 Li Gao
Li Gao Song Wang2,3†
Song Wang2,3† Miaomiao Yang
Miaomiao Yang Lei Yang
Lei Yang Guangpeng Li
Guangpeng Li Tong Wen
Tong Wen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 17 January 2023
Sec. Veterinary Infectious Diseases
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.1084945
Myostatin (MSTN) regulates muscle development and body metabolism through a variety of pathways and is a core target gene for gene editing in livestock. Gut fungi constitute a small part of the gut microbiome and are important to host health and metabolism. The influence of MSTN mutations on bovine gut fungi remains unknown. In this study, Internal Transcribed Spacer (ITS) high-throughput sequencing was conducted to explore the composition of gut fungi in the MSTN mutant (MT) and wild-type (WT) cattle, and 5,861 operational taxonomic units (OTUs) were detected and classified into 16 phyla and 802 genera. The results of the alpha diversity analysis indicated that no notable divergence was displayed between the WT and MT cattle; however, significant differences were noticed in the composition of fungal communities. Eight phyla and 18 genera were detected. According to the prediction of fungal function, saprotroph fungi were significantly more abundant in the MT group. The correlation analysis between gut fungal and bacterial communities revealed that MSTN mutations directly changed the gut fungal composition and, at the same time, influenced some fungi and bacteria by indirectly regulating the interaction between microorganisms, which affected the host metabolism further. This study analyzed the role of MSTN mutations in regulating the host metabolism of intestinal fungi and provided a theoretical basis for the relationship between MSTN and gut fungi.
The gut microbiota consists of microbial cells, including bacteria, fungi, archaea, protozoa, and viruses, and their relevant genetic material exists in the gastrointestinal (GI) tract of the host (1). Although fungi constitute only a small part of the gut microbiome, they play a key role in the life activities of the host. Fungal colonization facilitates host resistance to pathogenic infections and modulates the immune system (2, 3). Gut fungi can also maintain the barrier function of the intestinal mucosal lining through synergistic, antagonistic, or symbiotic interactions with the gut bacteria (4, 5) and play important roles in host health and metabolism (6, 7). Fungi are the most effective microbial group for degrading lignocellulose and are the core microorganisms for excavating high-efficiency fiber-degrading enzymes (7). The diversity of fungal flora in the gut, including the gut bacteria, is related to the diet, age, physical condition, and environment of the host.
Myostatin (MSTN) is an important negative regulatory factor in skeletal muscle development (8). It is a core target gene for gene editing in livestock to improve meat quality with a lower fat percentage and a more lean meat yield. Luxi cattle with the MSTN gene mutations prepared by the Inner Mongolia University through the clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/CAS9) technology have more developed muscles and a higher lean meat yield (9). Previous studies had analyzed the changes in gut bacteria composition in MSTN-mutated cattle and found that the MSTN mutation affects the metabolism of the body by regulating the composition of gut bacteria, thereby affecting the growth traits of beef cattle (10, 11). However, there are no reports on the changes in MSTN mutant bovine gut fungi and their correlation with host metabolism.
In this study, the gut fungal compositions of the MSTN gene mutant (MT) and wild-type (WT) cattle were investigated, and the association between gut fungal composition and bacterial composition was analyzed. These findings are of great significance to the metabolism-regulating mechanisms of MSTN and the interactions between bacteria and fungi.
This study was performed according to animal care and ethics in China and was approved by the Animal Ethics and Welfare Committee (AEWC) of Inner Mongolia University and Baotou Teacher's College.
The cattle used in the present study were presumably 24-month-old females raised as common beef cattle in a local ranch. Sample collection was performed as described in an earlier study (10). Total DNA was extracted from fecal samples using the QlAamp Fast DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. The fungal Internal Transcribed Spacer 2 (ITS2) region was obtained by applying the universal primers ITS (ITS1F: 5'-ACTTGGTCATTTAGAGGAAGTAA-3' and ITS2R: 5'-GCTGCGTTCTTCATCGATGC-3') and sequenced using the Illumina NovaSeq platform. Trimmomatic v0.33 software was used to leach the raw reads acquired by sequencing. Next, Cutadapt 1.9.1 software was applied to acquire clean reads. Clean reads were spliced by overlapping, and the acquired spliced data were leached based on the range length of different regions. The final non-chimeric reads were obtained using the dada2 (12) method in the QIIME2 2020.6 (13) software to denoise and remove chimeric sequences. The resulting reads were then clustered into operational taxonomic units (OTUs) based on 97% sequence similarity. The diversity of the fungal flora within the samples was studied using the Shannon, Simpson, Chao1, and abundance-based coverage estimator (ACE) indices, and a non-metric multidimensional scaling (NMDS) analysis by binary Jaccard distances was performed to compare the differences in fungal composition in different samples. The linear discriminant analysis (LDA) effect size (LEfSe) analysis was conducted to identify functional biomarkers in WT and MT samples (LDA ≥4.0) (14). Fungal functions were predicted using the FUNGuild software, and Spearman's correlations were conducted to prioritize indicator species linking fungi and bacteria.
A total of 492,564 (MT = 205,684, WT = 286,880) raw sequences of the ITS2 regions in the six sequenced samples (three MT and three WT cattle), with 35,429–102,161 (mean 74,452 ± 27,801) non-chimeric reads and 5,861 OTUs based on 97% sequence similarity, were obtained. These data were classified into 16 phyla, 64 classes, 159 orders, 372 families, and 802 genera. Both the Shannon diversity index and rarefaction curve tended to flatten, demonstrating that the depth and quantity of the sequencing met the requirements for further analysis (Figures 1A, B).
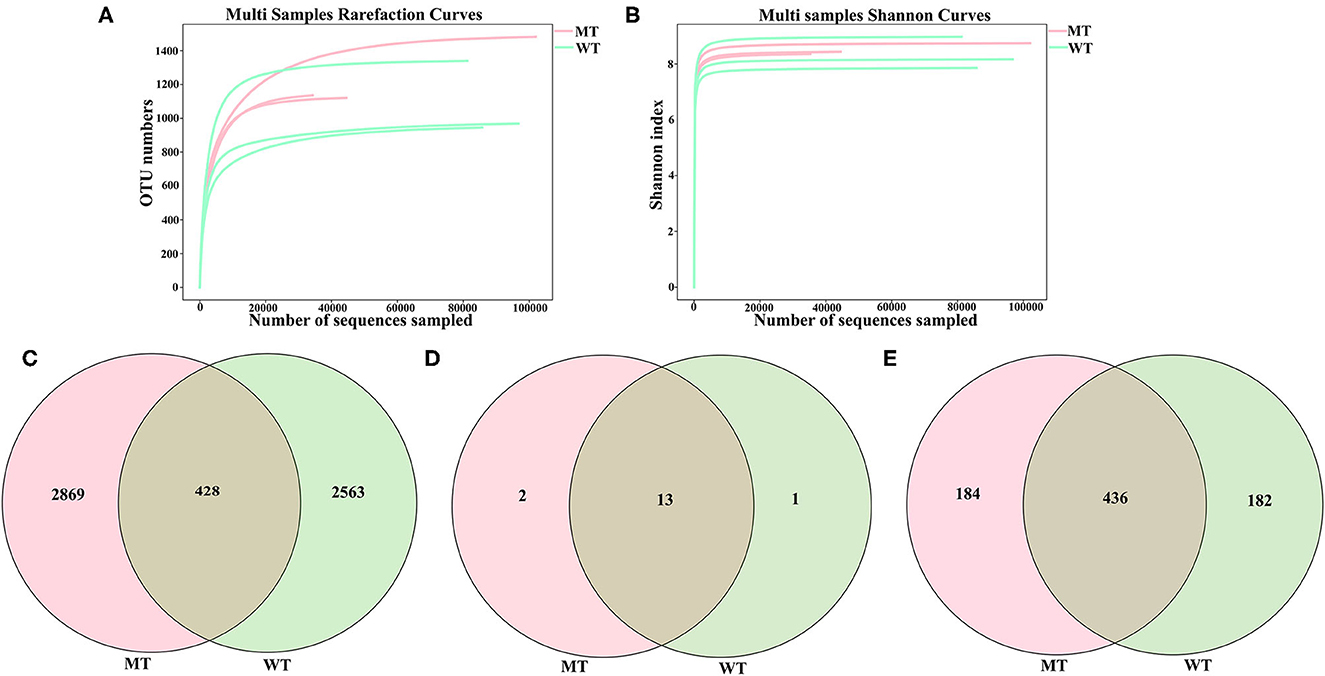
Figure 1. Feasibility analysis and operational taxonomic units (OTUs) distribution of the sequencing data. (A) Fungal rarefaction curves for all the samples; (B) Shannon curves for all the samples; (C) Gut fungal OTUs distribution in each group; (D) Gut fungal phylum distribution in each group; (E) Gut fungal genus distribution in each group.
