
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 04 January 2023
Sec. Veterinary Pharmacology and Toxicology
Volume 9 - 2022 | https://doi.org/10.3389/fvets.2022.1066048
This article is part of the Research Topic Toxicology of Heavy Metals and Intestinal Microbiome View all 4 articles
 Yiran Zhu1†
Yiran Zhu1† Kaimin Song2†
Kaimin Song2† Guodong Cheng1
Guodong Cheng1 Huiling Xu1
Huiling Xu1 Xiaozhou Wang2
Xiaozhou Wang2 Changxi Qi1
Changxi Qi1 Pu Zhang3*
Pu Zhang3* Yongxia Liu2*
Yongxia Liu2* Jianzhu Liu1*
Jianzhu Liu1*Introduction: Hexavalent chromium or Cr(VI) is essential to various industries, such as leather manufacturing and stainless steel production. Given that inevitable leakage from industries pollutes the soil and thereby affects the soil environment. Microbial communities could improve the quality of the soil. Abundant bacterial communities would significantly enhance the soil richness and resist external pressure, benefiting agriculture. But the pollution of heavy metal broke the balance and decrease the abundance of bacterial communities, which weak the self-adjust ability of soil. This study aimed to explore changes in the diversity of soil bacterial communities and to identify the influences of soil bacterial communities on enzymes in soil polluted by Cr(VI).
Methods: The target soils were sampled quickly and aseptically. Their chromium content was detected through inductively coupled plasma-mass spectrometry, and bacterial microbiome communities were explored through MiSeq high-throughput sequencing. Then, the content of nitrite reductase and catalases were investigated through enzyme-linked immunosorbent assay (ELISA).
Results: Chromium content in polluted soils was higher than that in the control soils at all depths. Sobs, Chao1, Ace, and Shannon diversity estimators in the control were higher, whereas Simpson's diversity estimators in the control soils were lower than those of contaminated samples at all depths. Contaminants affected the composition of the bacterial community. The soil microbial species were relatively single and inhomogeneous in the polluted soils. The bacterial phyla in polluted and controlled soils include Proteobacteria, Actinobacteria, Chloroflexi, and Acidobacteria, which differ markedly in abundance.
Discussion: The results of these observations provide insights into the ecotoxicological effects of Cr(VI) exposure to soil microorganisms. To sum up these results are critical for evaluating the stabilized state of microbial community structures, contributing to the assessment of the potential risk of metal accumulation in soils.
Heavy metals are naturally occurring elements, including non-essential elements, such as lead (Pb), cadmium (Cd), and mercury (Hg), and biologically necessary elements, such as copper (Cu), manganese (Mn), and zinc (Zn). Some heavy metals in soil are physiologically necessary for plants and animals, exerting indirect or direct effect on agricultural output and public health. To date, concern about heavy metal pollution of soil has grown throughout the world (1).
Heavy metal pollution is a serious problem because threatens the natural environment, especially greenhouse soils, and human health by generating toxicity and promoting bioaccumulation (2). Soil contaminations threaten consumer health through food chain accumulation (3, 4). Large quantities of heavy metals negatively impact soils by reducing their productivity, especially percentage of seed germination, and bacterial biomass (5, 6). Previous research employing fingerprinting techniques demonstrated that heavy metal contamination has immediate and long-term consequences on terrestrial microbial communities (7, 8). The variety and structure of microorganisms are considerably altered after heavy metal pollution (8–10). However, few studies have revealed interactions among microorganisms in heavy metal-contaminated soils, and the energy and nutrition cycling of soil microbes are unknown.
In heavy metal-polluted soil, exceeding threshold levels of heavy metals, such as Cd and Pb, may have a deleterious impact on the quantity and quality (function and diversity) of the microbiota. Specifically, the high accumulation of significant metals might decrease microorganism community diversity, reducing resistance and resilience to environmental stress. Cr(VI) is the seventh most abundant element and is abundant in the Earth's crust (11). It is one of the most highly carcinogenic and soluble elements that exist as oxyanions, such as CrO and Cr2O (12). Cr(VI) was widespread use in some industries, such as leather manufacturing and stainless steel production; inevitable leakage through industrial wastewater leads to soil pollution, which further affects the soil environment (13).
Soil microbial communities improve soil quality (14, 15). The diversity of soil microbial communities is related to soil properties (16). However, how soil bacterial communities respond to changes in heavy metals, especially Cr(VI), remains uncertain. Soil bacterial communities are crucial indicators for soil pollution and can be investigated through Illumina high-throughput sequencing. In this study, Cr(VI)-polluted soil samples were examined via high-throughput sequencing technology, and the responses of soil bacterial communities to Cr(VI) were determined. The richness and evenness of microbial composition were analyzed in uncontaminated and polluted soils collected from three depths.
In the current study, we used the Illumina MiSeq technique to analyze microbial communities in abandoned and heavy metal-contaminated soils. We postulated that interactions between microbes and heavy metal contamination in abandoned factory soils impact microbial communities and that these altered interactions may have enabled bacteria to adapt to heavy metals.
This study was conducted in an abandoned factory (42° 03′N, 123° 29′E) in Shenyang, Liaoning Province, China. This region typically has a north temperate continental monsoon climate, characterized by a dry and cold winter, mild and rainy summer, and strong winds in spring and autumn. Two land types (uncontaminated and polluted) and three depths (0–20, 20–40, and 40–60 cm) were used in exploring the effects of Cr(VI) on soil bacterial communities. Using the five-point sampling approach and soil samplers, we collected 18 soil samples (50 g each). The samples were separated into two portions after homogenization and then placed in the dark. One portion was stored at 4°C; and the other, at room temperature. Each land type had three replicated sites (Table 1).
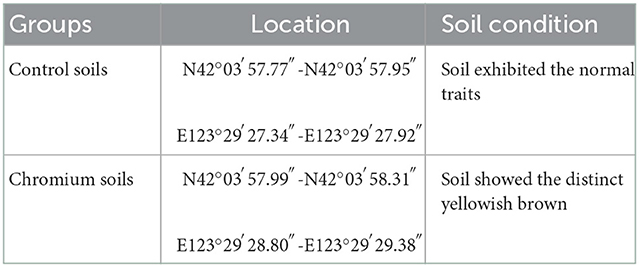
Table 1. Attribute of samples collected in the abandoned factory.
Microwave digestion and inductively coupled plasma-mass spectrometry (ICP-MS) were conducted. In brief, soils (0.2500 g, dried, filtered through a 0.15-mm nylon screen) were digested in a disintegration tank containing 11 mL of a 6:3:2 mixture of nitric, hydrochloric, and hydrofluoric acid. Then, the mixture was supplemented with ultrapure water and heated again to discharge acid. Finally, the solution was placed in a 50 mL volumetric flask, and the volume was determined. The chromium levels of the soils were analyzed through ICP-MS (Hewlett-Packard, HP-4500, Avondale, PA, USA) according to previous methods (17).
Soil microbial genomic DNA was isolated using an E.Z.N.A. soil DNA kit (Omega Bio-tek, Norcross, GA, U.S.) according to the manufacturer's instructions. DNA extracts were checked through 1% (w/v) agarose gel electrophoresis and stored for PCR amplification. The different regions of the 16S rRNA were amplified using special primers (V3–V4 regions for forward primers containing the sequence 5′-ACTCCTACGGGAGGCAGCAG-3′ and reverse primers 5′-GGACTACHVGGGTWTCTAAT-3′). The PCR protocol was as follows: 3 min denaturation at 95°C for the initial amplification, followed by 27 cycles at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s, and a final step at 72°C for 10 min. PCR products were purified using the AxyPrep DNA gel extraction kit (Axygen Biosciences, USA) according to the manufacturer's instructions and were quantified using the QuantiFluor-ST sensitive fluorometer (Promega, USA). High-throughput sequencing was performed on an Illumina MiSeq sequencing platform (Illumina, San Diego, USA) according to standard protocols. The obtained raw sequences were filtered according to quality.
The catalase (CAT) and nitrate reductase (NR) levels of the soils were determined using commercially available kits (Cat: ml076929 and ml076879, Mlbio, Shanghai, China). Other procedures were performed by strictly following the manufacturer's instructions.
Operational taxonomical units (OTUs) with 97% similarity were clustered using the algorithm UPARSE version 7.1 (http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. Alpha indices were acquired via Mothur 1.30.2 (https://www.mothur.org/wiki/Download_mothur). Tables for each taxonomy and beta diversity distance calculation were analyzed with Quiime 1.9.1 (http://qiime.org/install/index.html). The data were analyzed on the online platform of Majorbio Cloud Platform (www.majorbio.com) (18–21). The results are presented as the mean ± standard deviation and were checked by one-way ANOVA or student's t-test (t-test) using Statistical Package for the Social Sciences (version 19.0; SPSS Inc., Chicago, IL, USA). P < 0.05 indicated statistical significance.
The measured soil chromium contents varied substantially between the two land types (uncontaminated and polluted) and among the three depths (0–20, 20–40, and 40–60 cm). The concentrations of chromium increased in polluted soils at all depths compared with those in reference soils. Meanwhile, chromium content decreased with increasing depth (Table 2).
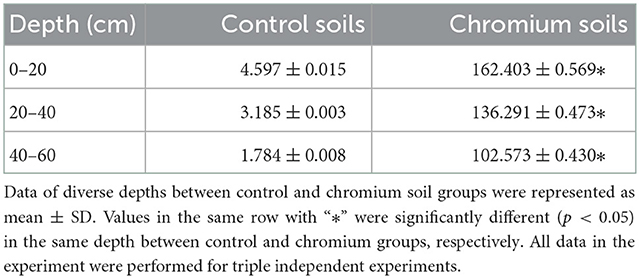
Table 2. The contents of Cr (mg/L) in different groups.
