 Taís Ramalho dos Anjos1
Taís Ramalho dos Anjos1 Vinícius Silva Castro2
Vinícius Silva Castro2 Edson Silva Machado Filho3
Edson Silva Machado Filho3 Philip Noel Suffys3*
Philip Noel Suffys3* Harrison Magdinier Gomes3Rafael Silva Duarte4
Harrison Magdinier Gomes3Rafael Silva Duarte4 Eduardo Eustáquio de Souza Figueiredo5
Eduardo Eustáquio de Souza Figueiredo5 Ricardo César Tavares Carvalho1*
Ricardo César Tavares Carvalho1*- 1Programa de Pós-graduação em Biociência Animal - Stricto Sensu, Faculdade de Medicina Veterinária, Universidade de Cuiabá (UNIC), Cuiabá, Brazil
- 2Programa de Pós-graduação em Ciência Animal, Faculdade de Agronomia e Zootecnia, Universidade Federal de Mato Grosso (UFMT), Cuiabá, Brazil
- 3Laboratório de Biologia Molecular Aplicado a Micobactérias, Instituto Oswaldo Cruz (IOC), Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil
- 4Laboratório de Micobactérias, Departamento de Microbiologia Médica, Instituto de Microbiologia Paulo de Góes, Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil
- 5Laboratório de Microbiologia Molecular de Alimentos, Faculdade de Nutrição, Universidade Federal de Mato Grosso (UFMT), Cuiabá, Brazil
The species Mycobacterium tuberculosis variant bovis (M. tuberculosis var. bovis) is associated with tuberculosis, mainly in cattle and buffaloes. This pathogen has the potential to infect other mammals, including humans. Tuberculosis caused by M. tuberculosis var. bovis is a zoonosis clinically identical to tuberculosis caused by Mycobacterium tuberculosis, and the recommended treatment in humans results in the use of antibiotics. In this study, we used the whole genome sequencing (WGS) methodology Illumina NovaSeq 6000 System platform to characterize the genome of M. tuberculosis var. bovis in cattle circulating in Mato Grosso, identify mutations related to drug resistance genes, compare with other strains of M. tuberculosis var. bovis brazilian and assess potential drug resistance. Four isolates of M. tuberculosis var. bovis of cattle origin representing the main livestock circuits, which had been more prevalent in previous studies in the state of Mato Grosso, were selected for the genomic study. The genome sizes of the sequenced strains ranged from 4,306,423 to 4,332,964 bp, and the GC content was 65.6%. The four strains from Mato Grosso presented resistance genes to pncA (pyrazinamide), characterized as drug-resistant strains. In addition to verifying several point mutations in the pncA, rpsA, rpsL, gid, rpoB, katG, gyrB, gyrA, tlyA, embA, embB, embC, fgd, fbiB, and fbiC genes, these genes were similar to antibiotic resistance in more than 92% of the Brazilian strains. Therefore, our results indicated a high genetic diversity between our isolates and other M. tuberculosis var. bovis isolated in Brazil. Thus, multiple transmission routes of this pathogen may be present in the production chain. So, to achieve a bovine tuberculosis-free health status, the use of the WGS as a control and monitoring tool will be crucial to determine these transmission routes.
Introduction
Mycobacterium tuberculosis variant bovis (M. tuberculosis var. bovis) is the etiological agent of bovine tuberculosis (bTB) (1). This pathogen belongs to the group of mycobacteria belonging to the Mycobacterium tuberculosis Complex (MTBC), which share genomic similarities of ~99.9% (2). M. tuberculosis var. bovis has zoonotic potential and can cause tuberculosis in humans (1). Transmission to humans can happen directly through the air, in which the pathogen is inhaled in aerosols from infected animals or carcasses, as well as indirectly through contamination through the consumption of contaminated animal products, such as raw milk and other dairy products without heat treatment, and through the consumption of undercooked or raw meat (3, 4).
This zoonosis mainly affects vulnerable people, such as HIV (human immunodeficiency virus) but also immunocompetent people (5), and is mainly related to factors such as poverty, unemployment, and a low level of education (6); it is also a public health problem and is clinically identical to tuberculosis caused by the bacterium Mycobacterium tuberculosis (M. tuberculosis), making differential diagnosis difficult (7, 8).
According to Sisco et al. (9), there is the possibility of acquiring tuberculosis through the BCG vaccine, especially in immunocompromised patients. They found a strain resistant to ethambutol, rifampicin, and isoniazid. With intrinsic or acquired antibiotic resistance, there is a need to perform drug susceptibility screening of the strain before or during patient treatment (9).
Bovine tuberculosis presents a chronic evolution characterized by the development of nodular lesions called tubercles, which can be located in any organ or tissue of the animal (10). In cases of tuberculosis in humans, treatment is carried out with the use of combinations of antibiotics, mainly rifampicin, isoniazid, ethambutol, streptomycin, ethionamide, pyrazinamide, and fluoroquinolones (6). Unlike M. tuberculosis, M. tuberculosis var. bovis is naturally resistant to pyrazinamide, one of the drugs used in the treatment of tuberculosis (11).
M. tuberculosis var. bovis have reference genes that encode proteins with known or unknown functions, such as catalase/peroxide activity (katG gene); encoding the DNA gyrase A subunit (gyrA); in the rpoB gene, which encodes a β subunit of RNA polymerase; RNA loops in the loops of RNA, which is encoded by the gene, related to changes in cell wall permeability (12); another example are bacteria related in genes of the emb region (embA, embB, and embC) with the biosynthesis of arabinogalactan and lipoarabinomannan, structural components of the tamarin wall (target ethambutol) (13).
According to Vázquez-Chacón et al. (14), there are highly reliable mutations in M. tuberculosis var. bovis that confer drug resistance. For example, mutations in the katG S315T genes; rpsL K43R or K88R; rrs A1401G and gyrA D94G confer resistance to isoniazid, streptomycin, aminoglycosides and fluoroquinolones, and these were identified in M. tuberculosis var. bovis (14). All M. tuberculosis var. bovis strains are naturally resistant to pyrazinamide (pncA), mutations were found at position C169G, and Mycobacterium bovis bacillus Calmette-Guérin (BCG) and Mycobacterium canetti strains are also resistant to this drug (15). The circulation of drug-resistant strains in cattle, especially those used in the treatment of tuberculosis in humans, represents a great risk for the occurrence of multidrug-resistant strains in the population (14).
Multidrug resistance (MDR) has increased worldwide, which is considered a threat to public health. Several recent investigations have reported the emergence of multidrug-resistant bacterial pathogens of different origins that increase the need for the appropriate use of antibiotics. In addition, routine application of antimicrobial susceptibility testing to detect the antibiotic of choice, as well as screening for emerging MDR strains (16–19).
The aim of this study was to characterize the genome of M. tuberculosis var. bovis in cattle circulating in Mato Grosso, identify mutations related to drug resistance genes, compare with other strains of M. tuberculosis var. bovis brazilian and assess potential drug resistance.
Materials and methods
Geographic area of study
This study was carried out in the state of Mato Grosso, located in midwestern Brazil (Latitude: 15° 35′ 56″ South, Longitude: 56° 5′ 42″ West), which is categorized by four livestock circuits (20), called CP 01—Pantanal; CP 02—Milk production; CP 03—Fattening and CP 04—Breeding, according to the predominant animal production characteristics, local ecosystem, and animal transit network (Figure 1).
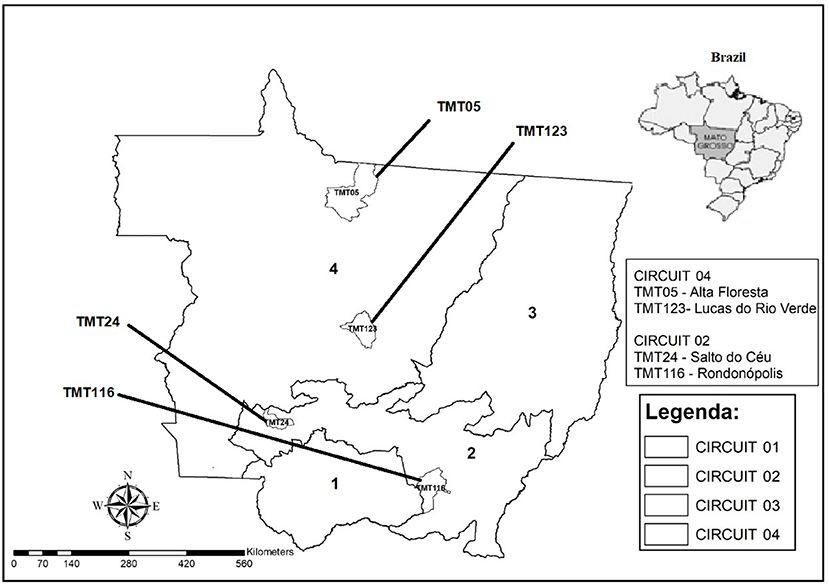
Figure 1. Map of Mato Grosso state with the municipalities demarcating the origins of the strains selected for genome sequencing.
