 Jiaxin Chen1†
Jiaxin Chen1† Peng Shang
Peng Shang Riaz Hussain
Riaz Hussain Dayou Shi
Dayou Shi- 1College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
- 2College of Animal Science, Tibet Agriculture and Animal Husbandry College, Linzhi, China
- 3Department of Pathology, Faculty of Veterinary and Animal Sciences, The Islamia University of Bahawalpur, Bahawalpur, Pakistan
Diquat (DQ) is an effective herbicide and is widely used in agriculture. Due to persistent and frequent applications, it can enter into aquatic ecosystem and induce toxic effects to exposed aquatic animals. The residues of DQ via food chain accumulate in different tissues of exposed animals including humans and cause adverse toxic effects. Therefore, it is crucial and important to understand the mechanisms of toxic effects of DQ in exposed animals. We used ducks as test specimens to know the effects of acute DQ poisoning on mechanisms of apoptosis and autophagy in liver tissues. Results on comparison of various indexes of visceral organs including histopathological changes, apoptosis, autophagy-related genes, and protein expression indicated the adverse effects of DQ on the liver. The results of our experimental trial showed that DQ induces non-significant toxic effects on pro-apoptotic factors like BAX, BAK1, TNF-α, caspase series, and p53. The results revealed that anti-apoptotic gene Parkin was significantly upregulated, while an upward trend was also observed for Bcl2, suggesting that involvement of the anti-apoptotic factors in ducklings plays an important role in DQ poisoning. Results showed that DQ significantly increased the protein expression level of the autophagy factor Beclin 1 in the liver. Results on key autophagy factors like LC3A, LC3B, and p62 showed an upward trend at gene level, while the protein expression level of both LC3B and p62 reduced that might be associated with process of translation affected by the pro-apoptotic components such as apoptotic protease that inhibits the occurrence of autophagy while initiating cell apoptosis. The above results indicate that DQ can induce cell autophagy and apoptosis and the exposed organism may resist the toxic effects of DQ by increasing anti-apoptotic factors.
Introduction
Diquat (1,10-ethylene-2,20-bipyridinium, DQ) is a widely used non-selective herbicide that belongs to bipyridine (Figure 1) (1, 2). The toxicity of DQ is less different than that of other herbicides like paraquat (PQ) due to its rapid degradation in the environment. Although DQ is less toxic than other herbicides, there are still numerous adverse effects that are related to DQ. A previous report has indicated that accidental exposure to this herbicide during applications induces different toxic effects (3). In addition, a previous study has shown that long-term exposure to DQ can increase the risk of Parkinson's disease in animals including public health (4). DQ can easily enter into aquatic ecosystem via direct discharge from production industry, agricultural sector, and runoff (5) and causes serious threats to fish (6), freshwater snails (7), ducks (8), and other aquatic animals. Peking ducks are mainly raised in rural areas of China, which are meat-based species. We choose the Peking ducks as our experiment animals because these animals have the closest connection with humans.
Figure 1. The molecular structure of DQ.
The liver is the target organ of DQ toxicity. The mechanism of liver injury induced by DQ is complex, but it is mainly related to reactive oxygen species (ROS). DQ can induce increased oxidative stress to produce a large number of superoxides and can alter different liver functional activities such as metabolism, detoxification, and immune response resulting in the production of a large number of ROS in the liver (9). The liver toxicity leads to hepatocyte degeneration, inflammation, mitochondrial dysfunction, and even apoptosis (10, 11). Oxidative stress can reduce glutathione (GSH) in the liver and can enhance the production of ROS to promote the activation of pro-apoptotic factors such as p53 and Bax leading to apoptosis (12). In the liver, the mitochondria are also affected by overproduction of ROS ultimately leading to oxidative stress. Studies have indicated that DQ can decrease the activities of mitochondrial complex I, II, III, and V in the liver and decrease the abundance of mitochondrial biosynthesis its related genes (SIRT1, PGC1ATP, and TfAM). Furthermore, oxidative stress can affect the activities of different antioxidant enzymes and interfere with the antioxidant defense mechanisms (13) in the liver and blood. It is reported that infants or piglets with underdeveloped antioxidant systems are more vulnerable to oxidative damage and liver diseases (14). The mechanisms of toxicity of DQ are still under debate. However, DQ induces its toxic effects in the liver through a variety of liver injuries.
Apoptosis is a process of cell death regulated by different genes, while autophagy is a self-protective process of degradation of its own components through the lysosomal system. Herbicides such as DQ and PQ can induce abnormal mechanisms of apoptosis and autophagy. A previous study has shown that different toxins can induce oxidative stress in mice and produce a large amount of ROS to promote DNA damage, upregulation of genes regulating cell cycle (CDKN1A, CDKN2C, and CDKN2D), and pro-apoptotic genes (caspase-3 and Bax) eventually leading to promote apoptosis (15). Nitrite interferes with the expression of anti-apoptosis gene Bcl2 and leads to apoptosis (16) by enhancing the expression of caspase-3 and Bax genes in tissues. It has been recorded that aloe emodin can activate abnormal signals of apoptosis in the liver of zebrafish, promote the expression of p65, upregulate the proteases caspase-3 and Bax in p53 pathways while can downregulate Bcl2 in p53 pathway, and upregulate the expression of TNF-α and JNK in NF-κB that can induce inflammation and apoptosis (17). Different insecticides like fipronil can also promote the production of peroxisome that damages the DNA and mitochondria, causes the overexpression of Bax, and aggravates hepatocyte apoptosis (18). Furthermore, studies have reported that various drugs can cause hepatocyte apoptosis. The most commonly used anti-tuberculosis drug activates inflammatory corpuscles NLRP3 to upregulate the expression of p53, Bax, and Cleaved-Cas3, inhibit the expression of Bcl2, induce inflammation and apoptosis, and damage hepatocytes in rats (19). High concentrations of acetaminophen can reduce GSH resulting in increased oxidative stress and severe mitochondrial dysfunction, promoting the expression of key apoptotic factors, and promoting apoptosis (20, 21). A previous study has shown that increased concentrations of manganese (Mn) can induce neurotoxicity, increase the levels of glutathione peroxidase (GSH-Px), superoxide dismutase (SOD), and malondialdehyde (MDA), and promote the expression of p53, Bax, Bak, fas, and caspase-3 to induce apoptosis (22). A previous study has reported that cadmium affects the expression of IP3R1 receptor through Ca2+ channels, induces DNA damage, and promotes the abnormal process of autophagy or apoptosis (23). Arsenic (As) and its methylation metabolites not only affect regulatory enzymes such as 2 (ERK2), p38, or c-jun to induce neuronal and neuroblastoma cell apoptosis through MAPK signaling pathways but also induce apoptosis through AMP-dependent protein kinase (AMPK)/mTOR signal pathways (24). The morphology of the mitochondria is also important in apoptosis (25, 26). The related proteins Drp1, Mfn1, and Mfn2, mitochondrial a-KGDH, that affect cell division and fusion are all related to the mitochondria (27). The mitochondria play a central role in the integration and circulation of intracellular death signals, such as oxidative stress and DNA damage (28, 29).