In total, the number of OTUs from the gut fungi shared by cattle in both the MT and WT groups was 428, while 2,869 and 2,563 OTUs existed only in the MT and WT cattle (Figure 1C), respectively. Thirteen phyla were shared by both MT and WT cattle, whereas two and one phyla existed only in MT and WT cattle, respectively (Figure 1D). At the genus level, 436 genera were shared by both MT and WT cattle, whereas 184 and 182 genera existed only in MT and WT cattle, respectively (Figure 1E).
No notable divergence was displayed in the Shannon (8.3326 ± 0.5766 vs. 8.5139 ± 0.2029, P = 0.65), Simpson (0.9871 ± 0.0063 vs. 0.9918 ± 0.0004, P = 0.33), ACE (1088.8220 ± 219.3710 vs. 1250.8343 ± 203.2833, P = 0.4), and Chao1 (1086.5587 ± 220.6933 vs. 1249.1780 ± 202.7929, P = 0.4) indices between the MT and WT cattle (P > 0.05) (Figures 2A–D), suggesting that alpha diversity had no notable divergence between the WT and MT cattle.
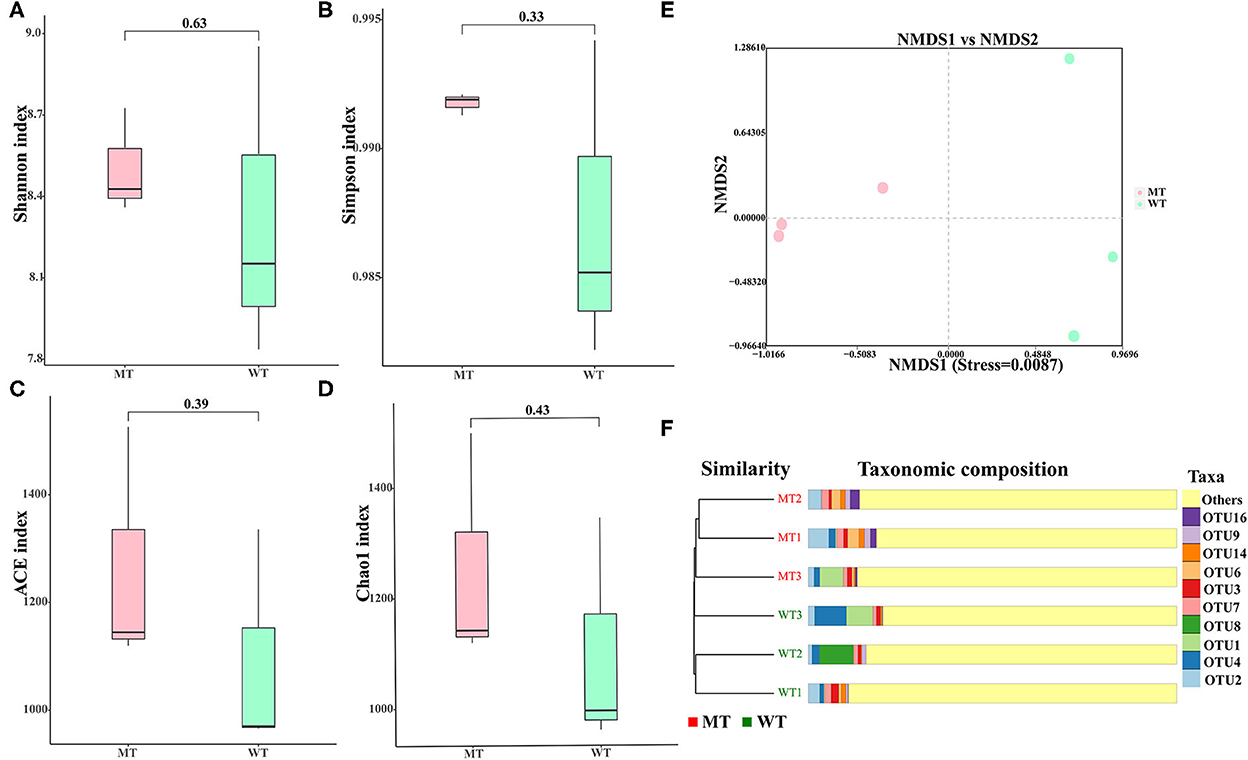
Figure 2. Gut fungal diversities of the wild-type (WT) and MSTN mutant (MT) cattle. (A) Shannon diversity; (B) Simpson indices; (C) abundance-based coverage estimator (ACE) index; (D) Chao1 diversity; (E) non-metric multidimensional scaling (NMDS) plots using Jaccard binary distance; (F) gut fungal clustering analysis.
The divergence in the fungal composition between the WT and MT cattle was assessed using the NMDS based on binary Jaccard distances to generate scatter plots (Figure 2E) and the cluster tree bar plot (Figure 2F). The samples in the same group of cattle showed a significant clustering trend (stress < 0.05), suggesting that a significant difference was noticed in the composition of fungal communities between the WT and MT cattle.
The relative abundance of dominant fungi at the phylum and genus levels was analyzed, and visible changes in the fungal composition were noticed between the WT and MT cattle. Among the 16 phyla identified in the six fecal samples, the average relative abundance of eight phyla was >1% (Figure 3A), including Ascomycota (50.66%), Basidiomycota (17.12%), Neocallimastigomycota (15.05%), unclassified fungi (7.41%), Mortierellomycota (3.92%), Rozellomycota (1.82%), Chytridiomycota (1.79%), and Glomeromycota (1.48%). Three phyla were the most predominant in the MT group: Ascomycota (54.16%), Basidiomycota (16.86%), and unclassified fungi (9.31%); together they comprised 80.33% of the total fungal composition (Figure 3A). Moreover, Ascomycota (47.18%) was the most predominant fungal phylum in the WT group, followed by Neocallimastigomycota (22.40%) and Basidiomycota (17.38%), which comprised ~86.96% of all the fungal taxa in the WT group.

