 Lulu Han
Lulu Han Hui Tao1
Hui Tao1 Qiyu Diao
Qiyu Diao Kai Cui
Kai Cui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 25 April 2022
Sec. Animal Nutrition and Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.809188
This article is part of the Research Topic Nutritional Management for the Development and Gut Health of Young Ruminants View all 8 articles
Early feeding regime has a substantial lifelong effect on lambs and weaning ewe's milk can lead to the intestinal injury of lambs. To explore the molecular regulatory mechanism of intestinal injury of lambs under weaning stress, the jejunum was conducted transcriptome and then integrated analyzed with our previous proteome data. A total of 255 upregulated genes and 285 downregulated genes were significantly identified. These genes showed low overlapping with differentially expressed proteins identified by isobaric tags for relative and absolute quantification (iTRAQ). However, according to their functions, the differentially expressed genes (DEGs) and proteins with the same expression trend were enriched for the similar Gene Ontology (GO) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, such as intestinal lipid absorption, urea cycle, peroxisome proliferator-activated receptor (PPAR) signaling pathway, and ferroptosis. Furthermore, the DEGs, including FABP2, ACSL3, APOA2, APOC3, and PCK1, might play essential roles in intestinal lipid absorption and immune response through the PPAR signaling pathway and ferroptosis. This study could provide new insights into early lamb breeding at the molecular level.
In the recent years, house feeding and intensive breeding have become the primary trend of the sheep industry due to the limitations of resources and the environment. To improve the utilization rate of ewes, the implementation of an artificial milk replacer feeding program to substitute ewe lactation has become a common practice of lamb breeding in many countries (1, 2). However, weaning is the most severe stimulating factor for the animals during early development, which is shown as the decreased feed intake, low nutrition absorption, aberrant mucosal immunity, and higher mortality (3–5). Young ruminants' intestinal histology and function would significantly change after the colonization of residential microorganisms, invasion of exogenous pathogens, and deletion of maternal antibodies (6–8), closely related to their growth, inflammatory response, and susceptibility to some diseases (5).
The small intestine, a critical multifunctional organ, plays crucial roles in digesting and absorbing nutrients, regulating immune response, and secreting various enzymes and immunoglobulins (8, 9). The intestinal mucosa is the largest interface between the host and the external environment, so intestinal cells are continuously exposed to the coexistence of symbiotic bacteria, diets, pathogenic microorganisms, and metabolites. For this reason, the intestinal epithelium and lamina propria form a multilayer and complex barrier mechanism to maintain intestinal homeostasis. However, the structure and function of the intestine are affected by weaning, which is essential for lambs whose digestive and immune systems are not yet mature (10). With the intestinal microflora disturbance, the metabolic imbalance, and the inflammatory reaction induced by pathogen invasion after weaning, the homeostasis of the intestine is broken. Therefore, it is urgent to understand the molecular mechanism of lamb intestinal injury caused by weaning ewe's milk, which could help us to draw better methods to minimize this effect.
Due to the high throughput and quantifiable advantage, RNA sequencing (RNA-seq) could detect the bulk differentially expressed genes (DEGs) (11), widely used to identify gene expressions and modulated pathways. Therefore, in this study, RNA-seq was performed to analyze the transcriptome of lamb jejunum. After screening the differentially expressed genes, we annotated their function to investigate the regulation of intestinal injury under weaning stress. At the same time, our previous proteomic data were integrated with the transcriptomic data to determine the same Gene Ontology (GO) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway items enriched in the two data sets. Those results provide more basis for exploring the molecular mechanism of lamb intestinal injury induced by weaning ewe's milk, which could help us to better understand intestinal function regulation after weaning.
The same animals were used according to our previous study (6). In summary, eight pairs of twin neonatal ram Hu lambs born on the same day with almost the same body weight (2.75 ± 0.2 kg) were randomly divided into the two groups. One group was artificially reared (AR) with milk replacer fed four times a day after sucking colostrum for 3 days and the other group was ewe-reared (ER) during the entire 15 days experiment. Each pair of twin samples could be monozygotic and dizygotic twins and siblings to reduce the influence of inherent genes in exploration of the differences of gene expression after AR or ER feeding procedures. The commercial milk replacer was acquired from Precision Animal Nutrition Research Center (Beijing, China; CP: 25.08%, DE: 18.38%, EE: 11.18%, Ash: 5.29%, Ca: 1.13%, and P: 0.51% of DM) and the feed amount of milk replacer was adjusted in direct accordance with 2% of the lamb's body weight.
On 15 days of the experiment, three pairs of healthy twins with similar weight and same gender were slaughtered with a compressed air pistol to cause a cerebral concussion, followed by exsanguination by cutting the jugular and carotid veins. After slaughter, the middle part of the jejunum with 2 cm was sampled quickly and frozen in liquid nitrogen.
The frozen jejunum samples were ground with liquid nitrogen and then weighted 1.0 g for each sample. Total RNA of each jejunum was extracted and purified using the Total RNA Extraction Kit (Invitrogen, Carlsbad, California, USA), according to the manufacturer's standard protocol. RNA integrity number (RIN) and accurate yield were initially measured by spectrophotometry on the Nanodrop 1000 (Nanodrop Technologies, Wilmington, Delaware, USA). Only high-quality RNAs (RIN > 8 and RNA yield > 2 μg) could be advanced to the next step. Individual complementary DNA (cDNA) libraries were generated from 0.5 μg of total RNA per sample using the Illumina TruSeq RNA Library Preparation Kit version 2 (Illumina Incorporation, San Diego, California, USA). Then, sequencing libraries were sequenced on the Illumina HiSeq 2500 (Illumina Incorporation) and generated 100 bp paired-end reads.
The raw reads were filtered and trimmed using Trimmomatic software (12) to remove low-quality reads and adapter sequences. In other words, the reads whose number of low-quality (sQ ≤ 5) bases contained in a single-ended sequencing read exceeded 50% of the total number of bases and N content > 10% were removed. The generated clean reads were aligned against sheep genome reference sequences (OARv4.0) by applying STAR aligner (13) with default parameters. Mapped reads were sorted by the Sequence Alignment/Map tools (SAM tools) (14) and then counted by gene using featureCounts (15) embedded in Subread (16).