Rank abundance curves were obtained by ranking the number of species (or OTUs) at a certain taxonomic level as the horizontal coordinate and the relative percentages of the number of species at that taxonomic level as the vertical coordinates. Low-quality reads were removed from the OTUs, and investigation was conducted using rank abundance curves for the analysis of bacterial abundance and evenness (Figure 1). The curve width represented bacterial abundance, and the curve shape indicated the evenness of soil bacteria. Changes in total species and core species richness were described through Pan–Core species analysis (Figures 2A, B).
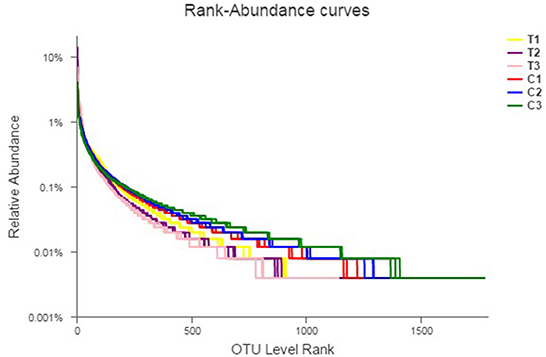
Figure 1. Rank-abundance curves describing the bacterial abundance and evenness. All data in the experiment represent three independent experiments.
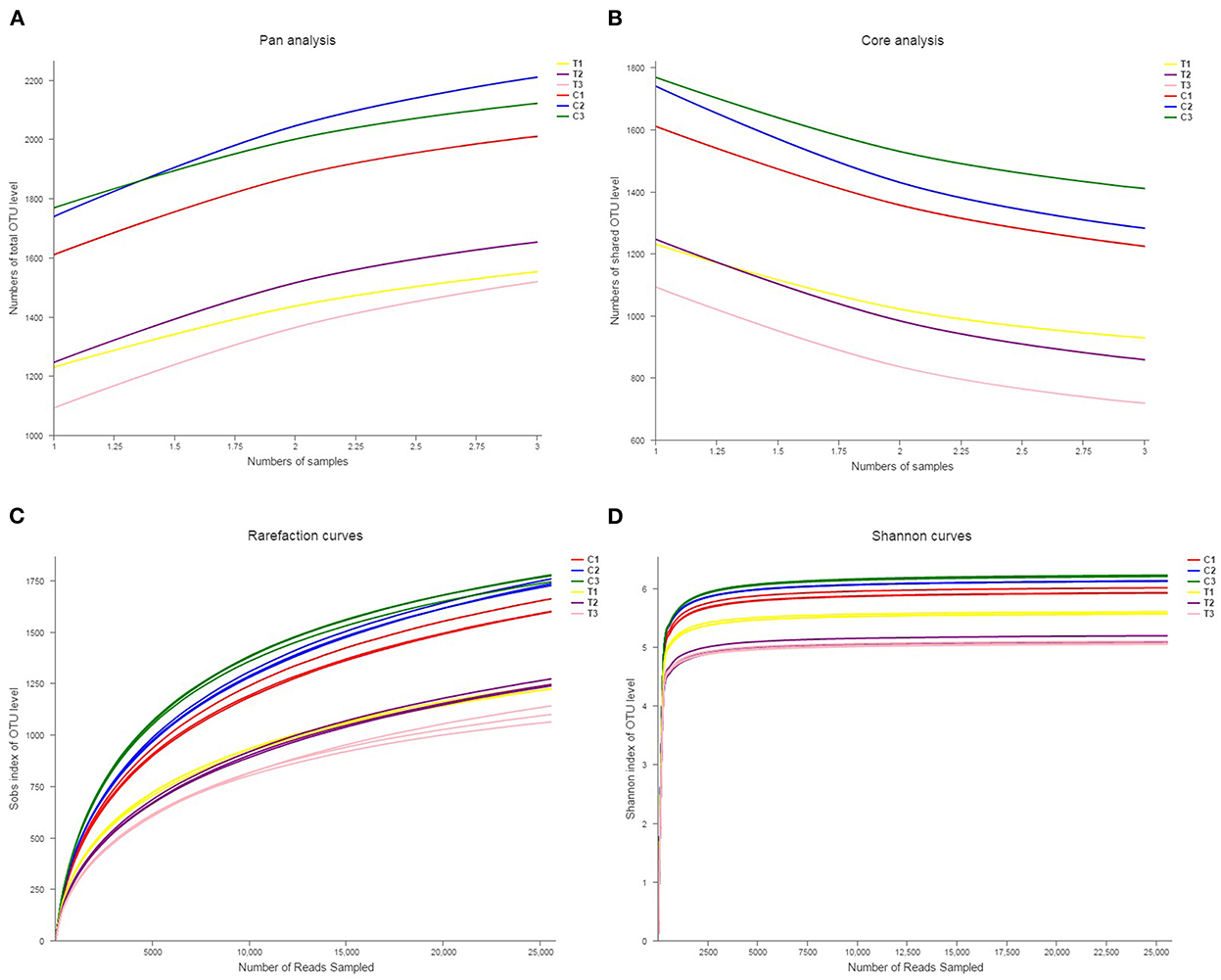
Figure 2. Pan-core species analysis was used to analyze total species and core species richness (A, B). The rarefaction curve was drawn to quantify the soils' OTU richness (C, D). All data in the experiment represent three independent experiments.
Alpha (α) diversity measurements were calculated to describe microbial diversity, such as OTU Sobs, Chao, Ace, Shannon, Simpson's, and coverage diversity estimators (Table 3). Initially, the coverage diversity indicator of each sample was over 98%, thereby indicating that the identified 16s rDNA sequences can represent the majority of bacteria present in the samples. Similarly, the uncontaminated samples had the highest Sobs, Shao, Ace, and Shannon diversity estimators but had the lowest Simpson's diversity estimator in all depths compared with polluted samples. Table 3 shows that the bacterial microbiomes ranged from 1,062 to 1,777 in the Sobs diversity estimator, 1,253.3 to 2,190.3 in the Chao diversity estimator, 5.04 to 6.22 Shannon diversity estimator, and 0.00501 to 0.0288 in the Simpson diversity estimator.
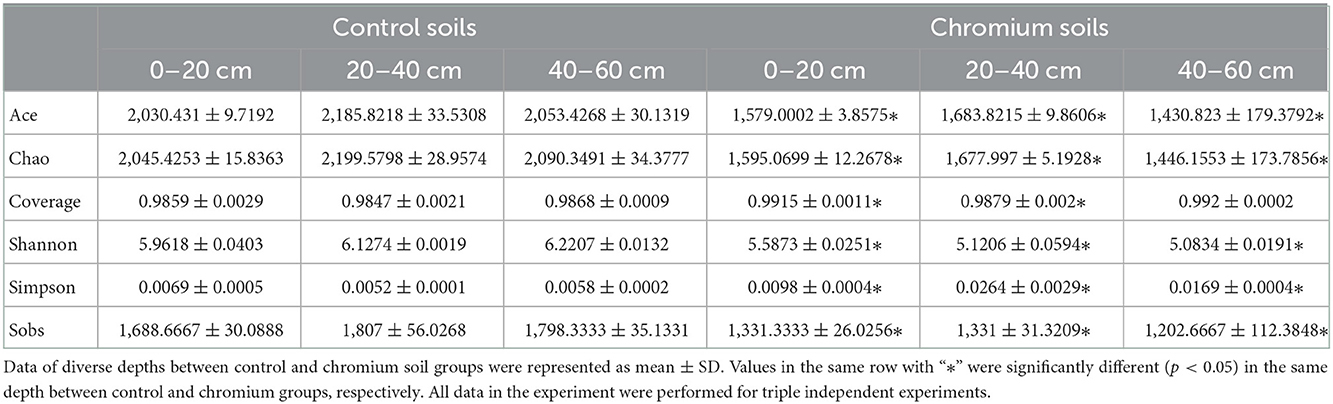
Table 3. Alpha diversity analysis for microbial diversity.
Rarefaction curve analysis was used in comparing the OTU indicators at different sequencing depths and taxon richness, and whether the acquired sequence amounts were sufficiently quantified was determined. The rarefaction curves in Figures 2C, D showed similar patterns for all soil samples, showing that soils were equally sampled and sufficiently sequenced.
As shown in Figure 3A, the Venn diagram used to evaluate out distribution in different treatments at the genus level reflected that 263 OTUs were common in all soil samples. In addition, up to 400 and 433 OTUs belong to uncontaminated and polluted soils, respectively. C1 and T1 had 366 OTUs, C2 and T2 had 377, and C3 and T3 had 367. However, C1, C2, C3, T1, T2, and T3 had 8, 4, 23, 10, 11, and 15 unique OTUs, respectively. At the genus levels, Acidobacteria was the dominant bacterium in all soil groups (C1–C3 and T1–T3, 8.06%, Figure 3B), control groups (C1–C3, 10.26%, Figure 3C), C1 and T1 groups (6.84%, Figure 3E), C2 and T2 groups (5.03%, Figure 3F), and C3 and T3 groups (7.80%, Figure 3G). However, Silanimonas was the dominant bacterium in the polluted soil groups (T1–T3, 8.28%, Figure 3D).
