 Zhenbiao Zhang
Zhenbiao Zhang Liu Zhang1
Liu Zhang1 Jianzhong Wang
Jianzhong Wang Zhaofei Xia
Zhaofei Xia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 10 March 2022
Sec. Veterinary Infectious Diseases
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.816415
This article is part of the Research TopicAntimicrobial Use, Antimicrobial Resistance, and the Microbiome in Animals, volume IIView all 13 articles
Klebsiella pneumoniae complex is an increasingly important bacterial pathogen that is capable of causing severe organs and life-threatening disease. This study aimed to investigate the multidrug resistance, phylogroups, molecular characterization, and hypervirulence-associated determinants of the complex, which were isolated from clinical diseased dogs and cats. A total of 35 K. pneumoniae complex (2.3%; 95% confidence interval, 1.6–3.2) isolates were identified from 1,500 samples, all of which were collected randomly from veterinary hospitals in the 12 regions across China. Antimicrobial susceptibility testing showed that isolates were extremely resistant to amoxicillin–clavulanate (82.9%) and trimethoprim–sulfamethoxazole (77.1%). The rate of multidrug-resistant reached an astonishing 82.9% and found a carbapenemase-producing strain carrying IncX3-blaNDM−5 derived a cat from Zhejiang. The prevalence rates of extended-spectrum β-lactamase gene blaCTX−M and plasmid-mediated quinolone resistance gene aac(6')Ib-cr were 51.4% and 45.7%, respectively. The resistance gene aph(3')-Ia of isolates from cats was more significantly (p < 0.05) prevalent than that from dogs. Likewise, K. pneumoniae complex harbored hypervirulence-associated genes ybt (11.4%), iuc (5.7%), and iroB (2.9%). Three (8.6%) of the 35 isolates were determined as hypermucoviscous by the string test. Lipopolysaccharide serotype O1v2 had the highest percentage of 25.7%, but capsular serotypes presented diversity distribution among the isolates. The core–genome phylogenetic tree demonstrated most of the isolates belonged to the KpI phylogroup (91.4%). Multilocus sequence typing analysis identified 25 different STs; ST15 and ST37 were the most abundant accounting for isolates, followed by ST307, ST656, ST1408, and ST4566. In addition, the prevalence of IncFIB-type plasmid for cat isolates was significantly higher (p < 0.05) than that for dogs. Sequences of IncX3 in blaNDM−5-positive strain contained regions showing >99% nucleotide sequence identity to the reference plasmid pNDM-MGR194 from the human.
Klebsiella species, gram-negative opportunistic pathogens, commonly caused acquired antimicrobial-resistant infections in hospitals or communities. They are belonging to the Enterobacteriaceae, which includes Escherichia, Salmonella, and Shigella. In companion animals, K. pneumoniae has been reported to colonize hosts and causes extraintestinal infections, such as urinary tract infections, pyometra, upper respiratory tract infections, and bloodstream infection (septicemia) (1, 2). The treatment of these infections was often difficult because of the emergence of antibiotic resistance, which may be associated with high morbidity and mortality rate (3). In recent years, multidrug-resistant (MDR), carbapenem-resistant K. pneumonia (CRKP), and hypervirulent strains (hvKP) spread widely as a critical public health threat in China and even the world (4).
The high incidence of K. pneumoniae antimicrobial resistance rate (AMR) received increasing attention, mainly due to the continuous increase in deaths associated with AMR clone produced by CRKP and hvKP. Most of the new AMR genes discovered in the past two decades were first detected and then spread widely among gram-negative bacterial pathogens, including the extended-spectrum β-lactamase (ESBL) forms of blaCTX−M and blaSHV, the carbapenemases blaKPC and blaNDM, and most recently mcr-1, the first plasmid-borne gene associated with colistin resistance (5). The emergence of these AMR genes from K. pneumoniae not only increases the risk of failure for human antibacterial treatment but also affects that for companion animals. Unfortunately, if the bacteria were transmitted from the pets to their owners, the antimicrobial bacteria from companion animals may have an important impact on human public health (6).
The new technology of molecular strain typing based on DNA sequencing provides various opportunities for elucidating the structure of the K. pneumoniae population (7). Multilocus sequence typing (MLST) provided a standardized and replicable system for K. pneumoniae identification and naming, which is based on chromosomally encoded seven housekeeping genes (rpoB, gapA, mdh, pgi, phoE, infB, and tonB) (8). Whole-genome sequencing (WGS) could identify closely related species in clinical and research laboratories that have an average nucleotide homology of 95–96% with K. pneumoniae through biochemical or proteomic analysis (9, 10). Sequencing of wzi alleles was a marker of capsule serotype (KL), which is highly predictive of capsule (K) serotype and had a strong correlation with KL/K type (11, 12). While O antigen of lipopolysaccharide (LPS) has been defined by sequence identity in the conserved wzm and wzt genes (13).
The members of the K. pneumoniae complex were first distinguished based on the gyrA and were designated as the phylogenetic group of K. pneumoniae. WGS confirmed that the average nucleotide consistency of the whole genome is ≥3%, which is sufficient to specify new species and help identify other member species: Klebsiella pneumoniae (Kp1/KpI), Klebsiella quasipneumoniae subspecies quasipneumoniae (Kp2/KpIIa), K. quasipneumoniae subspecies similipneumoniae (Kp4/KpIIb), Klebsiella variicola subspecies variicola (Kp3/KpIII), K. variicola subspecies tropica (Kp5), Klebsiella quasivariicola (Kp6), and Klebsiella africana (Kp7) (14, 15). K. variicola, an emerging pathogen in humans and had been reported numerous infections worldwide but with a lower frequency in wild and companion animals, can also display the hypermucoviscous (hmKv) and/or hypervirulent (hvKv) phenotypes (16, 17). K. quasipneumoniae is a new species discovered in recent years. It has extensive kinship with K. pneumoniae and K. variicola. K. quasipneumoniae is also viscous and easy to acquire resistance and has certain high virulence characteristics (18). In this study, we represented a molecular characterization of K. pneumoniae complex isolates collected from diseased companion animals as part of a national surveillance program from different regions in China and examined their epidemiological relatedness.
Between November 2017 and October 2019, a total of 1,500 clinical specimens with suspicious bacterial infections were collected from dogs (n = 835) and cats (n = 665) in veterinary hospitals. These hospitals were distributed in the following 26 regions in China: Guangdong, Shandong, Shanghai, Tianjin, Liaoning, Jiangsu, Hubei, Henan, Hebei, Sichuan, Hunan, Zhejiang, Fujian, Heilongjiang, Inner Mongolia, Shanxi, Shaanxi, Chongqing, Ningxia, Anhui, Jilin, Guangxi, Jiangxi, Hainan, Gansu, and Xinjiang. The sampling sites were not disinfected before collection, and the hosts of all samples came to the veterinary hospital for the first time, or they had been more than 2 months since the last antibiotic administration. In addition, the sampling of companion animals was conducted following the principles of the China Agricultural University Animal Ethics Committee document (no. AW01017102-2). All samples consisted of urine (37.1%, 557/1,500), abscess (14.3%, 215/1,500), skin (8.8%, 132/1,500), ear swabs (5.9%, 88/1,500), nasal swabs (5.4%, 81/1,500), coelomic fluid (5.2%, 78/1,500), throat swabs (4.7%, 71/1,500), surgical infection (4.7%, 70/1,500), tracheal lavage (2.7%, 41/1,500), pyometra (1.2%, 18/1,500), oral swabs (1.2%, 18/1,500), blood (1.1%, 16/1,500), vaginal swabs (0.9%, 14/1,500), eye secretion (0.9%, 13/1,500), and other samples (5.86%, 88/1,500) that contained anal swabs, prostatic fluid, synovial fluid, foreskin swabs, cerebrospinal fluid, and bile.
