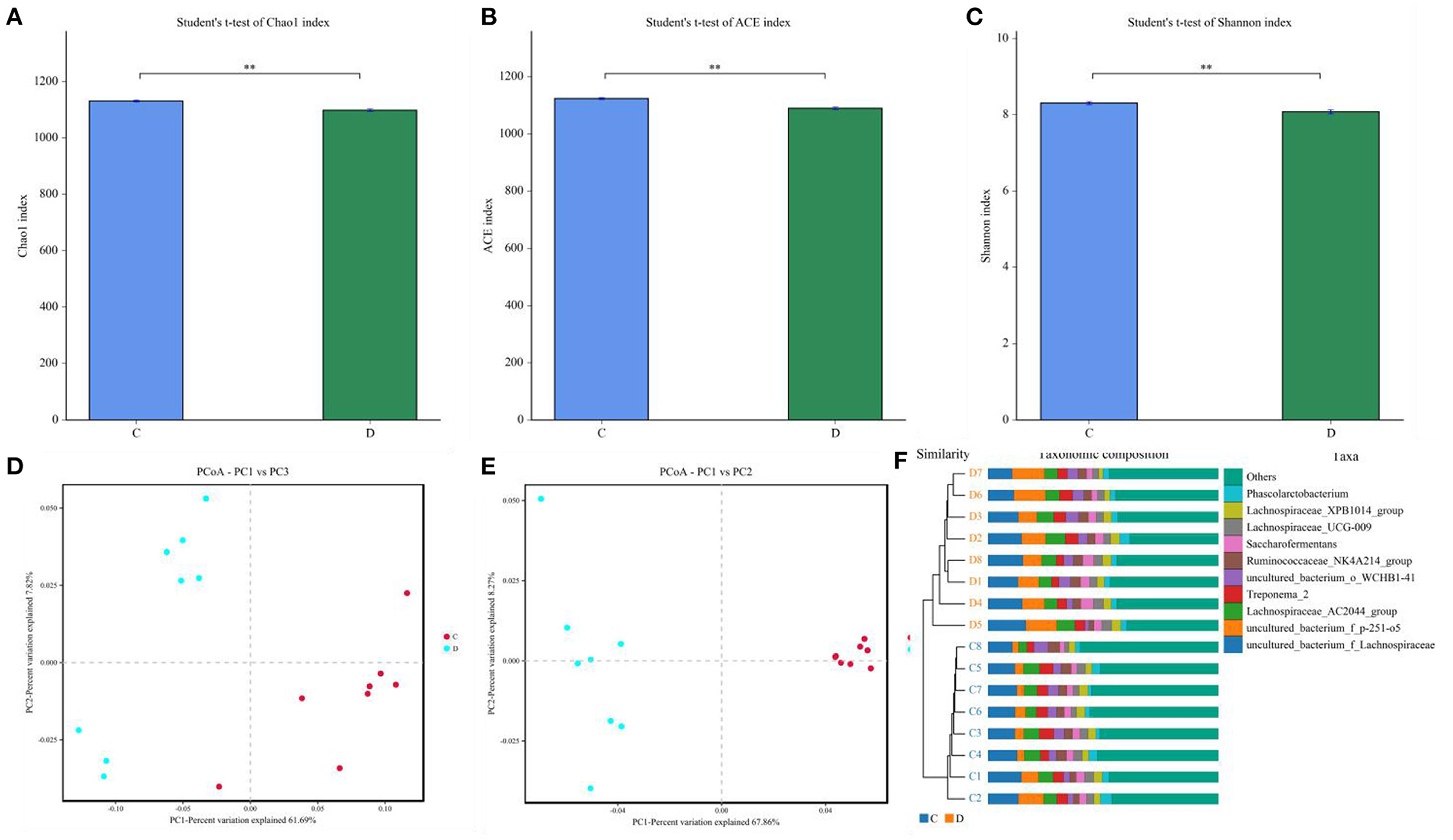
Figure 2. Comparative analysis of alpha and beta diversity of gut microbiota between the healthy and diarrheic horses. (A–C) represent Chao, ACE, and Shannon indices, respectively. (D,F) indicate PCoA map based on (E) weighted and (F) unweighted UniFrac distance. (F) Clustering analysis map.
Composition Analysis of the Gut Microbial Community in the Healthy and Diarrheic Horses
Gut microbial community composition in the healthy and diarrheic horses was assessed at different taxonomical levels. At the phylum level, Firmicutes (61.07, 68.87%), Bacteroidetes (25.77, 16.29%), Spirochaetes (4.48, 4.72%), and Kiritimatiellaeota (4.01, 3.88%) were dominant in the healthy and diarrheic horses regardless of health statuses (Figure 3A). Moreover, other phyla such as Actinobacteria (1.23, 1.1%), Fibrobacteres (1.25, 0.47%),Tenericutes (0.33, 1.05%), and Patescibacteria (0.4, 0.75%) in both groups were represented with a lower abundance. At the level of genus, uncultured_bacterium_f_Lachnospiraceae (13.67, 12.36%), uncultured_bacterium_f_p-251-o5 (10.79, 4.94%), Lachnospiraceae_AC2044_group (6.61, 5.88%), and Treponema_2 (4.47, 4.7%) were the predominant bacteria in both groups (Figure 3B). The heatmap also displayed the distribution and variability of the bacterial genera in the diarrheic horses (Figure 4).
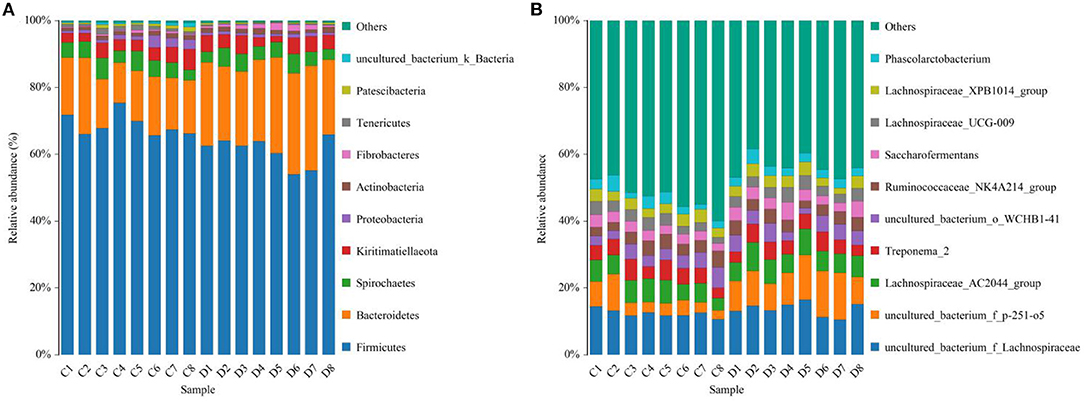
Figure 3. Proportion of preponderant bacterial (A) phyla and (B) genera in the healthy and diarrheic horses.