The mitochondria can cause cell injury or apoptosis by producing ROS, pro-inflammatory signals, or through mitochondrial membrane permeability. A little electron may escape from the mitochondrial electron transport chain resulting in the production of superoxide. The increased oxidant load also promotes the extra ROS produced by mitochondrial complex I to further enhance cell oxidative stress and promote cell death (30). In addition, ROS produced by the mitochondria can activate NLRP3, adaptation proteins ASC, and caspase-1 to form inflammatory bodies, and the accumulation of damaged mitochondria aggravates inflammation and leads to cell damage. The change of mitochondrial permeability is also an important cause of cell death, which leads to the dissipation of mitochondrial transmembrane potential and the cessation of oxonase. Furthermore, it leads to rapid necrosis of apoptotic (31). When apoptosis is induced by mitochondrial damage, the decrease of ATP produced by the mitochondria through respiratory chain or the increase of ROS will lead to the increase of autophagy (32). Moreover, the induction of autophagy affects the circuit in which the mitochondria transmit lethal signals, protecting cells from other deadly stimuli (33). However, the relationship between autophagy and apoptosis affected by the mitochondria is complex. These two pathways are activated at the same time and can be regulated by the same factors such as Bcl-2 family proteins, cystatin (caspase), ATG proteins, and p53. On the other hand, autophagy and apoptosis are antagonistic to each other, and autophagy of damaged mitochondrial cells can reduce apoptotic signal transmission and protect normal cells.
The pathways through which DQ induces toxic effects are not clear. Therefore, it is interesting to speculate that DQ may induce harmful effects via induction of oxidative stress and increased level of apoptosis factors by regulating apoptosis and autophagy-related signal pathways like NF-κB, MAPK, and mTOR (34). Autophagy is beneficial to inhibit DQ-induced apoptosis and alleviate the effects of DQ poisoning (35). Therefore, the purpose of this study was to explore the relationship between liver injury and apoptosis and autophagy genes and proteins in ducks induced by acute DQ poisoning.
Materials and Methods
Animals and Treatment
All the experiments were approved by the Animal Ethics Committee of South China Agricultural University (License Number: 2017A087) and were conducted following the ethical code of conduct for animal care and use. After 7 days of acclimatization, a total of 60 1-day-old Peking ducks were randomly divided into two groups including the control and treatment groups (feeding 100 mg/kg DQ on the first day). The ducks in the treatment group were exposed to DQ for 11 days. After weighing at day 11, ducks were anesthetized by injecting chloral hydrate through the intraperitoneal route. Blood was collected from the jugular vein for biochemical profile. The heart, liver, kidney, thymus, spleen, and bursa of fabric of the ducks were removed, weighed separately, and photographed, and the coefficient of each organ was calculated.
Histopathological Examination
Liver tissue of 1 × 1 cm was cut and fixed in 4% paraformaldehyde solution. After that, all the tissues were processed and then embedded in paraffin for 24 h. Approximately 0.4 μm thick liver slices were obtained with the help of KEDEE KD2258 manual rotary microtome and were then stained with hematoxylin and eosin (H&E). Briefly, the paraffin slices were dewaxed in xylene solution for 20 min and then sequentially soaked in benzene alcohol, anhydrous ethanol I, anhydrous ethanol II, 95% alcohol I, 95% alcohol II, and 80% alcohol solution each for 10 min to remove xylene. After that, the slices were soaked in hematoxylin for 8 min, rinsed with water for 15 min, then shaken in the differentiation solution for ~2 s, rinsed with water for 20 min again, and dyed again with eosin for 8 min. The dehydration and transparency procedures used in paraffin sections preparation were repeated. The residual liquid on the slice was removed, and neutral resin was dropped on slice, then sealed, and baked for more than 6 h. Finally, all the prepared sections were observed under light microscope (Y-TV55; NiKon, Japan) and photographed.
RT-qPCR Analysis
The primer and sequences used for real-time PCR (RT-PCR) are shown in Table 1. Total RNA was lysed from 100 mg of the liver using RNA isolate reagents (Vazyme, China). Total RNA was separated with chloroform and precipitated with isopropanol. Ethanol was used to wash the residual isopropanol. Total RNA concentration was measured by Microvolume UV–Vis spectrophotometer (Nanodrop™ One; Thermo Fisher Scientific, Madison, WI, USA). Approximately 5 μg total RNA was reverse-transcribed into cDNA using HiScript III RT SuperMix for qPCR (Vazyme, China). The mixture contained 1 μl cDNA primer, and ChamQ University SYBR qPCR Master Mix (Vazyme, China) was used to perform RT-qPCR on the real-time PCR detection system (QuantStudio™ 5; Thermo Fisher Scientific, Waltham, MA, USA). The 2−ΔΔ(Ct) method was used to calculate the relative gene expression level, and GAPDH was used as the internal reference gene. The results are expressed as normalize mRNA levels by reference gene.
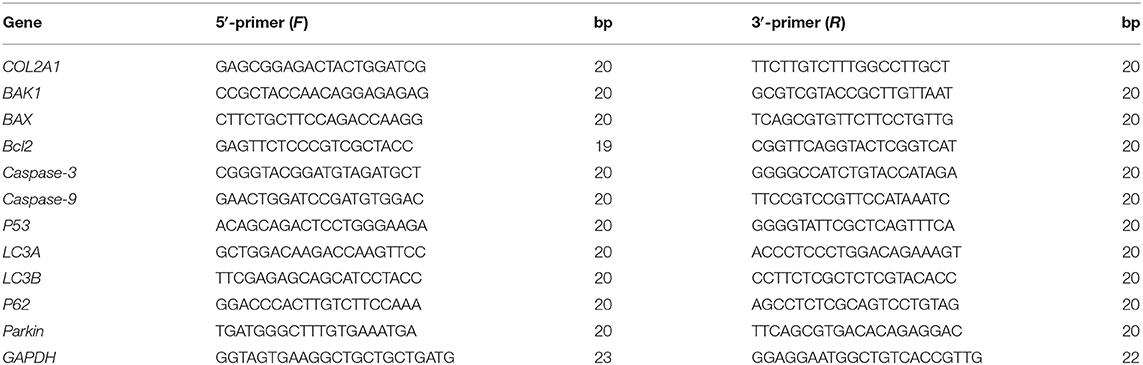
Table 1. Primer sequences used for real-time PCR.
Western Blot Analyses
The antibodies used for Western blot are shown in Table 2. Approximately 80 mg of the liver was lysed in RIPA lysis buffer (Meilunbio, China) and 1 mM protease inhibitor (PMSF) (Meilunbio, China) at 4°C, and the concentration was determined using the BCA protein concentration determination kit (Beyotime, China). After that, samples were diluted with 5 × SDS-PAGE loading buffer and boiled for 8 min. An equal amount of protein sample (10 μg) was added and electrophoresed on a 12.5% SDS-polyacrylamide denaturing gel and then transferred to polyvinylidene fluoride membranes. After blocking with Tris buffered saline Tween (TBST) containing 5% skimmed milk powder for 1 h, primary antibodies were incubated with membranes for 16 h. After washing with TBST for three times, the membranes were blocked with secondary antibody for 1 h. Finally, an electrochemiluminescent liquid (ECL) (Meilunbio, China) was prepared to measure the signal imprinting. ImageJ software was used to calculate the gray value of each band and perform normalization processing.