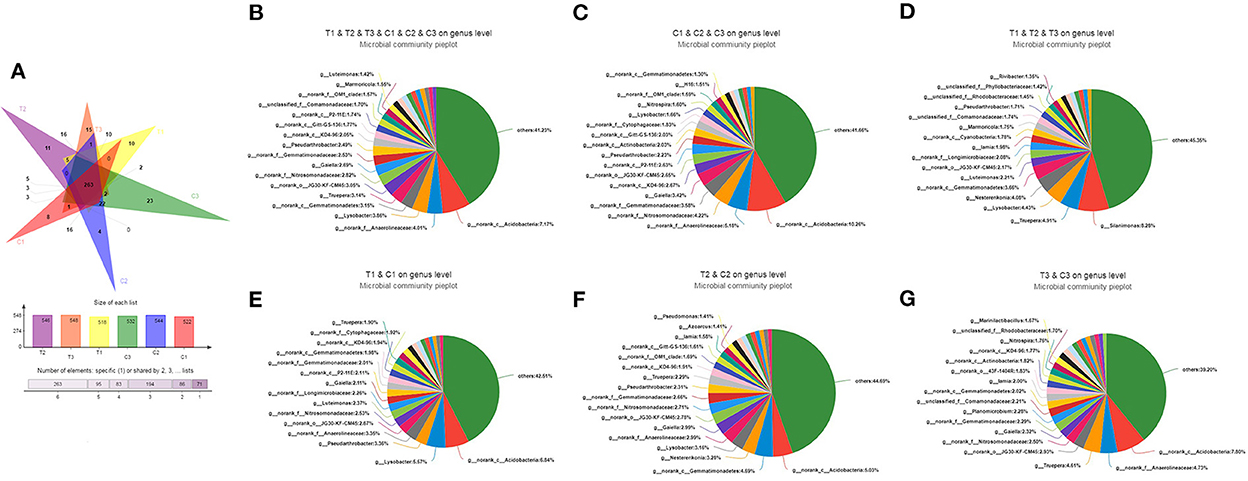
Figure 3. Venn diagram evaluating the distribution of OTUs among the different treatments to reflect the differences and similarities of soil samples (A). Pie diagrams were used to evaluate the distribution of genus among the C1, C2, C3, T1, T2, and T3 (B), C1, C2, and C3 (C), T1, T2, and T3 (D), C1 and T1 (E), C2 and T2 (F), and C3 and T3 (G), respectively. All data in the experiment represent three independent experiments.
The composition and structure of bacterial communities in the samples were analyzed at different taxonomic levels. Uncontaminated and contaminated groups showed analogous bacterial diversity but differed in terms of abundance to a certain extent (Figures 4, 6). Bacterial community analysis revealed relative abundance at the phylum level (Figure 4). Proteobacteria was the predominant phylum, and Actinobacteria was the secondary phylum. The relative abundance of bacterial phyla changed in the two soil types. In the C1 samples, Proteobacteria was 28.95% of the total bacteria, and the percentages of Chloroflexi in C1, C2, C3, T1, T2, and T3 were 21.55, 17.78, 22.24, 8.50, 6.37, and 4.97%, respectively. Acidobacteria and Chloroflexi decreased, whereas Proteobacteria and Deinococcus-Thermus were enriched in the Cr(VI)-polluted soils. At the phylum level, significance analysis of the relative abundance indicated that the significance levels of Actinobacteria, Gemmatimonadetes, Firmicutes, Deinococcus-Thermus, Cyanobacteria, and Nitrospirae in the chromium groups were higher than those in the control groups, whereas the significance levels of Chloroflexi, Acidobacteria, Bacteroidetes, and Planctomycetes were lower than those of the control groups. Additionally, Latescibacteria and Ignavibacteriae were unique in the control groups (Figure 5).
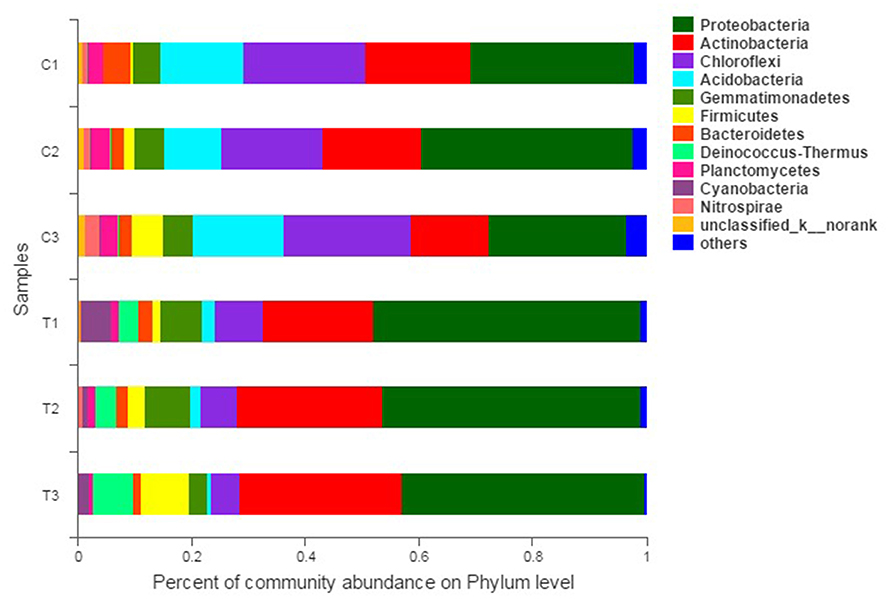
Figure 4. Bacterial community compositions and structures of soil samples. Bar diagrams were made to present the relative abundance levels at the phylum level of all samples. All data in the experiment represent three independent experiments.
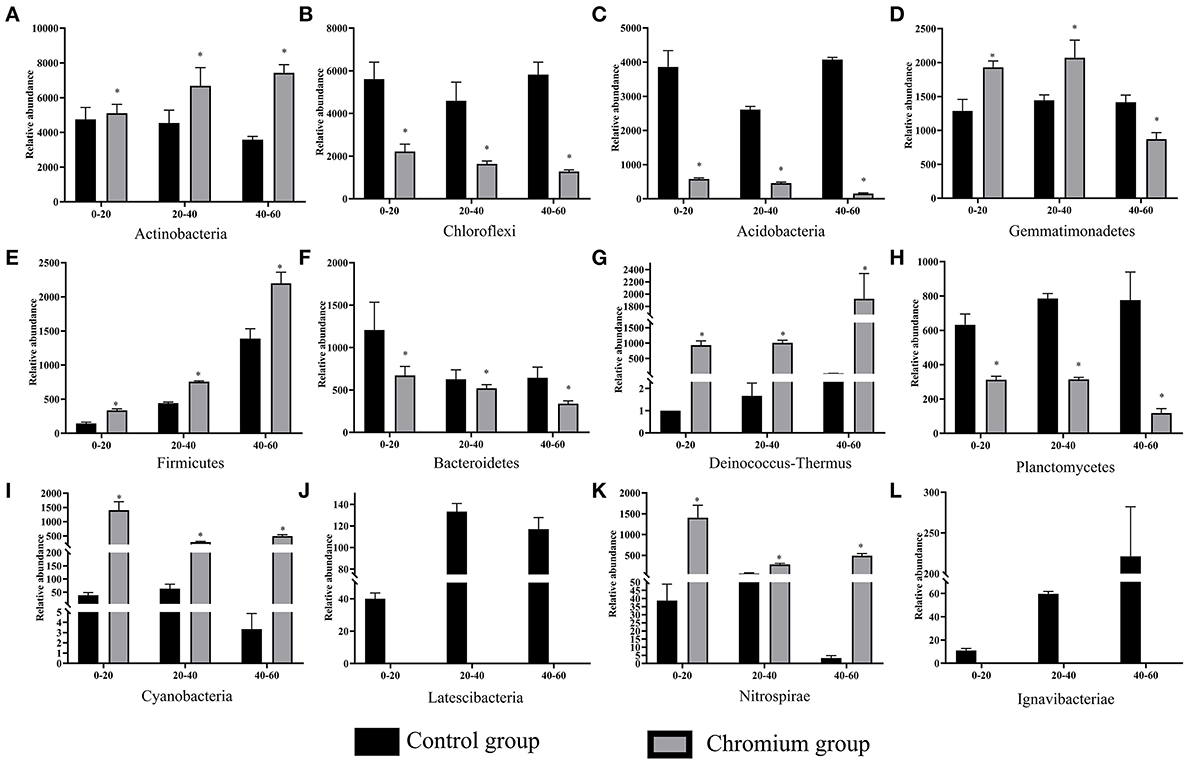
Figure 5. Significant differences in bacterial community composition (A–L) were analyzed at the phylum level. Values in the same row with “*” were significantly different (p < 0.05) at the same depth between the control and chromium groups, respectively. All data in the experiment represent three independent experiments.