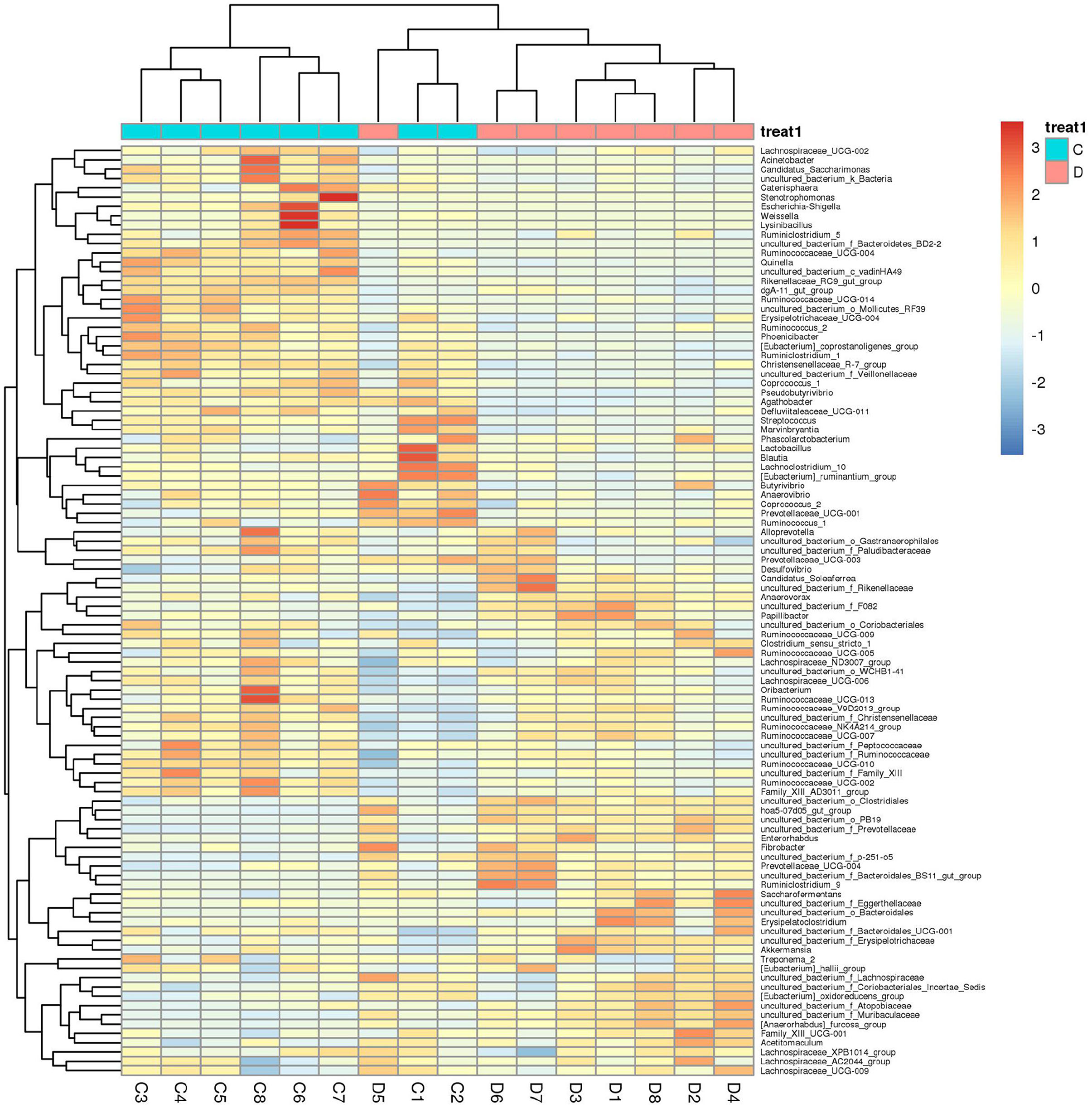
Figure 4. Heatmap of the genus-level hierarchical clustering of the microbial community in the healthy and diarrheic horses.
A comparison of gut microbiota at the levels of phylum and genus was also conducted between the healthy and diarrheic horses. At the level of phylum, the relative abundances of Bacteroidetes, Fibrobacteres, and Verrucomicrobia in the diarrheic horses were significantly higher than in the healthy populations, while the Planctomycetes, Tenericutes, Firmicutes, Patescibacteria, and Proteobacteria contents were lower (Table 2). Moreover, a comparison of the diarrheic and healthy horses showed a significant increase in the abundance of 14 genera (Breznakia, Enterorhabdus, Mailhella, Oscillospira, Proteus, Anaerorhabdus_furcosa_group, Erysipelotrichaceae_UCG-009, Prevotellaceae_UCG-004, Fibrobacter, Parvibacter, Acetitomaculum, Pygmaiobacter, Succinivibrionaceae_UCG-002, andCandidatus_Soleaferrea) as well as an obvious reduction in the abundance of 31 genera (Anaerofustis, Christensenellaceae_R-7_group, Defluviitaleaceae_UCG-011, Glutamicibacter, Lysinibacillus, Phoenicibacter, Pseudobutyrivibrio, Quinella, Ruminiclostridium_1, Ruminiclostridium_6, Ruminococcus_2, Selenomonas_1, Shuttleworthia, Solibacillus, Weissella, Eubacterium_coprostanoligenes_group, Marvinbryantia, Rikenellaceae_RC9_gut_group, Erysipelotrichaceae_UCG-004, Blautia, Candidatus_Saccharimonas, Coprococcus_1, Ruminococcaceae_UCG-014, Agathobacter, Ruminococcaceae_UCG-004, Vagococcus, Kurthia, Lachnospiraceae_UCG-002, Eubacterium_ruminantium_group, Brevibacterium, and Ruminococcaceae_UCG-002). LEfSe combined with LDA scores was conducted to further dissect the shifts in gut microbiota. Besides the above-mentioned differential taxa, the diarrheic horses also showed significantly higher abundances of Acinetobacter, Ruminococcaceae_UCG_010, and Streptococcus, and low abundances of Fibrobacter (Figures 5A,B).