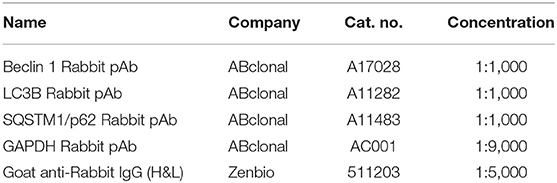
Table 2. Antibodies used for Western blot.
Statistical Analysis
Statistical analysis was performed on all data using GraphPad Prism 8.0 (GraphPad Inc., La Jolla, CA, USA) and SPSS for Windows (version 22; SPSS Inc., Chicago, IL, USA). The independent sample t-test was used to analyze the differences of the data between each group. Data were expressed as the mean ± standard deviation (SD). The data between different groups were analyzed by one-way analysis of variance (ANOVA) (n = 2, each repeated three times). Significance level was considered as p < 0.05, p < 0.01, and p < 0.001.
Results
The Influence of DQ on Organ Index and Serum Biochemistry
The indexes of the heart, liver, kidney, and spleen were significantly higher in treated ducks (Figure 2) than in the control group (p <0.05). The results showed no significant difference in the index of the cloacal bursa and thymus (p > 0.05) as compared with ducks of the control group.
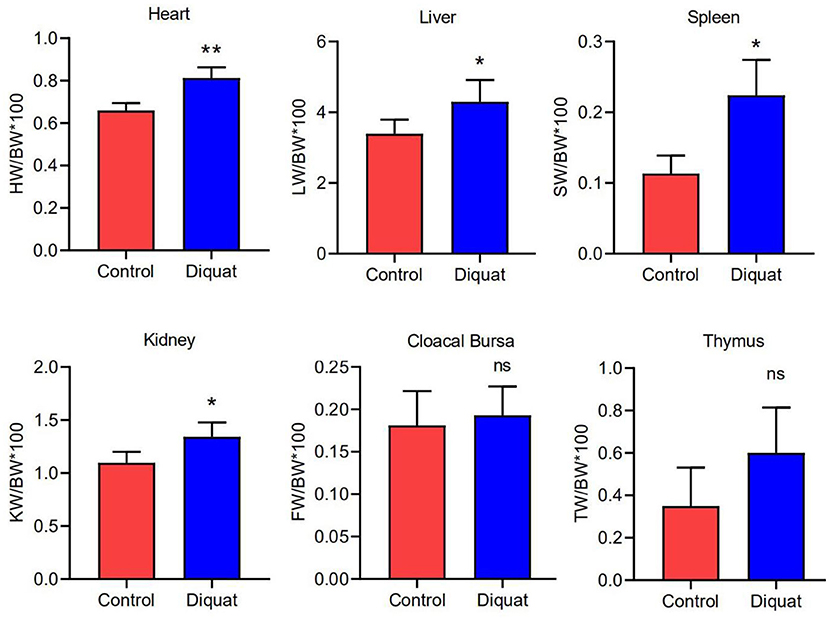
Figure 2. The organ index of the heart, liver, spleen, kidney, cloacal bursa, and thymus. Error bars indicate standard error of the mean (n = 4). “ns” and “*” indicate the level of significance. (“ns” means no significant difference, *p < 0.05, **p < 0.01 compared with the control conditions).
The results of serum biochemical examination are shown in Table 3. The results showed that DQ reduces the serum levels of calcium (Ca), phosphorus (P), triglyceride (TG), total protein (TP), total cholesterol (TC), albumin (ALB), γ-glutamyl transpeptidase (γ-GT), alkaline phosphatase (ALP), aspartate aminotransferase (AST), and total bilirubin (T-Bil) as compared with the control group. Among them, Ca, TG, ALB, ALP, and T-Bil are significantly reduced (p < 0.05), and AST is extremely significantly reduced (p < 0.01).
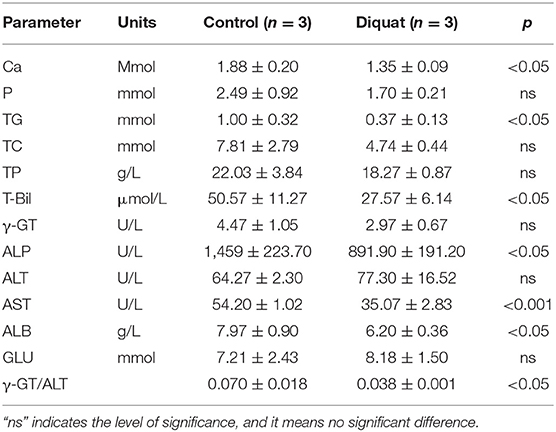
Table 3. Serum biochemical index.
The Effect of DQ on Liver Histopathology
In general, there were no obvious pathological changes in the liver of treated and untreated control groups (Figure 3). The basic liver structure of the control group and the DQ treatment group was normal and clear. No inflammatory response was observed in the portal area and liver parenchyma. Histologically, the liver cells of the control group were slightly enlarged, and the accumulation of eosinophilic glycogen material was observed in the cytoplasm. In the DQ treatment group, a large number of vacuoles (shown by black arrows) were observed in the liver cells, which were diffusely distributed. In liver sections of DQ, treated duck's fatty infiltration was obvious indicating that the drug may cause liver fat metabolism disorders.
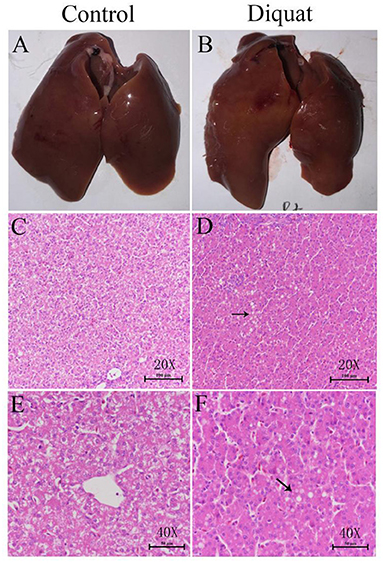
Figure 3. The effect of diquat on the liver. (A) The liver of the control group. (B) The liver of the DQ treated group. (C) Control group (HE, 20×). (D) DQ treated group (HE, 40 ×). (E) Control group (HE, 40 ×). (F) DQ treated group (HE, 40 ×).
The Effect of DQ on Cell Apoptosis
Compared with the control group, the expression of apoptosis-related genes is recorded as indicated in Figure 4. The expression level of caspase-3 decreased significantly, and the expression of Bax increased significantly. In addition, there was no significant difference in the expression of Bak1, Bcl2, p53, and Caspase9 genes. In these genes, only Caspase9 showed an upward trend, and the others showed a downward trend.