Gene expression levels in each library were normalized to fragments per kilobase of exon per million mapped reads (FPKM). The significant DEGs in pairwise comparison between the AR and ER groups were determined using the DESeq2 R package (17). The selected model for identifying the DEGs incorporated group and maternal effect (design = ~ group effect + maternal effect). The maternal effect is mainly related to the lamb individuals and were collected from different ewes, which could infect the performance during lactation. Wald inferences tests were used to assign false discovery rate (FDR)-adjusted p-values (q-values) for each gene in each pairwise contrast. Both the significance level (q-value < 0.05) and magnitude of expression change [|log2(fold-change)| ≥ 1] were used to select the significant DEGs. The functional analysis, including the KEGG and GO, for DEGs was used ClueGo (version 2.8.5) in Cytoscape (version 3.7.2) (18). All the pathways were shown with a q-value < 0.05 and the kappa score was used to define term–term interrelation and the functional groups based on shared genes between the terms. Meanwhile, the network specificity was set to “medium.” Finally, STRING version 11 (http://string-db.org/) was used to predict the protein–protein interactions (PPIs) of DEGs. A PPI network was drawn using Cytoscape and the cytoHubba application was used to identify the top 10 hub genes (19, 20). The isobaric tags for relative and absolute quantification (iTRAQ) data of the same samples has been published in the previous study (6). In this integrative study, DEGs were identified by RNA-seq. In addition, the differentially expressed proteins (DEPs) from iTRAQ were compared and overlapped for consistent results. In brief, direct and indirect correlations were combined to get a complete relationship between the two omics. The specific operation methods are as follows: geninfo identifier (GI) of the proteome is mapped to gene symbol through the gene2accession in the National Center for Biotechnology Information (NCBI) and then compared with the gene number of the transcriptome. Then, for the unrelated genes, the database (transcript sequence of transcriptome) and query (identified protein sequence) were used for the Basic Local Alignment Search Tool (BLAST) sequence alignment, the e-value threshold was selected as 1e−5, and the identity (sequence similarity) was guaranteed to be more than 95%. Next, the Pearson correlation method was used to calculate the correlation of sample pair ratio in transcriptome and proteome. To show the correlation between two omics in detail, we further classified the DEGs revealed by RNA-seq and the DEPs identified by the previous report minutely via the threshold of expression difference: fold-change ≥ 1.5 in transcriptome and fold-change ≥ 1.2 in the proteome.
RNA-seq produced more than 18,195,848 raw reads (Table 1). After data filtering, 18,136,753–27,932,670 clean reads were generated to quantify transcripts and single nucleotide polymorphism (SNP) calling. The Guanine and cytosine (GC) content in the libraries ranged from 48.24 to 50.56%. The six samples had at least 91.91% reads with ≥Q20 and 85.81% reads with ≥Q30. The majority of reads in each library were mapped to the sheep genome reference sequences and the average mapping rates were 78.96 and 77.15% for the AR and ER groups, respectively. A total of 18,590 transcripts and 396,743 SNPs were identified in the samples.
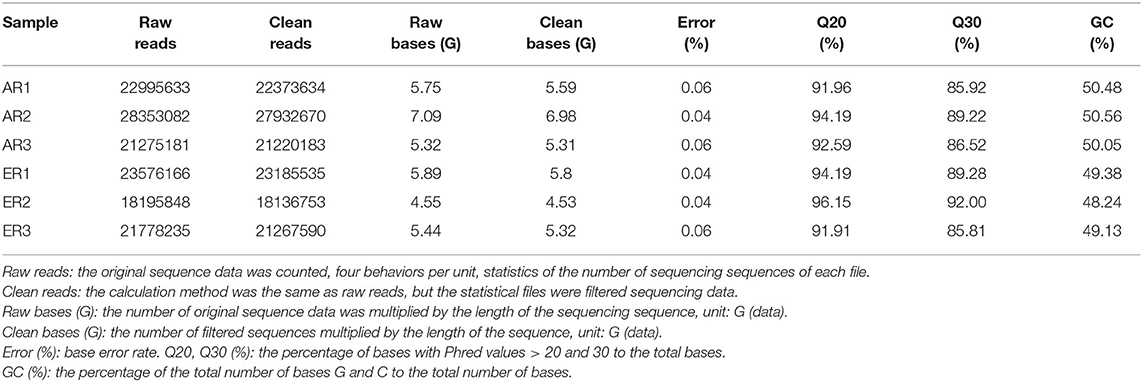
Table 1. The quality of data output.
The principal component analysis (PCA) of all the genes from intestinal samples showed separations between the two groups (Figure 1). Besides, samples were significantly divided into the two groups as experimental design (Supplementary Figure S1A). They were clustered into three branches as their ewe origination (Supplementary Figure S1B).
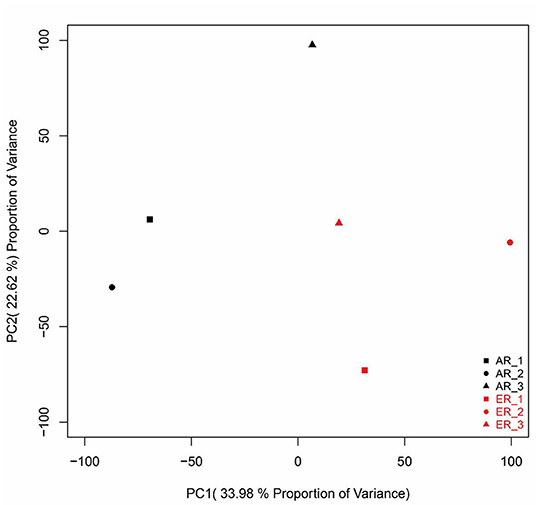
Figure 1. The principal component analysis (PCA) analysis of gene expressions in the intestinal samples. The black patterns represent the artificially reared (AR) group and the red patterns represent the ewe reared (ER) group.
After annotation, 255 upregulated DEGs and 285 downregulated DEGs in the AR group were compared with the ER group (Figures 2A,B). In this study, the genes related to lipid metabolic processes, defense response, regulation of immune response, and intestinal epithelium morphology verified by quantitative reverse transcription-PCR (qRT-PCR) in the proteome were consistent with their expression trends in the previous study (6). The GO enrichment analysis was carried out to explore the biological mechanisms underlying the two different feeding regimes. The 9 GO terms were enriched, including five biological processes (BPs) and four cellular components (CCs) on the DEGs (Figure 3). The significantly enriched BP terms were mainly related to the small molecule catabolic process, hexose transmembrane transporter activity, chemotaxis, and leukocyte activation. The enriched CC terms considerably were primarily involved in transaminase activity, extracellular space, extracellular matrix, extracellular organelle, and plasma lipoprotein particle.