Sampling and zootechnical data
A total of 41 suspected bTB lesion samples of the same number of animals were collected between September 2017 and May 2018, stored in sterile packaging and frozen during the postmortem inspection procedure. The sampling was carried out in slaughterhouses in the state of Mato Grosso by Federal (SIF) and state (SISE/MT) inspection authorities using the criteria established by the regulation of inspection industrial and sanitary of animal products (RIISPOA) (21). Simultaneously, sanitary inspection and collection of suspicious lesions were carried out following normal routine at slaughterhouses in the state.
Nucleic acid extraction from bovine tissue fragments containing suspicious lesions
Samples were divided into two aliquots, one for DNA extraction and another for culture. Sample preparation and DNA extraction from bovine tissue fragments containing suspicious lesions were performed according to Furlanetto et al. (22) using a commercial Dneasy Blood and Tissue kit (Qiagen®, Hilden, Germany). After extraction, the DNA was quantified by the fluorometric method using the QUBIT TM 2.0 Kit (Invitrogen®, Carlsbad, CA, United States).
Nested real-time PCR (nested qPCR) of DNA extracted from animal tissue fragments
Nested q-PCR was performed using TaqMan PCR Master Mix (Applied Biosystems®, Foster City, CA, United States), primers and probes for the Rv2807 gene (Applied Biosystems®, Foster City, CA, United States) specific for species belonging to MTBC and primers and probes for the TbD1 region (Applied Biosystems®, Foster City, CA, United States) of M. tuberculosis var. bovis based on the method described by Araújo et al. (23, 24) and modified by Carvalho et al. (25).
Bacterial isolation and identification
After detection of MTBC and M. tuberculosis var. bovis by nested q-PCR, samples positive for Rv2807 and TbD1 were submitted to microbiological culture, undergoing a decontamination process with 0.75% hexadecyl pyridinium chloride (HPC) and 10% sulfuric acid (H2SO4) (22, 26). After processing, the material was inoculated in duplicate in tubes with Stonebrink culture medium (Reagents: Malachite Green- C23H25CIN2/VETEC Rio de Janeiro, Brazil; Disodium Phosphate- NaHPO4/ISOFAR Rio de Janeiro, Brazil; Sodium pyruvate- C3H3NaO3/VETEC Rio de Janeiro, Brazil; monopotassium phosphateresiste—KH2PO4/SYNTH São Paulo, Brazil) a 2%, and incubated at 37°C under aerobic conditions for 90 days, with weekly control to record the multiplication rate, shape and recognition of characteristic colonies of mycobacteria (27–29).
The colonies suggestive of mycobacteria were the presence of bacilli identified by the technique of alcohol-acid resistant bacilli (AARB), by the Ziehl-Neelsen technique. Small, white colonies with irregular borders and a granular surface typical of M. tuberculosis var. bovis and after performing Ziehl-Neelsen, a massive presence of AARB was observed, thus, the colonies were identified as belonging to M. tuberculosis var. bovis, confirming what had been previously identified by nested q-PCR (30, 31).
DNA extraction from Mycobacterium tuberculosis variant bovis isolates
After microbial isolation, the process of extracting DNA from the isolates was carried out by the cetyl trimethyl ammonium bromide (CTAB) enzymatic method, following the protocol described by Van Soolingen et al. (32), and quantified by a NanoDrop® 2000 spectrophotometer (Thermo Scientific) to obtain a concentration of ≥ 50 ng/μL.
Genome sequencing and data analysis
Of the 41 samples analyzed, they were submitted to nested qPCR for MTBC (Rv2807 gene) and for M. tuberculosis var. bovis (TbD1 gene) and were submitted to microbiological culture, through which 23 isolates with characteristic colonies of M. tuberculosis var. bovis were obtained. Of these 23 isolates, four were selected as representatives of the regions studied, adopting criteria referring to representing the main livestock circuits that had shown higher prevalence in previous studies in the state of Mato Grosso (33, 34).
Four strains were selected for genome sequencing based on each different livestock circuits in the state of Mato Grosso (20). These livestock circuits were previously established due to the higher prevalence of M. bovis in the state (33, 34). The state of Mato Grosso is divided into four regions, called livestock circuits (CP), according to the bovine production that is predominant in that region. Circuit 1 represents the municipalities that have a predominant production of extensive breeding; circuit 2 is representative of milk production; circuit 3 are municipalities with a fattening cycle and circuit 4 with predominant production for the rearing system (18).
Of the 41 samples analyzed, after cultivation, 23 isolates were obtained, including 17 strains of CP 2 and six strains of CP 4. The four strains were then selected for sequencing, identified as TMT24 and TMT116 (CP 2) and TMT05 and TMT123 (CP 4), which represent the municipalities of Alta Floresta (TMT05—CP4), Lucas do Rio Verde (TMT123—CP4), Salto do Céu (TMT24—CP2), and Rondonópolis (TMT116—CP2).
Sequencing was performed using the Illumina NovaSeq 6000 System platform (Illumina, Inc., San Diego, CA, USA) and S4 Flow Cell Type PE150 (Illumina, Inc., San Diego, CA, USA) and operated by GenOne Biotechnologies (Rio de Janeiro, Brazil). To assess the raw sequence quality we used a FastQC (Version 0.11.8) (35). Thereafter, we used the Trimmomatic (version 0.36) to cut Illumina adapters. The parameters used were sliding window trimming applied as operator, 4 bases to average across, and 20 bases required for average quality. To carry out the assembly, we used Shovill version 1.1.0 (https://github.com/tseemann/shovill), with Spades used as an assembler, a depth of 100, a minimum contig length of 200 bp and a minimum coverage of 5 (36). After the generation of contigs, genomic annotation was obtained using Prokka version 1.14.5, with default parameters applied (37), and the presence of resistance and virulence genes was analyzed in silico. Nevertheless, other specific analyses for TB, such as the determination of the strain's lineage, spoligotyping in silico, and detection of resistance genes and mutations, we used TB profiler version 2.8.14 (https://github.com/jodyphelan/TBProfiler) (38). To check the assembly sequencing quality we used Quast (version 5.0.2) (39) with default parameters applied.
Data collection and construction of the phylogenetic tree
A total of 76 M. tuberculosis var. bovis genomes were used to construct the phylogenetic tree. Of these 76 genomes, four are the genomes obtained in the present study, another 49 SRA were obtained from the NCBI (Keywords: Mycobacterium bovis AND Brazil), and another 23 sequences were identified in scientific articles published but not covered by the NCBI Search (Supplementary material 1). Among the sequences, we selected the first M. tuberculosis var. bovis strain sequenced in Brazil, isolated from a bovine in 2010 in the state of São Paulo, of identification SRA: SRR6705904 (40). However, in a recent study carried out by Rodrigues et al. (33), 74 M. tuberculosis var. bovis genomes were made available at NCBI under BioProject PRJEB39667. However, as a convenience, we divided the 74 samples into eight different clades, as specified in the authors' phylogenetic tree, and analyzed one strain belonging to each clade (41). Additionally, 19 sequences were included belonging to the study performed by Conceição et al. (42) in a study of M. tuberculosis var. bovis isolated from Ilha de Marajó (42). A total of 19 genomes came from a study on M. tuberculosis var. bovis in animals raised in Amazonas under BioProject PRJNA675550. Another 17 genomes were derived from M. tuberculosis var. bovis isolated in deer in a safari park in southern Brazil under the BioProject PRJNA675476 (35). Finally, other genomes isolated from wild animals were included, such as 3 genomes from the isolation of M. tuberculosis var. bovis in llamas (SRR6865435, SRR7693877, SRR9850830) and 1 genome in capybara (SRR9850824). Another 3 strains described as BCG (Bacillus Calmette Guerin) were also inserted into the phylogenetic tree (9). Finally, 1 single genome (SRR12511761) from a bovine isolate was inserted into the construction of the phylogenetic tree. We added three outgroups, M. africanum, M. caprae, and M. tuberculosis, for better visualization of the clades. To construct the phylogenetic tree, the data were submitted to the Snippy package version 4.6.0 (https://github.com/tseemann/snippy). This tool was used to perform variant calling and core-genome alignment. For this, paired-end reads of all strains were analyzed using a minimum mapping quality of 60, with a minimum coverage of 10x, and a minimum proportion for variant evidence of 0.9. Mycobacterium tuberculosis H37Rv (NC_000962.3) was used as a reference strain for creating the core alignment. Subsequently, all multiple Snippy outputs from the core alignment with variant calling were combined into a SNP core alignment using Snippy-core version 4.6.0 (https://github.com/tseemann/snippy). Thus, the core-genome SNP alignment was used to perform a phylogenetic tree using the approximately maximum likelihood phylogenetic trees method with FastTree v. 2.1.11, with a site rate of 20, and turn off the minimum-evolution nearest-neighbor interchanges (NNIs) and minimum-evolution subtree-pruning-regrafting (SPRs). The phylogenetic tree image was obtained using Geneious Prime (v2021.2.2) and annotated using FigTree v1.4.4 (Figure 2) (39). An important point was that we removed four strains (ERR3445501, ERR3445502, ERR3445503, and ERR3445504) after generating the phylogenetic tree because possible DNA contamination may occur, one time that RDscan and the phylogenetic tree showed an unusual pattern.