Figure 3. A bar chart showing the relative abundance of the fungi in MSTN mutant (MT) and wild-type (WT) cattle. (A) The relative abundance of the fungi at the phylum level; (B) The relative abundance of the fungi at the genus level.
A total of 802 genera were detected, 18 of which had an average relative abundance of more than 1% (Figure 3B), including unclassified_fungi (7.41%), Caecomyces (5.02%), Mortierella (3.53%), unclassified_Ascomycota (3.24%), Alternaria (3.21%), Comoclathris (2.94%), Fusarium (2.76%), unclassified_Basidiomycota (2.53%), Orpinomyces (2.42%), Debaryomyces (2.42%), Cladosporium (2.35%), Penicillium (2.14%), unclassified_Sordariomycetes (2.11%), Cyllamyces (1.99%), Piromyces (1.74%), Thermomyces (1.59%), Filobasidium (1.52%), and Thermoascus (1.05%). Unclassified_fungi (9.31%) was the most predominant genus in the MT group, followed by Mortierella (4.20%) and Alternaria (4.10%), which together comprised ~50.74% of the total taxonomic groups identified. Caecomyces (7.85%), unclassified_fungi (5.50%), and Orpinomyces (4.08%) were the three most predominant genera in the WT group, comprising about 49.23% of the total taxonomic groups identified.
The gut fungal composition in MT and WT cattle samples was identified further to analyze how MSTN mutations interfere with the gut fungal communities of the cattle, and a relative abundance of more than 1% was determined at different classification levels (Figure 4A). Ascomycota, unclassified_fungi, Mortierellomycota, and Rozellomycota were more dominant in the MT group compared to those in the WT group at the phylum level, whereas the relative abundance of Neocallimastigomycota was lower (P < 0.05) (Figure 4A and Supplementary Table S1). At the genus level, unclassified_fungi, Mortierella, unclassified_Sordariomycetes, and Penicillium were more predominant in the MT group than in the WT group, whereas Caecomyces was lower in the MT group compared to that in the WT group (P < 0.05) (Figure 4A and Supplementary Table S2). Considering that the discriminant analysis may not be able to detect the entire fungus, LEfSe plus LDA scores were employed to distinguish the specific fungi related to the MSTN mutant (Figures 4B, C). The results showed that unclassified_fungi was the most notable genus in the MT group compared to that in the WT group, whereas Caecomyces and Cyllamyces were dramatically more abundant in the WT group compared to those in the MT group.
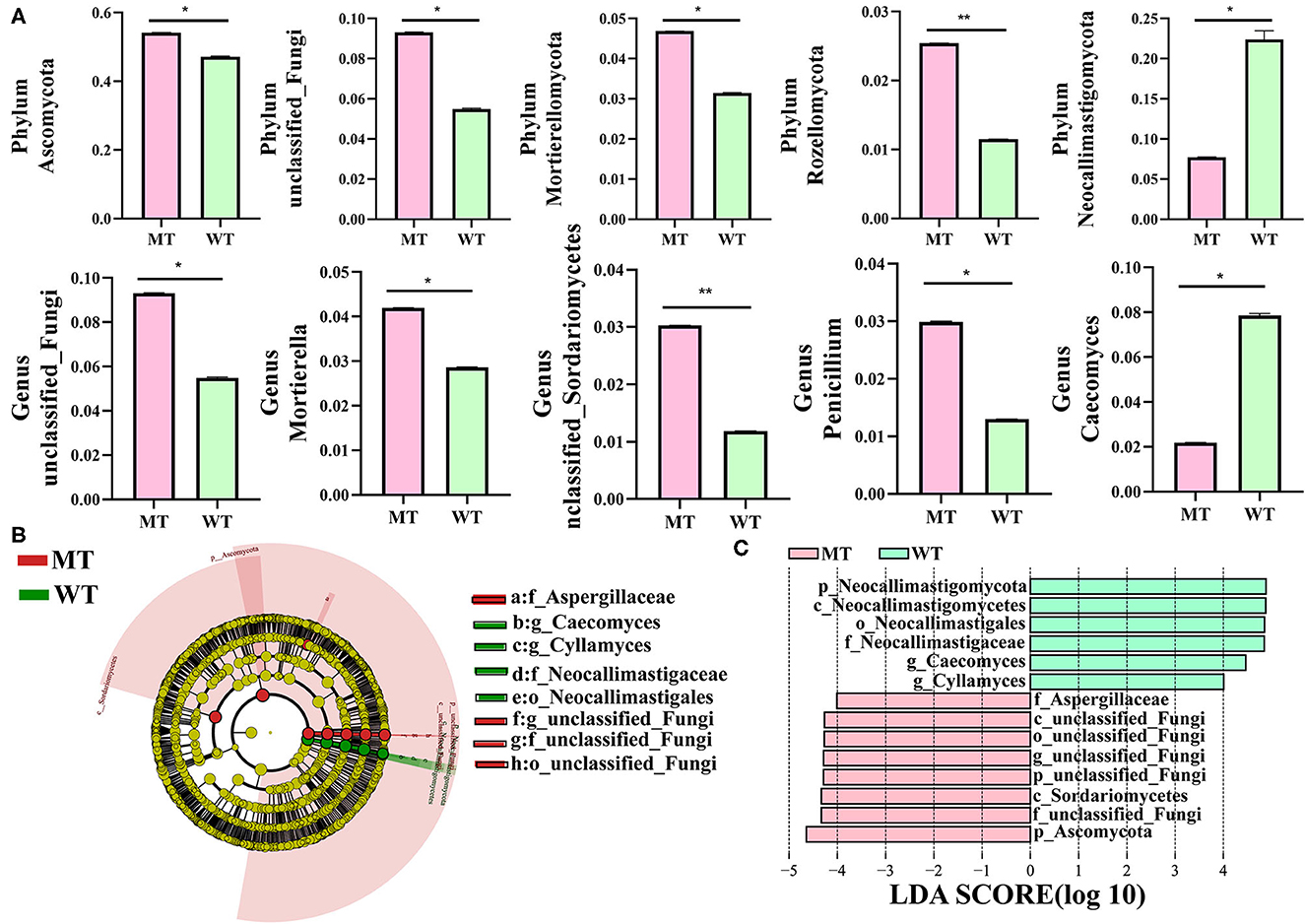
Figure 4. Comparisons of the gut fungal communities in MSTN mutant (MT) and wild-type (WT) cattle. (A) Metastatic analysis of those fungi whose relative abundance was more than 1%, and all the data stand for means ± SD. *P < 0.05, **P < 0.01. (B) Cladogram demonstrating the phylogenetic distribution of fungi related to the MSTN mutant. (C) Plot from linear discriminant analysis (LDA) effect size (LEfSe) analysis, linear discriminant analysis (LDA) scores >4.
The functions of fungi whose relative abundance was >1% were predicted by FUNGuild. According to the classification of nutrition, pathotroph fungi were more dominant in the MT group compared to those in the WT group but showed no significant differences (14.24 ± 2.74 vs. 11.52 ± 4.56%, P > 0.05), and saprotroph fungi were markedly more abundant in the MT group compared to those in the WT group(56.33 ± 2.62% vs. 49.10 ± 2.24%, P < 0.05), while the relative abundance of symbiotrophs was less in the MT group compared to that in the WT group, and no significant differences were observed (29.43 ± 0.87% vs. 39.38 ± 6.80%, P > 0.05) (Figure 5A).