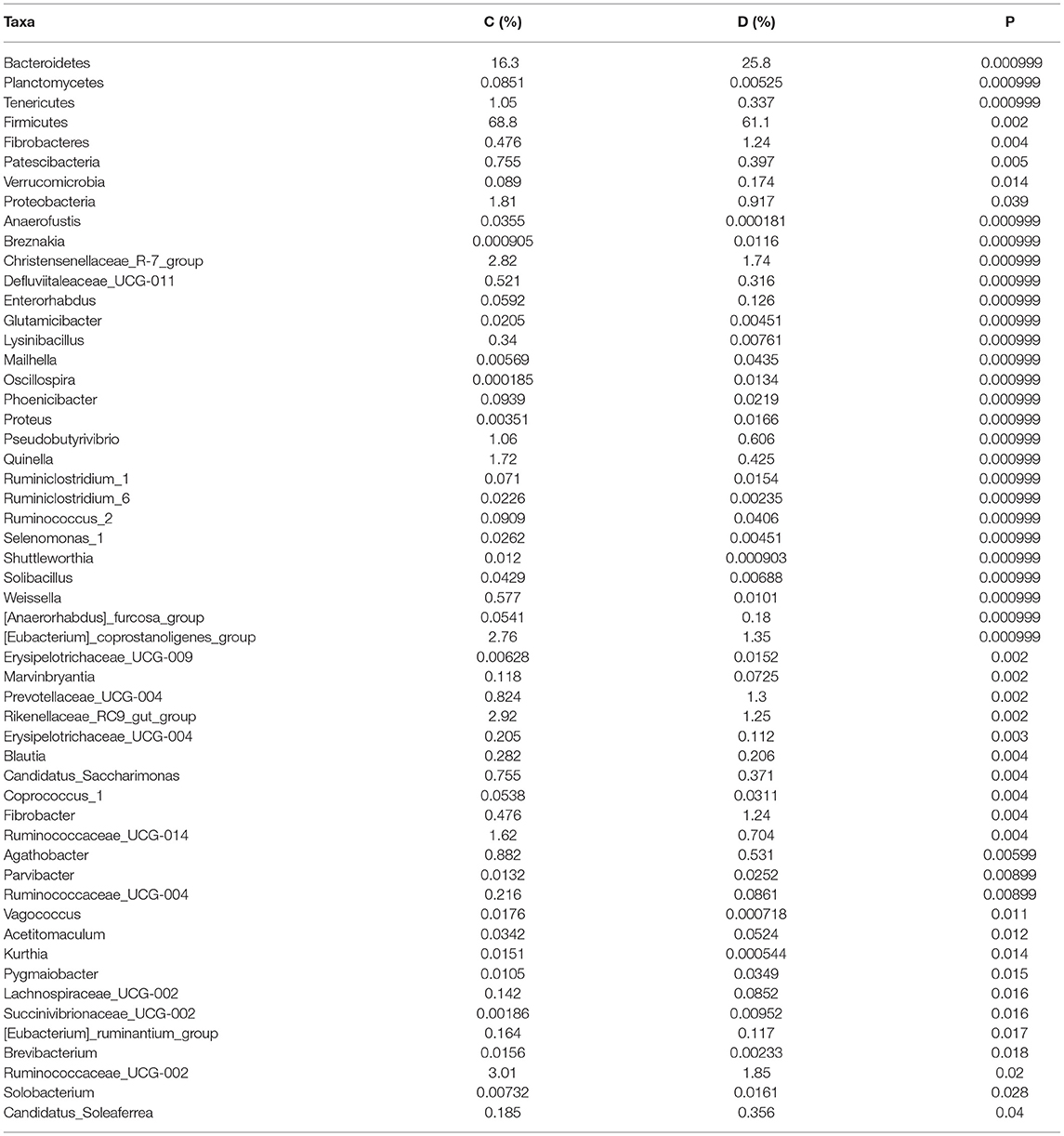
Table 2. Statistical comparison of differential taxa between the healthy and diarrheic horses. All the data are represented as mean ± SD.