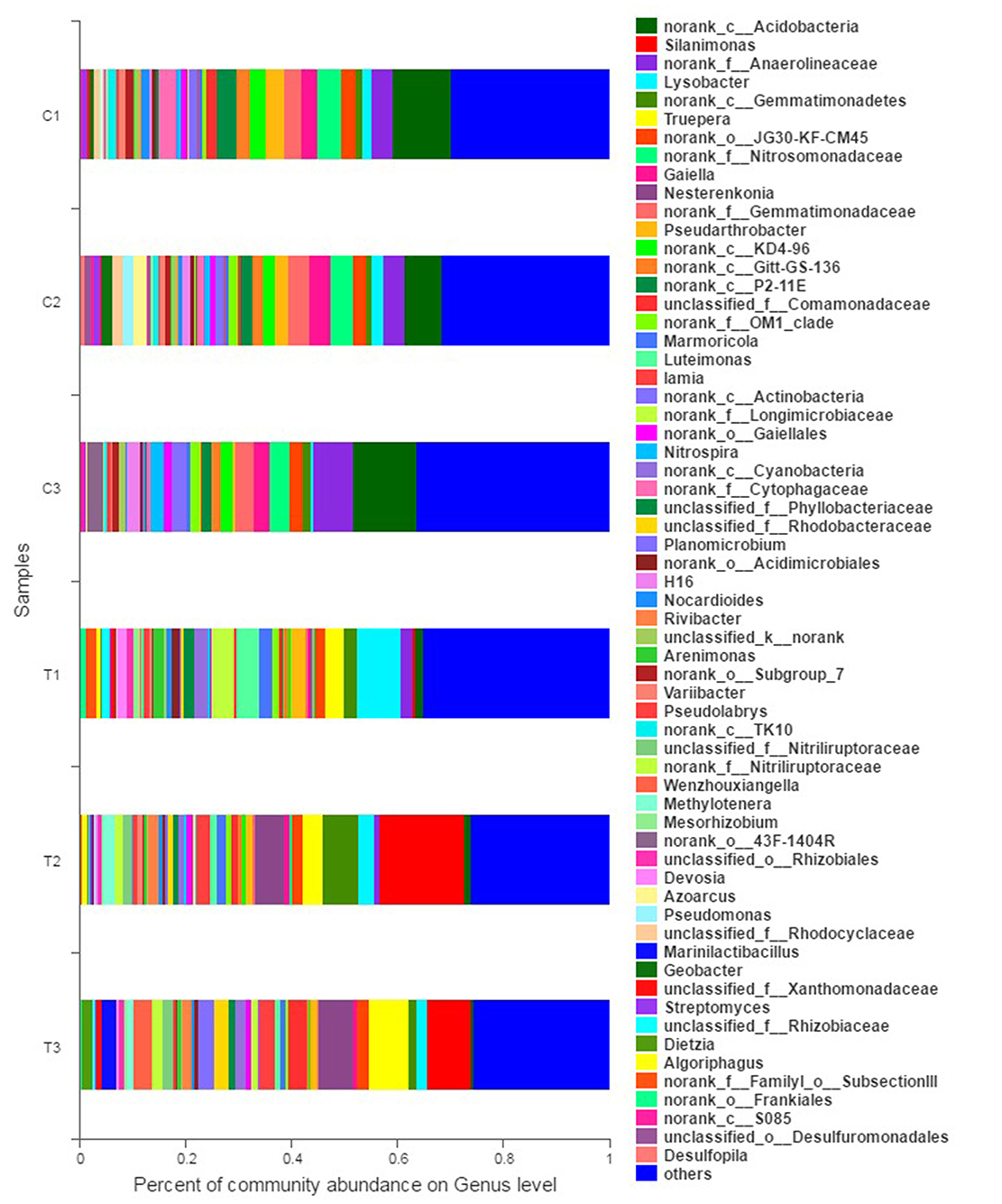
Figure 6. Bacterial community analysis was performed to present the relative abundance at the genus level. All data in the experiment represent three independent experiments.
The abundance of relative bacterial community at the genus level is illustrated in Figure 6. Norank Acidobacteria, norank Anaerolineaceae, and norank Nitrosomonadaceae were the dominant genera in the control groups (C1–C3). However, the dominant genera in polluted soils were diverse when the contaminants were input. Specifically, Lysobacter (8.33%), Luteimonast (4.36%), and norank Longimicrobiaceae (4.04%) were dominant in T1; Silanimonas (15.94%), norank Gemmatimonadetes (6.71%), and Nesterenkonia (5.45%) were dominant in T2; and Silanimonas (8.27%), Truepera (7.28%), and Nesterenkonia (6.52%) were dominant in T3.
A heatmap of microbial communities was further analyzed for the identification of the similarity and differences among bacterial community structures in the soil samples. The genera of the control groups (C1–C3) had a cluster pattern of bacterial community composition different from the pattern observed in polluted soils (T1–T3; Figure 7). At the genus level, the relative abundance of Methylotenera, Rivibacter, Nitriliruptoraceae, Wenzhouxiangella, Luteimonas, Silanimonas, Truepera, and Nesterenkonia in the chromium groups were higher than those in the control groups, whereas the relative abundance of Rhodocyclaceae, 43F-1404R, and H16 were lower than those in the control groups.
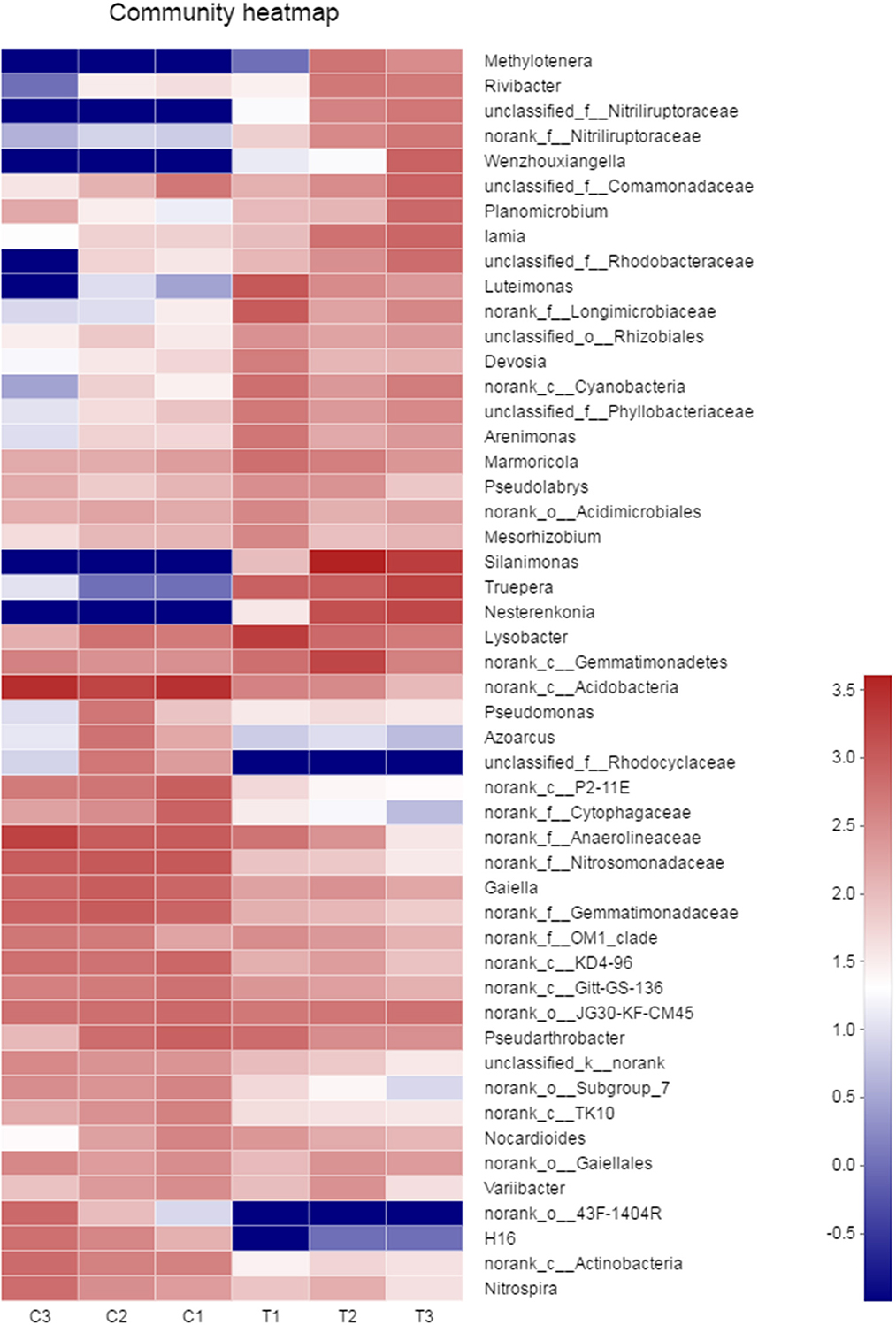
Figure 7. The similarity and differences in bacterial community structures in the soil samples were analyzed by heatmap. All data in the experiment represent three independent experiments.
Bacterial community composition varied by chromium content and soil depth. Therefore, the samples were separated by principal coordinate analysis (PCoA) and hierarchical clustering analysis, as illustrated in Figure 8B. At the OTU level, the PCoA indicated the clear separation of the bacterial communities in different groups. The microbiota of the T1, T2, and T3 samples was distinct from that of the C1, C2, and C3 samples. The first axis explained the 58.91% cumulative percentage variance of species, and the second axis explained 15.83%. The two axes explained a total of 74.74% variance of species. Meanwhile, further analysis of microbial communities was executed through a hierarchical clustering tree, which was used in identifying similarities and differences among bacterial community structures (Figure 8A). The microbial community structures in uncontaminated and polluted soils substantially differed, and these structures were divided into two clusters.