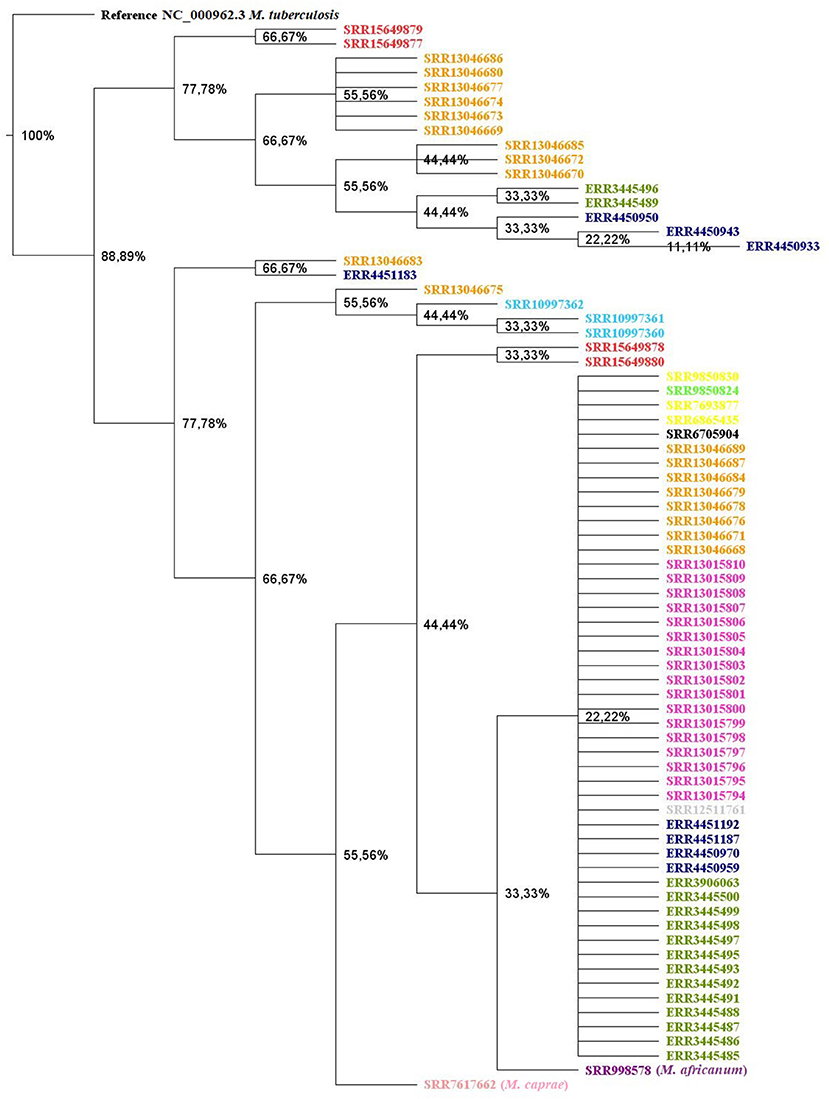
Figure 2. Maximum likelihood phylogenetic tree of Mycobacterium tuberculosis variant bovis genomes sequenced in the state of Mato Grosso grouped with other genomes sequenced in other states of Brazil. Four Mycobacterium tuberculosis variant bovis genomes isolated from bovine TMT05 (SRR15649880), TMT116 (SRR15649878), TMT24 (SRR15649879), and TMT123 (SRR15649877) from the state of Mato Grosso. Identifications in red: Mycobacterium tuberculosis variant bovis genomes obtained from cattle and sequenced in this study; green: Mycobacterium tuberculosis variant bovis genomes from cattle or buffalo from Marajó Island, Pará; orange: genomes of Mycobacterium tuberculosis variant bovis obtained from cattle or buffaloes from the Amazon; dark blue: Mycobacterium tuberculosis variant bovis genomes obtained from cattle from southern Brazil; pink: Mycobacterium tuberculosis variant bovis genomes obtained from deer from a safari in southern Brazil; yellow: llama; light blue: BCG strain genome; light green: capybara; gray: Mycobacterium tuberculosis variant bovis isolated from cattle; black: first Mycobacterium tuberculosis variant bovis strain sequenced in Brazil; outgroups: Mycobacterium africanum (purple), Mycobacterium caprae (light pink), and Mycobacterium tuberculosis (black).
Identification of regions of difference in Brazilian Mycobacterium tuberculosis variant bovis strains
RDscan (https://github.com/dbespiatykh/RDscan) was used to verify the deletion regions (RDs) in the analyzed genomes to understand the differentiation of M. tuberculosis var. bovis strains from Mato Grosso with the reference strain (Mbovis_AF212297) and the other Brazilian strains.
Results
Nested qPCR and microbiological culture
From the 41 suspected bTB lesions collected according to Brazilian official judgment criteria, 100% (41/41) positive for MTBC, 70.7% (29/41) positive for M. tuberculosis var. bovis in nested qPCR were submitted to cultivation, and 56% (23/41) generated colonies suggestive of M. tuberculosis var. bovis in Stonebrink medium. All were submitted and confirmed by this q-PCR, and four strains were selected for sequencing to represent two livestock circuits in the state of Mato Grosso, not only within a single circuit, in addition to selecting strains of M. tuberculosis var. bovis, which in previous studies were more prevalent (33, 34).
Sequencing, assembly, and genome annotation of the sequenced M. tuberculosis var. bovis strains
M. tuberculosis var. bovis genomic DNA was sequenced on an Illumina NovaSeq 6000 System platform with 28x to 31x coverage. DNA sequencing and assembly generated 4,306,423 to 4,332,964 nucleotides and a GC content of 65.6. As a result of the genome reading, the number of contig values obtained ranged from 70 to 95. With the Quast version 5.0.2 tools, we obtained N50 values that ranged from 137.022 to 205.012. Table 1 describes the size of the genomes, the number of CDSs that varied from 3,981 to 4,010, and other assembly and annotation results.
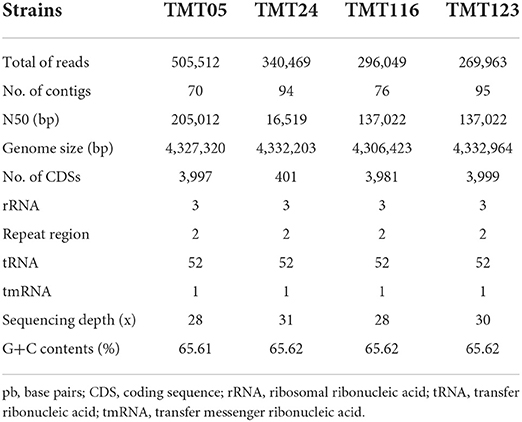
Table 1. Assembly and annotation of sequenced Mycobacterium tuberculosis variant bovis genomes.
Lineage, resistance genes, and mutations found in Mycobacterium tuberculosis variant bovis strains
To characterize the strains to the lineage level, spoligotyping in silico, and verification of the presence of resistance-associated genes, we used TB profiler version 2.8.14 software. Our results show that all strains in this study TMT05 (SRR15649880), TMT116 sample (SRR15649878), TMT24 (SRR15649879), and TMT123 (SRR15649877) belonged to the M. tuberculosis var. bovis lineage BOV AFRI and the spoligotype BOV 1; BOV 2. Another 71 genomes belong to the BOV 1 spoligotype, BOV 11, with the exception of two genomes (ERR3445502, ERR3445504).
The pncA gene, which is responsible for pyrazinamide resistance, was detected in all sequenced strains in this study, as expected. As in the other 71 Brazilian strains, with the exception of the two strains (ERR3445502, ERR3445504). Using Prokka version 1.14.5, it was possible to detect other genes that are associated with resistance to several antimicrobials, pncA and rpsA (pyrazinamide); katG, inhA, ahpC and glf (isoniazid); rpoB (rifampicin); rpsL and gid (streptomycin); embB, embB and embC (ethambutol); inhA, ethA and ethR (ethionamide); gyrA and gyrB (florquinolones); eis (kanamycin/amikacin); tlyA (capreomycin); thyA and ribD (paraminosalicylic acid); alr, ddl and cycA (cycloserine); atpE and mmpR5 (bedaquiline); rplC (linezolid); ddn, fgd1, fbiA, fbiB, fbiC, and fbiD (delamanid) (6, 43–48), according to Table 2.