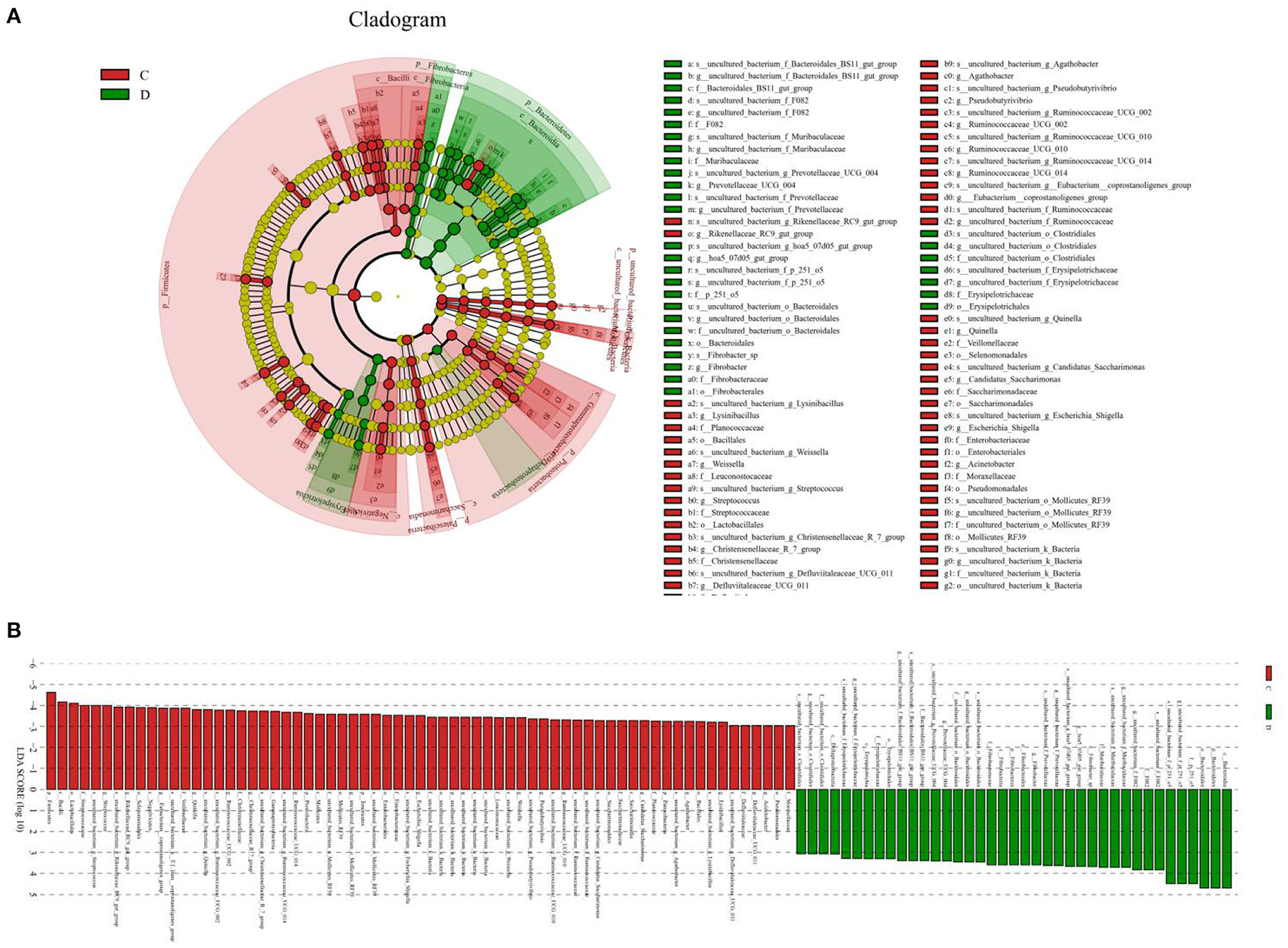
Figure 5. Differential biomarkers in gut microbiota of the horses associated with diarrhea. (A) Phylogenetic distribution of taxa with significant differences are visualized through the cladogram. (B) The criterion of significance was determined at LDA scores > 3.
Correlation Network Analysis
The results indicated that Christensenellaceae_R-7_group was positively associated with Weissella (0.8206), Phoenicibacter (0.8206), Quinella 0.8412), Defluviitaleaceae_UCG-011 (0.8088), and Ruminococcus_2 (0.8382) (Figure 6). Defluviitaleaceae_UCG-011 was positively correlated with Weissella (0.8059). Rikenellaceae_RC9_gut_group was positively related to Lysinibacillus (0.8344) and Stenotrophomonas (0.803). Ruminococcaceae_UCG-002 was positively correlated with Ruminococcaceae_UCG-014 (0.8647), Ruminococcaceae_UCG-010 (0.9324), and Mogibacterium (0.8471). Ruminococcaceae_UCG-014 was positively associated with Phoenicibacter (0.8618), Ruminococcaceae_UCG-004 (0.8206), and Ruminiclostridium_1 (0.8176). Weissella was positively correlated with Coprococcus_1 (0.8647) and Stenotrophomonas (0.8608).
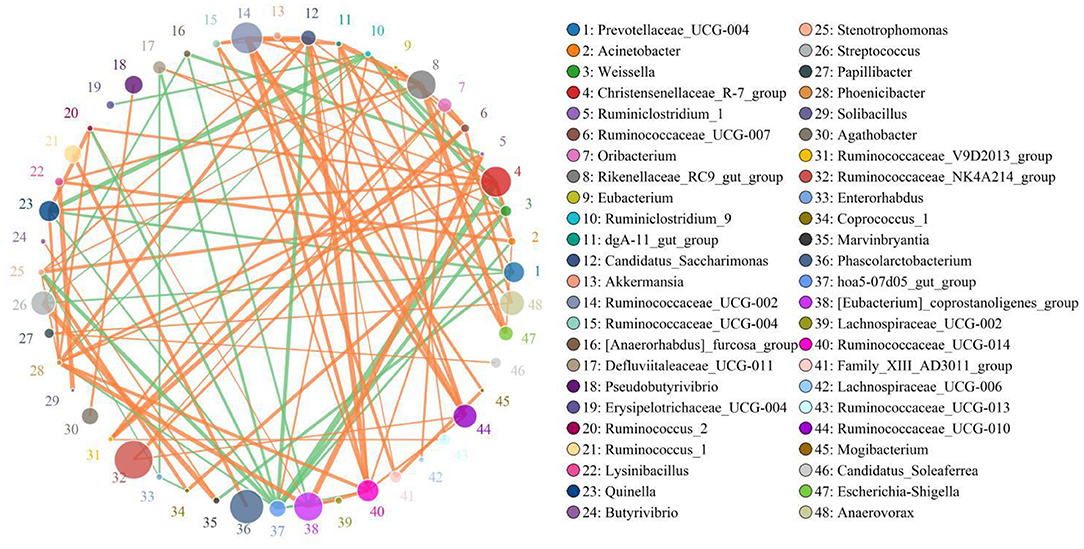
Figure 6. Network analysis indicates a connection among different bacteria. The orange lines indicate positive correlation and the green lines indicate negative correlation.
Discussion
Gut microbiota are a complicated and interactive ecosystem involving trillions of microbes (27, 28). Gut microbial interaction plays a key role in host health (29, 30). Gut microbiota could decrease the invasion and colonization of pathogens by regulating the intestinal barrier and environment, indicating its vital role in gastrointestinal diseases (31, 32). Consequently, the investigation of gut microbiota has attracted widespread attention. However, only a few studies have been conducted to investigate the gut microbiota in horses with different health statuses. In this study, we compared and analyzed gut microbial differences between healthy and diarrheic horses.