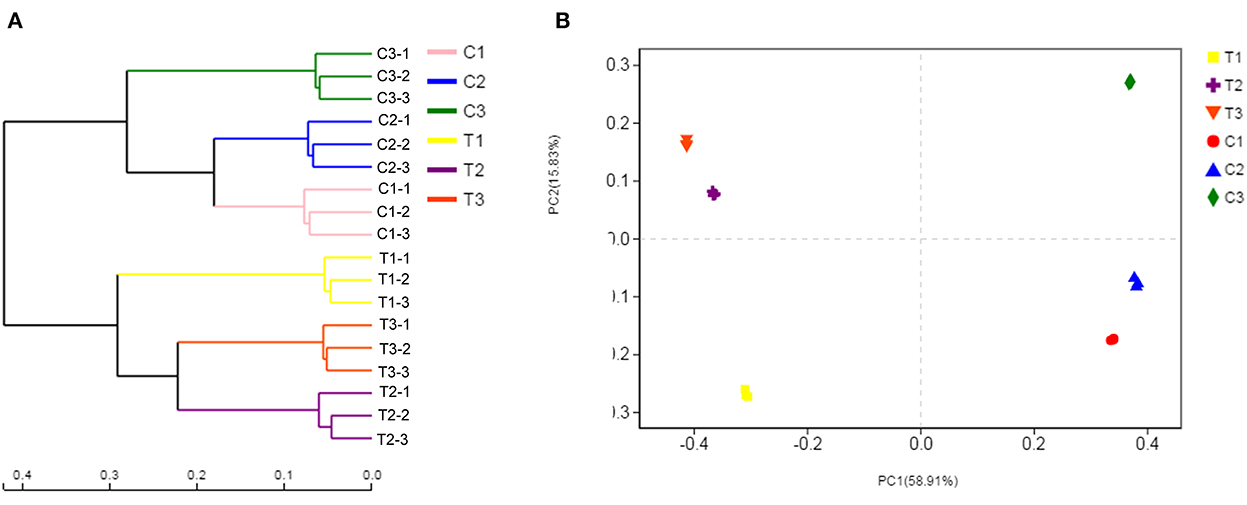
Figure 8. Soil samples were separated by hierarchical clustering analysis and PCoA. The similarity and differences in the bacterial community structures were determined by hierarchical clustering tree analysis (A). PCoA analysis of the bacterial communities at the OTU level (B). All data in the experiment represent three independent experiments.
A Kruskal-Wallis H-test was used for detecting richness exhibited by different samples of microbial communities at the phylum level. Proteobacteria, Actinobacteria, Chloroflexi, and Acidobacteria were the dominant bacterial phyla in all samples, but the C1–C3 and T1–T3 samples differed in the abundance of these bacteria (Figure 9). In addition, LDA effect size analysis (LEfSe) was used to discriminate between microbial communities in uncontaminated soils and those in polluted soils. Figures 10, 11 show differences in bacterial taxa. The dominant bacterial phyla were Bacteroidetes, Verrucomicrobia, Armatimonadetes, Chlorobi, and WS2 in C1; Planctomycetes and Latescibacteria in C2; Chloroflexi, Acidobacteria, and Nitrospirae in C3; Proteobacteria, Cyanobacteria, and BRC1 in T1; Gemmatimonadetes, BJ_169, M6__Dependentiae, and Parcubacteria in T2; and Actinobacteria, Deinococcus_Thermus, and Firmicutes in T3.
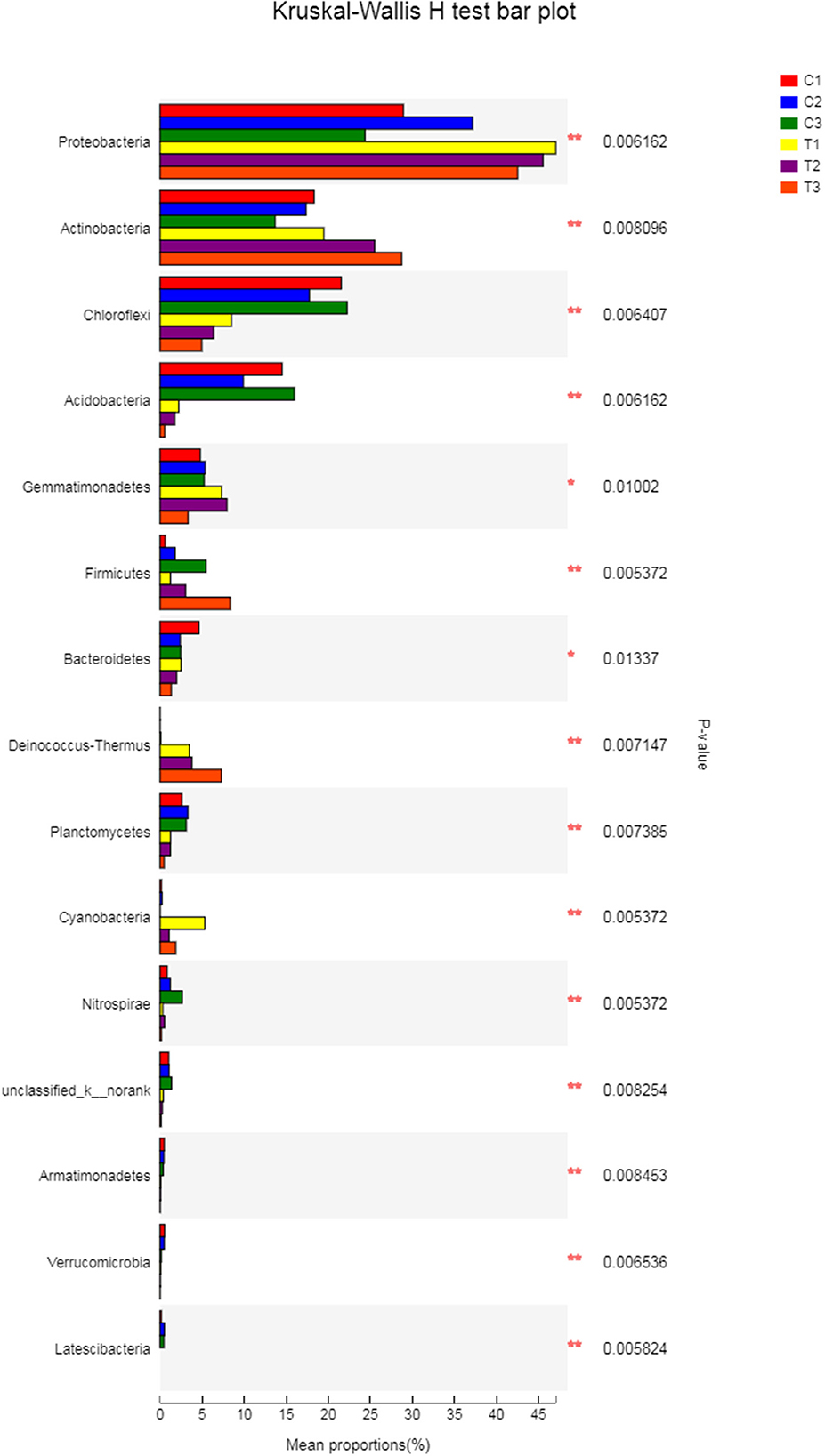
Figure 9. The Kruskal-Wallis H-test was chosen to detect the richness of microbial communities exhibited by different samples at the phylum level. All data in the experiment represent three independent experiments.
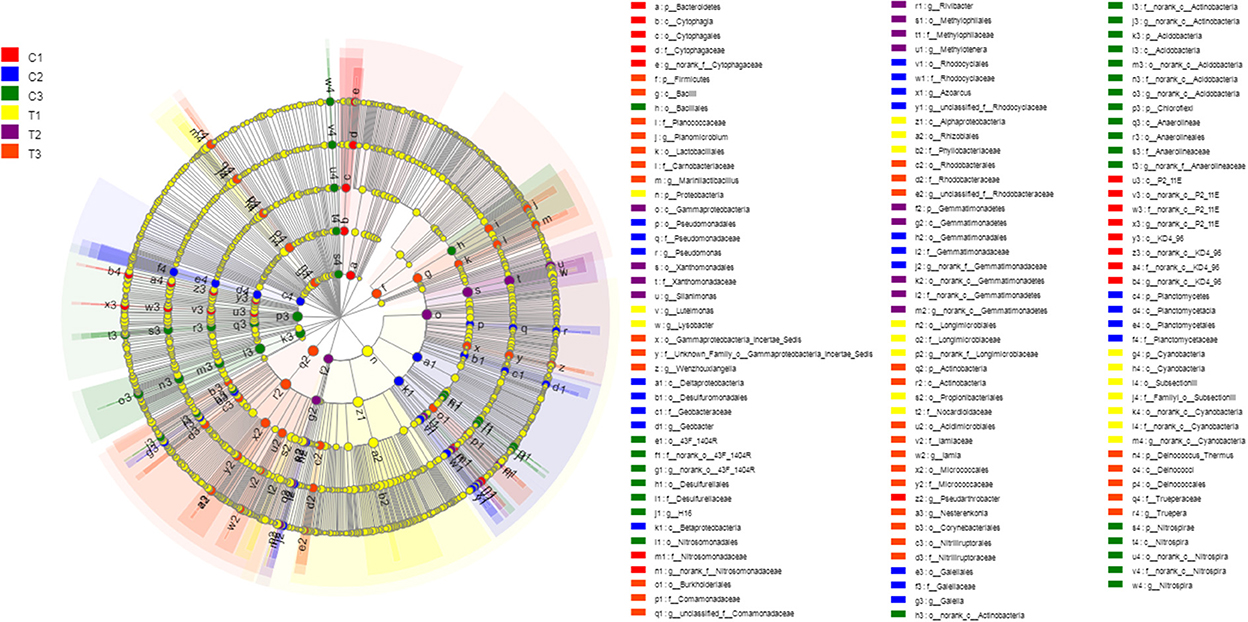
Figure 10. LEfSe was used to discriminate the microbial communities of all soil samples. Significantly affected microbial taxa were shown in LEfSe multi-level species hierarchy tree diagram. All data in the experiment represent three independent experiments.