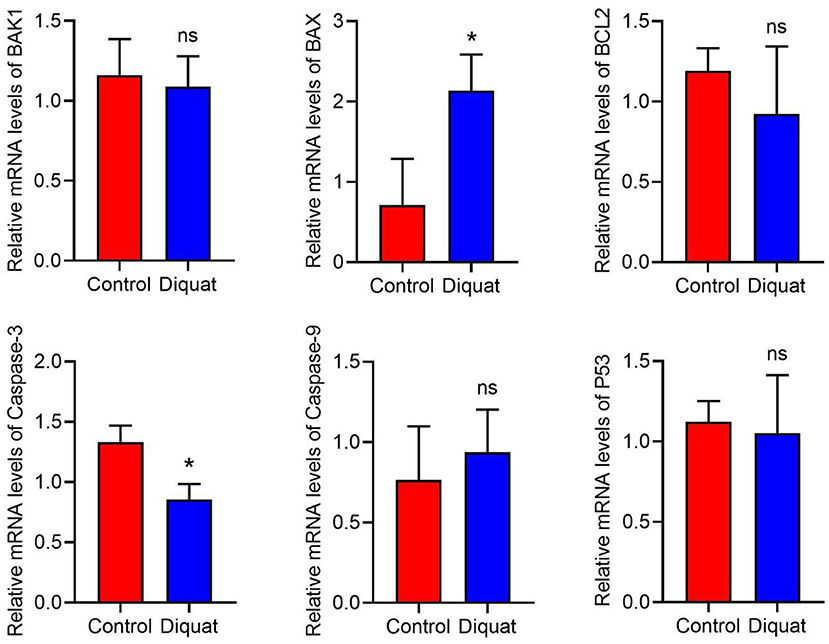
Figure 4. The expression of apoptotic genes in the liver. DQ affects the mRNA expression levels of apoptosis-related genes in liver, including BAK1, BAX, Bcl2, Caspase-3, Caspase-9, and P53. “ns” and “*” indicates the level of the Significance. (“ns” means no significant difference, *p < 0.05 compared to the control conditions).
The Effect of DQ on Autophagy
The changes of autophagy-related genes and proteins in the ducks treated with DQ are shown in Figure 5. As compared with the control group, there was no significant difference in the expression of LC3A, LC3B, and p62 genes, in which LC3A and LC3B increased non-significantly, and E3 ubiquitin ligase gene Parkin increased significantly. The protein expression like LC3B decreased significantly, while Beclin 1 increased significantly.
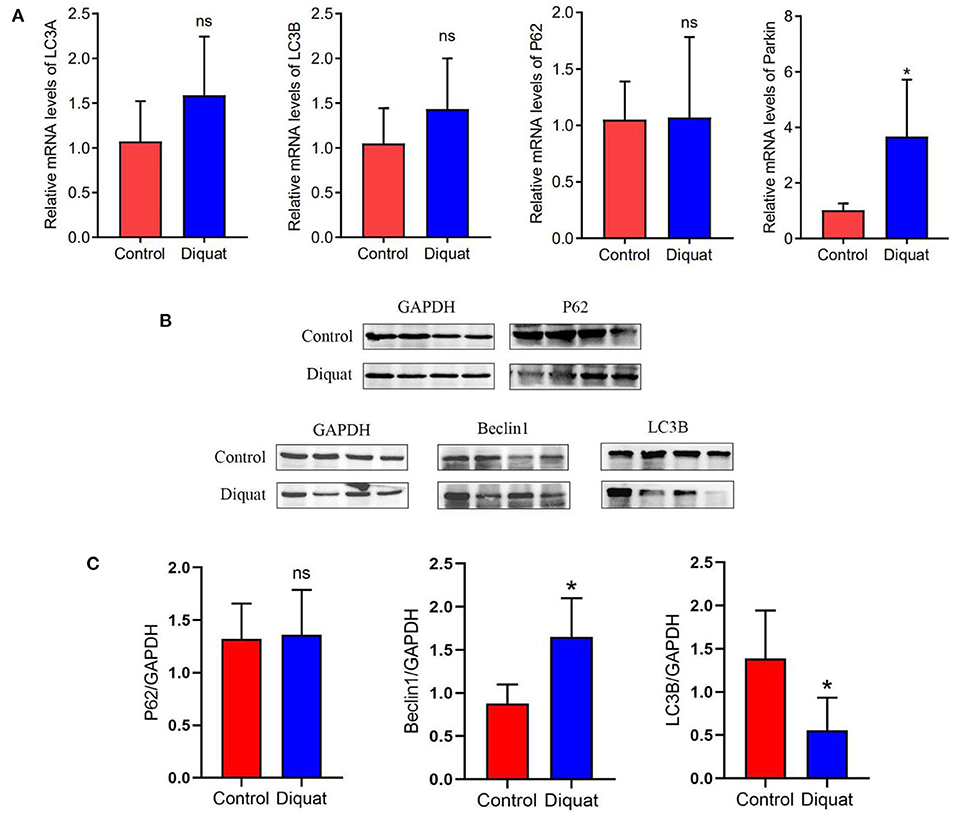
Figure 5. The expression of autophagy genes and proteins in the liver. (A) Autophagy mRNA gene expression: LC3A, LC3B, and P62. (B) Western blot detects protein expression bands: P62, Beclin 1, and LC3B. (C) The level of the protein expression: P62, Beclin 1, and LC3B. “ns” and * indicates the level of the significance. (“ns” means no significant difference, *p < 0.05 compared to the control conditions).
Discussion
Different histopathological changes in liver sections of treated ducks were observed in our experimental study. Previously, various pathological lesions such as widening of the alveolar septum with inflammatory cells, narrowing and atrophy of the alveolar sac and proliferation of collagen fiber in the lungs, and swelling and necrosis of renal tubular epithelial cells and telangiectasia in the kidneys have been reported (36). Inflammation of cardiomyocytes accompanied by inflammatory cell infiltration in the heart and focal inflammatory cell infiltration in the brain tissue have also been reported (37). In DQ exposed patients, the values of serum creatine and urea nitrogen were significantly increased suggesting severe renal function abnormalities (38). Mild bilateral patchy consolidation at the hilar in the lungs has been observed (39). The lungs mainly involve the alveolar epithelium, which may initially cause acute alveolitis and then pulmonary fibrosis (34). The inflammatory spots around the pleura gradually expand into plaques over time and developed into pulmonary fibrosis (40). Studies have found that the injection of DQ in pigs can increase the expression of mitochondria-related proteins including Pink1, Parkin, and LC3B in the intestine. It is suggested that DQ can induce mitochondrial autophagy (41). Previously, it has also been found that DQ can reduce the activity of SOD and GSH-Px in the intestinal mucosa and increase the contents of MDA (42, 43), suggesting that PQ causes oxidative stress in the intestine. In vivo and in vitro studies have shown that the oxidative stress caused by DQ can damage the integrity of the intestinal epithelial barrier that is manifested by the destruction of tight junctions (TJ) and reduction of epithelial cell viability (44–47). Different studies have indicated manifestations of DQ toxicity as decreased TER and increased FD4 flux (48), high serum lipopolysaccharide levels, and diamine oxidase activity in piglets (43). DQ reduces the activity of AHR in the spleen, causing oxidative damage to the spleen. The decreased activity of SOD and CAT suggests decreased antioxidant functions in the spleen (49). Long-term exposure to DQ had toxic effects on the reproductive system (50–52) that is manifested by reducing the quality of germ cells affecting early embryo development (53). Long-term ingestion of DQ in rats and dogs can induce cataracts (54). Dose-dependent axonal degeneration has been observed in dorsal root ganglion neurons (55). In addition, the production of nitric oxide and superoxide anion free radicals was significantly increased, and lipid peroxidation increased, suggesting that PQ causes neurotoxicity (56).