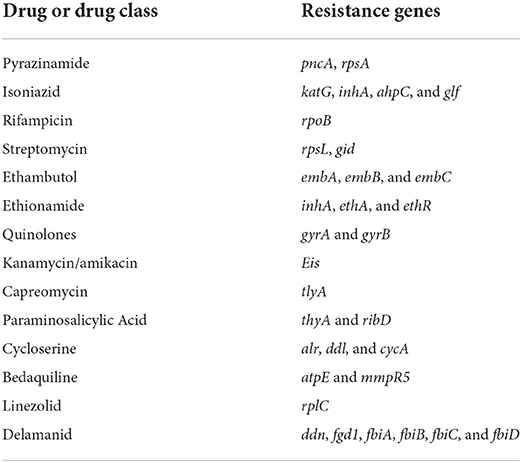
Table 2. Resistance genes in Mycobacterium tuberculosis variant bovis strains isolated in Mato Grosso associated with antibiotics routinely used in the treatment of human TB.
Mutations were detected in the genes pncA, rpsA, rpsL, gid, rpoB, katG, gyrB, gyrA, tlyA, embA, embB, embC, fgd1, fbiB, and fbiC, as described in Table 3 and Supplementary material 2.
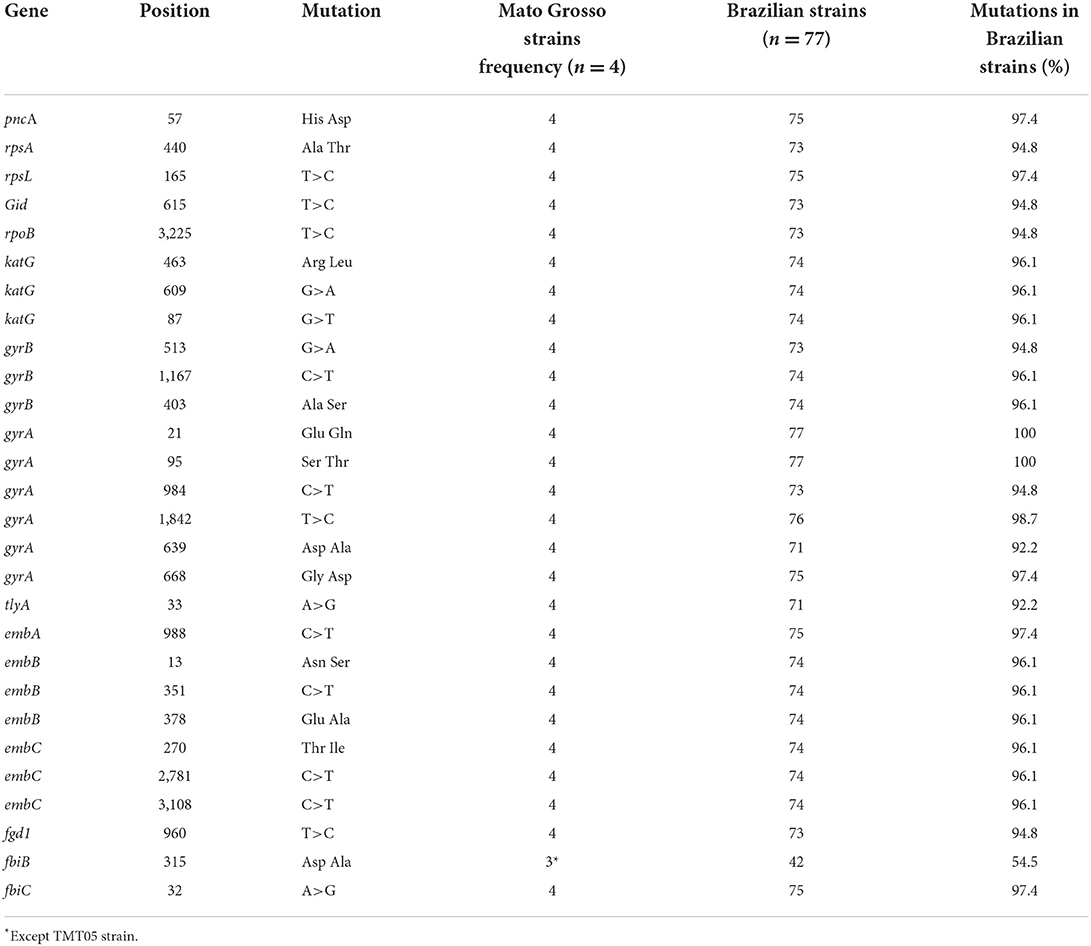
Table 3. Mutations present in Mycobacterium tuberculosis variant bovis strains isolated from caseous lesions obtained from bovine carcasses in the state of Mato Grosso, Brazil, with other Brazilian strains.
The strains sequenced in this work SRR15649878 (TMT116), SRR15649879 (TMT24), SRR15649877 (TMT123), and SRR15649880 (TMT05) as well as 68 Brazilian M. tuberculosis var. bovis strains were classified as drug resistant using the TBProfiler. Another three BCG strains (SRR10997360, SRR10997362, and SRR10997361) were classified as multidrug resistant (MDR), and another strain, ERR3445487, was also classified as MDR (Table 4).

Table 4. Drug-resistant TBProfiler report.
Genomic analysis of M. tuberculosis var. bovis strains
Illumina raw data [sequence read archive (SRA)] and the contigs genome with respective annotations were deposited with the NCBI and are described in Table 4. All SRA and genome annotation data were included under BioProject number PRJNA756983 (Table 5).

Table 5. Data on strains deposited at the NCBI.
In the phylogenetic tree (Figure 2), two large branches were determined, with a bootstrap value of 77.7% for each branch, called a clade. Through the phylogenetic tree, it can be observed that the lineages of M. tuberculosis var. bovis from the state of Mato Grosso are distributed in both clades.
Deletion regions (RDs) were identified in the four strains sequenced in this study as well as in the other 71 genomes analyzed, except for two genomes (ERR3445502; ERR3445503), through RDscan, including RD4, RD5, RD6, RD7, RD8, RD9, RD10, RD12, and RD13 (Supplementary material 4).
Some regions were deleted (Rv0095c, Rv0867c, and Rv1563c), and others were preserved (Rv3798) in the four strains sequenced in this study compared to the reference strain (Mbovis_AF212297). The Mato Grosso strain SRR15649880 (TMT05) showed the absence of the RDoryx_1 region and the Rv3508 gene, which differs from the reference strain.
The SRR15649878 (TMT116) genome showed deletion of the Rv0578c, RD145, Rv3511, Rv3512, Rv3513c, and Rv3514 regions and preservation of the RDoryx_4, RD182, Rv1361c, Rv1758, Rv1757c, Rv1756c, Rv2168c, and Rv2167c regions of the reference.
Discussion
The M. tuberculosis var. bovis lineage was identified by in silico spoligotyping in the four strains sequenced in this work TMT05 (SRR15649880), sample TMT116 (SRR15649878), TMT24 (SRR15649879), and TMT123 (SRR15649877) and belonged to the lineage BOV AFRI and BOV1 BOV2, the same lineage identified in another study of M. tuberculosis var. bovis in cattle from Bahia, Brazil (49). Another 71 genomes belong to the BOV1 spoligotype, BOV11, with the exception of two genomes (ERR3445502, ERR3445504).
According to Smith (50), the African 1 and African 2 clonal complexes are never or rarely found in cattle outside Africa. Carneiro et al. (51) pointed out that the current clonal complexes may not represent all the diversity of the M. tuberculosis var. bovis lineage and identified for the first time in Brazil the Lb1 lineage, and some strains identified in this lineage carry the Af2 clonal complex marker, frequently found in East Africa, demonstrating the importance of connections of this lineage with the African continent (51).
Due to Africa's proximity to countries such as Portugal and its colonies, hypothetically, strains may have been introduced for the first time in the Amazon region during the colonization of Brazil (51) and have been dispersed to other states in the country.
In addition to the presence of M. tuberculosis var. bovis, other important point is the emergence of drug resistance as serious threat to the control of human tuberculosis, as strains that are resistant to multiple drugs severely limit treatment options (52). With the results obtained in this work, it is worrisome because strains have the potential for resistance to many antibiotics, and there are few treatment options left in cases of zoonotic tuberculosis.
According to Mota et al. (53), there are 16 antibiotics known to have an effective action on any bacillus capable of causing tuberculosis in humans, and of these, six are of preferential use in the treatment of tuberculosis in humans, namely, streptomycin, rifampicin, ethambutol, pyrazinamide, ethionamide and isoniazid.