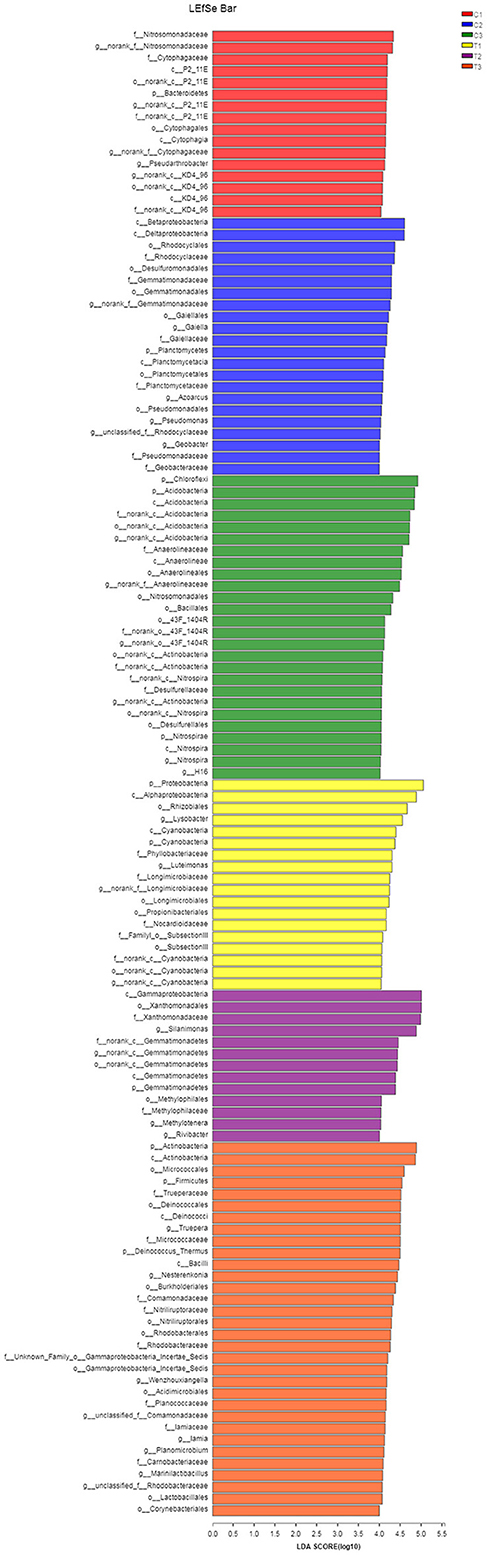
Figure 11. LEfSe was used to discriminate the microbial communities of all soil samples. LDA discriminant bar charts were used to count microbial taxa that differed significantly in all groups. All data in the experiment represent three independent experiments.
The correlation of environmental factors, soil samples, and bacterial floras were further assessed by redundancy analysis (RDA) (Figure 12). RDA results illustrated that the sequence of contributors to the diversification was as follows: bacterial community's chromium content > NR > depth > CAT (P < 0.05). The factors involved in chromium content, soil depths, and enzyme levels explained 85.79% diversity of the bacterial communities. The RDA1 axis explained 59.52% of the total variance separating C1–C3 groups from T1–T3 groups.
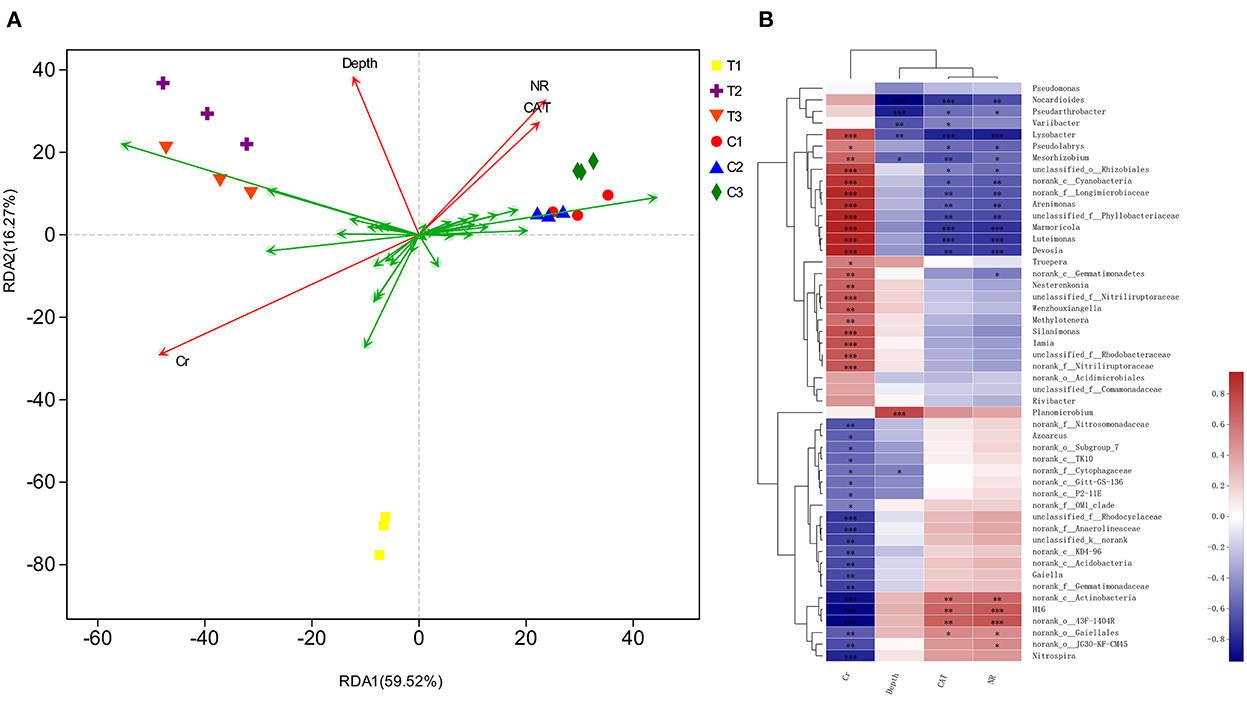
Figure 12. Based on different environmental factors, RDA result of bacterial communities (A) and heatmap of correlations (B) in soil samples. Values in the same row with “*, **, and ***” were significantly different (p < 0.05, p ≤ 0.01 or p ≤ 0.01). All data in the experiment are from three independent experiments.
The content of antioxidant enzymes (NR and CAT) decreased in Cr(VI)-polluted soils at all depths compared with those in C1–C3. Moreover, the NR and CAT content increased with depth (Figure 13).
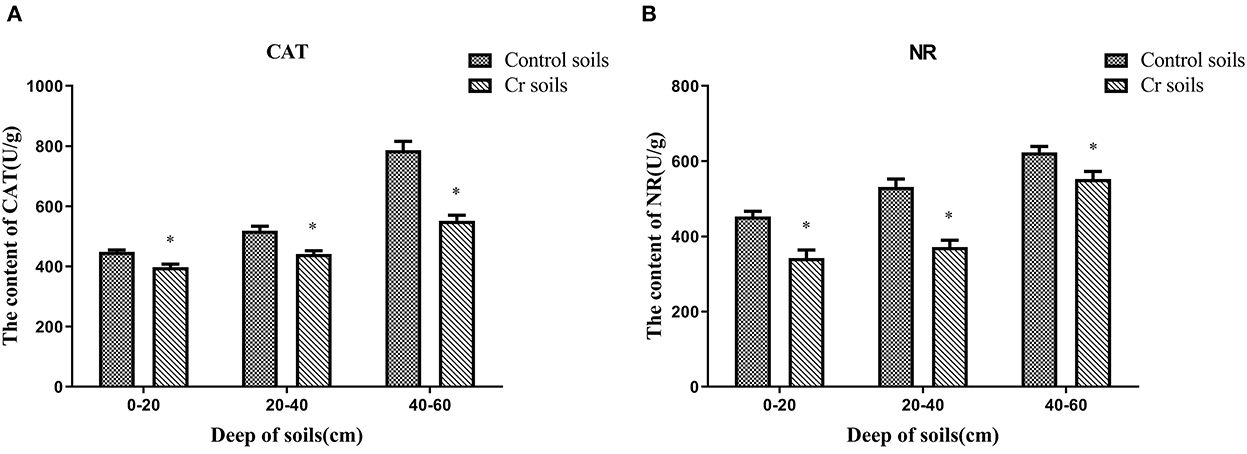
Figure 13. Antioxidant enzyme (NR and CAT) contents of soils (A, B). Data showing the diverse depths between Control and Chromium soil groups are presented as the mean ± SD. Values in the same row with “*” were significantly different (p < 0.05) in the same depth between the Control and Chromium groups. All data in the experiment represent three independent experiments.