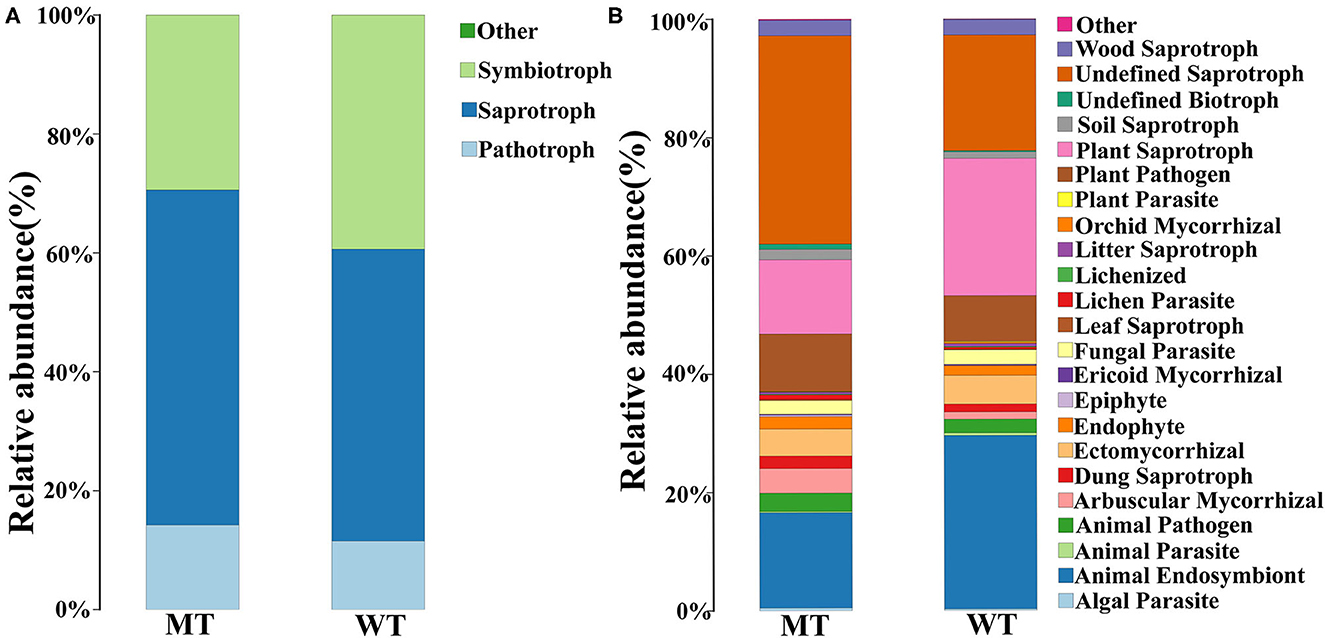
Figure 5. Fungal community functional prediction. (A) Function predicted in the trophic level. (B) Function predicted in the guild level.
When classified by guilds, the most important functions of both the MT and WT groups were undefined saprotrophs (27.41 ± 9.61%), animal endosymbionts (22.72 ± 9.24%), and plant saprotrophs (17.88 ± 6.84%). The most important functions in the MT group were undefined saprotrophs (27.41 ± 9.61%), animal endosymbionts (16.12 ± 3.12%), and plant saprotrophs (12.57 ± 2.64%). The most important functions in the WT group were animal endosymbionts (29.33 ± 8.54%), plant saprotrophs (23.20 ± 5.01%), and undefined saprotrophs (19.55 ± 5.66%), sequentially. The functions that were significantly different between the MT and WT groups were undefined saprotrophs and plant saprotrophs (P < 0.05) (Figure 5B).
The correlations between the different gut fungi were analyzed using the network analysis (Figure 6). The results indicated that unclassified_Agaricales was negatively correlated with Comoclathris (-1) and Naganishia (−0.8857) but positively correlated with Saccharomyces (0.8286). Tausonia was negatively correlated with Penicillium (-1) and positively correlated with Caecomyces (0.8286), Cyllamyces (0.9429), and unclassified_Leotiomycetes (0.9429). Talaromyces was negatively correlated with Filobasidium (-1), Naganishia (−0.9429), Thermomyces (−0.8857), and Vishniacozyma (−0.8857) and positively correlated with Saccharomyces (0.8286) and Inocybe (0.8286). Chaetomium was negatively correlated with Inocybe (-1), Debaryomyces (−0.9429), and Talaromyces (−0.8286) and positively correlated with Thermomyces (0.8286), Filobasidium (0.8286), and Thermoascus (0.8857). Caecomyces was negatively correlated with unclassified_fungi (−0.9429). Penicillifer was negatively correlated with Caecomyces (−0.9429) and positively correlated with unclassified_fungi (0.8857) and unclassified_Sordariomycetes (0.9429). Alternaria was negatively correlated with unclassified_Ascomycota (−0.9429). Cyllamyces was negatively correlated with Penicillium (−0.9429) and unclassified_fungi (−0.8286). Unclassified_Leotiomycetes was negatively correlated with Penicillium (−0.9429) and positively correlated with Cyllamyces (0.8286). Saccharomyces was negatively correlated with Thermomyces (−0.9429), Comoclathris (−0.8286), Piromyces (−0.8286), and Filobasidium (−0.8286) and positively correlated with unclassified_Ascomycota (0.8286). Limnoperdon was negatively correlated with Caecomyces (−0.9411), Tausonia (−0.9411), Cyllamyces (−0.8804), and unclassified_Leotiomycetes (−0.8804) and positively correlated with unclassified_fungi (0.8804), unclassified_Sordariomycet (0.8804), Penicillium (0.9411), Penicillifer (0.9411), and Helicobasidium (0.9411). Unclassified_Sordariomycetes was negatively correlated with Caecomyces (−0.8857) and positively correlated with Mortierella (0.8857) and unclassified_fungi (0.9429). Archaeorhizomyces was negatively correlated with Fusarium (−0.8857). Piromyces was negatively correlated with Debaryomyces (−0.8857). Inocybe was negatively correlated with Thermoascus (−0.8857), Thermomyces (−0.8286), and Filobasidium (−0.8286) and positively correlated with Debaryomyces (0.9429). Naganishia was negatively correlated with Saccharomyces (−0.8857) and positively correlated with Comoclathris (0.8857), Thermomyces (0.9429), and Filobasidium (0.9429). Mortierella was negatively correlated with Caecomyces (−0.8286) and positively correlated with unclassified_fungi (0.9429). Penicillium and Thermomyces were negatively correlated with Caecomyces (−0.8286). Helicobasidium was negatively correlated with Caecomyces (−0.8286) and Tausonia (−0.8286) and positively correlated with Penicillium (0.8286), unclassified_Sordariomycetes (0.8857), and Penicillifer (0.9429). Saitozyma was negatively correlated with Comoclathris (−0.8286) and positively correlated with unclassified_Agaricales (0.8286). Orpinomyces was negatively correlated with unclassified_Basidiomycota (−0.8286). Thermoascus and Plectosphaerella were negatively correlated with Debaryomyces (−0.8286). Plectosphaerella was positively correlated with Piromyces (0.8857). Trichoderma was negatively correlated with Piromyces (−0.8286) and positively correlated with unclassified_Ascomycota (0.8857) and Saccharomyces (0.8857). Filobasidium was positively correlated with Thermomyces (0.8857). Vishniacozyma was positively correlated with Filobasidium (0.8857), and Cladosporium was positively correlated with unclassified_Basidiomycota (0.9429).