The collected samples were evenly inoculated on sterile MacConkey inositol adonitol agar (HopeBio, Qingdao, China) containing 100 mg/L carbenicillin and then incubated at 37°C overnight. Clones with the red center were inoculated into a 1-mL volume of brain–heart infusion broth medium (Land Bridge, Beijing, China) and cultured 12 h at 37°C with shaking (200 revolutions/min). DNA of the bacterial solution was extracted by TIANamp Bacteria DNA Kit (Tiangen, Beijing, China) according to the manufacturer's instructions. Subsequently, the DNA was used as the template for polymerase chain reaction amplification of 16S rDNA gene as previously described (19), and amplicons were sequenced to confirm bacterial genus using the BLAST algorithm.
All K. pneumoniae complex isolates were subjected to antimicrobial susceptibility testing using the agar/broth dilution method with 16 clinically relevant antibiotics of 10 categories (amoxicillin–clavulanate, piperacillin–tazobactam, ceftazidime–avibactam, cefotaxime, cefepime, meropenem, imipenem, aztreonam, ciprofloxacin, enrofloxacin, gentamicin, amikacin, doxycycline, colistin, florfenicol, and trimethoprim–sulfamethoxazole). Escherichia coli ATCC 25922 was used as a quality control organism. Results of minimum inhibitory concentrations (MICs) were explained according to the breakpoints of Clinical and Laboratory Standards Institute documents VET08-ED4:2018 /M100-ED30:2020 and European Commission on Antimicrobial Susceptibility Testing (EUCAST) documents (version 9.0, 2019).
String test was conducted to define hypermucoviscous phenotype as previously described (20). All tested strains were cultured on 5% sheep blood agar plates and incubated overnight at 37°C. A standard bacteriological inoculation loop was used to touch the colonies lift gently. A viscous string ≥5 mm in length by stretching bacterial colonies was defined as string test positive.
Genomic DNA was extracted from all K. pneumoniae complex using TIANamp Bacteria DNA Kit according to the manufacturer's instruction. DNA library was established using KAPA HyperPrep kit (Roche, Basel, Switzerland), and sequencing was performed on the HiSeq X Ten platform (Illumina) with 150-bp paired-end reads by Annoroad Genomics Co., Ltd. The draft genomes were assembled using SPAdes (version 3.9.0) (21). All WGS data for this work were deposited in the GenBank and under BioProject accession no. PRJNA685900. Plasmid Inc types, antibiotic resistance genes, and virulence genes were identified using abricate (https://github.com/tseemann/abricate). Sequence types (STs) of K. pneumoniae were determined by the bioinformatics tool at https://cge.cbs.dtu.dk/services/MLST, and K. variicola was evaluated through the professional K. variicola MLST system (http://mlstkv.insp.mx) (22). Harvest package (version 1.1.2) (23) was used to demonstrate a phylogenetic tree of core–genome alignments for all assembled genomes and visualized using Interactive Tree of Life (http://itol.embl.de/) with the corresponding features of each isolate. Capsule serotype and O antigen were analyzed using Kleborate (https://github.com/katholt/Kleborate). A minimum spanning tree of all STs was generated by BioNumerics version 7.6 (Applied Maths, Belgium) using the BURST algorithm. BLAST Ring Image Generator (24) was used to compare the genetic background of different blaNDM-carrying plasmids.
Statistical significance was determined using χ2 test and Fisher exact test in SPSS Statistics (version 22; IBM Corporation), and the level of significance was set at p < 0.05.
Overall, 35 K. pneumoniae complex [35 of 1,500; 2.3%; 95% confidence interval (CI), 1.6–3.2] isolates were identified from 1,500 samples, which collected randomly from veterinary hospitals in the 12 regions across China (Figure 1), including 19 from dogs [19 of 835 (2.3%); 95% CI, 1.4–3.5] and 16 from cats [16 of 665 (2.4%); 95% CI, 1.4–3.9]; no significant difference among animals (p = 0.868) (Table 1). Two [2 of 35 (0.06%); 95% CI, 0.01–0.19] of K. variicola (Kp34 and Kp87) were, respectively, isolated in urine (cat from Shanghai) and skin swab (dog from Shandong). The K. quasipneumoniae (Kp36) was obtained from urine of dog from Guangdong (Figure 1). In female companion animals, the isolated rate of K. pneumoniae complex from cats was significantly higher than that from dogs (p = 0.038). Among samples from cats, most K. pneumoniae complex was isolated from urine [8 of 325 (2.5%); 95% CI, 1.1–4.8]. The most strains were isolated from throat swabs of dogs [6 of 56 (10.7%); 95% CI, 4.0–21.9]. Regarding the regions, majority of K. pneumoniae complex was isolated from Guangdong (Figure 1), where the largest number of samples was collected. The complex isolates were six of 306 (2.0%; 95% CI, 0.7–4.2) from dogs and 10 of 348 (2.9%; 95% CI, 1.4–5.2) from cats (p = 0.451), respectively. No significant difference was found in the prevalence of K. pneumoniae complex from different sources and regions between dogs and cats (p > 0.05) (Table 1).
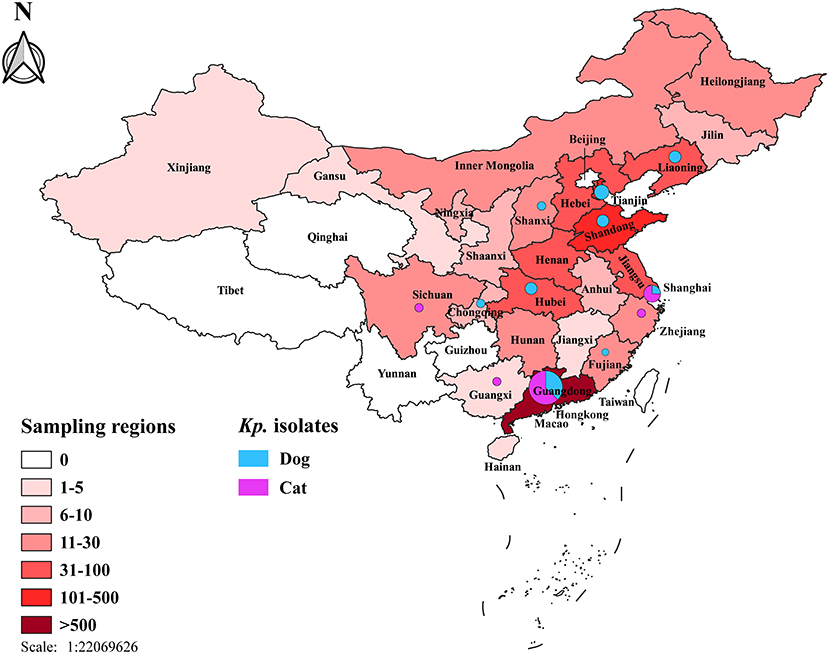
Figure 1. Map of the sampling locations and the prevalence of K. pneumoniae complex isolates among dogs and cats in the 26 regions across China. The color gradation stands for the number of samples in different regions. Numbers and the percentages of K. pneumoniae complex were demonstrated using isometric pie charts.