The central vein around the liver cells showed vacuolar degeneration with punctate necrosis due to exposure to DQ (36). The activity of total antioxidant capacity, SOD, and GSH-Px in the liver of piglets injected with DQ decreased suggesting lower status of the liver's antioxidant capacity. There were significant differences in the expression of liver mRNA and lncRNA. GNMT is highly expressed, and GCK is downregulated. These results indicate that DQ can affect glucose metabolism in the liver and reduce weight gain (57). It can also increase the accumulation of glutathione peroxidase (GPX) activity and MDA in the plasma and liver; increase the activity of AST, alanine aminotransferase, and T-Bil concentrations; and increase the relative liver weight, indicating that DQ causes liver damage (10). DQ increases Ca efflux due to the weakened ATP-dependent Ca chelation of liver microsomes (58). DQ-induced liver lipid peroxidation is manifested as increase in 11-, 12-, and 15-hydroxyeicosatetraenoic acid. DQ can also activate inflammatory cells, leading to the synthesis and release of certain pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6 (59).
As a commonly used herbicide, DQ mainly destroys plant cells to achieve the purpose of weeding by inducing redox cycle, releasing ROS and nitric oxide and inhibiting the effect of NADPH. However, it is the herbicidal principle that causes serious damage to the nervous system, liver, kidney, heart, and lungs of animals or humans, while the liver is the main source of ROS, and it has become the target of DQ (60). Liver injury has a prodigious impact on the organism, which can directly lead to an increase in morbidity and mortality of terrestrial and aquatic animals, especially birds (61).
Many studies have been conducted on DQ-induced liver injury, which mainly focus on oxidative stress, apoptosis, and autophagy. Previous studies have shown that DQ induces liver redox cycle to produce superoxide while inhibit the production of antioxidant enzymes, so that antioxidant enzymes are not enough to resist liver damage caused by oxidation. Secondly, the endoplasmic reticulum and mitochondria can regulate apoptosis and autophagy, while damaged mitochondria initiate autophagy mechanism to inhibit the release of cytochrome c (Cyt-c) and induce apoptosis. Damaged mitochondria can also reduce the accumulation of ROS, which inhibit the division of the mitochondria and prevent it from degradation by autophagy (62).
In the specific effect of DQ on the liver, some scholars have found that the content of glutathione disulfide (GSSG) can reflect the liver injury induced by DQ, while previous studies have shown that DQ leads to a sharp increase in the content of GSSG in the liver (12, 63). DQ can also increase the level of serum ALT and AST and decrease the level of anti-apoptosis factor Bcl2, which indicates hepatocyte apoptosis and injury (64). When DQ is put into the waters of fish fry, it was found that biosynthesis of protein and RNA was increased, while ATK/mTOR signal, SREBP pathway, and caspase pathway were activated. It was considered that this change was closely related to the increase of protein and mRNA, which further confirmed the occurrence of oxidative stress and apoptosis in fish (65–67). It is also found that intraperitoneal injection of DQ in piglets affects the MAPK signal pathway that leads to acute oxidative stress and increase lipids, antioxidant metabolites, and peroxide MDA in the liver (68), so it indicates that DQ can destroy cell lipids and induce cell death. Some scholars have found that DQ-induced oxidative stress can promote the production of H2O2 in the mitochondria, depolarize the mitochondria, and inactivate iron and sulfur in the mitochondria containing aconitase and other proteins, resulting in mitochondrial damage (69–72). Previous studies have found that mitochondrial damage and induced mitochondrial release of apoptosis and oxidation-related proteins can promote apoptosis. Oxidative stress induces the release of Cyt-c from the mitochondria that is the key factor of apoptosis, while the antioxidant enzyme Gpx4 can eliminate lipid peroxides in the mitochondria to inhibit the production of Cyt-c and reduce apoptosis (73). DQ can also affect the caspase signal pathway by inducing oxidative stress to produce ROS and enhance the activities of caspase-3 and caspase-9 in the liver tissue (74–78). It is found that there is a relationship between Bax/Bcl-2 and caspase. With the increase of Bax/Bcl-2 ratio, caspases are also gradually activated, which can promote apoptosis (79). It was also found that the activity of mitochondrial complex I was inhibited and ATP was consumed, which proved the destructive effect of DQ on the mitochondria. In addition, it is found that mitochondrial dysfunction and reduced apoptosis by inhibiting NF-KB and p53 signal pathways are the main pathways for DQ to induce inflammation and apoptosis. Secondly, some studies have found that the release of Smac/Diablo, endonuclease G, and other intermembrane space proteins after permeation of mitochondrial outer membrane can promote cell apoptosis by activating cystatin. A large amount of H2O2 is produced by the mitochondria, which further induces the production of ROS to aggravate the oxidative stress of cells that leads to further apoptosis (80).
Our research results are somewhat consistent with the above research results. In our study, we did not find that DQ had significant effects on pro-apoptotic factors BAX, BAK1, TNF-α, caspase series, and p53, but they showed an upward trend all together. The anti-apoptotic genes Parkin and Bcl2 were significantly upregulated, indicating that anti-apoptotic factors in ducklings play an important role in the period of acute DQ poisoning. It is well-known that the production of a large number of ROS can destroy the function of the mitochondria. Some studies have suggested that it may convert the apoptosis pathway into necrosis, thus showing the increase of inhibitory apoptosis factors. Previous studies have shown that Parkin gene not only plays an anti-apoptotic role but also inhibits the pro-apoptotic pathway of p53 (81, 82). In our study of autophagy factor, we found that DQ significantly upregulated the protein expression of autophagy factor Beclin 1 in the liver. The key autophagy factors LC3A, LC3B, and p62 showed an upward trend at the gene level, but the protein expression of LC3B and p62 decreased; it may be due to the effect of pro-apoptotic components such as apoptotic proteases during translation, which initiated apoptosis and inhibited the occurrence of autophagy at the same time (62). Most evidence shows that autophagy is the protective mechanism of cell initiation. Autophagy can inhibit apoptosis when autophagy is upregulated; similarly, apoptosis can also reduce autophagy. Therefore, we believe that acute duck poisoning has a significant effect on the liver. On the one hand, it activates the apoptosis and anti-apoptosis system, which proves that there is apoptosis in hepatocytes. On the other hand, the autophagy protection system was activated, and it was found that the autophagy and apoptosis system inhibited each other.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Animal Ethics Committee of South China Agricultural University (License Number: 2017A087).