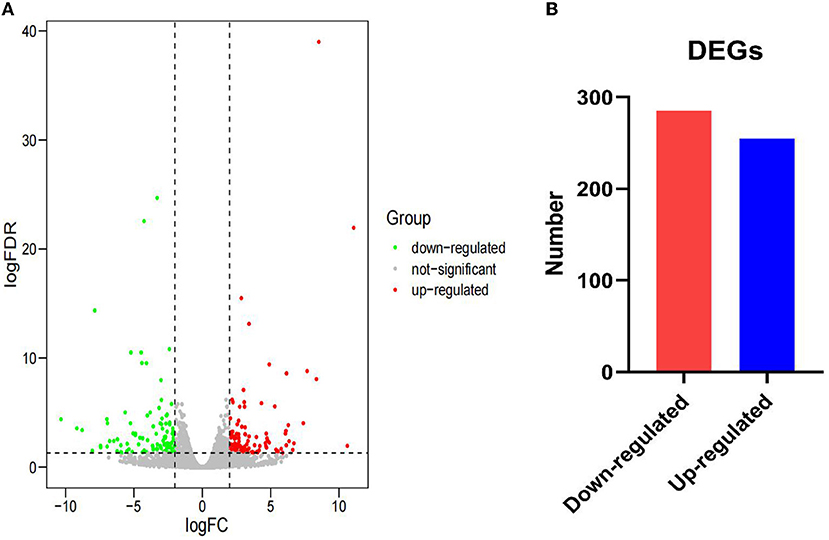
Figure 2. Gene expression between the AR and ER groups. (A) The expression of differentially expressed genes (DEGs) between the two groups. The red dots indicate upregulated DEGs and the green dots indicate downregulated DEGs. The gray dots reveal that there is no difference in the expression of genes between the two groups. (B) Number of significantly DEGs.
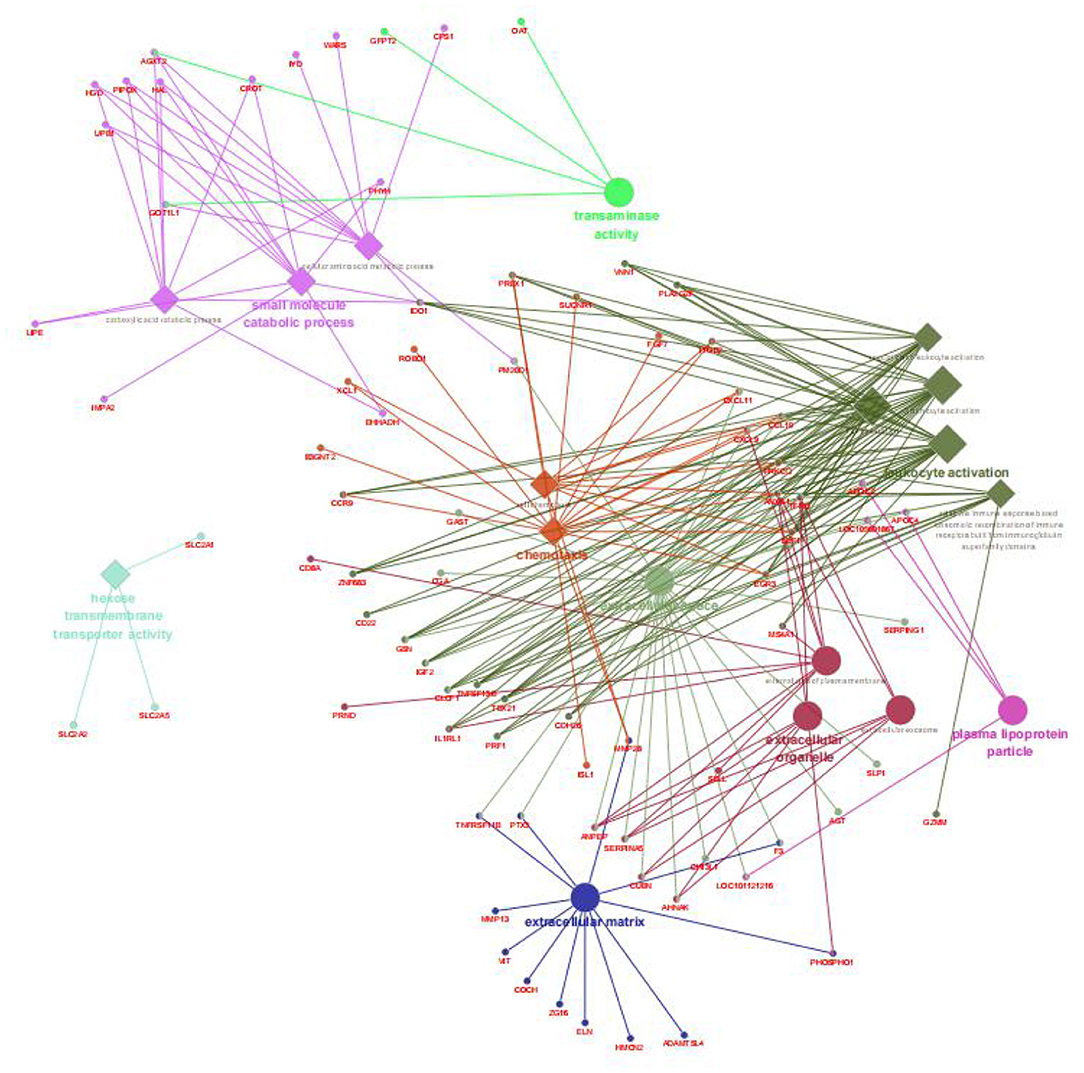
Figure 3. Enriched biological processes, cellular components, and molecular functions assigned to the DEGs between the AR and ER groups. The diamond showed the biological processes; the big circle showed the cellular components; the small circle showed the genes; the different colors represented the different functional groups, respectively, based on kappa score; all the DEGs were not enriched molecular functions.