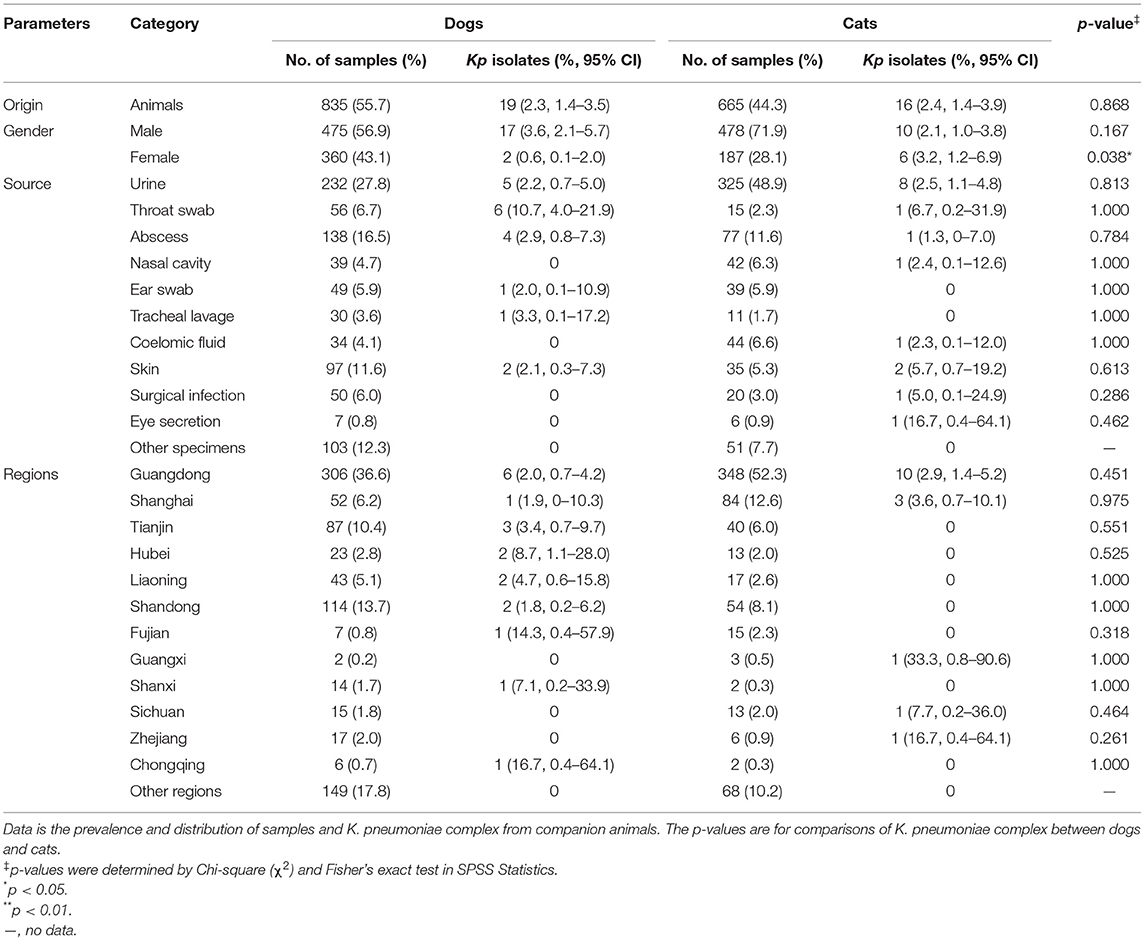
Table 1. The prevalence and distribution of K. pneumoniae complex from dogs and cats.
Of these K. pneumoniae complex from companion animals, resistance detection rate for dogs (1.9%; 95% CI, 1.1–3.1; n = 16/835) was not statistically different from the rate of cats (2.4%; 95% CI, 1.4–3.9; n = 16/665) (p = 0.514). Antimicrobial susceptibility testing of the 35 isolates showed that they were extremely resistant to amoxicillin–clavulanate (82.9%, n = 29/35) and trimethoprim–sulfamethoxazole (77.1%, n = 27/35) (Table 2). There were 29 isolates resistant to more than three categories of antibiotics; the rate of MDR reached an astonishing 82.9%, and the MDR rate was no different between dogs and cats (p = 0.187). MIC50 and MIC90 of strains from cats were generally greater than or equal to dogs. Among them, the MIC50 of amoxicillin and that of cefepime to cat strains were twice that of dog strains; cefotaxime and amikacin were four times, and aztreonam and enrofloxacin were 32 times. However, the florfenicol-MIC50 of dog isolates was 16 times higher than that of cats. Besides, the MIC90 values of ciprofloxacin and amoxicillin–clavulanate of strains from cats were two and four times higher than those of dog strains, respectively. K. pneumoniae complex from cats showed higher resistance frequencies against all antibiotics than that from dogs (p > 0.05) (Table 2). Unfortunately, there was one CRKP isolated from a cat in Zhejiang, which was concurrently resistant to all tested antibiotics except aztreonam and colistin. Also, two strains were resistant (MIC = 32 mg/L and MIC = 8 mg/L) to colistin according to EUCAST.
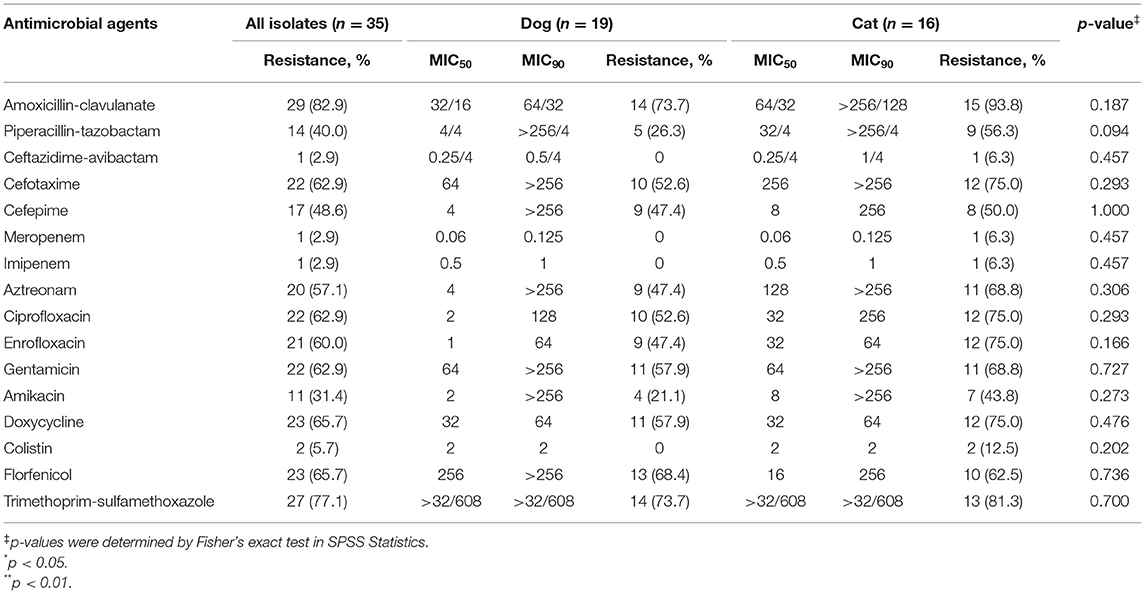
Table 2. Antimicrobial resistance of clinical K. pneumoniae complex isolates from dogs and cats.