The main genes detected were pncA, katG, inhA, ahpC, glf, rpoB, rpsL, embA, embB, embC, inhA, ethA, ethR, gyrA, gyrB, eis, tlyA, thyA, ribD, alr, ddl, cycA, atpE, mmpR5, rplC, ddn, fgd1, fbiA, fbiB, fbiC, and fbiD, according to Table 2. We detected point mutations in the genes pncA, rpsA, rpsL, gid, rpoB, katG, gyrB, gyrA, tlyA, embA, embB, embC, fgd1, fbiB, and fbiC, as described in Table 3. A limitation of the study is that the phenotypic analysis for comparison with the genotypic results has not yet been possible, but it is a future project to be done.
To improve successful treatment outcomes in cases of tuberculosis caused by multidrug resistant strains, 68 countries started using the drug bedaquiline, and 42 countries started using the drug delamanid at year-end 2017 (54); however, both drugs are not available in Brazil (6), and the strains sequenced presently demonstrated the presence of atpE and ddn genes that can confer resistance to these two drugs, respectively.
Multidrug-resistant strains of M. tuberculosis var. bovis hamper TB control and exhaust treatment options in humans (55–57). Gómez-Gonzales et al. (58) found variants in genes that can confer resistance to bedaquiline and delamanid in more than 33,000 M. tuberculosis isolates collected from humans even before the launch of these new drugs, suggesting an intrinsic resistance of the strains. This fact constitutes new obstacles that threaten the global control of TB (58).
Acquired resistance to anti-TB drugs occurs due to spontaneous mutations, including single nucleotide polymorphisms (SNPs) and insertions and deletions (indels), in genes encoding drug targets (58). Antibiotic resistance can occur spontaneously, even without antimicrobial exposure. As a result, drug sensitivity tests, including rapid molecular techniques, are needed for the accurate diagnosis and treatment of TB (59).
Vázquez-Chacón et al. (14) identified drug resistance mutations among M. tuberculosis var. bovis strains in the Americas. Their results showed that M. tuberculosis var. bovis isolates from animal strains harbor mutations that confer resistance to first- and second-line antibiotics, resistance to isoniazid, fluoroquinolones, streptomycin and aminoglycosides. These results highlight the importance of molecular surveillance to monitor the emergence of mutations that are associated with multidrug drug resistance in cattle and other non-human mammals (14). However, they did not find mutations in genes related to next-generation drugs such as delamid and bedaquiline.
According to Miotto et al. (60), there are high confidence mutations that confer antibiotic resistance in M. tuberculosis. The frequency of mutations in resistant and susceptible strains was calculated using statistical measures to classify mutations as high, moderate, minimal or indeterminate confidence to predict resistance. They found resistance-associated mutations for rifampicin, isoniazid, ofloxacin/levofloxacin, moxifloxacin, amikacin, kanamycin, capreomycin, streptomycin, ethionamide/prothionamide, and pyrazinamide (60). With the mutations identified in the M. tuberculosis var. bovis strains sequenced in this work, only the gyrA gene was associated with the results of Miotto et al. (60), in which one of the mutations in the gyrA gene was associated with not being related to resistance to the antibiotic moxifloxacin; that is, this mutation does not interfere with resistance, but the other mutations could not be associated.
The most common target genes associated with streptomycin resistance are rrs, rpsL, and gidB (11). The rpsL and gid genes were detected, and mutations were found in all isolates sequenced in this study as well as in most Brazilian strains of M. tuberculosis var. bovis. Studies carried out by Djeman et al. (11) also detected a streptomycin-resistant M. tuberculosis var. bovis isolate; according to the authors, their study was the first to describe a streptomycin-resistant M. tuberculosis var. bovis isolate of animal origin. Strains capable of causing multidrug resistance tuberculosis are a threat to human medicine, as they cause treatment failures and, in cases where it is not possible to treat the infection, lead to the death of the patient.
With studies in which good results were obtained, that is, cure of treated animals (53, 61), on the action of isoniazid in the treatment of bTB intermittently, using high doses for prolonged periods, the use of this drug in the treatment of bTB in herds infected with M. tuberculosis var. bovis has become routine, mainly in the dairy basins of Minas Gerais and São Paulo. The use of isoniazid in the routine treatment of bTB has become common, and milk cooperatives make this drug available in large quantities without any control, characterizing its indiscriminate use (53). This fact may be one of the reasons why M. tuberculosis var. bovis becomes resistant to isoniazid.
Antibiotics from the quinolone group are increasingly used in the treatment of respiratory infectious diseases in humans, which has led the strains of M. tuberculosis to be resistant to this drug (62). Mutations in the gyrA and gyrB genes are associated with quinolone resistance in M. tuberculosis, and these residues are thought to play a role in drug binding and quinolone resistance (62). Mutations in gyrA confer high-level resistance, while mutations in gyrB confer low-level resistance (52, 63).
In addition, there is an aggravating factor, and cases of patients with M. tuberculosis var. bovis and M. tuberculosis coinfection have been reported in urban areas (64). National studies have been restricted to urban areas, but rural areas should be investigated, since the occupational character of the disease mainly affects individuals who deal directly with live animals, handlers, breeders, veterinarians, and workers in meat, dairy and laboratories (6).
The strains currently show the same patterns in the detection of genes and mutations that can confer resistance to antibiotics; they are part of the same geographic area, which is the state of Mato Grosso, but they belong to very different livestock circuits. We also verified that the same conditions in more than 92% of the Brazilian strains of M. tuberculosis var. bovis presented similar patterns to those of Mato Grosso.
With the phylogenetic tree, we can verify two clades using a bootstrap above 77.7% and that the genomes sequenced in this work are distributed in both clades. The genomes ERR3445501, ERR3445502, ERR3445503, ERR3445504 were external to the clades, with ERR3445502 and ERR3445504 probably belonging to another species of the MTC complex that was inserted in the NCBI as M. tuberculosis var. bovis, so we removed these four genomes when generating the phylogenetic tree because they distort the branches of the tree. The explanation of these four genomes being external to the clades can be explained by the results of the RDscan, which did not identify the deletion regions preserved in M. tuberculosis var. bovis (RD4-RD10; RD12; RD13) in the ERR3445502 and ERR3445504 genomes and in the ERR3445503 genome RD9 deletion region identified (Supplementary material 4). The strains external to the clades ERR3445502, ERR3445503 and ERR3445504 were classified as MRD or pre-MDR and showed resistance to the drugs ciprofloxacin, fluoroquinolones, levofloxacin, moxifloxacin, ofloxacin (mutation in gyrB) and streptomycin (mutation in rrs), with the strains ERR3445504; pyrazinamide (pncA), isoniazid (kasA), and ethambutol (embB) strains ERR3445501 and ERR3445503; and isoniazid (kasA) strain (ERR3445502; Supplementary material 3).
The three BCG strains (SRR10997360, SRR10997362, and SRR10997361) were grouped together in a branch and were classified as MDR (Table 4). In addition to being resistant to pyrazinamide, they were resistant to other drugs. The BCG strains (SRR10997360 and SRR10997361) showed mutations in the rpoB (rifampicin), katG (isoniazid) and pncA (pyrazinamide) genes; the BCG strain (SRR10997362) showed mutations in rpoB and pncA; strain SRR13046675 showed mutation only in pncA. The other Brazilian strains showed only drug resistance to pncA. These results were reported by TB profiler, who compares the results of mutations already recognized in the literature with the confirmation that that particular mutation is truly linked to drug resistance. Strain ERR3445487 showed mutations in rrs (streptomycin) and pncA, also classified as MDR.
Although we selected four genomes from four different municipalities in the state of Mato Grosso, coming from different properties, a limitation of our study is the small sample size, which makes it difficult to interpret the reality of bTB in the state. A small number of animal carcasses with lesions suggestive of bTB were included in the genomic study. However, they are the first lineages sequenced in the state of Mato Grosso and, therefore, the first data on the genomics of M. tuberculosis var. bovis in this region, with the potential to help the epidemiological surveillance system in the state.
Regarding livestock circuits (CP), the genomes did not remain in the same clade, and there was no association of livestock circuits in this sample, with samples TMT05 and TMT123 representing the livestock circuit (CP4) and the livestock circuits TMT24 and TMT116.
In this study, we provide the first sequencing-based description of the population structure of M. tuberculosis var. bovis in the state of Mato Grosso, central-west region of Brazil. Other studies in the future may sequence a greater number of isolates from more herds, focusing on different livestock circuits and relating them to genetic diversity within the state, with the obtainment of available bovine movement data.
According to Carneiro et al. (51), in the Amazonas region, no significant difference was observed in the distribution of genetic diversity between the hosts (bovine and buffalo). In the study carried out by Conceição et al. (42) from Ilha de Marajó, it was not possible to verify these data because the information on the hosts is not available in the database.