According to the national survey bulletin on soil pollution in China, 34.9% of all wastelands surveyed exceeded the standard in terms of the amount of pollutants. The involved pollutants were zinc, chromium, mercury, lead, arsenic, and polycyclic aromatic hydrocarbons, which were produced by chemical and metal manufacturing industries (22). In the present study, chromium content in T1–T3 notably exceeded the highest national control standard value of 1,300 mg/kg (GB 15618-2018). The high risk of potentially toxic metals migrating to the environment can lead to the frequent migration of potentially toxic metals into the soil (23).
As a result of the influences of soil microbes and soil environments, differences in soil microbes are inconspicuous even after long-term succession (24, 25). Hence, bacterial microbiome communities were used in identifying changes in Cr(VI)-polluted soils.
In this study, T1–T3 curves were steeper than C1–C3 curves, indicating that the species uniformity of the control groups was more consistent than T1–T3. Furthermore, OTU levels increased with depth in C1–C3, whereas those of T1–T3 were the opposite. The results obtained from the rank abundance curve highlighted a significant fraction of variations in C1–C3 and T1–T3 diversities (in horizontal and vertical axes) that can be attributed to the content, mobility, and permeability of Cr(VI). Table 2 shows that the chromium content of T1–T3 was higher than that of C1–C3 and had a decisive effect on the microbiome. Meanwhile, chromium content at different depths influenced bacterial taxa distribution and microbiome evenness.
Biodiversity is evaluated by the richness, evenness, and α diversity indices of the microbial composition (26). In the present research, Sobs, Chao, Ace, Shannon, and Simpson's diversity indices and coverage indices were assessed to represent the richness and evenness of a microbial community. The results of microbial richness in Sobs, Chao, and Ace indices were similar in different land types but varied by soil depth. The results of microbial evenness in Shannon and Simpson's diversity indices indicated an analogous phenomenon. These results revealed that soil microbial diversity was stable across soil environments; therefore, soil microbial diversity can reflect soil properties (27). Moreover, the diversity indices of the microbial composition revealed that the microbial community is crucial to the preservation of the function and stability of soil ecosystems (28–30). The results in Figures 2C, D indicate that the microbial community significantly decreased after being polluted with Cr(VI).
Proteobacteria and Actinobacteria were the dominant phyla in all soil samples, consistent with other reports (31, 32). The properties of Proteobacteria, a slightly acidic and metabolically diverse species, can explain the high presence of these in soil samples (33). In addition, Actinomycetale are common in metal-impacted soils, especially chromium-polluted soils (34–36). Analysis of the similarity tree of multiple samples indicated the similar community structures of C1–C3 and Cr(VI)-polluted soils. By contrast, the relative abundance of strains markedly differed between the control and polluted soil samples.
The findings revealed that changes in microbial diversity and abundance can affect soil microbial biomass and activity (37). Therefore, soil microbial community (diversity and structure) integrated with microbial biomass and activity might be used in developing an approach for evaluating the risk of contaminated soils. Thus, a potential method for the bioremediation of contaminated soil can involve the addition of uncontaminated soil bacteria and removal of contaminated soil bacteria for soil microbial stability. Such bacteria include Latescibacteria (which only existed in control soils), which can degrade organics (38).
The composition of soil bacterial communities is closely related to soil properties, such as antioxidation enzymes; hence, information regarding shifts in the antioxidation enzymes of soil may be used in predicting changes in the bacterial community after Cr(VI) pollution. In addition, soil microbial biomass and activity can sensitively assess soil quality. Pb can bind with NH2 and SH enzyme groups, thereby causing enzymes to lose their activity and preventing them from harming soil microorganisms (39, 40). Pb might underlie the mechanism for decreasing CAT and NR levels in Cr-polluted soils.
Previous reports have demonstrated that pollutants, enzyme levels, and spatial depth can reveal the diversity of soil bacterium (41, 42). Among the influencing parameters, chromium was closely related to bacterial communities' composition and structure (r2 = 0.9412, P = 0.001), whereas depth was weaker linked to diversity (r2 = 0.3728, P = 0.03). Soils were collected at three depths, but the phyla were dominated by Proteobacteria, Actinobacteria, Chloroflexi, and Acidobacteria. The depth-specific differences were not the main factors for bacterial diversity in this study.
MiSeq sequence results showed a significant difference between Cr(VI)-polluted soils and control soils, regardless of the depths of Cr(VI)-polluted soils. The data of soil microbial biomass indicated that the soil polluted with Cr(VI) suppressed the microbial diversity and antioxidized enzyme levels, changing bacterial community composition and structure significantly, revealing the distribution of Latescibacteria and Ignavibacteriae. The depths values and CAT levels had minor contributions to bacterial community changes. By contrast, Cr(VI) contamination was the main factor that affected the balance of bacterial microbial communities by inhibiting bacterial diversity, microbial community structure complexity, and antioxidant enzyme levels. Collectively, differences in bacterial microbiome communities and antioxidant enzyme levels of soils can facilitate the assessment of the degree of chromium contamination in soil samples. These results are critical for stabilizing the state of microbial community structures in view of the potential risk of metal accumulation in soils.
The data presented in the study are deposited in the Dryad repository, accession number is https://doi.org/10.5061/dryad.dr7sqvb2f.
YL and JL: conceptualization. XW and YZ: data curation. KS: formal analysis. YL: funding acquisition. PZ: investigation. XW and HX: methodology. JL, YL, and PZ: project administration and supervision. XW and CQ: resources. HX: software. GC and YL: validation. YZ: visualization. YZ and KS: writing—original draft. YZ, YL, and JL: writing—review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the National Nature Science Foundation of China (No. 31872535) and the Shandong Key R&D Program (GG201809160113).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Liu P, Yang Y, Li M. Responses of soil and earthworm gut bacterial communities to heavy metal contamination. Environ Pollut. (2020) 265:114921. doi: 10.1016/j.envpol.2020.114921
2. Diaconu M, Pavel LV, Hlihor RM, Rosca M, Fertu DI, Lenz M, et al. Characterization of heavy metal toxicity in some plants and microorganisms-a preliminary approach for environmental bioremediation. Nat Biotechnol. (2020) 56:130–9. doi: 10.1016/j.nbt.2020.01.003
3. Fagnano M, Agrelli D, Pascale A, Adamo P, Fiorentino N, Rocco C, et al. Copper accumulation in agricultural soils: risks for the food chain and soil microbial populations. Sci Total Environ. (2020) 734:139434. doi: 10.1016/j.scitotenv.2020.139434
4. Si L, Zhang J, Hussain A, Qiao Y, Zhou J, Wang X. Accumulation and translocation of food chain in soil-mulberry (Morus alba L.)-silkworm (Bombyx mori) under single and combined stress of lead and cadmium. Ecotoxicol Environ Saf. (2021) 208:111582. doi: 10.1016/j.ecoenv.2020.111582
5. Montes De Oca-Vasquez G, Solano-Campos F, Vega-Baudrit JR, Lopez-Mondejar R, Vera A, Moreno JL, et al. Organic amendments exacerbate the effects of silver nanoparticles on microbial biomass and community composition of a semiarid soil. Sci Total Environ. (2020) 744:140919. doi: 10.1016/j.scitotenv.2020.140919
6. Ali J, Ali F, Ahmad I, Rafique M, Munis MFH, Hassan SW, et al. Mechanistic elucidation of germination potential and growth of Sesbania sesban seedlings with Bacillus anthracis PM21 under heavy metals stress: an in vitro study. Ecotoxicol Environ Saf. (2021) 208:111769. doi: 10.1016/j.ecoenv.2020.111769
7. Bouskill NJ, Handy RD, Ford TE, Galloway TS. Differentiating copper and arsenic toxicity using biochemical biomarkers in Asellus aquaticus and Dreissena polymorpha. Ecotoxicol Environ Saf. (2006) 65:342–9. doi: 10.1016/j.ecoenv.2005.07.027
8. Lorenz N, Hintemann T, Kramarewa T, Katayama A, Yasuta T, Marschner P, et al. Response of microbial activity and microbial community composition in soils to long-term arsenic and cadmium exposure. Soil Biol Biochem. (2006) 38:1430–7. doi: 10.1016/j.soilbio.2005.10.020
9. Zhao X, Huang J, Zhu X, Chai J, Ji X. Ecological effects of heavy metal pollution on soil microbial community structure and diversity on both sides of a river around a mining area. Int J Environ Res Public Health. (2020) 17:5680. doi: 10.3390/ijerph17165680