Carbapenem resistance gene blaNDM−5 was harbored in one isolate of abscess from cat in Zhejiang, but it was not detected in dogs, and other blaNDM variants were not found (Figure 2). The blaSHV (91.4%, n = 32/35) were the most prevalent resistant genes in companion animals. ESBL gene blaCTX−M was the second commonly present in dogs (22.9%, n = 8/35) and cats (28.6%, n = 10/35), respectively (p = 0.315). There were seven CTX-M genotypes (−3, −14, −15, −27, −55, −65, −122), dominated by blaCTX−M−15 (22.9%, n = 8/35) and blaCTX−M−55 (10.3%, n = 4/35). There was one strain with coexisting blaCTX−M−55 and blaCTX−M−122, which were isolated from tracheal lavage of a dog in Guangdong. The other ESBL and AmpC-containing isolates harbored blaOXA (25.7%, n = 9/35) and blaDHA (5.7%, n = 2/35). The blaLEN (n = 2) and blaOKP (n = 1) were only harbored by K. variicola (Kp34 and Kp87) and K. quasipneumoniae (Kp36). Aminoglycoside-non-susceptible isolates frequently harbored aph(3″)-Ib (45.7%, n = 16/35) and along with aph(6)-Id. The resistance gene aph(3')-Ia of isolates from cats was more significantly (p = 0.042) prevalent than that from dogs. Among the assessed plasmid-mediated quinolone resistance genes, aac(6')Ib-cr, qnrB, qnrS, and oqxAB were 16 (45.7%, n = 16/35), 11 (31.4%, n = 11/35), 11 (31.4%, n = 11/35), and 35 (100%, n = 35/35), respectively. The prevalence of other resistance genes that we obtained was also severely resistant gene; fosA, dfrA, and sul1 were prevalent in isolates (Figure 2).
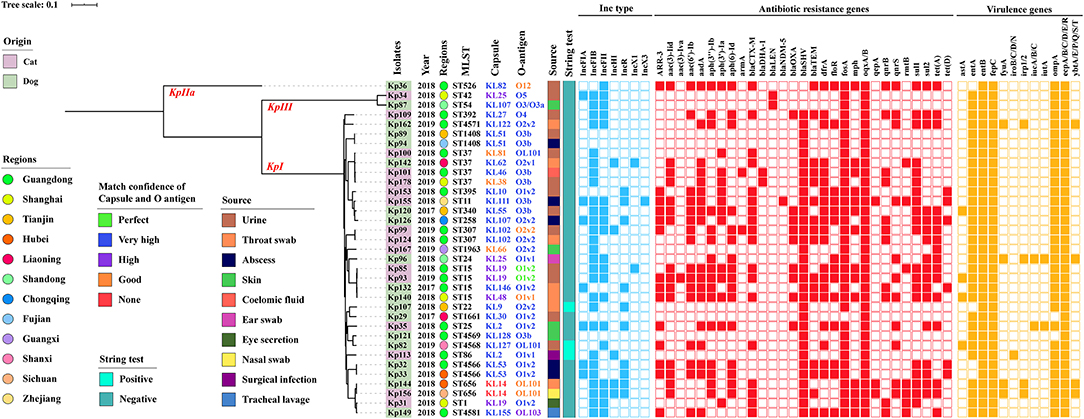
Figure 2. Core genome-based phylogenetic tree and distribution of K. pneumoniae complex phylogroups, origins, isolated years, collected regions, MLST, capsular serotype, LPS O antigen, isolate sources, string test, Inc-type plasmids, antibiotic resistance genes, and virulence genes among isolates from companion animals. The origins, regions, KL/O match confidence, sources, and string test of K. pneumoniae complex are differentiated by color. Inc-type plasmids, antibiotic resistance genes, and virulence genes are denoted by filled squares for the presence and empty squares for absence.
In addition, K. pneumoniae complex harbored 11.4% (n = 4/35) yersiniabactin (ybtA/E/P/Q/S/T), 5.7% (n = 2/35) aerobactin (iucA/B), and 2.9% (n = 1/35) salmochelin (iroB/C/D/N), but other key virulence genes such as colibactin (clb) and regulators of mucoid phenotype genes (rmpA/A2) were not detected. Gene ybt and iuc coexisted in strain Kp96 (ST24), which was from an ear swab of a dog in Shandong (Figure 2). Besides, virulence-associated genes such as enterotoxins (astA, entA/B), ferrienterochelin receptor (fepC), yersiniabactin receptor (fyuA), yersiniabactin biosynthesis (irp1/2), aerobactin receptor (iutA), outer membrane protein (ompA), and common pili (ecpA/B/C/D/E/R) were identified using abricate and demonstrated in Figure 2, and there was no significant difference in virulence-associated genes between dogs and cats (p > 0.05).
Three of the 35 isolates (8.6%) were determined to be hypermucoviscous K. pneumonia (hmKp) by the string test. One of the hmKp that indicated strong virulence had a KL2 capsular serotype and O1v1 LPS serotype, and the remaining two strains were serotype KL9/O2v2 and KL127/OL101 (Figure 2). Isolates covered 26 capsular serotypes and presented diversity distribution. KL19 was the most abundant (8.6%, n = 3/35), accounting for three isolates, followed by KL2, KL25, KL51, KL53, KL102, and KL107, and two K. pneumonia strains with KL14 but match confidence was none (Figures 2, 3A). While O1v2 had the highest percentage of 25.7% (n = 9/35) and then O3b, O2v2, O1v1, and OL101 (Figure 3B). The O antigens of three-string test–positive isolate were O1v1, O2v2, and OL101, respectively. O antigen named OL101 onward was defined on the basis of gene content and was not yet associated with a specific serologically defined O type.