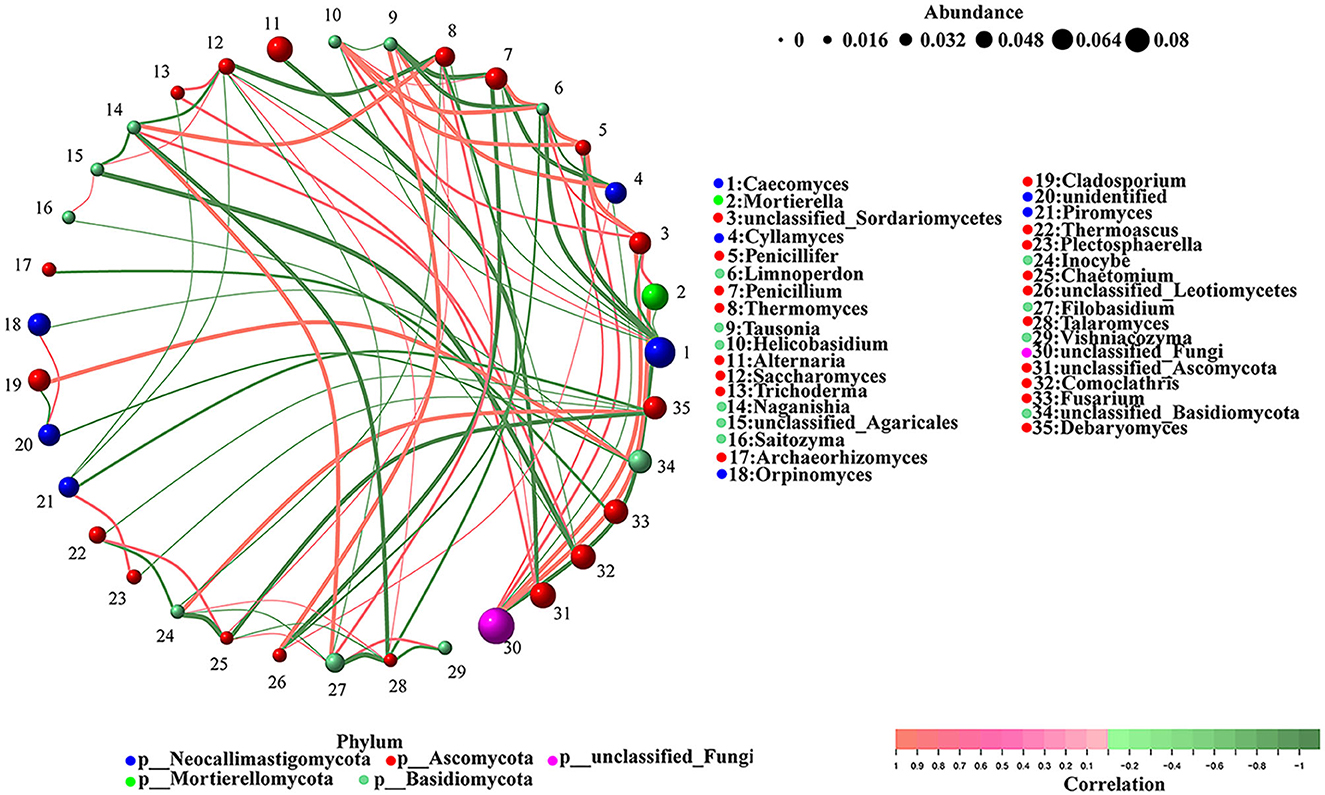
Figure 6. The correlations between different gut fungi. The color of the node indicates fungal taxa, and the relative abundance is represented by the weighted node size. Red lines show a positive correlation, while the green lines show a negative correlation.
The correlation between fungal and bacterial species was analyzed to distinguish the marker species linking fungi and bacteria. Spearman's correlation between multiple fungal species (>1% in each group) and bacterial species (>1% in each group) of the WT and MT cattle was determined. At the phylum level (Figure 7A), Mortierellomycota, Rozellomycota, and unclassified_fungi were positively correlated with Bacteroidetes (P < 0.05), and Rozellomycota and Mortierellomycota were negatively correlated with Firmicutes (P < 0.05).
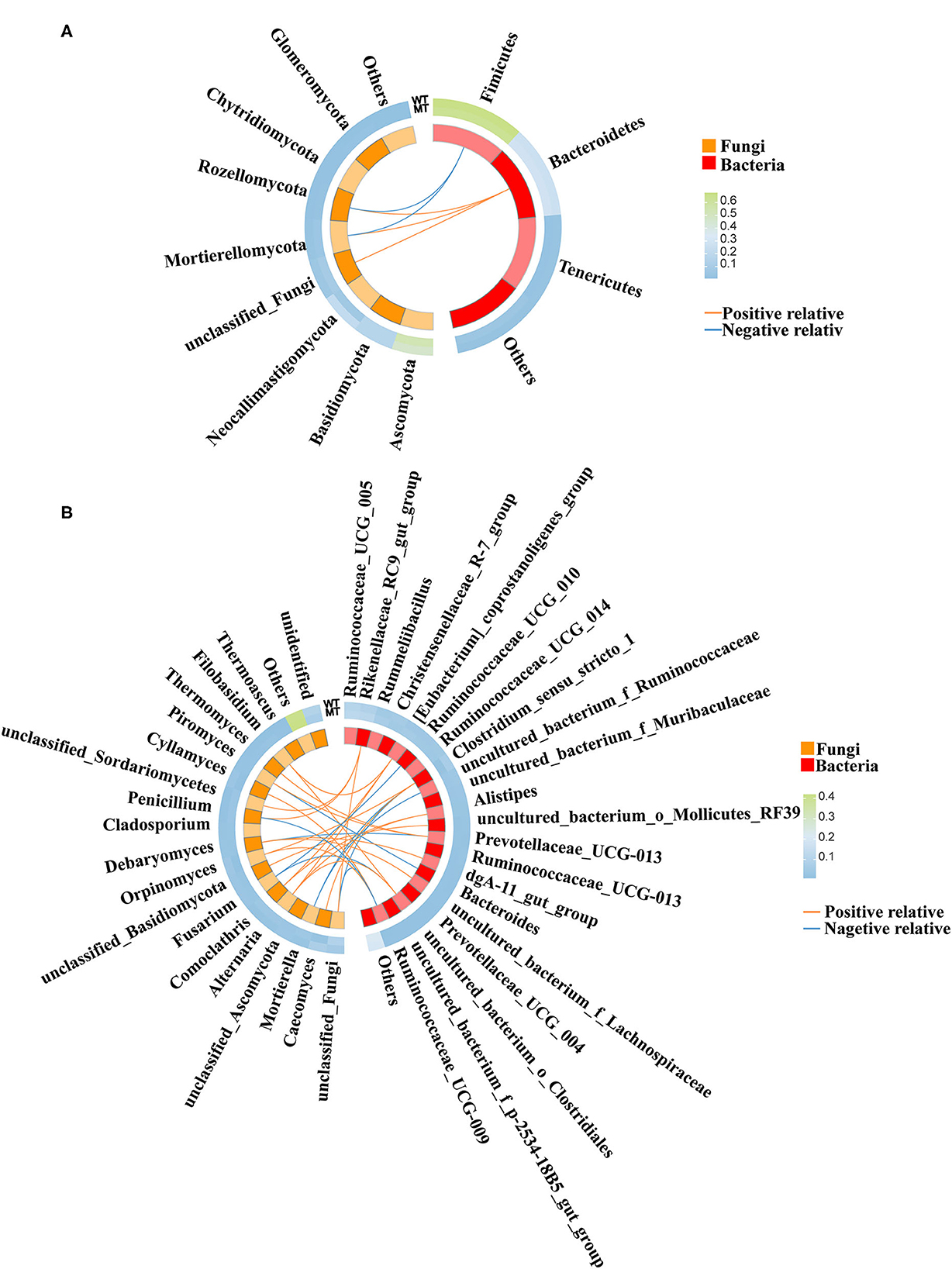
Figure 7. The correlation between the gut fungal communities and gut bacterial communities. (A) Correlation between gut fungal communities and gut bacterial communities at the phylum level. (B) Correlation between gut fungal communities and gut bacterial communities at the genus level.