10. Zhang M, Sun Q, Chen P, Wei X, Wang B. How microorganisms tell the truth of potentially toxic elements pollution in environment. J Hazard Mater. (2022) 431:128456. doi: 10.1016/j.jhazmat.2022.128456
11. Zheng R, Li J, Zhu R, Wang R, Feng X, Chen Z, et al. Enhanced Cr(VI) reduction on natural chalcopyrite mineral modulated by degradation intermediates of RhB. J Hazard Mater. (2022) 423:127206. doi: 10.1016/j.jhazmat.2021.127206
12. Wen Q, Pan C, Qin X, Ma Q, Feng S. One-pot synthesis of novel water-dispersible fluorescent silicon nanoparticles for selective Cr2O7(2-) sensing. Anal Methods. (2021) 13:390–8. doi: 10.1039/D0AY01977A
13. Pei Y, Tao C, Ling Z, Yu Z, Ji J, Khan A, et al. Exploring novel Cr(VI) remediation genes for Cr(VI)-contaminated industrial wastewater treatment by comparative metatranscriptomics and metagenomics. Sci Total Environ. (2020) 742:140435. doi: 10.1016/j.scitotenv.2020.140435
14. Bertola M, Ferrarini A, Visioli G. Improvement of soil microbial diversity through sustainable agricultural practices and its evaluation by -omics approaches: a perspective for the environment, food quality and human safety. Microorganisms. (2021) 9:1400. doi: 10.3390/microorganisms9071400
15. Meier S, Moore F, Gonzalez ME, Medina J, Campos P, Khan N, et al. Effects of three biochars on copper immobilization and soil microbial communities in a metal-contaminated soil using a metallophyte and two agricultural plants. Environ Geochem Health. (2021) 43:1441–56. doi: 10.1007/s10653-019-00436-x
16. Wang Y, Liu L, Yang J, Duan Y, Luo Y, Taherzadeh MJ, et al. The diversity of microbial community and function varied in response to different agricultural residues composting. Sci Total Environ. (2020) 715:136983. doi: 10.1016/j.scitotenv.2020.136983
17. Zhu Y, Chen P, Wan H, Wang Y, Hao P, Liu Y, et al. Selenium-chromium(VI) interaction regulates the contents and correlations of trace elements in chicken brain and serum. Biol Trace Elem Res. (2018) 181:154–63. doi: 10.1007/s12011-017-1038-7
18. He Q, Wang H, Chen L, Gao S, Zhang W, Song J, et al. Elevated salinity deteriorated enhanced biological phosphorus removal in an aerobic granular sludge sequencing batch reactor performing simultaneous nitrification, denitrification and phosphorus removal. J Hazard Mater. (2020) 390:121782. doi: 10.1016/j.jhazmat.2019.121782
19. Liu L, She X, Chen X, Qian Y, Tao Y, Li Y, et al. Microbiota succession and chemical composition involved in the radish fermentation process in different containers. Front Microbiol. (2020) 11:445. doi: 10.3389/fmicb.2020.00445
20. Zhang Y, Yan C, Liu H, Pu S, Chen H, Zhou B, et al. Bacterial response to soil property changes caused by wood ash from wildfire in forest soils around mining areas: relevance of bacterial community composition, carbon and nitrogen cycling. J Hazard Mater. (2021) 412:125264. doi: 10.1016/j.jhazmat.2021.125264
21. Ren Y, Yu G, Shi C, Liu L, Guo Q, Han C, et al. Majorbio cloud: a one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta. (2022) 1:e12. doi: 10.1002/imt2.12
22. Ma Y, Wang L, Cao Y, Liang T, Wang P, Luo H, et al. Stabilization and remediation of heavy metal-contaminated soils in China: insights from a decade-long national survey. Environ Sci Pollut Res Int. (2022) 29:39077–87. doi: 10.1007/s11356-021-18346-w
23. Hou R, Wang L, Shen Z, Alessi DS, Hou D. Simultaneous reduction and immobilization of Cr(VI) in seasonally frozen areas: remediation mechanisms and the role of ageing. J Hazard Mater. (2021) 415:125650. doi: 10.1016/j.jhazmat.2021.125650
24. Shu D, Guo Y, Zhang B, Zhang C, Van Nostrand JD, Lin Y, et al. Rare prokaryotic sub-communities dominate the complexity of ecological networks and soil multinutrient cycling during long-term secondary succession in China's Loess Plateau. Sci Total Environ. (2021) 774:145737. doi: 10.1016/j.scitotenv.2021.145737
25. Sun R, Wang X, Tian Y, Guo K, Feng X, Sun H, et al. Long-term amelioration practices reshape the soil microbiome in a coastal saline soil and alter the richness and vertical distribution differently among bacterial, archaeal, and fungal communities. Front Microbiol. (2021) 12:768203. doi: 10.3389/fmicb.2021.768203
26. Tan H, Liu T, Yu Y, Tang J, Jiang L, Martin FM, et al. Morel production related to soil microbial diversity and evenness. Microbiol Spectr. (2021) 9:e0022921. doi: 10.1128/Spectrum.00229-21
27. Rachwal K, Gustaw K, Kazimierczak W, Wasko A. Is soil management system really important? Comparison of microbial community diversity and structure in soils managed under organic and conventional regimes with some view on soil properties. PLoS ONE. (2021) 16:e0256969. doi: 10.1371/journal.pone.0256969
28. Cao C, Tao S, Cui Z, Zhang Y. Response of soil properties and microbial communities to increasing salinization in the meadow grassland of northeast China. Microb Ecol. (2021) 82:722–35. doi: 10.1007/s00248-021-01695-x
29. Chen L, Xu H, Wu S, Baoyin T. Plant and soil properties mediate the response of soil microbial communities to moderate grazing in a semiarid grassland of northern China. J Environ Manage. (2021) 284:112005. doi: 10.1016/j.jenvman.2021.112005
30. Yang N, Tian C, Lv Y, Hou J, Yang Z, Xiao X, et al. Novel primers for 16S rRNA gene-based archaeal and bacterial community analysis in oceanic trench sediments. Appl Microbiol Biotechnol. (2022) 106:2795–809. doi: 10.1007/s00253-022-11893-3
31. Li M, Wang J, Yao T, Zhang T, Zhou Q, Mihray M. Bacterial diversity and community structure in the rhizosphere of four halophytes. Curr Microbiol. (2021) 78:2720–32. doi: 10.1007/s00284-021-02536-3
32. Tang Z, Zhang L, He N, Gong D, Gao H, Ma Z, et al. Soil bacterial community as impacted by addition of rice straw and biochar. Sci Rep. (2021) 11:22185. doi: 10.1038/s41598-021-99001-9
33. Rogiers T, Claesen J, Van Gompel A, Vanhoudt N, Mysara M, Williamson A, et al. Soil microbial community structure and functionality changes in response to long-term metal and radionuclide pollution. Environ Microbiol. (2021) 23:1670–83. doi: 10.1111/1462-2920.15394
34. Rasool A, Nasim W, Xiao T, Ali W, Shafeeque M, Sultana SR, et al. Microbial diversity response in thallium polluted riverbank soils of the Lanmuchang. Ecotoxicol Environ Saf. (2020) 187:109854. doi: 10.1016/j.ecoenv.2019.109854
35. Liu M, Wang Y, Liu X, Korpelainen H, Li C. Intra- and intersexual interactions shape microbial community dynamics in the rhizosphere of Populus cathayana females and males exposed to excess Zn. J Hazard Mater. (2021) 402:123783. doi: 10.1016/j.jhazmat.2020.123783
36. Rajivgandhi G, Vimala RTV, Maruthupandy M, Alharbi NS, Kadaikunnan S, Khaled JM, et al. Enlightening the characteristics of bioflocculant of endophytic actinomycetes from marine algae and its biosorption of heavy metal removal. Environ Res. (2021) 200:111708. doi: 10.1016/j.envres.2021.111708
37. Liu YM, Cao WQ, Chen XX, Yu BG, Lang M, Chen XP, et al. The responses of soil enzyme activities, microbial biomass and microbial community structure to nine years of varied zinc application rates. Sci Total Environ. (2020) 737:140245. doi: 10.1016/j.scitotenv.2020.140245
38. Wang Y, Li T, Li C, Song F. Differences in microbial community and metabolites in litter layer of plantation and original Korean pine forests in north temperate zone. Microorganisms. (2020) 8:2023. doi: 10.3390/microorganisms8122023
39. Tang J, Zhang L, Zhang J, Ren L, Zhou Y, Zheng Y, et al. Physicochemical features, metal availability and enzyme activity in heavy metal-polluted soil remediated by biochar and compost. Sci Total Environ. (2020) 701:134751. doi: 10.1016/j.scitotenv.2019.134751
40. Muhammad Z, Ramzan R, Zhang R, Zhao D, Gul M, Dong L, et al. Assessment of in vitro and in vivo bioremediation potentials of orally supplemented free and microencapsulated Lactobacillus acidophilus KLDS strains to mitigate the chronic lead toxicity. Front Bioeng Biotechnol. (2021) 9:698349. doi: 10.3389/fbioe.2021.698349
41. Frey B, Walthert L, Perez-Mon C, Stierli B, Kochli R, Dharmarajah A, et al. Deep soil layers of drought-exposed forests harbor poorly known bacterial and fungal communities. Front Microbiol. (2021) 12:674160. doi: 10.3389/fmicb.2021.674160
Keywords: microbiome, chromium, microbial diversity and structure, soil enzymes, MiSeq high-throughput sequencing
Citation: Zhu Y, Song K, Cheng G, Xu H, Wang X, Qi C, Zhang P, Liu Y and Liu J (2023) Changes in the bacterial communities in chromium-contaminated soils. Front. Vet. Sci. 9:1066048. doi: 10.3389/fvets.2022.1066048
Received: 10 October 2022; Accepted: 01 December 2022;
Published: 04 January 2023.
Edited by:
Huabin Cao, Jiangxi Agricultural University, ChinaReviewed by:
Abraham Loera Muro, Centro de Investigación Biológica del Noroeste (CIBNOR), MexicoCopyright © 2023 Zhu, Song, Cheng, Xu, Wang, Qi, Zhang, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianzhu Liu,  bGl1anpAc2RhdS5lZHUuY24=; Yongxia Liu, bGl1eW9uZ3hpYUBzZGF1LmVkdS5jbg==; Pu Zhang, enA4MTk4NDIzQDE2My5jb20=
bGl1anpAc2RhdS5lZHUuY24=; Yongxia Liu, bGl1eW9uZ3hpYUBzZGF1LmVkdS5jbg==; Pu Zhang, enA4MTk4NDIzQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.