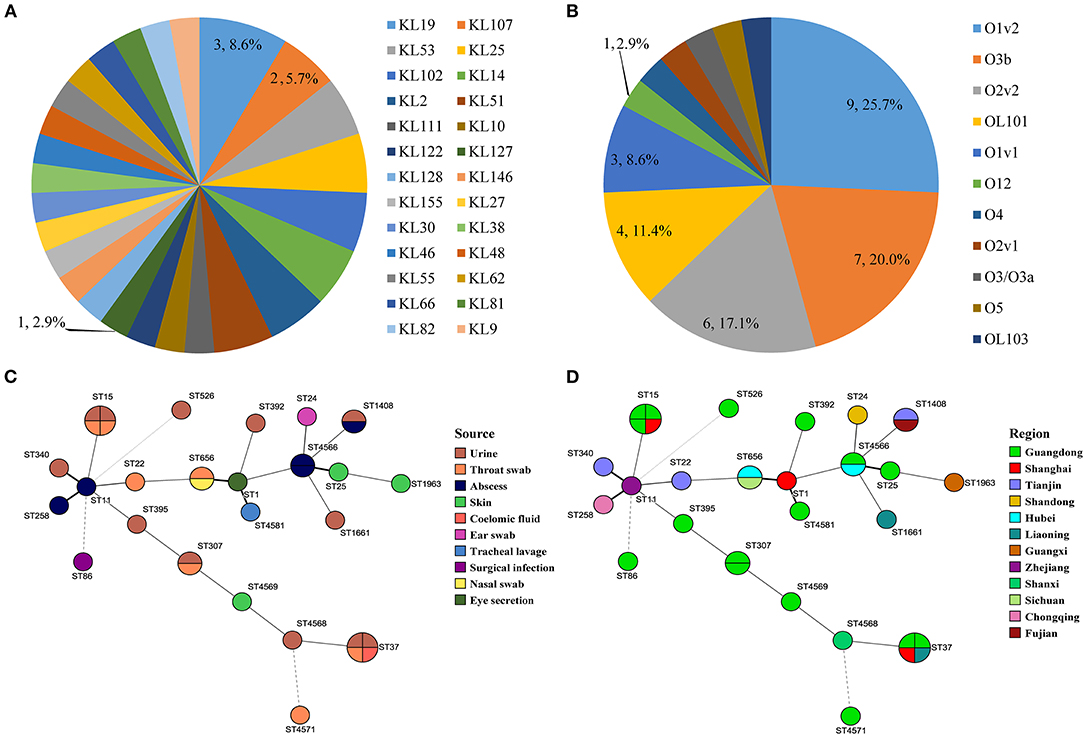
Figure 3. Serotype distribution of capsular serotype and O antigen (A,B). Minimum spanning trees of MLST data (except K. variicola) based on source and region (C,D).
Core–genome phylogenetic tree demonstrated most of the isolates belonged to KpI phylogroup (91.4%, n = 32/35), with only one being KpIIa and two appertained to the KpIII phylogroup (Figure 2). MLST analysis identified 25 different STs among the 35 K. pneumoniae complex; the diversity was similar to the capsular serotypes, and five of them were novel STs (ST4566, ST4568, ST4569, ST4571, ST4581). ST15 and ST37 (11.4%, n = 4/35) were the most abundant accounting for isolates and followed by ST307, ST656, ST1408, and ST4566. The STs of K. variicola Kp34 and Kp87 were ST42 and ST54; they were from the urine of cat and a skin swab of dog, respectively (Figure 2). ST of blaNDM−5-positive K. pneumoniae was ST11. Apart from the KpIIa phylogroup, each lineage comprised strains from dogs and cats. Minimum spanning tree analysis further supported the commonality of K. pneumoniae complex from different sources and regions with the same STs (Figures 3C,D).
The backbone sequences were assembled, and all contigs and gaps were identified by WGS analyses. Through comparison and monitoring, we obtained seven Inc-type plasmids, which were dominant by IncFIB (77.1%, n = 27/35), IncFII (54.3%, n = 19/35), IncR (28.6%, n = 10/35), followed by IncFIA (20%, n = 7/35), IncHI (14.3%, n = 5/35), IncX1 (5.7%, n = 2/35), and IncX3 (2.9%, n = 1/35) (Figure 2). The prevalence of IncFIB plasmid for cat isolates was significantly higher than that for dogs (p = 0.047). In addition, BLASTn results demonstrated that blaNDM−5 was harbored on IncX3-type in plasmid blaNDM−5-positive strains (Kp155), which also contains IncFIA, IncFIB, IncHI, and IncR (Figure 2). Sequences of IncX3 in Kp155 (ST11), which is a source of abscess derived from cat in Zhejiang, contained regions showing >99% nucleotide sequence identity to the reference plasmid pNDM-MGR194 (46253bp, GenBank accession no. KF220657). And blaNDM−5 was included in an insertion sequence (IS) cassette (ΔISAba125-IS5-blaNDM-ble-trpF-dsbC-IS26) compared by ISfinder, which was consistent with blaNDM−5-carrying plasmids originating from human (pQDE2-NDM, MH917280), dog (pP16NDM-502, MN701974), chicken (p1079-NDM, MG825384), and goose (pL65-9, CP034744) (Figure 4).
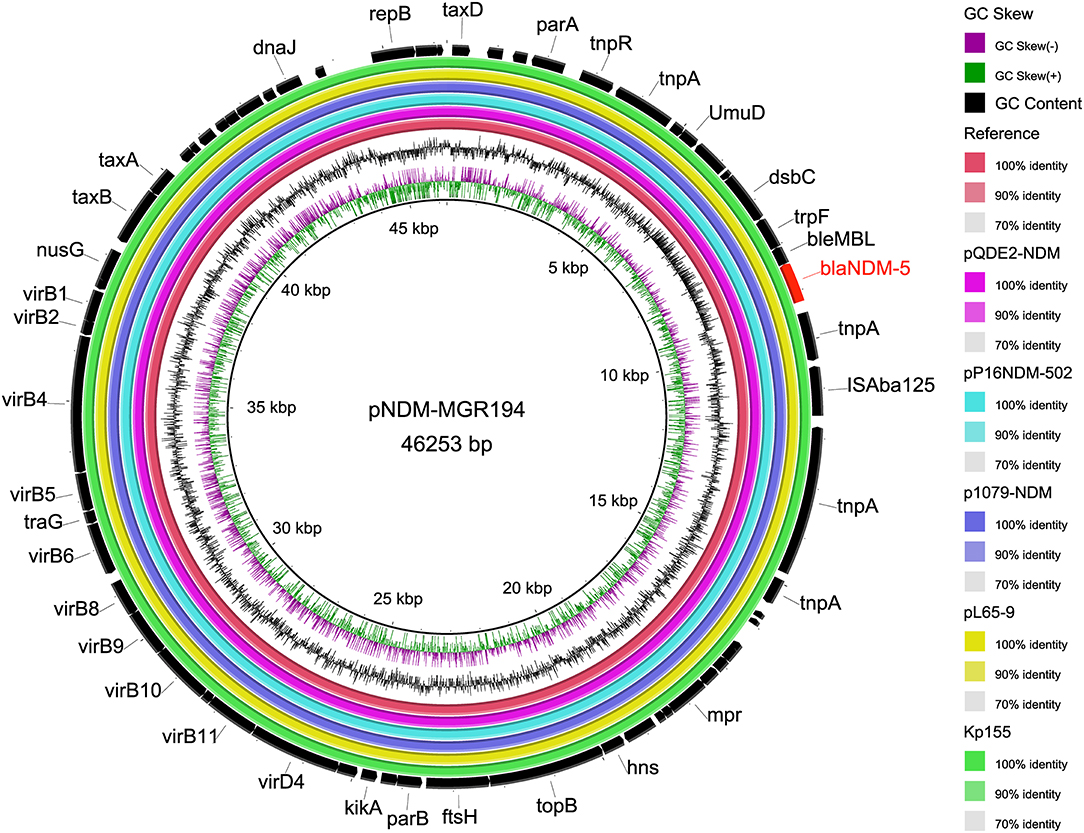
Figure 4. Comparison of whole-genome sequences of plasmid IncX3 carrying blaNDM−5. Each ring represents an isolated strain. The internal ring is the reference sequence of NDM-5 carrying plasmid pNDM-MGR194 (46253bp, GenBank accession no. KF220657), and the outside rings are the other five plasmids, including Kp155 from this study. From inside to outside, the rings were derived from human (pQDE2-NDM, MH917280), dog (pP16NDM-502, MN701974), chicken (p1079-NDM, MG825384), and goose (pL65-9, CP034744).