We also performed the same enrichment analysis based on the KEGG pathway database and identified five pathways overrepresented in the pathway annotations of the given DEGs. As shown in Figure 4, the significant enrichment pathways were mainly associated with the peroxisome proliferator-activated receptor (PPAR) signaling pathway (ACSL5, FABP1, EHHADH, OLR1, DBI, PLIN2, and SCD), hematopoietic cell lineage (CD8A, CD1E, CD22, CD55, TFRC, MS4A1, CR2, and ANPEP), ferroptosis (TFRC, SLC40A1, LPCAT3, 9LC39A14, LOC101115614, and ACSL5), retinol metabolism (LOC101104808, AOX1, CYP1A1, RDH12, LOC101118447, LOC101116729, and LOC101117764), and tryptophan metabolism (EHHADH, IDO1, CYP1A1, AOX1, LOC101104808, and AOC1). Furthermore, we identified the top 10 hub genes among the DEGs, including ORM1, HPX, TTR, APOA2, APOA3, CP, AFM, SERPINC1, LTF, and RBP4, using the cytoHubba application (Figure 5).
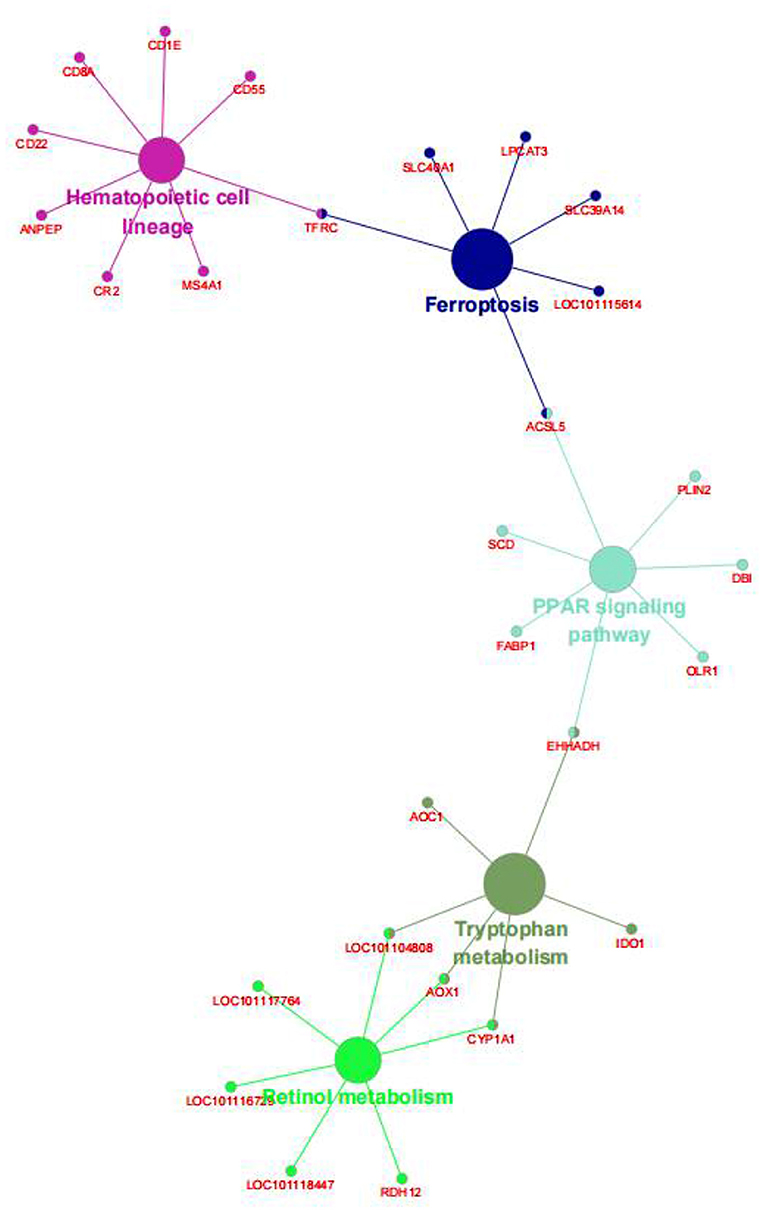
Figure 4. The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the DEGs between the AR and ER groups. The big circle showed the metabolic pathway; the small circle showed the genes; the different colors represented different pathways, respectively.
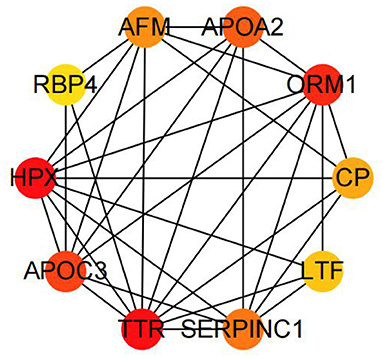
Figure 5. The top 10 hub genes by cytoHubba in the DEGs between the AR and ER groups.
According to Pearson's analysis of the overall correlation between proteome and transcriptome expression changes, we found that transcriptome and proteome had a low correlation (Figure 6A). Moreover, the results of most genes are concentrated near the coordinate center 0, indicating that the expression of most genes at the transcriptome and proteome levels changes the same. There is no significant change. However, the correlation was significantly higher when the expression variation was consistent and opposite (Figures 6B,C). Further, the specific expression correlation between transcriptome and proteome is given in Figure 6D. Based on this, we analyzed the screened DEGs using the KEGG and the GO databases in GlueGo, respectively (Figures 7A,B). Interestingly, the genes that were upregulated in both the transcriptome and proteome or upregulated in transcriptome but downregulated in the proteome could not be enriched in the KEGG and the GO databases. Nevertheless, genes that were upregulated in transcriptome but downregulated in proteome were increased in the functions of immunoglobulin-mediated immune response, Staphylococcus aureus infection, and negative regulation of leukocyte differentiation. Meanwhile, the genes having the same downregulated trend in transcriptome and proteome were enriched in the PPAR signaling pathway (APOA2, APOC3, ACSL3, CPS1, FABP2, and PCK1), intestinal lipid absorption (APOA2, FABP2, and CEL), urea cycle (CPS1, PCK1, SLC25A15, and ASS1), and ferroptosis (ACLS3, CP, and TFRC).

Figure 6. Differential correlation of proteome-transcriptome expression. (A) The correlation between all the associated protein and messenger RNA (mRNA). (B) The distribution of correlation of consistent-changed transcriptome and proteome. (C) The distribution of correlation of different-changed transcriptome and proteome. (D) Detailed classification of expression correlation.