Author Contributions
JC, YS, RL, and FL were responsible for the study conception and design. RH and DS revised the manuscript. JC, YS, PS, and DS were involved in the drafting of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by the Natural Science Foundation of Guangdong Province (2021A1515011010) and Science and Technology Major Project of Tibet Autonomous Region (XZ202101ZD0005N).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang XH, Souders CL, Zhao YH, Martyniuk CJ. Mitochondrial bioenergetics and locomotor activity are altered in zebrafish (Danio rerio) after exposure to the bipyridylium herbicide diquat. Toxicol Lett. (2018) 283:13–20. doi: 10.1016/j.toxlet.2017.10.022
2. Yastrub TO, Omelchuk ST, Yastrub AM. Dermal absorption of diquat and potential occupational risk. Wiad Lek. (2020) 73:1459–64. doi: 10.36740/WLek202007127
3. Fortenberry GZ, Beckman J, Schwartz A, Prado JB, Graham LS, Higgins S, et al. Magnitude and characteristics of acute paraquat- and diquat-related illnesses in the US: 1998-2013. Environ Res. (2016) 146:191–9. doi: 10.1016/j.envres.2016.01.003
4. Pouchieu C, Piel C, Carles C, Gruber A, Helmer C, Tual S, et al. Pesticide use in agriculture and Parkinson's disease in the AGRICAN cohort study. Int J Epidemiol. (2018) 47:299–310. doi: 10.1093/ije/dyx225
5. Magalhães N, Carvalho F, Dinis-Oliveira RJ. Human and Experimental Toxicology of Diquat Poisoning: Toxicokinetics, Mechanisms of Toxicity, Clinical Features, and Treatment. London: SAGE Publications (2018).
6. Souders CN, Liang X, Wang X, Ector N, Zhao YH, Martyniuk CJ. High-throughput assessment of oxidative respiration in fish embryos: advancing adverse outcome pathways for mitochondrial dysfunction. Aquat Toxicol. (2018) 199:162–73. doi: 10.1016/j.aquatox.2018.03.031
7. Bouetard A, Besnard AL, Vassaux D, Lagadic L, Coutellec MA. Impact of the redox-cycling herbicide diquat on transcript expression and antioxidant enzymatic activities of the freshwater snail Lymnaea stagnalis. Aquat Toxicol. (2013) 126:256–65. doi: 10.1016/j.aquatox.2012.11.013
8. Sewalk CJ, Brewer GL, Hoffman DJ. Effects of diquat, an aquatic herbicide, on the development of mallard embryos. J Toxicol Environ Health A. (2000) 62:33–45. doi: 10.1080/00984100050201659
9. Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. (2012) 19:4850. doi: 10.2174/092986712803341520
10. Chen YP, Gu YF, Zhao HR, Zhou YM. Dietary squalene supplementation alleviates diquat-induced oxidative stress and liver damage of broiler chickens. Poultry Sci. (2021) 100:100919. doi: 10.1016/j.psj.2020.12.017
11. Yang L, Yu Z, Hou J, Deng Y, Zhou Z, Zhao Z, et al. Toxicity and oxidative stress induced by T-2 toxin and HT-2 toxin in broilers and broiler hepatocytes. Food Chem Toxicol. (2016) 87:128–37. doi: 10.1016/j.fct.2015.12.003
12. Wu KC, Zhang Y, Klaassen CD. Nrf2 protects against diquat-induced liver and lung injury. Free Radic Res. (2012) 46:1220–9. doi: 10.3109/10715762.2012.700709
13. Zhang H, Chen Y, Chen Y, Jia P, Ji S, Xu J, et al. Comparison of the effects of resveratrol and its derivative pterostilbene on hepatic oxidative stress and mitochondrial dysfunction in piglets challenged with diquat. Food Funct. (2020) 11:4202–15. doi: 10.1039/D0FO00732C
14. Jia P, Ji S, Zhang H, Chen Y, Wang T. Piceatannol ameliorates hepatic oxidative damage and mitochondrial dysfunction of weaned piglets challenged with diquat. Animals (Basel). (2020) 10:239. doi: 10.3390/ani10071239
15. Huang B, Chen Q, Wang L, Gao X, Zhu W, Mu P, et al. Aflatoxin B1 induces neurotoxicity through reactive oxygen species generation, DNA damage, apoptosis, and S-phase cell cycle arrest. Int J Mol Sci. (2020) 21:517. doi: 10.3390/ijms21186517
16. El-Nabarawy NA, Gouda AS, Khattab MA, Rashed LA. Effects of nitrite graded doses on hepatotoxicity and nephrotoxicity, histopathological alterations, and activation of apoptosis in adult rats. Environ Sci Pollut Res Int. (2020) 27:14019–32. doi: 10.1007/s11356-020-07901-6
17. Quan Y, Gong L, He J, Zhou Y, Liu M, Cao Z, et al. Aloe emodin induces hepatotoxicity by activating NF-κB inflammatory pathway and P53 apoptosis pathway in zebrafish. Toxicol Lett. (2019) 306:66–79. doi: 10.1016/j.toxlet.2019.02.007
18. Sayed AA, El-Desouky MA, Ibrahim KA. Garlic and allopurinol attenuate hepatic apoptosis induced by fipronil in male albino rats. Regul Toxicol Pharmacol. (2019) 107:104400. doi: 10.1016/j.yrtph.2019.05.025
19. Zhang Y, Qu X, Gao H, Zhai J, Tao L, Sun J, et al. Quercetin attenuates NLRP3 inflammasome activation and apoptosis to protect INH-induced liver injury via regulating SIRT1 pathway. Int Immunopharmacol. (2020) 85:106634. doi: 10.1016/j.intimp.2020.106634
20. Feng Y, Cui R, Li Z, Zhang X, Jia Y, Zhang X, et al. Methane alleviates acetaminophen-induced liver injury by inhibiting inflammation, oxidative stress, endoplasmic reticulum stress, and apoptosis through the Nrf2/HO-1/NQO1 signaling pathway. Oxid Med Cell Longev. (2019) 2019:7067619. doi: 10.1155/2019/7067619
21. Cao P, Sun J, Sullivan MA, Huang X, Wang H, Zhang Y, et al. Angelica sinensis polysaccharide protects against acetaminophen-induced acute liver injury and cell death by suppressing oxidative stress and hepatic apoptosis in vivo and in vitro. Int J Biol Macromol. (2018) 111:1133–9. doi: 10.1016/j.ijbiomac.2018.01.139
22. Zhang K, Zhu Y, Wang X, Zhao X, Li S, Teng X. Excess manganese-induced apoptosis in chicken cerebrums and embryonic neurocytes. Biol Trace Elem Res. (2017) 180:297–305. doi: 10.1007/s12011-017-0992-4
23. Rani A, Kumar A, Lal A, Pant M. Cellular mechanisms of cadmium-induced toxicity: a review. Int J Environ Health Res. (2014) 24:378–99. doi: 10.1080/09603123.2013.835032
24. Garza-Lombo C, Pappa A, Panayiotidis MI, Gonsebatt ME, Franco R. Arsenic-induced neurotoxicity: a mechanistic appraisal. J Biol Inorg Chem. (2019) 24:1305–16. doi: 10.1007/s00775-019-01740-8
25. Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer. (2017) 3:857–70. doi: 10.1016/j.trecan.2017.10.006
26. Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics-the cancer connection. Biochim Biophys Acta Bioenerg. (2017) 1858:602–14. doi: 10.1016/j.bbabio.2017.01.004
27. Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol. (2020) 98:139–53. doi: 10.1016/j.semcdb.2019.05.022
28. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. (2004) 305:626–9. doi: 10.1126/science.1099320
29. Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. (2000) 256:42–9. doi: 10.1006/excr.2000.4838
30. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. (2013) 87:1157–80. doi: 10.1007/s00204-013-1034-4
31. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. (2011) 333:1109–12. doi: 10.1126/science.1201940
32. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. (2011) 13:589–98. doi: 10.1038/ncb2220
33. Wrighton KH. Autophagy: shaping the fate of mitochondria. Nat Rev Mol Cell Biol. (2011) 12:344–5. doi: 10.1038/nrm3116
34. Park A, Koh HC. NF-kappaB/mTOR-mediated autophagy can regulate diquat-induced apoptosis. Arch Toxicol. (2019) 93:1239–53. doi: 10.1007/s00204-019-02424-7
35. Choi SE, Park YS, Koh HC. NF-kappaB/p53-activated inflammatory response involves in diquat-induced mitochondrial dysfunction and apoptosis. Environ Toxicol. (2018) 33:1005–18. doi: 10.1002/tox.22552
36. Sun YQ, Yuan L, Gao HB, Yao DQ, Chen QS, Tian YP. Establishment and evaluation of acute diquat poisoning model in Wistar rats. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. (2019) 37:342–6. doi: 10.3760/cma.j.issn.1001-9391.2019.05.005
37. Wu YZ, Kan BT, Wang WJ, Zhang ZC, Jia JE, Li XQ, et al. The experimental study of diquat on the half-Lethal dose and pothological injuny of related organs in wistor rats. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. (2018) 36:813–18. doi: 10.3760/cma.j.issn.1001-9391.2018.11.004
38. Xing J, Chu Z, Han D, Jiang X, Zang X, Liu Y, et al. Lethal diquat poisoning manifesting as central pontine myelinolysis and acute kidney injury: a case report and literature review. J Int Med Res. (2020) 48:300060520943824. doi: 10.1177/0300060520943824
39. Safaei AA, Dadashzadeh P. Acute kidney injury in patients with paraquat intoxication; a case report and review of the literature. J Renal Inj Prev. (2016) 5:203–6. doi: 10.15171/jrip.2016.43
40. Yu G, Kan B, Jian X, Wang J, Sun J, Song C. A case report of acute severe paraquat poisoning and long-term follow-up. Exp Ther Med. (2014) 8:233–6. doi: 10.3892/etm.2014.1727
41. Cao S, Wu H, Wang C, Zhang Q, Jiao L, Lin F, et al. Diquat-induced oxidative stress increases intestinal permeability, impairs mitochondrial function, and triggers mitophagy in piglets. J Anim Sci. (2018) 96:1795–805. doi: 10.1093/jas/sky104
42. Lv M, Yu B, Mao XB, Zheng P, He J, Chen DW. Responses of growth performance and tryptophan metabolism to oxidative stress induced by diquat in weaned pigs. Animal. (2012) 6:928–34. doi: 10.1017/S1751731111002382
43. Yin J, Liu M, Ren W, Duan J, Yang G, Zhao Y, et al. Effects of dietary supplementation with glutamate and aspartate on diquat-induced oxidative stress in piglets. PLoS ONE. (2015) 10:e0122893. doi: 10.1371/journal.pone.0122893
44. Jiao N, Wu Z, Ji Y, Wang B, Dai Z, Wu G. L-glutamate enhances barrier and antioxidative functions in intestinal porcine epithelial cells. J Nutr. (2015) 145:2258–64. doi: 10.3945/jn.115.217661
45. Liu F, Cottrell JJ, Furness JB, Rivera LR, Kelly FW, Wijesiriwardana U, et al. Selenium and vitamin E together improve intestinal epithelial barrier function and alleviate oxidative stress in heat-stressed pigs. Exp Physiol. (2016) 101:801–10. doi: 10.1113/EP085746
46. Liu J, Zhang Y, Li Y, Yan H, Zhang H. L-tryptophan enhances intestinal integrity in diquat-challenged piglets associated with improvement of redox status and mitochondrial function. Animals (Basel). (2019) 9:1045. doi: 10.3390/ani9050266
47. Zheng P, Yu B, He J, Yu J, Mao X, Luo Y, et al. Arginine metabolism and its protective effects on intestinal health and functions in weaned piglets under oxidative stress induced by diquat. Br J Nutr. (2017) 117:1495–02. doi: 10.1017/S0007114517001519
48. Wijtten PJ, van der Meulen J, Verstegen MW. Intestinal barrier function and absorption in pigs after weaning: a review. Br J Nutr. (2011) 105:967–81. doi: 10.1017/S0007114510005660
49. Wu X, Cao W, Jia G, Zhao H, Chen X, Wu C, et al. New insights into the role of spermine in enhancing the antioxidant capacity of rat spleen and liver under oxidative stress. Anim Nutr. (2017) 3:85–90. doi: 10.1016/j.aninu.2016.11.005
50. Gupta RK, Miller KP, Babus JK, Flaws JA. Methoxychlor inhibits growth and induces atresia of antral follicles through an oxidative stress pathway. Toxicol Sci. (2006) 93:382–9. doi: 10.1093/toxsci/kfl052
51. Gupta RK, Schuh RA, Fiskum G, Flaws JA. Methoxychlor causes mitochondrial dysfunction and oxidative damage in the mouse ovary. Toxicol Appl Pharmacol. (2006) 216:436–45. doi: 10.1016/j.taap.2006.06.013
52. Gupta RK, Aberdeen G, Babus JK, Albrecht ED, Flaws JA. Methoxychlor and its metabolites inhibit growth and induce atresia of baboon antral follicles. Toxicol Pathol. (2007) 35:649–56. doi: 10.1080/01926230701459960
53. Zhang JQ, Gao BW, Wang J, Wang XW, Ren QL, Chen JF, et al. Chronic exposure to diquat causes reproductive toxicity in female mice. PLoS ONE. (2016) 11:e0147075. doi: 10.1371/journal.pone.0147075
54. Clark DG, Hurst EW. The toxicity of diquat. Br J Ind Med. (1970) 27:51–5. doi: 10.1136/oem.27.1.51
55. Fischer LR, Glass JD. Oxidative stress induced by loss of Cu,Zn-superoxide dismutase (SOD1) or superoxide-generating herbicides causes axonal degeneration in mouse DRG cultures. Acta Neuropathol. (2010) 119:249–59. doi: 10.1007/s00401-009-0631-z
56. Djukic M, Jovanovic MD, Ninkovic M, Stevanovic I, Curcic M, Topic A, et al. Intrastriatal pre-treatment with L-NAME protects rats from diquat neurotoxcity. Ann Agric Environ Med. (2012) 19:666–72. doi: 10.1108/09653561211278761
57. Wang J, Li ZX, Yang DD, Liu PQ, Wang ZQ, Zeng YQ, et al. Diquat determines a deregulation of lncRNA and mRNA expression in the liver of postweaned piglets. Oxid Med Cell Longev. (2019) 2019:9148535. doi: 10.1155/2019/9148535
58. Tsokos-Kuhn JO, Smith CV, Hughes H, Mitchell JR. Liver membrane calcium transport in diquat-induced oxidative stress in vivo. Mol Pharmacol. (1988) 34:209–14.