Because the genomes of Ilha de Marajó are strongly related and form part of a clade and were probably introduced in the region during a single event still unknown (51), in contrast, the genomes of the Amazonas region were determined in three different events, not knowing the order of these events, which may have originated from neighboring cities, originated from Ilha de Marajó and even imported from other states (51). There are still many answers to be answered regarding the true epidemiological picture of bTB both in the state of Mato Grosso and other states in Brazil. It is necessary to know the genetic profile and understand the transmission routes of M. tuberculosis var. bovis, as it is essential to assist in the control and eradication of bTB.
The genetic profile of the genomes generated in this study should be further explored, including verification of possible specific genetic regions to strains from Mato Grosso, developing specific primers and probes to be used in the epidemiological investigation of the region.
Additionally, cultural aspects, such as consuming products derived from unpasteurized raw milk and consuming raw or undercooked meat, can pose a threat to human health (65). Mato Grosso is a large state and is strongly linked with livestock, and many rural workers have contact with animals and can be infected with M. tuberculosis var. bovis.
It needs to be investigated whether this zoonotic disease underestimates the risk of bTB for humans in the state of Mato Grosso. We believe that the information generated in this work is essential and that it can contribute to strategies for the control and eradication of bTB in the center-west of Brazil, reducing the risk to human health and ensuring food safety. These results indicate that epidemiological surveillance in Mato Grosso should invest in controlling import of cattle and/or buffaloes from neighboring states, such as Pará and Amazonas, which tested negative for bTB. Additionally, bTB control programs should invest in encouraging disease control, especially in the milk production region, which is represented by livestock circuit 2 (CP2), where there is a greater number of bTB outbreaks. In addition to having stricter public policies regarding the indiscriminate use of antimicrobials used in cattle, it can cause resistance to drugs used in the treatment of TB in humans.
The four strains sequenced in this study as well as 71 other genomes analyzed, except for two genomes (ERR3445502; ERR3445503), showed the deleted regions RD4, RD5, RD6, RD7, RD8, RD9, RD10, RD12, and RD13, which are highly conserved in M. tuberculosis var. bovis (30).
The genome that observed more deletion regions of Mato Grosso strains was SRR15649878 (TMT116). Three regions with deletions (RD) (Table 4), different from the reference strain, in the TMT116 strain in the milk region in the state of Mato Grosso, meaning a strain that had a greater number of mutations and more evolved in a region that has a greater number of bovine tuberculosis cases. The Mato Grosso strain SRR15649880 (TMT05) showed the absence of the Rv3508 gene that may be correlated with the response to the inhibition of aerobic respiration in mycobacteria (66).
The Rv0095c gene is present in the reference strain but deleted in the four strains sequenced in this study as well as in most Brazilian strains, except (SRR12511761; SRR9850830). The Rv0095c gene, a conserved protein of unknown function, has been associated with the successful transmission of a clade of M. tuberculosis in Peru (67).
The Rv0867c gene was not deleted in the four strains sequenced in this work or in another 60 Brazilian strains, different from the reference strain. This gene represents one of five Rpf (secreted growth factor)-like genes that are expressed in actively growing cells, stimulate bacterial growth at low concentrations and are related to dormant cell resuscitation (68). The four strains sequenced in this work do not have the Rv1563c gene deletion, as well as the other 72 Brazilian strains, which were deleted in the reference strain.
The Rv3798 gene is deleted in the four sequenced strains and in another 59 Brazilian strains and is preserved in the reference strain. It was not possible to identify a distinct regional genomic characteristic in the Mato Grosso strains compared to other Brazilian strains.
Through genome sequencing, it was possible to characterize M. tuberculosis var. bovis in samples of caseous lesions suggestive of bovine tuberculosis obtained during slaughter in slaughterhouses in the state of Mato Grosso, Brazil. The size of the genomes ranged from 4,306,423 to 4,332,964 bp; there was a number of CDs that ranged from 3,981 to 4,010 and identified that the sequenced strains of M. tuberculosis var. bovis are part of the BOV-AFRI strain. Comparing the M. tuberculosis var. bovis strains sequenced in this work with other strains sequenced in Brazil, it was possible to verify that the genomes were divided into the two clades (bootstrap above 77.7%) and that they are in different branches within these clades. The M. tuberculosis var. bovis strains from Mato Grosso sequenced in this work were determined to be drug-resistant strains. Future phenotypic analyses are needed to determine their real potential for resistance to other drugs, in addition to pyrazinamide resistance, as other mutations (rpsA, rpsL, gid, rpoB, katG, gyrB, gyrA, tlyA, embA, embB, embC, fgd, fbiB, and fbiC) were found in genes responsible for resistance to several antibiotics used in the treatment of tuberculosis in humans, similar to most brazilian M. tuberculosis var. bovis strains.
With these results, being the first sequencing of the genome of M. tuberculosis variant bovis in the State of Mato Grosso, we believe that these results are essential for the control and eradication of bTB in the State, reducing the risk to human health and ensuring food security. We emphasize the need to expand the use of the WGS in the Bovine Tuberculosis Control and Eradication Programs, with the objective of effectively achieving the eradication of the disease, not only in the State of Mato Grosso, which is the largest producer of cattle in the country, but also in other countries. brazilian states, in order to obtain bovine tuberculosis-free health status.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The animal study was reviewed and approved by Comissão de Ética no Uso de Animais da Universidade de Cuiabá (CEUA/UNIC). Written informed consent for participation was not obtained from the owners because notifiable zoonosis diagnosis.
Author contributions
Material preparations were performed by TA and RC. Data analysis was performed by TA, VC, EF, RC, EM, and PS. The first manuscript was written by TA. All authors contributed to the study conception and design, corrected the previous versions of the manuscript, read, and approved the final manuscript text.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001 and also by Foundation for Research Support in Mato Grosso State—FAPEMAT (Fundação de Amparo a Pesquisa do Estado de Mato Grosso) Grant No. 366047/2017. Conselho Nacional de Desenvolvimento Científico e Tecnológico (National Council for Scientific and Technological Development), Grant Number 310181/2021-6 and Fundação Oswaldo Cruz (Oswaldo Cruz Foundation).
Acknowledgments
The authors are grateful to Instituto de Defesa Agropecuaria de Mato Grosso (INDEA) for the partnership in providing suspected bovine tuberculosis samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.1006090/full#supplementary-material
References
1. Garnier T, Eiglmeier K, Camus JC, Medina N, Mansoor H, Pryor M, et al. The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci USA. (2003) 100:7877–82. doi: 10.1073/pnas.1130426100
2. Lasserre M, Fresia P, Greif G, Iraola G, Castro-Ramos M, Juambeltz A, et al. Whole genome sequencing of the monomorphic pathogen Mycobacterium bovis reveals local differentiation of cattle clinical isolates. BMC Genomics. (2018) 19:2. doi: 10.1186/s12864-017-4249-6
3. World Health Organization. Roadmap for Zoonotic Tuberculosis. (2017). Available online at: https://apps.who.int/iris/handle/10665/259229 (accessed November 3, 2021).
4. Garcia MS, Melo AF, Carvalho GF, Pomim GP, Neves PMS, Silva RAB, et al. Epidemiologia da tuberculose bovina na América do Sul. Res Soc Dev. (2021) 10:17936. doi: 10.33448/rsd-v10i9.17936
5. Ruggiero AP, Ikuno AA, Ferreira VCA, Roxo E. Tuberculose bovina alternativas para o diagnóstico. Arq Inst Biol. (2007) 74:552007. doi: 10.1590/1808-1657v74p0552007
6. BRASIL. Manual de Recomendações para o Controle da Tuberculose no Brasil. (2019). Available online at: https://portaldeboaspraticas.iff.fiocruz.br/wp-content/uploads/2021/04/manual_recomendacoes_controle_tuberculose_brasil_2_ed.pdf (accessed: September 20, 2021).
7. Carvalho RCT, Vasconcellos SEG, Issa MDA, Soares Filho PM, Mota PMPC, Araújo FRD, et al. Molecular typing of Mycobacterium bovis from cattle reared in Midwest Brazil. PLoS ONE. (2016) 11:162459. doi: 10.1371/journal.pone.0162459
8. Oliva HNP, Oliveira AG, Godinho ACVCQ, Alves BLR, Ramos MTBP, Galdino VAC, et al. Estudo epidemiológico da tuberculose no estado de Minas Gerais. Revista Eletrônica Acervo Saúde. (2019) 18:e78. doi: 10.25248/reas.e78.2019
9. Sisco MC, Silva MG, Lopez B, Arguelles C, Mendonça-Lima L, de Waard JH, et al. Newly sequenced genomes of four Bacillus Calmette Guerin vaccines. Mem Inst Oswaldo Cruz. (2020) 115:2760190401. doi: 10.1590/0074-02760190401
10. BRASIL. Tuberculose bovina em cervídeos. (2020). Available online at: https://www.infoteca.cnptia.embrapa.br/infoteca/bitstream/doc/1122940/1/Tuberculose-bovina-em-cervideos.pdf (accessed September 10, 2021).