Gut microbial diversity and abundance change dynamically within certain limits and affect by age, diet, and environment, but these normal changes cannot damage intestinal functions (33, 34). However, some intense stimuli and diseases such as heavy metals, antibiotics, and gastrointestinal diseases may destroy ecological balance and induce gut microbial dysbiosis (35–37). Li et al. revealed reduced alpha diversity of gut microbiota in giraffes during diarrhea (18). Furthermore, He et al. reported that the gut microbial diversity of piglets with diarrhea was significantly decreased (38). In this study, we observed that diarrhea results in a significant reduction in gut microbial diversity of horses, indicating gut microbial dysbiosis. Research showed that higher gut microbial abundance and diversity were conducive to maintaining intestinal homeostasis and functions (39). Conversely, gut microbial dysbiosis may impair intestinal barrier functions and mucosal immunity, which, in turn, increases morbidity caused by pathogenic bacteria and opportunistic pathogens (40, 41). Consequently, diarrheic horses suffering from gut microbial dysbiosis are at increased risk of bowel dysfunction and other diseases. A PCoA was conducted to dissect the effect of diarrhea on gut microbial main components of horses. The results demonstrated that the samples of healthy horses were clustered together and separated from the diarrheic samples, suggesting that the main components of gut microbiota changed significantly under the influence of diarrhea. Although all the selected horses possessed the same diet and environment, the gut microbiota changed during diarrhea. Consequently, we suspected that diarrhea was the main driving force of gut microbial dysbiosis in horses.
This research indicated that Firmicutes and Bacteroidetes were the most preponderant bacterial phyla in horses regardless of health status, which was consistent with previous findings on other mammals such as pigs, cattle, and goats, implying their key roles in intestinal ecology and function (42, 43). However, although the species of the dominant phyla were not altered, their abundances changed dramatically. In this study, we observed that the proportions of Bacteroidetes, Fibrobacteres, and Verrucomicrobia in the gut microbial community of the horses were significantly increased during diarrhea. Interestingly, Li et al. also reported that these bacterial phyla in the gut microbiota of diarrheic giraffes were significantly increased (18). For herbivores, Firmicutes participated in the degradation of cellulose, which is essential for nutrition and energy intake (44). Moreover, most members of Firmicutes are regarded as intestinal beneficial bacteria, showing positive regulation of intestinal homeostasis, disease resistance, and growth performance (45, 46). Proteobacteria exhibits multiple metabolic functions that contribute to meeting host nutrient and energy requirements (44).
We also found significant changes in some bacterial genera during diarrhea, which may play key roles in gut microbial balance and the development of diarrhea. Moreover, some dramatically decreased bacterial genera in the diarrheic horses including Ruminiclostridium, Ruminococcus, Rikenellaceae, Christensenellaceae, Pseudobutyrivibrio, Weissella, Eubacterium_coprostanoligenes, Ruminococcaceae, Lachnospiraceae, Blautia, Lachnospiraceae, and Coprococcusand Blautia are considered as intestinal beneficial bacteria and are critical for intestinal functions and host health. Ruminiclostridium, which mostly resides in the gastrointestinal tract, displayed the characteristics of decreasing gastrointestinal diseases and improving the growth performance of animals (47). Previous studies have reported that Ruminococcus participated in the degradation of starch and cellulose (48). Rikenellaceae has been previously demonstrated to degrade plant-derived polysaccharides as well as control colitis by stimulating the differentiation of T-regulatory cells (49). As a recognized beneficial bacterium, Christensenellaceae not only is associated with immunoregulation and host health but also contributes to the regulation of intestinal homeostasis and the environment (50). Moreover, Christensenellaceae can also produce several hydrolases including β-glucosidase, β-galactosidase, and α-arabinosidase (24). Pseudobutyrivibrio can produce butyrate, which is conducive to reducing angiocardiopathy and diabetes by activating brown adipose tissues (51). Moreover, recent investigations on butyrate-producing bacteria have provided evidence that they were potentially intestinal beneficial bacteria because of their important roles in alleviating inflammatory bowel disease and regulating immunologic functions (52, 53). Weissella exhibits the characteristics of antioxidation and anti-inflammatory, which contributes to maintaining intestinal homeostasis and improving disease resistance of the host (54). Additionally, Weissella has been reported to reduce fat accumulation and protect the liver in mice induced by a high-fat diet (55). Earlier research indicated that the relative abundance of Eubacterium_coprostanoligenes in the intestine was negatively correlated to the severity of anxiety (56). Eubacterium_coprostanoligenes also displayed the characteristics of reducing cholesterol (57). Numerous bodies of evidence demonstrated that Ruminococcaceae was primarily responsible for digesting starch and cellulose and showed positive regulation of intestinal homeostasis and environment (58). Notably, the higher abundance of Ruminococcaceae contributes to reducing intestinal permeability, non-alcoholic fatty liver, and liver cirrhosis (59, 60). Lachnospiraceae plays an important role in intestinal homeostasis by ameliorating intestinal inflammation (61). Remarkably, some of the above-mentioned bacteria such as Blautia, Lachnospiraceae, Coprococcus, Ruminococcaceae, Ruminococcus, and Ruminiclostridium were considered producers of short-chain fatty acids (SCFAs) (47). Consistent with this study, several previous research studies on other animals have also indicated a significant reduction in SCFA-producing bacteria during diarrhea (18, 62). Previous studies have indicated that SCFAs not only participated in the positive regulation of intestinal homeostasis, immunization, and barrier function but also play key roles in reducing inflammation and regulating energy intake (63, 64). Importantly, SCFAs can also inhibit the proliferation of pathogenic bacteria, showing significant effects of improving the intestinal environment to prevent diseases (65). These decreased beneficial bacteria in diarrheic horses play key roles in maintaining host health and intestinal homeostasis. Consequently, we speculated that these decreased bacteria may be important drivers of diarrhea in horses. Notably, we also observed that some decreased intestinal beneficial bacteria showed a significant correlation with other bacteria. It suggested that diarrhea can also indirectly impair other bacteria by interaction, which may further enhance the influence of diarrhea on the gut microbial community and induce gut microbial dysbiosis.
In summary, this study first explored changes in the gut microbiota in diarrheic horses. The results showed that diarrhea dramatically decreased the gut microbial diversity and altered the taxonomic composition, characterized by a reduced percentage of intestinal beneficial bacteria. This study fills in the gaps in the characteristics of gut microbiota in healthy and diarrheic horses and conveys a vital message that gut microbial dysbiosis may be one of the causes of diarrhea in horses. Importantly, this study contributes to the prevention and treatment of diarrheic horses from the gut microbial perspective.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA808959.