China Antimicrobial Surveillance Network (CHINET) has well-documented the antimicrobial resistance of humans in China (http://www.chinets.com/). However, investigations on companion animals are still lacking in this regard in China. In the current study, we collected 1,500 clinical samples of companion animals for the isolation of K. pneumoniae complex and investigated the prevalence of antibiotic resistance, virulence, and molecular typing through whole-genome analysis. Thirty-five K. pneumoniae (2.3%) complex was identified from 1,500 samples, which was a lower isolation rate compared with Italy (3.53%) (25). There was no significant difference among animals, 19 from dogs and 16 from cats; the isolation rate of K. pneumoniae complex from cats was significantly higher than that from dogs in female animals. But the rate of MDR, 82.9%, was higher than that reported in Singapore (50%) (26). MIC50 and MIC90 of antibiotics to K. pneumoniae complex from cats were generally greater than or equal to that from dogs, suggesting that the resistance of feline strains was more severe than that of dogs. The results were consistent with previous investigations in Iberian Peninsula (27) and China (28). K. pneumoniae complex from cats showed no significant difference (p > 0.05) against all antibiotics compared with that from dogs, which was consistent with the previous study in South Korea (29). In our study, the resistance of amoxicillin–clavulanate and trimethoprim–sulfamethoxazole were 82.9% and 77.1%, all generally higher than those reported in Portugal (30). One CRKP was resistant to meropenem and imipenem and harbored carbapenemase gene blaNDM−5, which has been reported in humans (31) and other animals (32), but rarely detected in companion animals.
We found a higher prevalence of ESBLs in K. pneumoniae complex clinical isolates (57.1%, n = 20/35), compared with those from companion animals in Japan (34.8%) (1), Italy (21.4%) (25), Germany, and other European countries (7.6%) (33). In this study, ESBLs were CTX-M-genotypes (−3, −14, −15, −27, −55, −65, −122) and SHV-genotypes (−41, −42), which was not quite the same as that in previous reports (34). Among CTX-M genotypes, which were dominant by blaCTX−M−15 (22.9%, n = 8/35) and blaCTX−M−55 (10.3%, n = 4/35), there was one strain with coexisting blaCTX−M−55 and blaCTX−M−122. In particular, the CTX-M ESBL genes were widely present in the field of human medicine, and the CTX-M-15–producing K. pneumoniae complex was the most frequently detected genotype associated with extended-spectrum antibiotic resistance in humans and animals (35). Because of the close contact between companion animals and humans, the genotypes of K. pneumoniae complex from companion animals in this study were similar to those of humans in China. Otherwise, blaSHV (91.4%) was the most prevalent resistant genes, which were constituted by SHV-187 (28.6%, n = 10/35), −106 (17.1%, n = 6/35), −110 (14.3%, n = 5/35), −182 (11.4%, n = 4/35), −145 (5.7%, n = 2/35), and SHV-11,−28,−41,−42, and−62 (each 2.9%, 1/35) in our study. The other ESBL and AmpC-containing isolates harbored blaOXA (25.7%, n = 9/35) and blaDHA (5.7%, n = 2/35). The blaLEN (n = 2) and blaOKP (n = 1) were only harbored by K. variicola (Kp34 and Kp87) and K. quasipneumoniae (Kp36). K. variicola has been widely recognized as an important opportunistic human pathogen commonly involved in hospital-acquired infections; multiple antibiotic resistance genes have been shown to exist, such as clinically relevant resistance determinants such as blaCTX−M, blaDHA, and blaLEN. The blaLEN gene corresponds to an intrinsic chromosomal β-lactamase in the K. variicola genome (36). Meanwhile, blaDHA has been indicated to cause the resistance of K. pneumoniae complex in companion animals from a veterinary hospital in Switzerland (37). Population diversity studies have shown that K. pneumoniae is phylogenetically closely related to K. variicola and K. quasipneumoniae (38). The blaOKP β-lactamases, closely related to blaSHV, and the OKP type enzyme were also clearly found in the phylogenetic group KpII of K. quasipneumoniae (39). Some variant K. variicola has been identified from multiple sources, including environments; humans; animals such as dogs, birds, monkeys, and cattle (mastitis); and plants such as coriander (food supply). As potential pathogens of zoonotic, K. variicola from companion animals could be transmitted to humans (16).
Hypervirulence and hypermucoviscosity are two different K. pneumoniae phenotypes; they could predict positive value by the string test and molecular markers such as the virulence genes ybt, clb, iuc, iro, rmpA, and rmpA2 (40). Three of (8.6%, n = 3/35) the 35 isolates were determined to be hypermucoviscous by string test in our study. Kleborate is a tool to screen genome assemblies of K. pneumoniae complex and the complex for integrative conjugative element–associated virulence loci (ybt, clb) and virulence plasmid–associated loci (iro, iuc, rmpA, rmpA2) (41). In this study, one isolate of ST24 harbored ybt and iuc, which derived an ear swab from a dog in Shandong, and its virulence gene score was evaluated by Kleborate as four. Also, one other strain scored 3 because it harbored iuc, and three isolates appeared ybt, so it was scored 1. The capsular polysaccharide is located outside the outer membrane; the most common hvKp capsule types are K1, K2, K5, K20, K54, and K57, of which K1 and K2 account for ~70% of hvKp isolates. Otherwise, hvKp strain also has the O antigen, which is part of LPS. K1 and K2 capsule types are usually related to the O1 O antigen type (42). In the current study, capsular presented diversity distribution and covered 26 serotypes, which were predominated by KL19. One of the hmKp that indicated strong virulence had a KL2 capsular serotype and O1v1 LPS serotype. While LPS serotype O1v2 had the highest percentage of 25.7%, the O antigens of three-string test–positive isolate were O1v1, O2v2, and OL101, respectively.
Global problem clones have been isolated from a series of animals, such as ST11 in poultry, ST15 in companion animals, ST23 in non-human primates and horses, and ST25 in pigs (15). Not surprisingly, ST15 and ST37 (11.4%, n = 4/35) were the most abundant accounting for isolates in this study. Previous researches on dairy farms in the United States (43), canals or farms in Thailand (44), and farms in the North of England (45) had revealed a large amount of diversity between clinical isolates, and non-human samples. MLST analysis identified 25 different STs among the 35 K. pneumoniae complexes in our research, which diversity was similar to the capsular serotypes, and five of them were novel STs (ST4566, ST4568, ST4569, ST4571, ST4581). The STs of K. variicola Kp34 and Kp87 from companion animals were ST42 and ST54, respectively. ST42 was mainly prevalent in humans of Mexico; ST54 was dominantly found in the United States and Vietnam according to previous research (22). In China, K. variicola from both humans and plants has been described, with ST65 and ST92 corresponding to human isolates described in different reports (22). However, the report of K. variicola from the source of companion animals is the first. CRKP or MDR ST11 K. pneumoniae harboring KL64 or KL47 and virulence plasmids carrying iuc (or rmpA2) is widely spread in China (46). Coincidentally, the ST of plasmid IncX3-blaNDM−5-positive and MDR K. pneumoniae was ST11 in our results. Otherwise, some researchers believe that hvKP could accumulate plasmids carrying virulence and resistance genes, which can continuously enhance its resistance to major antibiotics (47). Inc-type plasmids were analyzed and suggested that there was some correlation between MDR and plasmid type. These findings indicated that the variants of K. pneumoniae complex resistance genes, virulence genes, and mobile plasmid elements are not limited to certain hosts, emphasizing the need for coordinated control in the concept of One Health.