At the genus level (Figure 7B), Filobasidium and Alternaria were positively correlated with uncultured_bacterium_o_Clostridiales (P < 0.05). Unclassified_Basidiomycota was positively correlated with uncultured_bacterium_o_Mollicutes_RF39 (P < 0.05). Comoclathris and Alternaria were positively correlated with Alistipes (P < 0.05). Debaryomyces was positively correlated with uncultured_bacterium_f_Lachnospiraceae (P < 0.05). Thermomyces and Piromyces were positively correlated with Prevotellaceae_UCG-003 (P < 0.05). Caecomyces and Orpinomyces were positively correlated with Clostridium_sensu_stricto_1 (P < 0.05). Alternaria, Comoclathris, and Filobasidium were positively correlated with [Eubacterium]_coprostanoligenes_group (P < 0.05). Cyllamyces and Orpinomyces were positively correlated with Ruminococcaceae_UCG-009 (P < 0.05). Unclassified_fungi, Penicillium, and unclassified_Sordariomycetes were positively correlated with Rikenellaceae_RC9_gut_group (P < 0.05). Debaryomyces was positively correlated with dgA-11_gut_group (P < 0.05). Fusarium was negatively correlated with Ruminococcaceae_UCG-010 (P < 0.05). Piromyces was negatively correlated with Bacteroides (P < 0.05). Penicillium and unclassified_fungi were negatively correlated with Ruminococcaceae_UCG-009 (P < 0.05), Mortierella was negatively correlated with uncultured_bacterium_f_Ruminococcaceae (P < 0.05), Fusarium was negatively correlated with uncultured_bacterium_o_Clostridiales (P < 0.05), Mortierella and unclassified_fungi were negatively correlated with Clostridium_sensu_stricto_1 (P < 0.05), and Debaryomyces was negatively correlated with Prevotellaceae_UCG-003 (P < 0.05).
MSTN is a negative regulator of muscle development in several species (15–20) and MSTN has been used as a target gene in animals for high-yield meat production. Many studies have been conducted to inhibit its expression through various strategies (21–23). Studies showed that MSTN can regulate multiple growth and metabolic pathways, such as muscle development, bone development (24), glucose metabolism (25, 26), and fat metabolism (27).
Fungi make up a relatively small proportion of the intestinal tract but are essential for the homeostasis of the intestinal flora (4, 5, 28). The gut flora participates in the maintenance of host health by influencing the metabolism and synthesis of some nutrients, vitamins, and hormones; by removing drugs and toxic metabolites; and contributing to the development and maturation of host immune cells to protect against pathogens (29, 30). Additionally, intestinal fungi are closely related to bacteria, and their stable antagonistic and mutually beneficial symbiosis is fundamental to the homeostatic balance of intestinal microorganisms. The present study previously detected bacterial communities in the MSTN mutant cattle and found that MSTN mutations affected the metabolism of the host by affecting the composition of the bacteria (10). However, knowledge of the characteristics of the gut fungal community composition in the MSTN mutation cattle is still limited. In the current study, ITS high-throughput sequencing was performed to investigate the fungal community composition of MT cattle. Furthermore, the relevance of the gut fungal community composition and bacterial composition was analyzed.
According to the alpha diversity analysis, no notable difference between the richness and diversity of the observed OTUs in the fecal samples of the WT and MT cattle was found, suggesting that the MSTN mutation had no remarkable effect on the gut fungal richness and diversity. The NMDS results based on the binary Jaccard analysis indicated that there were notable differences in the composition of the fungal community between the MT and WT cattle.
At the phylum level, Ascomycota, Basidiomycota, and Neocallimastigomycota were the predominant fungal phyla in the intestinal tracts of both the WT and MT cattle. This fact is consistent with previous studies on the dominant fungi of herbivores such as cattle (31), sheep (32), Tibetan piglets (33), and highland animals such as horses, yaks (34), and adult beach sheep (35). However, the relative abundance of Ascomycota and Neocallimastigomycota between the WT and MT cattle showed dramatic differences; Ascomycota was significantly higher in the MSTN mutation cattle compared to that in WT cattle, while Neocallimastigomycota was significantly lower in MSTN mutation cattle compared to that in WT cattle. Fungi at the phylum level in the digestive tract of ruminants help to depolymerize complex molecular structures such as lignocellulosic biomass, produce biogas and bioethanol, improve animal feed digestibility, and perform various other functions (36, 37). The direct feeding of anaerobic fungal cultures, such as Ascomycota, to ruminants can improve feed intake, milk quality, and milk production (38, 39). Members of Neocallimastigomycota usually live in the digestive tract of mammals (40, 41), and they are considered the most effective microbial group for degrading lignocellulose in the digestive tract of herbivores (42, 43). In the present study, the increased Ascomycota phylum and decreased Neocallimastigomycota phylum in MSTN mutation cattle may be one of the important reasons for the muscular development of MSTN gene-mutated cattle, but a detailed mechanism needs to be explored further.
At the genus level, unclassified_fungi, Mortierella, and Alternaria were the most predominant genera in the MT group, whereas Caecomyces, unclassified_fungi, and Orpinomyces were the most predominant genera present in the WT group. Unclassified_fungi, Mortierella, unclassified Sordariomycetes, and Penicillium were more predominant in the MT group compared to that in the WT group, while Caecomyces was lower in the MT group compared to that in the WT group (P < 0.05). All Alternaria, unclassified_Sordariomycetes, and Penicillium belong to the phylum Ascomycota, which are associated with improved animal production traits (38, 39). Mortierella is one of the most important fungi that produces polyunsaturated fatty acids (44, 45) and has various physiological functions, such as anti-inflammatory, anti-cancer, anti-coagulation, lowering blood lipids, and preventing cardiovascular diseases (46). Caecomyces and Orpinomyces belong to the phylum Neocallimastigomycota and function to decompose cellulose (40, 41). These results indicate that the mutation of MSTN in cattle not only improves the composition and proportion of microorganisms associated with meat quality production traits but also increases the content of beneficial fungi in the gut, which in turn regulates the metabolism of the body.