11. Djemal SE, Camperio C, Armas F, Siala M, Smaoui S, Messadi-Akrout F, et al. Detection of a streptomycin-resistant Mycobacterium bovis strain through antitubercular drug susceptibility testing of Tunisian Mycobacterium tuberculosis complex isolates from cattle. BMC Vet Res. (2018) 14:1–8. doi: 10.1186/s12917-018-1623-9
12. Dixit A, Freschi L, Vargas R, Calderon R, Sacchettini J, Drobniewski F, et al. Whole genome sequencing identifies bacterial factors affecting transmission of multidrug-resistant tuberculosis in a high-prevalence setting. Sci Rep. (2019) 9:5602. doi: 10.1038/s41598-019-41967-8
13. Campos HS. Mycobacterium tuberculosis resistente: de onde vem a resitência? Bol Pneumol Sanit. (1999) 7:51–4. doi: 10.5123/S0103-460X1999000100006
14. Vázquez-Chacón CA, Rodríguez-Gaxiola FJ, López-Carrera CF, Cruz-Rivera M, Martínez-Guarneros A, Parra-Unda R, et al. Identification of drug resistance mutations among Mycobacterium bovis lineages in the Americas. PLoS Negl Trop Dis. (2021) 15:9145. doi: 10.1371/journal.pntd.0009145
15. Bouzouita I, Draoui H, Mahdhi S, Essalah L, Saidi LS. Amplification of genomic regions of difference for the identification of. Afr Health Sci. (2021) 21:5. doi: 10.4314/ahs.v21i3.4
16. Algammal AM, Hashem HR, Al-Otaibi AS, Alfifi KJ, El-Dawody EM, Mahrous E, et al. Emerging MDR-Mycobacterium avium subsp. avium in house-reared domestic birds as the first report in Egypt. BMC Microbiol. (2021) 21:237. doi: 10.1186/s12866-021-02287-y
17. Kareem SM, Al-Kadmy IMS, Kazaal SS, Mohammed Ali AN, Aziz SN, Makharita RR, et al. Detection of gyrA and parC mutations and prevalence of plasmid-mediated quinolone resistance genes in Klebsiella pneumoniae. Infect Drug Resist. (2021) 14:555–63. doi: 10.2147/IDR.S275852
18. Hetta HF, Al-Kadmy IMS, Khazaal SS, Abbas S, Suhail A, El-Mokhtar MA, et al. Antibiofilm and antivirulence potential of silver nanoparticles against multidrug-resistant Acinetobacter baumannii. Sci Rep. (2021) 11:10751. doi: 10.1038/s41598-021-90208-4
19. Algammal AM, Mabrok M, Ezzat M, Alfifi KJ, Esawy AM, Elmasry N, et al. Prevalence, antimicrobial resistance (AMR) pattern, virulence determinant and AMR genes of emerging multi-drug resistant Edwardsiella tarda in Nile tilapia and African catfish. Aquaculture. (2022) 548:737643. doi: 10.1016/j.aquaculture.2021.737643
20. Negreiros RL, Dias RA, Ferreira F, Ferreira Neto JS, Gonçalves VSP, Silva MCP, et al. Situação epidemiológica da brucelose bovina no Estado de Mato Grosso. Arq Bras Med Veterinária e Zootec. (2009) 61:56–65. doi: 10.1590/S0102-09352009000700008
21. BRASIL. Decreto n° 9.013, de 29 de março de 2017. (2017). Available online at: https://www.in.gov.br/materia/-/asset_publisher/Kujrw0TZC2Mb/content/ id/20134722/do1-2017-03-30-decreto-n-9-013-de-29-de-marco-de-2017-20134698 (accessed September 10, 2021).
22. Furlanetto LV, Figueiredo EES, Conte Júnior CA, Carvalho RCT, Silva FGS, Silva JT, et al. Uso de métodos complementares na inspeção post mortem de carcaças com suspeita de tuberculose bovina. Pesqu Veterinária Bras. (2012) 32:1138–44. doi: 10.1590/S0100-736X2012001100011
23. Araújo CP, Osório ALAR, Jorge KSG, Ramos CAN, Filho AFS, Vidal CES, et al. Detection of Mycobacterium bovis in bovine and bubaline tissues using nested-PCR for TbD1. Inacio J, organizador. PLoS ONE. (2014) 9:1023. doi: 10.1371/journal.pone.0091023
24. Araújo CP, Osório ALAR, Jorge KSG, Ramos CAN, Souza Filho AF, Vidal CES, et al. Direct detection of Mycobacterium tuberculosis complex in bovine and bubaline tissues through nested-PCR. Braz J Microbiol. (2014) 45:35. doi: 10.1590/S1517-83822014000200035
25. Carvalho RCT, Furlanetto LV, Maruyama FH, de Araújo CP, Barros SLB. do Nascimento Ramos CA. Evaluation of the efficiency of nested q-PCR in the detection of Mycobacterium tuberculosis complex directly from tuberculosis-suspected lesions in post-mortem macroscopic inspections of bovine carcasses slaughtered in the state of Mato Grosso, Brazil. Meat Sci. (2015) 106:11–5. doi: 10.1016/j.meatsci.2015.03.017
26. Ambrosio SR, Oliveira EM de D, Rodriguez CAR, Ferreira Neto JS, Amaku M. Comparison of three decontamination methods for Mycobacterium bovis isolation. Braz J Microbiol. (2008) 39:241–4. doi: 10.1590/S1517-83822008000200008
27. Cousins DV, Francis BR, Gow BL. Advantages of a new agar medium in the primary isolation of Mycobacterium bovis. Vet Microbiol. (1989) 20:89–95. doi: 10.1016/0378-1135(89)90010-2
28. OIE. Bovine tuberculosis. (2018). Available online at: https://www.oie.int/en/disease/bovine-tuberculosis/ (accessed November 1, 2021).
29. BRASIL. Manual Nacional de Vigilância Laboratorial da Tuberculose e outras Micobactérias. (2008). Available online at: https://bvsms.saude.gov.br/bvs/publicacoes/manual_vigilancia_laboratorial_tuberculose.pdf (accessed November 3, 2021).
30. Ramos JM, Heinemann MB, Ferreira Neto JS, Souza Filho AFD, Cárdenas NC, Dantas AFM, et al. Isolation and identification of Mycobacterium bovis in bovines with positive reaction to the tuberculin test in the state of Paraíba, northeast Brazil. Arq do Instit Biológ. (2018) 85:1657000842016. doi: 10.1590/1808-1657000842016
31. Mota PMPC, Lobato FCF, Assis RA, Lage AP, Parreiras PM. Isolamento de Mycobacterium bovis em cão. Arqu Bras de Med Vet e Zoot. (2001) 53:1–3. doi: 10.1590/S0102-09352001000400003
32. Van Soolingen D, Hermans PW, de Haas PE, Soll DR, Van Embden JD. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J Clin Microbiol. (1991) 29:1991. doi: 10.1128/jcm.29.11.2578-2586.1991
33. Néspoli JMB, Negreiros RL, Amaku M, Dias RA, Ferreira F, Telles EO, et al. Epidemiological situation of bovine tuberculosis in the State of Mato Grosso, Brazil. Semina Ciênc Agrár. (2016) 37(5Supl.2):3589. doi: 10.5433/1679-0359.2016v37n5Supl2p3589
34. Barcelos FG, dos Anjos TR, de Arruda Alencar LA, de Freitas MJS, Espinosa MM, Carvalho RCT. Risk factors associated with the presence of Mycobacterium bovis in macroscopic lesions suspected as being caused by bovine tuberculosis detected in slaughterhouses. Semina Ciênc Agrár. (2022) 2022:v1. doi: 10.21203/rs.3.rs-581494/v1
35. Seemann T, Shovill. (2022). Available online at: https://github.com/tseemann/shovill (accessed May 13, 2022).
36. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
37. Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. (2015) 7:51. doi: 10.1186/s13073-015-0164-0
38. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST quality assessment tool for genome assemblies. Bioinformatics. (2013) 29:1072–5. doi: 10.1093/bioinformatics/btt086
39. Matin MA, Nahid AA, Sarkar B, Ullah MA, Araf Y, Adnan N. Genome sequences of two novel coronavirus (SARS-CoV-2) isolates from Dhaka, Bangladesh. Microbiol Resour Announcement. (2021) 10:21. doi: 10.1128/MRA.00511-21
40. Guimaraes AMS, Zimpel CK, Ikuta CY, do Nascimento NC, dos Santos AP, Messick JB, et al. Draft genome sequence of Mycobacterium bovis strain SP38, a pathogenic bacterium isolated from a bovine in Brazil. Genome Announc. (2015) 3:15. doi: 10.1128/genomeA.00511-15
41. Rodrigues RA, Ribeiro Araújo F, Rivera Dávila AM, Etges RN, Parkhill J, van Tonder AJ. Genomic and temporal analyses of Mycobacterium bovis in southern Brazil. Microb Genomics. (2021) 7:569. doi: 10.1099/mgen.0.000569
42. Conceição ML, Conceição EC, Furlaneto IP, Silva SP, Guimarães AES, Gomes P, et al. Phylogenomic perspective on a unique Mycobacterium bovis clade dominating bovine tuberculosis infections among cattle and buffalos in Northern Brazil. Sci Rep. (2020) 10:1747. doi: 10.1038/s41598-020-58398-5
43. Alexander DC, Ma JH, Guthrie JL, Blair J, Chedore P, Jamieson FB. Gene sequencing for routine verification of pyrazinamide resistance in Mycobacterium tuberculosis: a role for pncA but not rpsA. J Clin Microbiol. (2012) 50:3726–8. doi: 10.1128/JCM.00620-12
44. BRASIL. Manual de recomendações para o controle da tuberculose no Brasil. (2011). Available online at: https://bvsms.saude.gov.br/bvs/publicacoes/manual_recomendacoes_controle_tuberculose_brasil.pdf (accessed November 1, 2021).