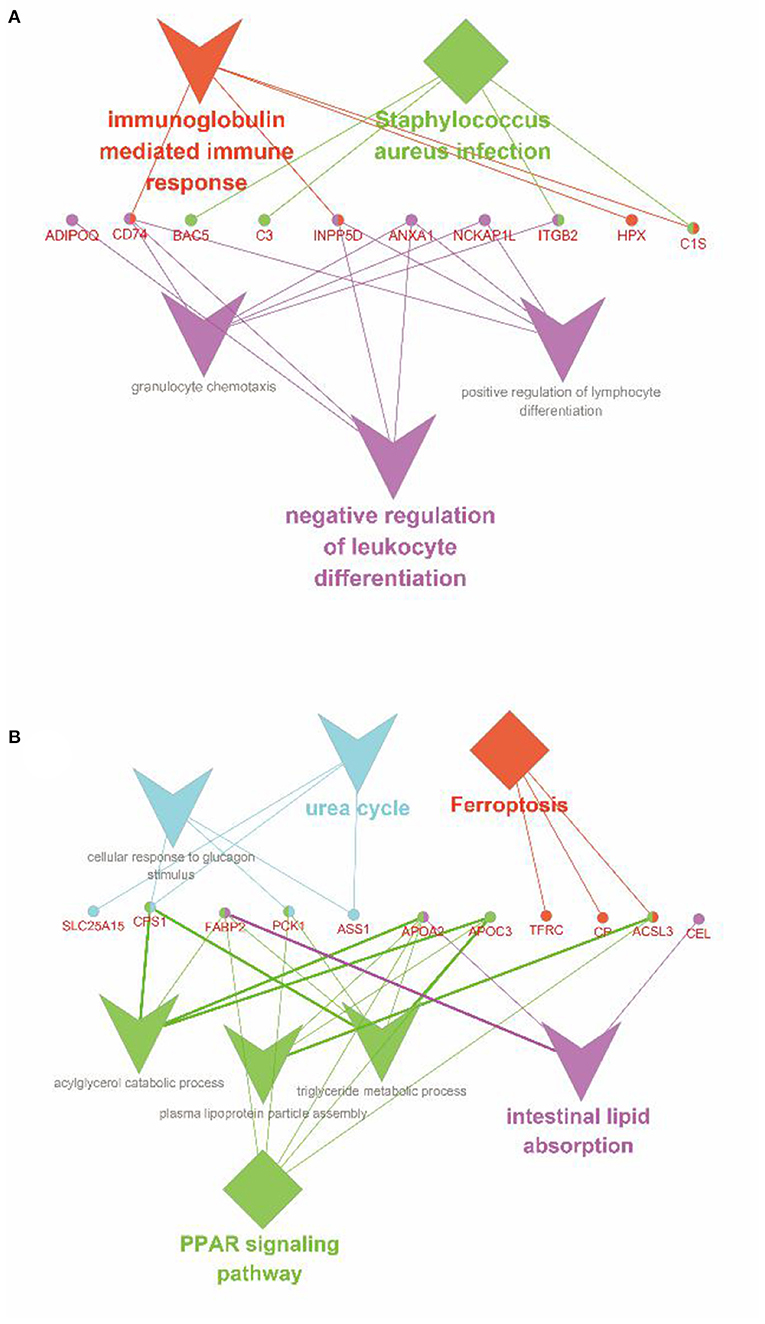
Figure 7. Enrichment analysis of the co-expressed DEGs between transcriptome and proteome. (A) Enrichment analysis of the genes, which were upregulated in transcriptome but downregulated in proteome. (B) Enrichment analysis of the genes, which were downregulated in transcriptome and proteome. The diamond represented the metabolic pathways in the KEGG database; the triangle represented the functions in the GO database; the circle showed the genes; the different colors represented the different functional groups, respectively, based on kappa score.
The intestine is the largest immune and digestive organ of mammals, suffering from any damage, which will lead to a series of health problems. Its mucous membrane is the largest interface between the body and the external environment, digesting and metabolizing nutrients, and resisting the invasion of foreign substances. Therefore, in this study, twin samples from three ewes were selected to explore the effects of AR and ER feeding regimes on the intestine health of lambs. Early intestine tissue sections have shown that the stress of separation from the dam at weaning destroyed the inherent morphological structure of the intestine, resulting in intestinal villus atrophy and crypt depth deepening (6). To further explore the intestinal dysfunction of lambs caused by the stress of weaning ewe's milk, we carried out the transcriptome analysis of intestinal tissues of lambs after AR or ER feeding procedures by RNA-seq technique and the DEGs were screened. Based on the comprehensive analysis of the DEGs and DEPs, it was found that the two key KEGG pathways were mainly enriched—the PPAR signaling pathway and ferroptosis.
The DEGs mainly enriched the 9 GO terms in the transcriptome, which primarily mediated the metabolism of nutrients, the immune response, the cellular composition, and the integrin signaling of lambs under weaning stress. Transaminase activity and plasma lipoprotein particles are closely related to intestinal fat digestion and absorption, supported by a study of Yokoyama (21). Yokoyama concludes that the level of transaminase in the small intestine is positively correlated with the synthesis of enzyme proteins and the transport of fatty acids to intestinal cells. In addition, the hexose transmembrane transporter activity is closely related to the digestion and metabolism of glucose and other monosaccharides and the small molecule catabolic process. Its significantly enrichment indicated that the absorption and transportation of nutrients have changed significantly after separating from the ewe at weaning. At the same time, the leukocyte activation and chemotaxis reflected the changes in intestinal immunity in lambs.
Furthermore, the greatly enriched KEGG pathways mainly included the PPAR signaling pathway and ferroptosis. Notably, these pathways are closely related to immune response, cellular process, and nutrient metabolism, which indicated that weaning stress most likely disturbed these processes. For example, the solute carrier (SLC) group of membrane transporters is mainly distributed on the cell membrane (22). It is the main intestinal transporter of many amino acids and peptides (23). Meanwhile, according to Liao and Gaowa's study, it was found that changing the nutrient intake ratio of young ruminants would lead to an increased expression level of nutrient transporters in jejunal enterocytes (24, 25).
Although in this study, the correlation between the transcriptome and proteome of the same samples was low and it is a common phenomenon (26, 27). The main reasons for this phenomenon might be: (1) the process of transcription and translation is very complex and there are RNA-mediated transcription regulation and posttranslational protein modification, so transcriptome analysis cannot completely reveal protein expression and (2) both the RNA and protein quantitative methods have their limitations, such as: RNA-seq may involve in overestimation or underestimation of the expression level, while iTRAQ will show a significant underestimation of the degree of protein upregulation or downregulation (28). Therefore, in the recent years, comprehensive transcriptomic and proteomic methods have been conducted in diverse study fields. The integrating proteomic and transcriptomic analysis showed that the two GO terms (intestinal lipid absorption and urea cycle) and the 2 KEGG pathways (PPAR signaling pathway and ferroptosis) were significantly downregulated after weaning stress.