59. Smith CV, Hughes H, Lauterburg BH, Mitchell JR. Oxidant stress and hepatic necrosis in rats treated with diquat. J Pharmacol Exp Ther. (1985) 235:172–7.
60. Karuppagounder SS, Ahuja M, Buabeid M, Parameshwaran K, Abdel-Rehman E, Suppiramaniam V, et al. Investigate the chronic neurotoxic effects of diquat. Neurochem Res. (2012) 37:1102–11. doi: 10.1007/s11064-012-0715-3
61. Avanzo JL, de Mendonca CJ, Pugine SM, de Cerqueira CM. Effect of vitamin E and selenium on resistance to oxidative stress in chicken superficial pectoralis muscle. Comp Biochem Physiol C Toxicol Pharmacol. (2001) 129:163–73. doi: 10.1016/s1532-0456(01)00197-1
62. Kaminskyy VO, Zhivotovsky B. Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal. (2014) 21:86–102. doi: 10.1089/ars.2013.5746
63. Madhu C, Gregus Z, Klaassen CD. Marked interanimal differences in susceptibility of Sprague-Dawley rats to diquat-induced oxidative stress in the liver: correlation with hepatic uptake of diquat. J Pharmacol Exp Ther. (1992) 263:1003–8.
64. Chen Y, Chen Y, Zhang H, Wang T. Pterostilbene as a protective antioxidant attenuates diquat-induced liver injury and oxidative stress in 21-day-old broiler chickens. Poult Sci. (2020) 99:3158–67. doi: 10.1016/j.psj.2020.01.021
65. Luu W, Sharpe LJ, Stevenson J, Brown AJ. Akt acutely activates the cholesterogenic transcription factor SREBP-2. Biochim Biophys Acta. (2012) 1823:458–64. doi: 10.1016/j.bbamcr.2011.09.017
66. McCuaig LM, Martyniuk CJ, Marlatt VL. Morphometric and proteomic responses of early-life stage rainbow trout (Oncorhynchus mykiss) to the aquatic herbicide diquat dibromide. Aquat Toxicol. (2020) 222:105446. doi: 10.1016/j.aquatox.2020.105446
67. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. (2014) 4:64. doi: 10.3389/fonc.2014.00064
68. Doan N, Liu Y, Xiong X, Kim K, Wu Z, Bravo DM, et al. Organic selenium supplement partially alleviated diquat-induced oxidative insults and hepatic metabolic stress in nursery pigs. Br J Nutr. (2020) 24:1–11. doi: 10.1017/S0007114520000689
69. Chen L, Yoo SE, Na R, Liu Y, Ran Q. Cognitive impairment and increased Abeta levels induced by paraquat exposure are attenuated by enhanced removal of mitochondrial H(2)O(2). Neurobiol Aging. (2012) 33:432.e15–26. doi: 10.1016/j.neurobiolaging.2011.01.008
70. Cantu D, Fulton RE, Drechsel DA, Patel M. Mitochondrial aconitase knockdown attenuates paraquat-induced dopaminergic cell death via decreased cellular metabolism and release of iron and H(2)O(2). J Neurochem. (2011) 118:79–92. doi: 10.1111/j.1471-4159.2011.07290.x
71. Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE. (2009) 4:e7095. doi: 10.1371/journal.pone.0007095
72. Czerniczyniec A, Karadayian AG, Bustamante J, Cutrera RA, Lores-Arnaiz S. Paraquat induces behavioral changes and cortical and striatal mitochondrial dysfunction. Free Radic Biol Med. (2011) 51:1428–36. doi: 10.1016/j.freeradbiomed.2011.06.034
73. Liang H, Ran Q, Jang YC, Holstein D, Lechleiter J, McDonald-Marsh T, et al. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic Biol Med. (2009) 47:312–20. doi: 10.1016/j.freeradbiomed.2009.05.012
74. Zhang H, Chen Y, Chen Y, Jia P, Ji S, Xu J, et al. Comparison of the effects of resveratrol and its derivative pterostilbene on hepatic oxidative stress and mitochondrial dysfunction in piglets challenged with diquat. Food Funct. (2020) 11:4202–15. doi: 10.1039/d0fo00732c
75. Liu B. Role of oxidative stress and antioxidants in thiram-induced tibial dyschondroplasia. Pak Vet J. (2021) 41:1–6. doi: 10.29261/pakvetj/2020.094
76. Qamar H. Recovery of chickens affected with tibial dyschondroplasia by application of grape seed extract through downregulating ca2 gene and enhancing liver functions. Pak Vet J. (2019) 39:527–33. doi: 10.29261/pakvetj/2019.076
77. Qamar H. Effect of grape seed extract on tibial dyschondroplasia incidence, liver weight, and tibial angiogenesis in chickens. Pak Vet J. (2020) 40:187–94. doi: 10.29261/pakvetj/2019.109
78. Bakr AF, Abdelgayed SS, EL-Tawil OS, Bakeer AM. Assessment of ginger extract and ginger nanoparticles protective activity against acetaminophen-induced hepatotoxicity and nephrotoxicity in rats. Pak Vet J. (2019) 39:479–86. doi: 10.29261/pakvetj/2019.060
79. Luo Z, Zhu W, Guo Q, Luo W, Zhang J, Xu W, et al. Weaning induced hepatic oxidative stress, apoptosis, and aminotransferases through MAPK signaling pathways in piglets. Oxid Med Cell Longev. (2016) 2016:4768541. doi: 10.1155/2016/4768541
80. Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. (2004) 24:7771–8. doi: 10.1523/JNEUROSCI.1842-04.2004
81. Kanapathipillai M. Treating p53 mutant aggregation-associated cancer. Cancers (Basel). (2018) 10:154. doi: 10.3390/cancers10060154
Keywords: diquat, liver, apoptosis, autophagy, ducks
Citation: Chen J, Su Y, Lin R, Lin F, Shang P, Hussain R and Shi D (2021) Effects of Acute Diquat Poisoning on Liver Mitochondrial Apoptosis and Autophagy in Ducks. Front. Vet. Sci. 8:727766. doi: 10.3389/fvets.2021.727766
Received: 19 June 2021; Accepted: 12 July 2021;
Published: 11 August 2021.
Edited by:
Yung-Fu Chang, Cornell University, United StatesReviewed by:
Fazul Nabi, Lasbela University of Agriculture, Water and Marine Sciences, PakistanFan Yang, Jiangxi Agricultural University, China
Copyright © 2021 Chen, Su, Lin, Lin, Shang, Hussain and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Shang, bmVtb3NocG1oQDEyNi5jb20=; Dayou Shi, c2hpZGF5b3VAc2NhdS5lZHUuY24=
†These authors have contributed equally to this work