Ethics Statement
The animal study was reviewed and approved by the Ethics Committee of the Wuhan Business University.
Author Contributions
YanL and YaoL conceived and designed the experiments. YaoL contributed to sample collection and preparation. YaoL analyzed the data. YaoL wrote the manuscript. SZ and XW revised the manuscript. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by the Wuhan Business University project (No. 2019KY003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Aoyun Li for uploading the data and the first phases of the animal experiment.
References
1. Liu J, Wang HW, Lin L, Miao CY, Zhang Y, Zhou BH. Intestinal barrier damage involved in intestinal microflora changes in fluoride-induced mice. Chemosphere. (2019) 234:409–18. doi: 10.1016/j.chemosphere.2019.06.080
2. Li A, Wang Y, He Y, Liu B, Iqbal M, Mehmood K, et al. Environmental fluoride exposure disrupts the intestinal structure and gut microbial composition in ducks. Chemosphere. (2021) 277:130222. doi: 10.1016/j.chemosphere.2021.130222
3. Cao Q, Li R, Fu R, Zhang X, Yue B, Wang J, et al. Intestinal fungal dysbiosis in mice induced by fluoride. Chemosphere. (2020) 245:125617. doi: 10.1016/j.chemosphere.2019.125617
4. Hu L, Geng S, Li Y, Cheng S, Fu X, Yue X, et al. Exogenous fecal microbiota transplantation from local adult pigs to crossbred newborn piglets. Front Microbiol. (2017) 8:2663. doi: 10.3389/fmicb.2017.02663
5. Wickramasuriya SS, Park I, Lee K, Lee Y, Kim WH, Nam H, Lillehoj HS. Role of physiology, immunity, microbiota, and infectious diseases in the gut health of poultry. Vaccines (Basel). (2022) 10. doi: 10.3390/vaccines10020172
6. Wang J, Zhu G, Sun C, Xiong K, Yao T, Su Y, et al. TAK-242 ameliorates DSS-induced colitis by regulating the gut microbiota and the JAK2/STAT3 signaling pathway. Microb Cell Fact. (2020) 19:158. doi: 10.1186/s12934-020-01417-x
7. Li A, Ding J, Shen T, Han Z, Zhang J, Abadeen ZU, et al. Environmental hexavalent chromium exposure induces gut microbial dysbiosis in chickens. Ecotoxicol Environ Saf. (2021) 227:112871. doi: 10.1016/j.ecoenv.2021.112871
8. Kong A, Zhang C, Cao Y, Cao Q, Liu F, Yang Y, et al. The fungicide thiram perturbs gut microbiota community and causes lipid metabolism disorder in chickens. Ecotoxicol Environ Saf. (2020) 206:111400. doi: 10.1016/j.ecoenv.2020.111400
9. Tanase DM, Gosav EM, Neculae E, Costea CF, Ciocoiu M, Hurjui LL, et al. Role of gut microbiota on onset and progression of microvascular complications of type 2 diabetes (T2DM). Nutrients. (2020) 12. doi: 10.3390/nu12123719
10. Wan H, Wang Y, Zhang H, Zhang K, Chen Y, Chen C, et al. Chronic lead exposure induces fatty liver disease associated with the variations of gut microbiota. Ecotoxicol Environ Saf. (2022) 232:113257. doi: 10.1016/j.ecoenv.2022.113257
11. Yan H, Qin Q, Chen J, Yan S, Li T, Gao X, et al. Gut microbiome alterations in patients with visceral obesity based on quantitative computed tomography. Front Cell Infect Microbiol. (2021) 11:823262. doi: 10.3389/fcimb.2021.823262
12. Dreisbach C, Morgan H, Cochran C, Gyamfi A, Henderson WA, Prescott S. Metabolic and microbial changes associated with diet and obesity during pregnancy: what can we learn from animal studies? Front Cell Infect Microbiol. (2021) 11:795924. doi: 10.3389/fcimb.2021.795924
13. Wang Y, Zhang H, Zhu L, Xu Y, Liu N, Sun X, et al. Dynamic distribution of gut microbiota in goats at different ages and health states. Front Microbiol. (2018) 9:2509. doi: 10.3389/fmicb.2018.02509
14. Gryaznova MV, Dvoretskaya YD, Syromyatnikov MY, Shabunin SV, Parshin PA, Mikhaylov EV, et al. Changes in the microbiome profile in different parts of the intestine in piglets with diarrhea. Animals (Basel). (2022) 12. doi: 10.3390/ani12030320
15. Han Z, Li K, Shahzad M, Zhang H, Luo H, Qiu G, et al. Analysis of the intestinal microbial community in healthy and diarrheal perinatal yaks by high-throughput sequencing. Microb Pathog. (2017) 111:60–70. doi: 10.1016/j.micpath.2017.08.025
16. Wang Y, Li A, Zhang L, Waqas M, Mehmood K, Iqbal M, et al. Probiotic potential of Lactobacillus on the intestinal microflora against Escherichia coli induced mice model through high-throughput sequencing. Microb Pathog. (2019) 137:103760. doi: 10.1016/j.micpath.2019.103760
17. Wang Y, Li A, Liu J, Mehmood K, Wangdui B, Shi H, et al. L. pseudomesenteroides and L johnsonii isolated from yaks in Tibet modulate gut microbiota in mice to ameliorate enteroinvasive Escherichia coli-induced diarrhea. Microb Pathog. (2019) 132:1–9. doi: 10.1016/j.micpath.2019.04.020
18. Li A, Liu B, Li F, He Y, Wang L, Fakhar-E-Alam KM, et al. Integrated Bacterial and Fungal Diversity Analysis Reveals the Gut Microbial Alterations in Diarrheic Giraffes. Front Microbiol. (2021) 12:712092. doi: 10.3389/fmicb.2021.712092
19. Han Z, Li A, Pei L, Li K, Jin T, Li F, et al. Milk replacer supplementation ameliorates growth performance and rumen microbiota of early-weaning yimeng black goats. Front Vet Sci. (2020) 7:572064. doi: 10.3389/fvets.2020.572064
20. Guo X, Liu S, Wang Z, Zhang XX Li M, Wu B. Metagenomic profiles and antibiotic resistance genes in gut microbiota of mice exposed to arsenic and iron. Chemosphere. (2014) 112:1–8. doi: 10.1016/j.chemosphere.2014.03.068