Peroxisome proliferator-activated receptors, belonging to the nuclear receptor family, are a lipid ligand-activated nuclear transcription factor, including three members: PPAR-γ, PPAR-α, and PPAR-β/δ (29), which play a vital role in growth and development, carbohydrate and lipid metabolism, immunity, and inflammation (30, 31). The main mechanism of the PPAR signaling pathway is that it binds to the peroxisome proliferator response element (PPRE) as a heterodimer with retinoid X receptor (RXR) after the PPAR activation to regulate the expression of the target gene, which is vital for the regulation of intestinal immune response and fat metabolism. The PPARs control intestinal immune response mainly by changing the phenotype of macrophages and combining with short-chain fatty acids to induce the expansion and differentiation of T lymphocytes (Treg cells) (32–35). Notably, butyric acid is the main carbon source of intestinal epithelial cells (36) and the ligand of the PPAR (33). In addition, if the PPAR-α pathway is inhibited, the expression of interleukin-22 (IL-22) secreted by Th1 and Th17 cells will decrease and the structural integrity of the intestinal mucosal barrier will be destroyed, resulting in an increased intestinal susceptibility to bacteria and endotoxin (37, 38). Therefore, the changes in milk replacer nutritional composition compared with ewe's milk may lead to the accumulation of intestinal microorganisms and their metabolites and the steady-state imbalance of lambs.
As shown in Figure 7B, APOA2, APOC3, ACSL3, CPS1, FABP2, and PCK1 were significantly enriched in the PPAR signaling pathway. Among these, PCK1 encodes a cytoplasmic enzyme in sheep intestines to promote the production of phosphoenolpyruvate (39) and control the production of glycerol and lipids (40), which plays an essential role in intestinal lipid metabolism. FABP2 encodes an intestinal fatty acid-binding protein and is only distributed in mature intestinal cells. Hence, the downregulation of its expression indicates the decrease of intestinal lipid metabolism and the blocked intestinal development of lambs (41). APOA2 mediates the activation of the PPAR signaling pathway in the intestine (42). Like APOC3, the proteins they encode are related to the metabolism of triglyceride-rich lipoproteins and there is a strong interaction between them (43, 44). Besides, APOA2 is also involved in the metabolism of free fatty acids, but APOC3 itself, such as lipopolysaccharide, can promote the release of interleukin-1β (IL-1β) from monocytes and induce inflammation. Several studies have shown that weaning, as the most severe stimulating factor in early life, can destroy intestinal mucosal barrier structure, reduce immune and antioxidant function, increase the expression of IL-1β and tumor necrosis factor-α (TNF-α), and induce intestinal inflammation (1, 5, 45). In summary, we speculated that weaning stress downregulated the expression of the PPAR signaling pathway, resulting in intestinal immune function maladjustment, the structural integrity of intestinal mucosal barrier destruction, increased susceptibility to endotoxin, and induced severe inflammatory response. Moreover, the downregulation of the PPAR signaling pathway led to the disorder of lipid metabolism and the decrease of the PPAR ligands, which further aggravated this process.
Ferroptosis is an iron-dependent regulatory form of cell death, different from apoptosis, necrosis, and autophagy, which involves fatal iron-catalyzed lipid damage (46). The main mechanism is that Fe2+/ester oxygenase catalyzes the high expression of polyunsaturated fatty acids (PUFAs) on the cell membrane, resulting in lipid peroxidation and reactive oxygen species (ROS) accumulating, further inducing cell death. In addition, PUFAs is more susceptible to lipid peroxidation (47) and free PUFAs inserted into extracellular phospholipids after esterification can be used as a signal of iron ptosis after oxidation (48). Previous studies have shown that the Acyl-CoA synthetase long-chain (ACSL) family is involved in PUFA-phosphatidylethanolamines (PUFA-PEs) biosynthesis in the cell membrane and is the substrate of lipid peroxidation (48). Interestingly, our combined transcriptome and proteome analysis showed that the expression of ACSL3 was significantly downregulated. Above all, we speculated that after separating from the ewe at weaning, the lambs downregulated the expression of ACSL3 and other relative genes to resist the intestinal injury caused by ROS, resulting in the remodeling of PUFAs on the cell membrane, the disorder of lipid metabolism and absorption, and the lack of lipid peroxidation substrate, thus enhancing the resistance to ferroptosis. Notably, it has been already found that the normal development of the human fetal immune system depends on adequate intake of PUFAs, so it may be related to ferroptosis, which provides a new idea for us to study the regulation of the intestinal immune system in weaned lambs (49). Above all, we infer that the cause of ferroptosis is the lack or the low concentration of transferrin in milk replacer, which results in the obstruction of absorption and transport of iron ions in the intestine and the accumulation of Fe2+ in the epithelial cells of the intestinal mucosa. This result is consistent with the exfoliation and death of epithelial cells observed in the previous section and the effects of epithelial cell proliferation and disturbance of nutrient absorption and transport in the transcriptional group.
The integrating proteomic and transcriptomic analysis also showed that immunoglobulin-mediated immune response, Staphylococcus aureus infection, and negative regulation of leukocyte differentiation were significantly upregulated in transcriptome, but downregulated in the proteome. Due to post-transcription regulation, there is a deviation between transcriptome analysis results and protein expression. Some previous studies have shown that microRNA (miRNA) mediates the regulation of cell response under stress (50, 51). It was found that miR-146b could directly act on the Toll-like receptor 4 (TLR4) of intestinal epithelial cells of piglets to regulate intestinal metabolism and immune response of piglets under weaning stress (52). Based on this, we speculate that miRNA-mediated post-transcriptional regulation of messenger RNA (mRNA) may be involved in regulating intestinal metabolism and immune response of lambs under the stress of separating from the ewe at weaning. Therefore, to reveal the molecular regulatory mechanism of intestinal injury of lambs under weaning stress, we will further explore miRNA and its target genes in our following study. In addition, this study has only been conducted for 15 days. The long-term effect of early weaning on intestinal gene expression in lambs is unknown, worthy of further exploration in future study.