According to the prediction of fungal function, saprotroph fungi were significantly more abundant in the MT group than in the WT group. As a well-known active decomposer in the ecosystem, saprophytic fungi can decompose organic compounds such as animal and plant residues (47). They live on various dead organic substances, secrete digestive enzymes into these substances, and decompose them into small molecules such as glucose, which can be used by saprophytic fungi and other competitors (48). In this study, we found that the abundance of saprophytic fungi in the intestines of MT cattle was significantly increased compared to a similar abundance in the intestines of the WT cattle. Therefore, it was speculated that complex macromolecular organics can be degraded more efficiently and fully in MT cattle, allowing more nutrients to be absorbed and utilized by the host through the intestines, thus leading to enhanced growth and metabolism of the host.
Microbes inhabiting the intestinal tract can form symbiotic, synergistic, or antagonistic relationships that maintain the stability of the gut environment (49). For this reason, alterations in gut fungi and bacteria can influence the functions of other fungi and bacteria by regulating the interactions between microorganisms. The correlation analysis between the gut fungal and bacterial communities in the present study suggested that significant correlations were noticed between some conspicuously or not significantly changed fungi and bacteria influenced by the MSTN mutation, which may additionally influence global gut functions. These results suggested that the MSTN mutation changed the gut fungal and bacterial compositions directly while indirectly influencing some fungi and bacteria through the interaction of microorganisms, which affected the host metabolism further.
The ITS gene high-throughput sequencing was conducted to explore the fungal community composition in the fecal samples of the MSTN mutant (MT) and wild-type (WT) cattle in this study. The gut fungi analysis revealed that the MSTN mutant had no effect on the richness and diversity of gut fungi. However, compared to wild type (WT) cattle, the composition of gut fungal community was changed in MSTN gene mutant (MT) cattle. According to the prediction of fungal function, the abundance of saprotroph fungi was significantly higher in the MT group compared to that in the WT group. The correlation analysis between the gut fungal and bacterial communities revealed that the MSTN mutation directly changed the gut fungal compositions and indirectly influenced some fungi and bacteria by regulating the interaction of microorganisms, which affect the host metabolism further. This study also analyzed the role of MSTN mutation in regulating the host metabolism of intestinal fungi and provided a theoretical basis for the relationship between the MSTN and gut fungi.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal study was reviewed and approved by Animal Ethics and Welfare Committee (AEWC) of Inner Mongolia University and Baotou Teachers College. Written informed consent was obtained from the owners for the participation of their animals in this study.
LG and TW designed the research and wrote the manuscript. SW, LW, ZL, and MY collected the samples. LG and LY analyzed the data. GL and LY revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the National Natural Science Foundation of China (No. 31860013), the Natural Science Foundation of China, Inner Mongolia Autonomous Region of China (Nos. 2021BS08013 and 2021MS03078), and the High-level Talents Introduced Scientific Research Startup Fund Project of Baotou Teacher's College (No. BTTCRCQD2018-001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.1084945/full#supplementary-material
ITS, Internal Transcribed Spacer; OTUs, operational taxonomic units; MT, MSTN mutant; WT, wild type; LEfSe, Linear discriminant analysis (LDA) effect size; NMDS, Non-metric multidimensional scaling; LDA, Linear discriminant analysis; PCR, polymerase chain reaction.
1. Ventura M, O'Flaherty S, Claesson MJ, Turroni F, Klaenhammer TR, van Sinderen D, et al. Genome-scale analyses of health-promoting bacteria:probiogenomics. Nat Rev Microbiol. (2009) 7:61–71. doi: 10.1038/nrmicro2047
2. Kong HH, Segre JA. Cultivating fungal research. Science. (2020) 368:365–6. doi: 10.1126/science.aaz8086
3. Moreno-Sabater A, Autaa G, Sterlin D, Jerbi A, Villette R, Holm JB, et al. Systemic anti-commensal response to fungi analyzed by flow cytometry is related to gut mycobiome ecology. Microbiome. (2020) 8:159. doi: 10.1186/s40168-020-00924-8
4. Qiu X, Zhang F, Yang X, Wu N, Jiang W, Li X, et al. Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Sci Rep. (2015) 5:10416. doi: 10.1038/srep10416
5. Koon HW, Su B, Xu C, Mussatto CC, Tran DH, Lee EC, et al. Probiotic Saccharomyces boulardii CNCM I-745 prevents outbreak-associated Clostridium difficile-associated cecal inflammation in hamsters. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G610–23. doi: 10.1152/ajpgi.00150.2016
6. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. (2021) 19:55–71. doi: 10.1038/s41579-020-0433-9
7. Suhr MJ, Hallen-Adams HE. The human gut mycobiome:pitfalls and potentials—A mycologist's perspective. Mycologia. (2015) 107:1057–73. doi: 10.3852/15-147
8. Beyer TA, Narimatsu M, Weiss A, David L, Wrana JL. The TGFβ superfamily in stem cell biology and early mammalian embryonic development. Biochim Biophys Acta. (2013) 1830:2268–79. doi: 10.1016/j.bbagen.2012.08.025
9. Li GP, Bai CL, Wei ZY, Su GH, Wu YX, Han LD, et al. Study on gene editing of yellow cattle myostatin. J Inner Mongolia Univ (Nat Sci Ed). (2020) 51:12–32. doi: 10.13484/j.nmgdxxbzk.20200102
10. Wen T, Mao C, Gao L. Analysis of the gut microbiota composition of myostatin mutant cattle prepared using CRISPR/Cas9. PLoS ONE. (2022) 17:e0264849. doi: 10.1371/journal.pone.0264849
11. Zhou X, Gu M, Zhu L, Wu D, Yang M, Gao Y, et al. Comparison of microbial community and metabolites in four stomach compartments of myostatin-gene-edited and non-edited cattle. Front Microbiol. (2022) 13:844962. doi: 10.3389/fmicb.2022.844962
12. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2:High-resolution sample inference from Illumina amplicon data. Nat Methods. (2016) 13:581–3. doi: 10.1038/nmeth.3869
13. Reproducible i. scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
14. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
15. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. (1997) 94:12457–61. doi: 10.1073/pnas.94.23.12457
16. Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. (2004) 350:2682–8. doi: 10.1056/NEJMoa040933
17. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. (2006) 38:813–8. doi: 10.1038/ng1810
18. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. (1997) 387:83–90. doi: 10.1038/387083a0
19. Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. (2007) 3:e79. doi: 10.1371/journal.pgen.0030079
20. Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. (1997) 7:910–6. doi: 10.1101/gr.7.9.910
21. Wang K, Tang X, Xie Z, Zou X, Li M, Yuan H, et al. CRISPR/Cas9-mediated knockout of myostatin in Chinese indigenous Erhualian pigs. Transgenic Res. (2017) 26:799–805. doi: 10.1007/s11248-017-0044-z
22. Yu B, Lu R, Yuan Y, Zhang T, Song S, Qi Z, et al. Efficient TALEN-mediated myostatin gene editing in goats. BMC Dev Biol. (2016) 16:26. doi: 10.1186/s12861-016-0126-9
23. Wang X, Niu Y, Zhou J, Zhu H, Ma B, Yu H, et al. CRISPR/Cas9-mediated MSTN disruption and heritable mutagenesis in goats causes increased body mass. Anim Genet. (2018) 49:43–51. doi: 10.1111/age.12626
24. Tang L, An S, Zhang Z, Fan X, Guo J, Sun L, et al. MSTN is a key mediator for low-intensity pulsed ultrasound preventing bone loss in hindlimb-suspended rats. Bone. (2021) 143:115610. doi: 10.1016/j.bone.2020.115610
25. Xin XB, Yang SP Li X, Liu XF, Zhang LL, Ding XB, et al. Proteomics insights into the effects of MSTN on muscle glucose and lipid metabolism in genetically edited cattle. Gen Comp Endocrinol. (2020) 291:113237. doi: 10.1016/j.ygcen.2019.113237
26. Yang S, Li X, Liu X, Ding X, Xin X, Jin C, et al. Parallel comparative proteomics and phosphoproteomics reveal that cattle myostatin regulates phosphorylation of key enzymes in glycogen metabolism and glycolysis pathway. Oncotarget. (2018) 9:11352–70. doi: 10.18632/oncotarget.24250
27. Lipina C, Kendall H, McPherron AC, Taylor PM, Hundal HS. Mechanisms involved in the enhancement of mammalian target of rapamycin signalling and hypertrophy in skeletal muscle of myostatin-deficient mice. FEBS Lett. (2010) 584:2403–8. doi: 10.1016/j.febslet.2010.04.039
28. Che Y, Liu K, Li QR. The development of fungal community in intestinal diseases. Parent Enteral Nutr. (2015) 22:378–81. doi: 10.16151/j.1007-810x.2015.06.016
29. Molloy MJ, Bouladoux N, Belkaid Y. Intestinal microbiota:shaping local and systemic immune responses. Semin Immunol. (2012) 24:58–66. doi: 10.1016/j.smim.2011.11.008
30. Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, et al. Immunological consequences of intestinal fungal dysbiosis. Cell Host Microbe. (2016) 19:865–73. doi: 10.1016/j.chom.2016.05.003
31. Richardson MJ. Diversity and occurrence of coprophilous fungi. Mycol Resm. (2001) 105:387–402. doi: 10.1017/S0953756201003884
32. Kittelmann S, Naylor GE, Koolaard JP, Janssen PH. A proposed taxonomy of anaerobic fungi (class Neocallimastigomycetes) suitable for large-scale sequence-based community structure analysis. PLoS ONE. (2012) 7:e36866. doi: 10.1371/journal.pone.0036866
33. Kong QH, Liu Y, Suolang SZ, Liu SZ, Tan ZH, Shang P, et al. Fungal diversity in Tibetan piglet fecal samples. Mycosystema. (2020) 39:1241–9. doi: 10.13346/j.mycosystema.190406
34. Yang X, Dai X, Liu L, Wang XW. Fungal diversity in herbivore feces in the Tibetan Plateau. Mycosystema. (2014) 33:621–31. doi: 10.13346/j.mycosystema.130105
35. Fu ZL, Chen L, Li N. Changes of rumen fungi diversity and microflora structure of adult Tan sheep under different feeding modes. Chin J Animal Sci. (2020) 56:209–13. doi: 10.19556/j.0258-7033.20191015-05
36. Mangiola F, Ianiro G, Franceschi F, Fagiuoli S, Gasbarrini G, Gasbarrini A. Gut microbiota in autism and mood disorders. World J Gastroenterol. (2016) 22:361–8. doi: 10.3748/wjg.v22.i1.361
37. Gomez DE, Arroyo LG, Costa MC, Viel L, Weese JS. Characterization of the fecal bacterial microbiota of healthy and diarrheic dairy calves. J Vet Intern Med. (2017) 31:928–39. doi: 10.1111/jvim.14695
38. Thareja A, Puniya AK, Goel G, Nagpal R, Sehgal JP, Singh PK, et al. In vitro degradation of wheat straw by anaerobic fungi from small ruminants. Arch Anim Nutr. (2006) 60:412–7. doi: 10.1080/17450390600884443
39. Zhang J, Shi H, Wang Y, Li S, Cao Z, Ji S, et al. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in holstein heifers. Front Microbiol. (2017) 8:2206. doi: 10.3389/fmicb.2017.02206
40. Saye LMG, Navaratna TA, Chong JPJ, O'Malley MA, Theodorou MK, Reilly M. The anaerobic fungi: challenges and opportunities for industrial lignocellulosic biofuel production. Microorganisms. (2021) 9:694. doi: 10.3390/microorganisms9040694
41. Wilken SE, Monk JM, Leggieri PA, Lawson CE, Lankiewicz TS, Seppälä S, et al. Experimentally validated reconstruction and analysis of a genome-scale metabolic model of an anaerobic neocallimastigomycota fungus. mSystems. (2021) 6:e00002–21. doi: 10.1128/mSystems.00002-21
42. Bauchop, T. The anaerobic fungi in rumen fibre digestion. Agricult Environ. (1981) 6:339–48. doi: 10.1016/0304-1131(81)90021-7
43. Wood TM, Wilson CA. Studies on the capacity of the cellulase of the anaerobic rumen fungus piromonas communis p to degrade hydrogen bond-ordered cellulose. Appl Microbiol Biotechnol. (1995) 43:572–8. doi: 10.1007/BF00218468
44. Zhang H, Cui Q, Song X. Research advances on arachidonic acid production by fermentation and genetic modification of Mortierella alpina. World J Microbiol Biotechnol. (2021) 37:4. doi: 10.1007/s11274-020-02984-2
45. Zhang H, Wang Z, Feng Y, Cui Q, Song X. Phytohormones as stimulators to improve arachidonic acid biosynthesis in Mortierella alpina. Enzyme Microb Technol. (2019) 131:109381. doi: 10.1016/j.enzmictec.2019.109381
46. Suresh Y, Das UN. Protective action of arachidonic acid against alloxan-induced cytotoxicity and diabetes mellitus. Prostaglandins Leukot Essent Fatty Acids. (2001) 64:37–52. doi: 10.1054/plef.2000.0236
47. Chen MR, Lin YH, He XB, Han GM. Colonization of endophytic fungi in Chinese fir leaves Effects on litter decomposition and microbial activity. Microbiol Bull. (2020) 47:1404–17. doi: 10.13344/j.microbiol.china.190867 (in Chinese)
48. Franc, P. Why do saprophytic fungi have excellent medicinal properties? Edible Med Fungi. (2019) 27:363–5.
Keywords: MSTN mutant, gut fungi, cattle, ITS, high-throughput sequencing, correlation
Citation: Gao L, Wang S, Yang M, Wang L, Li Z, Yang L, Li G and Wen T (2023) Gut fungal community composition analysis of myostatin mutant cattle prepared by CRISPR/Cas9. Front. Vet. Sci. 9:1084945. doi: 10.3389/fvets.2022.1084945
Received: 17 November 2022; Accepted: 23 December 2022;
Published: 17 January 2023.
Edited by:
Guillermo Tellez-Isaias, University of Arkansas, United StatesReviewed by:
Santiago Uribe-Diaz, University of Arkansas, United StatesCopyright © 2023 Gao, Wang, Yang, Wang, Li, Yang, Li and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Yang,  bXJrbm93YWxsQDEyNi5jb20=; Guangpeng Li, Z3BlbmdsaUBpbXUuZWR1LmNu; Tong Wen, V2VudG9uZzEwMjlAc2luYS5jb20=
bXJrbm93YWxsQDEyNi5jb20=; Guangpeng Li, Z3BlbmdsaUBpbXUuZWR1LmNu; Tong Wen, V2VudG9uZzEwMjlAc2luYS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.