21. Cao QQ, Lin LX, Xu TT, Lu Y, Zhang CD, Yue K, et al. Aflatoxin B1 alters meat quality associated with oxidative stress, inflammation, and gut-microbiota in sheep. Ecotoxicol Environ Saf. (2021) 225:112754. doi: 10.1016/j.ecoenv.2021.112754
22. Li Y, Zuo Z, Zhang B, Luo H, Song B, Zhou Z, et al. Impacts of early-life paraquat exposure on gut microbiota and body weight in adult mice. Chemosphere. (2022) 291:133135. doi: 10.1016/j.chemosphere.2021.133135
23. Zhou J, Shu R, Yu C, Xiong Z, Xiao Q, Li Z, et al. Exposure to low concentration of trifluoromethanesulfonic acid induces the disorders of liver lipid metabolism and gut microbiota in mice. Chemosphere. (2020) 258:127255. doi: 10.1016/j.chemosphere.2020.127255
24. Xin J, Chai Z, Zhang C, Zhang Q, Zhu Y, Cao H, et al. Comparing the microbial community in four stomach of dairy cattle, yellow cattle and three yak herds in qinghai-tibetan plateau. Front Microbiol. (2019) 10:1547. doi: 10.3389/fmicb.2019.01547
25. Xi L, Song Y, Qin X, Han J, Chang YF. Microbiome analysis reveals the dynamic alternations in gut microbiota of diarrheal giraffa camelopardalis. Front Vet Sci. (2021) 8:649372. doi: 10.3389/fvets.2021.649372
26. Li A, Yang Y, Qin S, Lv S, Jin T, Li K, et al. Microbiome analysis reveals gut microbiota alteration of early-weaned Yimeng black goats with the effect of milk replacer and age. Microb Cell Fact. (2021) 20:78. doi: 10.1186/s12934-021-01568-5
27. Liu Z, Li A, Wang Y, Iqbal M, Zheng A, Zhao M, et al. Comparative analysis of microbial community structure between healthy and Aeromonas veronii-infected Yangtze finless porpoise. Microb Cell Fact. (2020) 19:123. doi: 10.1186/s12934-020-01383-4
28. Li A, Yang Y, Zhang Y, Lv S, Jin T, Li K, et al. Microbiome analysis reveals the alterations in gut microbiota in different intestinal segments of Yimeng black goats. Microb Pathog. (2021) 155:104900. doi: 10.1016/j.micpath.2021.104900
29. Eberl C, Ring D, Munch PC, Beutler M, Basic M, Slack EC, et al. Reproducible colonization of germ-free mice with the oligo-mouse-microbiota in different animal facilities. Front Microbiol. (2019) 10:2999. doi: 10.3389/fmicb.2019.02999
30. Pilla R, Suchodolski JS. The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front Vet Sci. (2019) 6:498. doi: 10.3389/fvets.2019.00498
31. Zhou A, Yuan Y, Yang M, Huang Y, Li X, Li S, et al. Crosstalk between the gut microbiota and epithelial cells under physiological and infectious conditions. Front Cell Infect Microbiol. (2022) 12:832672. doi: 10.3389/fcimb.2022.832672
32. Wu Y, Nie C, Luo R, Qi F, Bai X, Chen H, et al. Effects of multispecies probiotic on intestinal microbiota and mucosal barrier function of neonatal calves infected with E. coli K99. Front Microbiol. (2021) 12:813245. doi: 10.3389/fmicb.2021.813245
33. Dias J, Marcondes MI, Motta DSS, Cardoso DMES, Fontes NM, Tassinari RR, et al. Bacterial community dynamics across the gastrointestinal tracts of dairy calves during preweaning development. Appl Environ Microbiol. (2018) 84. doi: 10.1128/AEM.02675-17
34. Li B, Zhang K, Li C, Wang X, Chen Y, Yang Y. Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (Capra hircus) during preweaning development. Front Microbiol. (2019) 10:2125. doi: 10.3389/fmicb.2019.02125
35. Jin C, Zeng Z, Fu Z, Jin Y. Oral imazalil exposure induces gut microbiota dysbiosis and colonic inflammation in mice. Chemosphere. (2016) 160:349–58. doi: 10.1016/j.chemosphere.2016.06.105
36. Mayneris-Perxachs J, Cardellini M, Hoyles L, Latorre J, Davato F, Moreno-Navarrete JM, et al. Iron status influences non-alcoholic fatty liver disease in obesity through the gut microbiome. Microbiome. (2021) 9:104. doi: 10.1186/s40168-021-01052-7
37. Chang X, Kang M, Shen Y, Yun L, Yang G, Zhu L, et al. Bacillus coagulans SCC-19 maintains intestinal health in cadmium-exposed common carp (Cyprinus carpio L) by strengthening the gut barriers, relieving oxidative stress and modulating the intestinal microflora. Ecotoxicol Environ Saf. (2021) 228:112977. doi: 10.1016/j.ecoenv.2021.112977
38. He K, Yan W, Sun C, Liu J, Bai R, Wang T, et al. Alterations in the diversity and composition of gut microbiota in weaned piglets infected with Balantioides coli. Vet Parasitol. (2020) 288:109298. doi: 10.1016/j.vetpar.2020.109298
39. Bui AT, Williams BA, Hoedt EC, Morrison M, Mikkelsen D, Gidley MJ. High amylose wheat starch structures display unique fermentability characteristics, microbial community shifts and enzyme degradation profiles. Food Funct. (2020) 11:5635–46. doi: 10.1039/D0FO00198H
40. Kosiewicz MM, Zirnheld AL, Alard P. Gut microbiota, immunity, and disease: a complex relationship. Front Microbiol. (2011) 2:180. doi: 10.3389/fmicb.2011.00180
41. Wang B, Deng B, Yong F, Zhou H, Qu C, Zhou Z. Comparison of the fecal microbiomes of healthy and diarrheic captive wild boar. Microb Pathog. (2020) 147:104377. doi: 10.1016/j.micpath.2020.104377
42. Zhang L, Jiang X, Li A, Waqas M, Gao X, Li K, et al. Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe. (2020) 61:102115. doi: 10.1016/j.anaerobe.2019.102115