In this study, we used transcriptome and proteome to enrich and analyze the DEGs and found that the PPAR signaling pathway and ferroptosis may be the key drivers damaging the small intestine of lambs. Significantly differentially expressed genes such as FABP2, ACSL3, APOA2, APOC3, and PCK1 may be the key genes. These results could provide new ideas for alleviating the intestinal injury of lambs caused by weaning ewe's milk and a theoretical basis for preparing the new and efficient milk replacer.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject, Accession Number PRJNA781073.
This trial was conducted at the Linqing Runlin Animal Husbandry Corporation Ltd., in Shandong, China. All the experiments were performed according to the Regulations for the Administration of Affairs Concerning Experimental Animals published by the Ministry of Science and Technology, China, in 2004. The Chinese Academy of Agricultural Sciences Animal Ethics Committee approved all the experiments and human animal care and handling procedures were followed throughout the experiment (AEC-CAAS-20160715).
LH contributed to study design, sample processing, data analyses, and writing the original draft. SW and DH contributed to data analyses, review, and editing. HT, LK, and QD contributed to the conception, study design, and sample collection. KC contributed to project administration, study design, and supervision. All authors contributed to writing the manuscript.
The National Natural Science Foundation of China (32172764) and the Cooperative Guidance Project of Prospering Inner Mongolia Through Science and Technology in 2021 (2021CG0024) supported this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge every partner in the Key Laboratory of Feed Biotechnology of the Ministry of Agriculture and Rural Affairs of Chinese Academy of Agricultural Sciences for their selfless help in the process of experiment and article writing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.809188/full#supplementary-material
1. Zhang Q, Li C, Niu X, Zhang Z, Li F, Li F. An intensive milk replacer feeding program benefits immune response and intestinal microbiota of lambs during weaning. BMC Vet Res. (2018) 14:366. doi: 10.1186/s12917-018-1691-x
2. McCoard SA, Cristobal-Carballo O, Knol FW, Heiser A, Khan MA, Hennes N, et al. Impact of early weaning on small intestine, metabolic, immune and endocrine system development, growth and body composition in artificially reared lambs. J Anim Sci. (2020) 98:skz356. doi: 10.1093/jas/skz356
3. Belanche A, Cooke J, Jones E, Worgan HJ, Newbold CJ. Short- and long-term effects of conventional and artificial rearing strategies on the health and performance of growing lambs. Animal. (2019) 13:740–9. doi: 10.1017/S1751731118002100
4. Campbell JM, Crenshaw JD, Polo J. The biological stress of early weaned piglets. J Anim Sci Biotechnol. (2013) 4:19. doi: 10.1186/2049-1891-4-19
5. Novais AK, Deschene K, Martel-Kennes Y, Roy C, Laforest JP, Lessard M, et al. Weaning differentially affects mitochondrial function, oxidative stress, inflammation and apoptosis in normal and low birth weight piglets. PLoS ONE. (2021) 16:e0247188. doi: 10.1371/journal.pone.0247188
6. Cui K, Wang B, Zhang N, Tu Y, Ma T, Diao Q. Itraq-based quantitative proteomic analysis of alterations in the intestine of hu sheep under weaning stress. PLoS ONE. (2018) 13:e0200680. doi: 10.1371/journal.pone.0200680
7. Pascual-Alonso M, Miranda-de la Lama GC, Aguayo-Ulloa L, Ezquerro L, Villarroel M, Marin RH, et al. Effect of postweaning handling strategies on welfare and productive traits in lambs. J Appl Anim Welf Sci. (2015) 18:42–56. doi: 10.1080/10888705.2014.941107
8. Moeser AJ, Pohl CS, Rajput M. Weaning stress and gastrointestinal barrier development: implications for lifelong gut health in pigs. Anim Nutr. (2017) 3:313–21. doi: 10.1016/j.aninu.2017.06.003
9. Wang J, Chen L, Li D, Yin Y, Wang X, Li P, et al. Intrauterine growth restriction affects the proteomes of the small intestine, liver, and skeletal muscle in newborn pigs. J Nutr. (2008) 138:60–6. doi: 10.1093/jn/138.1.60
10. Lessard M, Blais M, Beaudoin F, Deschene K, Verso LL, Bissonnette N, et al. Piglet weight gain during the first two weeks of lactation influences the immune system development. Vet Immunol Immunopathol. (2018) 206:25–34. doi: 10.1016/j.vetimm.2018.11.005
11. Hrdlickova R, Toloue M, Tian B. RNA-seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA. (2017) 8:e1364. doi: 10.1002/wrna.1364
12. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
13. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. Star: ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
14. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and samtools. Bioinformatics. (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
15. Liao Y, Smyth GK, Shi W. Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1201/b16589
16. Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. (2013) 41:e108. doi: 10.1093/nar/gkt214
17. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with Deseq2. Genome Biol. (2014) 15:ARTN 550. doi: 10.1186/s13059-014-0550-8
18. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
19. Brohee S, van Helden J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics. (2006) 7:488. doi: 10.1186/1471-2105-7-488
20. Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. Cytohubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. (2014) 8:S11. doi: 10.1186/1752-0509-8-S4-S11
21. Yokoyama H, Kobayashi A, Kondo K, Oshida S-i, Takahashi T, Masuyama T, et al. A pharmacologic increase in activity of plasma transaminase derived from small intestine in animals receiving an Acyl Coa: diacylglycerol transferase (Dgat) 1 inhibitor. J Toxicol Sci. (2018) 43:135–57. doi: 10.2131/jts.43.135
22. Perland E, Fredriksson R. Classification systems of secondary active transporters. Trends Pharmacol Sci. (2017) 38:305–15. doi: 10.1016/j.tips.2016.11.008
23. Zhu X, Jiao J, Zhou C, Tang S, Wang M, Kang J, et al. Effects of dietary methionine and lysine supplementation on nutrients digestion, serum parameters and mRNA expression of related amino acid sensing and transporting genes in growing goats. Small Ruminant Res. (2018) 166:1–6. doi: 10.1016/j.smallrumres.2018.07.002