We found a high prevalence of MDR K. pneumoniae complex isolates from sick dogs and cats in different regions of China. The abuse of combination medications was likely contributed to that, and especially widespread use of amoxicillin–clavulanate and trimethoprim–sulfamethoxazole in the veterinary hospital. These strains harbored blaSHV, blaCTX−M, and blaNDM, and most promoted an MDR profile. Meanwhile, the emergence of hvKP and epidemic clones has increased the risks of veterinarians. Diversity analysis of the core–genomes and STs of this clinical K. pneumoniae complex from different sources and regions suggested that the dissemination of Inc-type plasmids has broad reservoirs in K. pneumoniae complex. Relevant measures must be formulated to suppress or block transmission of high-risk K. pneumoniae complex clonal lineages to ensure the safety of companion animal practitioners and public health.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA685900.
The animal study was reviewed and approved by China Agricultural University Animal Ethics Committee document (No. AW01017102-2). Written informed consent was obtained from the owners for the participation of their animals in this study.
ZX and JW: conceived and designed study, collected, complied, and analyzed data. LZ, HD, and HZ: statistical analyses. YS and QA: collected the clinical samples. ZZ and JW: drafted and edited manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Beijing Science and Technology Planning Project (Z171100001517008), Project of Science and Technology Innovation Fund of Shanxi Agricultural University (2021BQ70 and 2021BQ06), Central Funds Guiding the Local Science and Technology Development In Shanxi Province (YDZJSX2021A034), Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20210012), Project of Scientific Research for Excellent Doctors, Shanxi Province, China (SXBYKY2021047), and Research Fund (Clinical Diagnosis and Treatment of Pet) for Young College Teachers in Ruipeng Commonweal Foundation (RPJJ2020021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Xiaokun Deng for her help to this study in the process of collecting samples. We thank the team of curators of the Institut Pasteur MLST system (Paris, France) for importing novel alleles, profiles, and isolates at: http://bigsdb.web.pasteur.fr.
1. Harada K, Shimizu T, Mukai Y, Kuwajima K, Sato T, Usui M, et al. Phenotypic and molecular characterization of antimicrobial resistance in Klebsiella spp. isolates from companion animals in Japan: clonal dissemination of multidrug-resistant extended-spectrum beta-lactamase-producing Klebsiella pneumoniae. Front Microbiol. (2016) 7:1021. doi: 10.3389/fmicb.2016.01021
2. Martin RM, Cao J, Brisse S, Passet V, Wu W, Zhao L, et al. Molecular epidemiology of colonizing and infecting isolates of Klebsiella pneumoniae. mSphere. (2016) 1:16. doi: 10.1128/mSphere.00261-16
3. Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. (2013) 11:297–308. doi: 10.1586/eri.13.12
4. Lee CR, Lee JH, Park KS, Jeon JH, Kim YB, Cha CJ, et al. Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front Cell Infect Microbiol. (2017) 7:483. doi: 10.3389/fcimb.2017.00483
5. Wyres KL, Holt KE. Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. (2016) 24:944–56. doi: 10.1016/j.tim.2016.09.007
6. Lloyd DH. Reservoirs of antimicrobial resistance in pet animals. Clin Infect Dis. (2007) 45(Suppl.2):S148–52. doi: 10.1086/519254
7. Diancourt L, Passet V, Verhoef J, Grimont PA, Brisse S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol. (2005) 43:4178–82. doi: 10.1128/JCM.43.8.4178-4182.2005
8. Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. (2006) 60:561–88. doi: 10.1146/annurev.micro.59.030804.121325
9. Long SW, Linson SE, Ojeda Saavedra M, Cantu C, Davis JJ, Brettin T, et al. Whole-genome sequencing of human clinical Klebsiella pneumoniae isolates reveals misidentification and misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. mSphere. (2017) 2:17. doi: 10.1128/mSphereDirect.00290-17
10. Rodrigues C, Passet V, Rakotondrasoa A, Brisse S. Identification of Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola and related phylogroups by MALDI-TOF mass spectrometry. Front Microbiol. (2018) 9:3000. doi: 10.3389/fmicb.2018.03000
11. Brisse S, Passet V, Haugaard AB, Babosan A, Kassis-Chikhani N, Struve C, et al. wzi Gene sequencing, a rapid method for determination of capsular type for Klebsiella strains. J Clin Microbiol. (2013) 51:4073–8. doi: 10.1128/JCM.01924-13
12. Wyres KL, Wick RR, Gorrie C, Jenney A, Follador R, Thomson NR, et al. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb Genom. (2016) 2:e000102. doi: 10.1099/mgen.0.000102
13. Wick RR, Heinz E, Holt KE, Wyres KL. Kaptive web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella genomes. J Clin Microbiol. (2018) 56:18. doi: 10.1128/JCM.00197-18
14. Rodrigues C, Passet V, Rakotondrasoa A, Diallo TA, Criscuolo A, Brisse S. Description of Klebsiella africanensis sp. nov, Klebsiella variicola subsp tropicalensis subsp nov and Klebsiella variicola subsp variicola subsp nov. Res Microbiol. (2019) 170:165–70. doi: 10.1016/j.resmic.2019.02.003
15. Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. (2020) 18:344–59. doi: 10.1038/s41579-019-0315-1
16. Rodriguez-Medina N, Barrios-Camacho H, Duran-Bedolla J, Garza-Ramos U. Klebsiella variicola: an emerging pathogen in humans. Emerg Microbes Infect. (2019) 8:973–88. doi: 10.1080/22221751.2019.1634981
17. Morales-Leon F, Opazo-Capurro A, Caro C, Lincopan N, Cardenas-Arias A, Esposito F, et al. Hypervirulent and hypermucoviscous extended-spectrum beta-lactamase-producing Klebsiella pneumoniae and Klebsiella variicola in Chile. Virulence. (2021) 12:35–44. doi: 10.1080/21505594.2020.1859274
18. Garza-Ramos U, Silva-Sanchez J, Catalan-Najera J, Barrios H, Rodriguez-Medina N, Garza-Gonzalez E, et al. Draft genome sequence of a hypermucoviscous extended-spectrum-beta-lactamase-producing Klebsiella quasipneumoniae subsp. Similipneumoniae. Clin Isolate Genome Announc. (2016) 4:16. doi: 10.1128/genomeA.00475-16
19. Su J-H, Zhu Y-H, Ren T-Y, Guo L, Yang G-Y, Jiao L-G, et al. Distribution and antimicrobial resistance of salmonella isolated from pigs with diarrhea in China. Microorganisms. (2018) 6:40117. doi: 10.3390/microorganisms6040117
20. Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. (2013) 4:107–18. doi: 10.4161/viru.22718
21. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
22. Barrios-Camacho H, Aguilar-Vera A, Beltran-Rojel M, Aguilar-Vera E, Duran-Bedolla J, Rodriguez-Medina N, et al. Molecular epidemiology of Klebsiella variicola obtained from different sources. Sci Rep. (2019) 9:10610. doi: 10.1038/s41598-019-46998-9
23. Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. (2014) 15:524. doi: 10.1186/s13059-014-0524-x
24. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. (2011) 12:402. doi: 10.1186/1471-2164-12-402
25. Donati V, Feltrin F, Hendriksen RS, Svendsen CA, Cordaro G, Garcia-Fernandez A, et al. Extended-spectrum-beta-lactamases, AmpC beta-lactamases and plasmid mediated quinolone resistance in Klebsiella spp. from companion animals in Italy. PLoS ONE. (2014) 9:e90564. doi: 10.1371/journal.pone.0090564
26. Hartantyo SHP, Chau ML, Fillon L, Ariff A, Kang JSL, Aung KT, et al. Sick pets as potential reservoirs of antibiotic-resistant bacteria in Singapore. Antimicrob Resist Infect Control. (2018) 7:106. doi: 10.1186/s13756-018-0399-9
27. Li Y, Fernandez R, Duran I, Molina-Lopez RA, Darwich L. Antimicrobial resistance in bacteria isolated from cats and dogs from the Iberian Peninsula. Front Microbiol. (2020) 11:621597. doi: 10.3389/fmicb.2020.621597
28. Zhang Z, Lei L, Zhang H, Dai H, Song Y, Li L, et al. Molecular investigation of Klebsiella pneumoniae from clinical companion animals in Beijing, China, 2017-2019. Pathogens. (2021) 10:30271. doi: 10.3390/pathogens10030271
29. Hong JS, Song W, Park HM, Oh JY, Chae JC, Shin S, et al. Clonal spread of extended-spectrum cephalosporin-resistant enterobacteriaceae between companion animals and humans in South Korea. Front Microbiol. (2019) 10:1371. doi: 10.3389/fmicb.2019.01371
30. Marques C, Menezes J, Belas A, Aboim C, Cavaco-Silva P, Trigueiro G, et al. Klebsiella pneumoniae causing urinary tract infections in companion animals and humans: population structure, antimicrobial resistance and virulence genes. J Antimicrob Chemother. (2019) 74:594–602. doi: 10.1093/jac/dky499
31. Krishnaraju M, Kamatchi C, Jha AK, Devasena N, Vennila R, Sumathi G, et al. Complete sequencing of an IncX3 plasmid carrying blaNDM-5 allele reveals an early stage in the dissemination of the blaNDM gene. Indian J Med Microbiol. (2015) 33:30–8. doi: 10.4103/0255-0857.148373
32. Wang Y, Zhang R, Li J, Wu Z, Yin W, Schwarz S, et al. Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry production. Nat Microbiol. (2017) 2:16260. doi: 10.1038/nmicrobiol.2016.260
33. Ewers C, Stamm I, Pfeifer Y, Wieler LH, Kopp PA, Schonning K, et al. Clonal spread of highly successful ST15-CTX-M-15 Klebsiella pneumoniae in companion animals and horses. J Antimicrob Chemother. (2014) 69:2676–80. doi: 10.1093/jac/dku217
34. Poirel L, Nordmann P, Ducroz S, Boulouis HJ, Arne P, Millemann Y. Extended-spectrum beta-lactamase CTX-M-15-producing Klebsiella pneumoniae of sequence type ST274 in companion animals. Antimicrob Agents Chemother. (2013) 57:2372–5. doi: 10.1128/AAC.02622-12
35. Dorado-Garcia A, Smid JH, Van Pelt W, Bonten MJM, Fluit AC, Van Den Bunt G, et al. Molecular relatedness of ESBL/AmpC-producing Escherichia coli from humans, animals, food and the environment: a pooled analysis. J Antimicrob Chemother. (2018) 73:339–47. doi: 10.1093/jac/dkx397
36. Fonseca EL, Ramos ND, Andrade BG, Morais LL, Marin MF, Vicente AC. A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn Microbiol Infect Dis. (2017) 87:315–7. doi: 10.1016/j.diagmicrobio.2017.01.005
37. Wohlwend N, Endimiani A, Francey T, Perreten V. Third-generation-cephalosporin-resistant Klebsiella pneumoniae isolates from humans and companion animals in Switzerland: spread of a DHA-producing sequence type 11 clone in a veterinary setting. Antimicrob Agents Chemother. (2015) 59:2949–55. doi: 10.1128/AAC.04408-14
38. Cuenod A, Wuthrich D, Seth-Smith HMB, Ott C, Gehringer C, Foucault F, et al. Whole-genome sequence-informed MALDI-TOF MS diagnostics reveal importance of Klebsiella oxytoca group in invasive infections: a retrospective clinical study. Genome Med. (2021) 13:150. doi: 10.1186/s13073-021-00960-5
39. Haeggman S, Lofdahl S, Paauw A, Verhoef J, Brisse S. Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother. (2004) 48:2400–8. doi: 10.1128/AAC.48.7.2400-2408.2004
40. Catalan-Najera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp. phenotypes? Virulence. (2017) 8:1111–23. doi: 10.1080/21505594.2017.1317412
41. Lam MMC, Wick RR, Wyres KL, Gorrie CL, Judd LM, Jenney AWJ, et al. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb Genom. (2018) 4:196. doi: 10.1099/mgen.0.000196
42. Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. (2019) 32:19. doi: 10.1128/CMR.00001-19
43. Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci USA. (2015) 112:E3574–81. doi: 10.1073/pnas.1501049112
44. Runcharoen C, Moradigaravand D, Blane B, Paksanont S, Thammachote J, Anun S, et al. Whole genome sequencing reveals high-resolution epidemiological links between clinical and environmental Klebsiella pneumoniae. Genome Med. (2017) 9:6. doi: 10.1186/s13073-017-0397-1
45. Ludden C, Moradigaravand D, Jamrozy D, Gouliouris T, Blane B, Naydenova P, et al. A one health study of the genetic relatedness of Klebsiella pneumoniae and their mobile elements in the East of England. Clin Infect Dis. (2020) 70:219–26. doi: 10.1093/cid/ciz174
46. Gu DX, Dong N, Zheng ZW, Lin D, Huang M, Wang LH, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. (2018) 18:37–46. doi: 10.1016/S1473-3099(17)30489-9
Keywords: Klebsiella pneumoniae complex, dog, cat, whole-genome sequence, multidrug resistance
Citation: Zhang Z, Zhang L, Dai H, Zhang H, Song Y, An Q, Wang J and Xia Z (2022) Multidrug-Resistant Klebsiella pneumoniae Complex From Clinical Dogs and Cats in China: Molecular Characteristics, Phylogroups, and Hypervirulence-Associated Determinants. Front. Vet. Sci. 9:816415. doi: 10.3389/fvets.2022.816415
Received: 16 November 2021; Accepted: 31 January 2022;
Published: 10 March 2022.
Edited by:
Patrick Rik Butaye, Ross University School of Veterinary Medicine, Saint Kitts and NevisReviewed by:
Ulises Garza-Ramos, National Institute of Public Health, MexicoCopyright © 2022 Zhang, Zhang, Dai, Zhang, Song, An, Wang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianzhong Wang, d2p6QHN4YXUuZWR1LmNu; amlhbnpob25nd2FuZ0BjYXUuZWR1LmNu; Zhaofei Xia, emhhb2ZlaXhpYWNhdUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.