43. Li K, Mehmood K, Zhang H, Jiang X, Shahzad M, Dong X, et al. Characterization of fungus microbial diversity in healthy and diarrheal yaks in Gannan region of Tibet Autonomous Prefecture. Acta Trop. (2018) 182:14–26. doi: 10.1016/j.actatropica.2018.02.017
44. Sun B, Wang X, Bernstein S, Huffman MA, Xia DP, Gu Z, et al. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci Rep. (2016) 6:26035. doi: 10.1038/srep26035
45. Li A, Wang Y, Li Z, Qamar H, Mehmood K, Zhang L, et al. Probiotics isolated from yaks improves the growth performance, antioxidant activity, and cytokines related to immunity and inflammation in mice. Microb Cell Fact. (2019) 18:112. doi: 10.1186/s12934-019-1161-6
46. Garneau JE, Tremblay DM, Moineau S. Characterization of 1706, a virulent phage from Lactococcus lactis with similarities to prophages from other Firmicutes. Virology. (2008) 373:298–309. doi: 10.1016/j.virol.2007.12.002
47. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. (2014) 121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9
48. Miller TL, Currenti E, Wolin MJ. Anaerobic bioconversion of cellulose by Ruminococcus albus, Methanobrevibacter smithii, and Methanosarcina barkeri. Appl Microbiol Biotechnol. (2000) 54:494–8. doi: 10.1007/s002530000430
49. Seshadri R, Leahy SC, Attwood GT, Teh KH, Lambie SC, Cookson AL, et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat Biotechnol. (2018) 36:359–67. doi: 10.1038/nbt.4110
50. Kong F, Hua Y, Zeng B, Ning R, Li Y, Zhao J. Gut microbiota signatures of longevity. Curr Biol. (2016) 26:R832–3. doi: 10.1016/j.cub.2016.08.015
51. Cai W, Xu J, Li G, Liu T, Guo X, Wang H, et al. Ethanol extract of propolis prevents high-fat diet-induced insulin resistance and obesity in association with modulation of gut microbiota in mice. Food Res Int. (2020) 130:108939. doi: 10.1016/j.foodres.2019.108939
52. Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. (2013) 498:99–103. doi: 10.1038/nature12198
53. Bui TP, Ritari J, Boeren S, de Waard P, Plugge CM, de Vos WM. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat Commun. (2015) 6:10062. doi: 10.1038/ncomms10062
54. Yu HS, Lee NK, Choi AJ, Choe JS, Bae CH, Paik HD. Antagonistic and antioxidant effect of probiotic Weissella cibaria JW15. Food Sci Biotechnol. (2019) 28:851–5. doi: 10.1007/s10068-018-0519-6
55. Choi SI, You S, Kim S, Won G, Kang CH, Kim GH. Weissella cibaria MG5285 and Lactobacillus reuteri MG5149 attenuated fat accumulation in adipose and hepatic steatosis in high-fat diet-induced C57BL/6J obese mice. Food Nutr Res. (2021) 65. doi: 10.29219/fnr.v65.8087
56. Chen YH, Bai J, Wu D, Yu SF, Qiang XL, Bai H, et al. Association between fecal microbiota and generalized anxiety disorder: Severity and early treatment response. J Affect Disord. (2019) 259:56–66. doi: 10.1016/j.jad.2019.08.014
57. Liu Z, Yin B. Alterations in the gut microbial composition and diversity of tibetan sheep infected with echinococcus granulosus. Front Vet Sci. (2021) 8:778789. doi: 10.3389/fvets.2021.778789
58. Zhao J, Yao Y, Li D, Xu H, Wu J, Wen A, et al. Characterization of the gut microbiota in six geographical populations of Chinese rhesus macaques (Macaca mulatta), implying an adaptation to high-altitude environment. Microb Ecol. (2018) 76:565–77. doi: 10.1007/s00248-018-1146-8
59. Rai R, Saraswat VA, Dhiman RK. Gut microbiota: its role in hepatic encephalopathy. J Clin Exp Hepatol. (2015) 5:S29–36. doi: 10.1016/j.jceh.2014.12.003
60. Huang C, Song P, Fan P, Hou C, Thacker P, Ma X. Dietary sodium butyrate decreases postweaning diarrhea by modulating intestinal permeability and changing the bacterial communities in weaned piglets. J NUTR. (2015) 145:2774–80. doi: 10.3945/jn.115.217406
61. Zhao L, Zhang Q, Ma W, Tian F, Shen H, Zhou M, et al. Combination of quercetin and resveratrol reduces obesity in high-fat diet-fed rats by modulation of gut microbiota. Food Funct. (2017) 8:4644–56. doi: 10.1039/C7FO01383C
62. Xi L, Song Y, Han J, Qin X. Microbiome analysis reveals the significant changes in gut microbiota of diarrheic Baer's Pochards (Aythya baeri). Microb Pathog. (2021) 157:105015. doi: 10.1016/j.micpath.2021.105015
63. Melbye P, Olsson A, Hansen TH, Sondergaard HB, Bang OA. Short-chain fatty acids and gut microbiota in multiple sclerosis. Acta Neurol Scand. (2019) 139:208–19. doi: 10.1111/ane.13045
64. Goverse G, Molenaar R, Macia L, Tan J, Erkelens MN, Konijn T, et al. Diet-derived short chain fatty acids stimulate intestinal epithelial cells to induce mucosal tolerogenic dendritic cells. J Immunol. (2017) 198:2172–81. doi: 10.4049/jimmunol.1600165
Keywords: diarrhea, horse, dysbiosis, gut microbiota, healthy
Citation: Li Y, Lan Y, Zhang S and Wang X (2022) Comparative Analysis of Gut Microbiota Between Healthy and Diarrheic Horses. Front. Vet. Sci. 9:882423. doi: 10.3389/fvets.2022.882423
Received: 23 February 2022; Accepted: 30 March 2022;
Published: 02 May 2022.
Edited by:
Fazul Nabi, Lasbela University of Agriculture, Water and Marine Sciences, PakistanReviewed by:
Waseem Ali, Sindh Agriculture University, PakistanAftab Shaukat, Huazhong Agricultural University, China
Copyright © 2022 Li, Lan, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanfang Lan, bGFueWFuZjA5MjZAc2luYS5jb20=