24. Liao SF, Vanzant ES, Harmon DL, McLeod KR, Boling JA, Matthews JC. Ruminal and abomasal starch hydrolysate infusions selectively decrease the expression of cationic amino acid transporter mRNA by small intestinal epithelia of forage-fed beef steers. J Dairy Sci. (2009) 92:1124–35. doi: 10.3168/jds.2008-1521
25. Gaowa N, Li W, Gelsinger S, Murphy B, Li S. Analysis of host jejunum transcriptome and associated microbial community structure variation in young calves with feed-induced acidosis. Metabolites. (2021) 11:414. doi: 10.3390/metabo11070414
26. Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, et al. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol Cell Proteomics. (2004) 3:960–9. doi: 10.1074/mcp.M400055-MCP200
27. Zhang HZ Li YY, An T, Huang FX, Wang MQ, Liu CX, et al. Comparative transcriptome and itraq proteome analyses reveal the mechanisms of diapause in aphidius gifuensis ashmead (Hymenoptera: Aphidiidae). Front Physiol. (2018) 9:1697. doi: 10.3389/fphys.2018.01697
28. Shirran SL, Botting CH, A. Comparison of the accuracy of itraq quantification by Nlc-Esi Msms and Nlc-Maldi Msms methods. J Proteomics. (2010) 73:1391–403. doi: 10.1016/j.jprot.2010.03.003
29. Wahli W, Michalik L. Ppars at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. (2012) 23:351–63. doi: 10.1016/j.tem.2012.05.001
30. Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. (2006) 55:1104–13. doi: 10.1136/gut.2005.081745
31. Deng K, Ren C, Fan Y, Pang J, Zhang G, Zhang Y, et al. Yap1 regulates Pparg and Rxr alpha expression to affect the proliferation and differentiation of ovine preadipocyte. J Cell Biochem. (2019) 120:19578–89. doi: 10.1002/jcb.29265
32. Are A, Aronsson L, Wang S, Greicius G, Lee YK, Gustafsson JA, et al. Enterococcus faecalis from newborn babies regulate endogenous ppargamma activity and Il-10 levels in colonic epithelial cells. Proc Natl Acad Sci USA. (2008) 105:1943–8. doi: 10.1073/pnas.0711734105
33. Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell. (2016) 64:982–92. doi: 10.1016/j.molcel.2016.10.025
34. Zenhom M, Hyder A, Kraus-Stojanowic I, Auinger A, Roeder T, Schrezenmeir J. Ppargamma-dependent peptidoglycan recognition protein 3 (Pglyrp3) expression regulates proinflammatory cytokines by microbial and dietary fatty acids. Immunobiology. (2011) 216:715–24. doi: 10.1016/j.imbio.2010.10.008
35. Dziarski R, Gupta D. Review: mammalian peptidoglycan recognition proteins (Pgrps) in innate immunity. Innate Immun. (2010) 16:168–74. doi: 10.1177/1753425910366059
36. Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
37. Manoharan I, Suryawanshi A, Hong Y, Ranganathan P, Shanmugam A, Ahmad S, et al. Homeostatic Pparalpha signaling limits inflammatory responses to commensal microbiota in the intestine. J Immunol. (2016) 196:4739–49. doi: 10.4049/jimmunol.1501489
38. Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, et al. Il-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol. (2013) 190:5306. doi: 10.4049/jimmunol.1300016
39. Moisa SJ, Shike DW, Faulkner DB, Meteer WT, Keisler D, Loor JJ. Central role of the ppargamma gene network in coordinating beef cattle intramuscular adipogenesis in response to weaning age and nutrition. Gene Regul Syst Biol. (2014) 8:17–32. doi: 10.4137/GRSB.S11782
40. Kamisaka Y, Kimura K, Uemura H, Ledesma-Amaro R. Modulation of gluconeogenesis and lipid production in an engineered oleaginous saccharomyces cerevisiae transformant. Appl Microbiol Biotechnol. (2016) 100:8147–57. doi: 10.1007/s00253-016-7662-x
41. Reisinger KW, Elst M, Derikx JP, Nikkels PG, de Vries B, Adriaanse MP, et al. Intestinal fatty acid-binding protein: a possible marker for gut maturation. Pediatr Res. (2014) 76:261–8. doi: 10.1038/pr.2014.89
42. Duval C, Muller M, Kersten S. Pparalpha and dyslipidemia. Biochim Biophys Acta. (2007) 1771:961–71. doi: 10.1016/j.bbalip.2007.05.003
43. Gong T, Zhou R. Apoc3: an 'Alarmin' triggering sterile inflammation. Nat Immunol. (2020) 21:9–11. doi: 10.1038/s41590-019-0562-3
44. van't Hooft FM, Ruotolo G, Boquist S, de Faire U, Eggertsen G, Hamsten A. Human evidence that the apolipoprotein a-ii gene is implicated in visceral fat accumulation and metabolism of triglyceride-rich lipoproteins. Circulation. (2001) 104:1223–8. doi: 10.1161/hc3601.095709
45. Cao ST, Wang CC, Wu H, Zhang QH, Jiao LF, Hu CH. Weaning disrupts intestinal antioxidant status, impairs intestinal barrier and mitochondrial function, and triggers mitophagy in piglets. J Anim Sci. (2018) 96:1073–83. doi: 10.1093/jas/skx062
46. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
47. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
48. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic pes navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
49. Enke U, Seyfarth L, Schleussner E, Markert UR. Impact of pufa on early immune and fetal development. Br J Nutr. (2008) 100:1158–68. doi: 10.1017/S000711450801413X
50. Engedal N, Zerovnik E, Rudov A, Galli F, Olivieri F, Procopio AD, et al. From oxidative stress damage to pathways, networks, and autophagy via microRNAs. Oxid Med Cell Longev. (2018) 2018:4968321. doi: 10.1155/2018/4968321
51. Li Q, Yang C, Du J, Zhang B, He Y, Hu Q, et al. Characterization of miRNA profiles in the mammary tissue of dairy cattle in response to heat stress. BMC Genomics. (2018) 19:975. doi: 10.1186/s12864-018-5298-1
Keywords: sheep, intestinal injury, weaning stress, transcriptome, proteome
Citation: Han L, Tao H, Kang L, Wang S, Diao Q, Han D and Cui K (2022) Transcriptome and iTRAQ-Based Proteome Reveal the Molecular Mechanism of Intestinal Injury Induced by Weaning Ewe's Milk in Lambs. Front. Vet. Sci. 9:809188. doi: 10.3389/fvets.2022.809188
Received: 04 November 2021; Accepted: 14 March 2022;
Published: 25 April 2022.
Edited by:
Morteza Hosseini-Ghaffari, University of Bonn, GermanyReviewed by:
Kejun Wang, Henan Agricultural University, ChinaCopyright © 2022 Han, Tao, Kang, Wang, Diao, Han and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deping Han, aGFuZGVwaW5nMTk4NEBjYXUuZWR1LmNu; Kai Cui, Y3Vpa2FpQGNhYXMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.