45. Jacobs WR, Hatfull GF. Molecular Genetics of Mycobacteria. Washington: John Wiley & Sons (2020).
46. Jugheli L, Bzekalava N, de Rijk P, Fissette K, Portaels F, Rigouts L. High level of cross-resistance between kanamycin, amikacin, and capreomycin among Mycobacterium tuberculosis isolates from Georgia and a close relation with mutations in the rrs gene. Antimicrob Agents Chemother. (2009) 53:5064–8. doi: 10.1128/AAC.00851-09
47. Maus CE, Plikaytis BB, Shinnick TM. Molecular analysis of cross-resistance to capreomycin, kanamycin, amikacin, and viomycin in Mycobacterium tuberculosis. Antimicrob Agents Chemother. (2005) 49:3192–7. doi: 10.1128/AAC.49.8.3192-3197.2005
48. MISAU. Manual de Diagnóstico e Tratamento de Tuberculose Resistente e Multi-Droga Resistente. (2009). Available online at: http://gard-cplp.ihmt.unl.pt/Documentos/Paises/Mocambique/Manual_tuberculose_MDR_2009_Mocambique.pdf (accessed October 15, 2021).
49. Santos ESV, Alzamora Filho F, Santos BR, Silva AV, Gomes HM, Suffys PN, et al. Spoligotyping, genotyping, and spatial distribution of Mycobacterium bovis in cattle in the state of Bahia, Brazil. Pesqui Veterinária Bras. (2021) 41:6729. doi: 10.1590/1678-5150-pvb-6729
50. Smith NH. The global distribution and phylogeography of Mycobacterium bovis clonal complexes. Infect Genet Evol. (2012) 12:857–65. doi: 10.1016/j.meegid.2011.09.007
51. Carneiro PA, Zimpel CK, Pasquatti TN, Silva-Pereira TT, Takatani H, Silva CBDG, et al. Genetic diversity and potential paths of transmission of Mycobacterium bovis in the amazon: the discovery of M. bovis lineage Lb1 circulating in South America. Front Vet Sci. (2021) 8:63. doi: 10.3389/fvets.2021.630989
52. Kabir S, Tahir Z, Mukhtar N, Sohail M, Saqalein M, Rehman A. Fluoroquinolone resistance and mutational profile of gyrA in pulmonary MDR tuberculosis patients. BMC Pulm Med. (2020) 20:138. doi: 10.1186/s12890-020-1172-4
53. Mota PMPC, Mota PMC, Lobato FCF, Lage AP, Ribeiro ACCL, Leite, RC. Avaliação do tratamento com isoniazida em bovinos infectados naturalmente pelo Mycobacterium bovis. Arquivo Brasileiro de Medicina Veterinária e Zootecnia. (2004) 56:581–8. doi: 10.1590/S0102-09352004000500003
54. World Health Organization. Global Tuberculosis Report. (2018). Available online at: https://apps.who.int/iris/handle/10665/330191 (accessed November 9, 2021).
55. Sagasti S, Millán-Lou MI, Soledad Jiménez M, Martín C, Samper S. In-depth analysis of the genome sequence of a clinical, extensively drug-resistant Mycobacterium bovis strain. Tuberculosis. (2016) 100:46–52. doi: 10.1016/j.tube.2016.06.005
56. Thoen C, LoBue P, de Kantor I. The importance of Mycobacterium bovis as a zoonosis. Vet Microbiol. (2006) 112:339–45. doi: 10.1016/j.vetmic.2005.11.047
57. Hughes VM, Skuce R, Doig C, Stevenson K, Sharp JM, Watt B. Analysis of multidrug-resistant Mycobacterium bovis from three clinical samples from Scotland. Int J Tuberc Lung Dis. (2003) 7:1191–8.
58. Gómez-González PJ, Perdigao J, Gomes P, Puyen ZM, Santos-Lazaro D, Napier G, et al. Genetic diversity of candidate loci linked to Mycobacterium tuberculosis resistance to bedaquiline, delamanid and pretomanid. Sci Rep. (2021) 11:19431. doi: 10.1038/s41598-021-98862-4
59. Yang JS, Kim KJ, Choi H, Lee SH. Delamanid, bedaquiline, and linezolid minimum inhibitory concentration distributions and resistance-related gene mutations in multidrug-resistant and extensively drug-resistant tuberculosis in Korea. Ann Lab Med. (2018) 38:563–8. doi: 10.3343/alm.2018.38.6.563
60. Miotto P, Tessema B, Tagliani E, Chindelevitch L, Starks AM, Emerson C, et al. A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur Respir J. (2017) 50:2017. doi: 10.1183/13993003.01354-2017
61. Langenegger J, Leite GO, Oliveira Junior J. Tratamento intermitente da tuberculose bovina com isoniazida. Pesqui Vet Bras. (1991) 1991:55–9.
62. Aubry A, Veziris N, Cambau E, Truffot-Pernot C, Jarlier V, Fisher LM. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: functional analysis of mutant enzymes. Antimicrob Agents Chemother. (2006) 50:104–12. doi: 10.1128/AAC.50.1.104-112.2006
63. Zhang Y, Yew WW. Mechanisms of drug resistance in Mycobacterium tuberculosis [state of the art series. Drug-resistant tuberculosis Edited by C-Y Chiang Number 1 in the series]. Int J Tuberc Lung Dis. (2009) 13:1320–30. doi: 10.1128/9781555817657.ch8
64. Silva MR, Rocha AS, Araújo FR, Fonseca-Júnior AA, Alencar AP, Suffys PN, et al. Risk factors for human Mycobacterium bovis infections in an urban area of Brazil. Mem Inst Oswaldo Cruz. (2018) 113:2760170445. doi: 10.1590/0074-02760170445
65. OMS. Hoja de ruta contra la tuberculosis zoonótica. (2017). Available online at: https://apps.who.int/iris/handle/10665/259231 (accessed January 14, 2022).
66. Yu G, Fu X, Jin K, Zhang L, Wu W, Cui Z, et al. Integrative analysis of transcriptome and genome indicates two potential genomic islands are associated with pathogenesis of Mycobacterium tuberculosis. Gene. (2011) 489:21–9. doi: 10.1016/j.gene.2011.08.019
67. Vargas R, Freschi F, Marin M, Epperson LE, Smith M, Oussenko I, et al. In-host population dynamics of Mycobacterium tuberculosis complex during active disease. Elife. (2021) 10:e61805. doi: 10.7554/eLife.61805
Keywords: Mycobacterium tuberculosis variant bovis, mutation, phylogeny, drug resistance, zoonosis
Citation: Anjos TRd, Castro VS, Machado Filho ES, Suffys PN, Gomes HM, Duarte RS, Figueiredo EEdS and Carvalho RCT (2022) Genomic analysis of Mycobacterium tuberculosis variant bovis strains isolated from bovine in the state of Mato Grosso, Brazil. Front. Vet. Sci. 9:1006090. doi: 10.3389/fvets.2022.1006090
Received: 28 July 2022; Accepted: 28 October 2022;
Published: 16 November 2022.
Edited by:
Abdelaziz Ed-Dra, Sultan Moulay Slimane University, MoroccoReviewed by:
Abdelazeem Algammal, Suez Canal University, EgyptSaid Oulghazi, Moulay Ismaïl University, Morocco
Copyright © 2022 Anjos, Castro, Machado Filho, Suffys, Gomes, Duarte, Figueiredo and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip Noel Suffys, cHN1ZmZ5c0BnbWFpbC5jb20=; Ricardo César Tavares Carvalho, cmljYXJkb19jYXJ2YWxobzg4QGhvdG1haWwuY29t