 Lisa Smart
Lisa Smart Dez Hughes
Dez Hughes
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Vet. Sci. , 07 May 2021
Sec. Comparative and Clinical Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fvets.2021.661660
This article is part of the Research Topic Fluid Therapy in Animals View all 26 articles
The goal of resuscitative fluid therapy is to rapidly expand circulating blood volume in order to restore tissue perfusion. Although this therapy often serves to improve macrohemodynamic parameters, it can be associated with adverse effects on the microcirculation and endothelium. The endothelial surface layer (ESL) provides a protective barrier over the endothelium and is important for regulating transvascular fluid movement, vasomotor tone, coagulation, and inflammation. Shedding or thinning of the ESL can promote interstitial edema and inflammation and may cause microcirculatory dysfunction. The pathophysiologic perturbations of critical illness and rapid, large-volume fluid therapy both cause shedding or thinning of the ESL. Research suggests that restricting the volume of crystalloid, or “clear” fluid, may preserve some ESL integrity and improve outcome based on animal experimental models and preliminary clinical trials in people. This narrative review critically evaluates the evidence for the detrimental effects of resuscitative fluid therapy on the ESL and provides suggestions for future research directions in this field.
The importance of microcirculatory function and health of the endothelium has become a large area of interest for criticalists in the last two decades. Over this time, fluid resuscitation strategies have experienced a shift in perspective. Aggressive “clear” fluid resuscitation was once considered vital for stabilization of macrohemodynamic variables. However, in certain patient cohorts this approach has now been shown to either not improve outcome or worsen outcome (1–4). Though fast administration of fluid often serves to normalize the commonly measured clinical parameters during shock, microcirculation may not necessarily benefit from this therapy. There is growing evidence that fluid resuscitation may actually harm the endothelium by modifying, or shedding, the endothelial surface layer (ESL).
The ESL includes a structural scaffold, the endothelial glycocalyx (EG), and associated molecules suspended within a plasma layer. We use the term EG when referring to specific components of the structure, or biomarkers of these components, whereas ESL is used when referring to the layer as a whole. In regards to shedding of the ESL, the term ESL will be used when the evidence supports loss of the whole layer and the term EG used when the evidence only supports that isolated components have been shed. Use of the term glycocalyx refers to any type of cell surface glycocalyx and is not restricted to the endothelium, such as during discussion of glycocalyx shedding biomarkers.
Resuscitative fluid therapy includes a range of fluid choices, including isotonic crystalloid fluid, synthetic colloid fluid, and hyperosmolar crystalloid fluid, with the former being the most common type of fluid used (5). In general, due to the pharmacodynamic properties of these fluids in either healthy subjects or shock models (6–9), the volume of fluid is “large” for isotonic crystalloid (at least a quarter to half of estimated blood volume), “moderate” for synthetic colloid (an eighth to quarter of estimated blood volume), and “small” for hyperosmolar crystalloids. All three types of fluid resuscitation will be covered in this review with a particular focus on isotonic “large-volume” crystalloid fluid therapy due to the evidence for its effects on the ESL. A summary of the evidence reviewed in this article is provided in Box 1.
Box 1. Summary of the evidence for effects of “clear” fluid therapy on endothelial surface layer (ESL) shedding or modification.
Proposed mechanisms of ESL shedding
Dilution of plasma proteins
Release of natriuretic peptides
Inflammatory cytokine release (certain fluid types)
Possible downstream effects
Exacerbation of inflammation
Microcirculatory dysfunction
Increased vascular permeability
Increased interstitial edema
Prothrombotic conditions
Proposed strategies that may mitigate ESL shedding*
Reduction in dose of clear fluids
Slowing down administration of resuscitative fluids
“Earlier” vasopressor therapy for vasodilatory shock
Adjunctive protein administration (such as plasma)
*These proposed strategies are not based on evidence from clinical veterinary research and require further investigation. These guidelines are opinion of the authors only, after considering the breadth of evidence available. It is currently unknown if “glycoprotective” strategies benefit patients. Therapy should always be tailored to individual patient needs, with prioritization of reestablishing adequate perfusion and a thorough cost to benefit analysis.
Most cells in the body are covered in a protective layer of carbohydrate scaffold, which houses many different molecules that serve a variety of functions. This surface layer is called a glycocalyx. The general structure of the glycocalyx has similarities between cell types, with only small variations in individual proteins or carbohydrate molecules. The EG coats the luminal surface of the endothelium and is a vital structure for cell signaling and transvascular permeability. It is composed of proteoglycans, glycosaminoglycans (GAGs), and glycoproteins (Figure 1). Together with mobile or soluble components, such as albumin, this compromises the ESL. Proteoglycans are large molecules that have a cytoplasmic, transcellular, and extracellular domain (syndecan) or are attached by a glycosylphosphatidylinositol anchor (glypican-1) (Figure 1) (10–12). Their extracellular component, or ectodomain, is covered by GAG side-chains and performs important roles that assist with cell to cell, or cell to matrix, interactions (13). These structures provide a structural scaffold for the ESL, in which many other molecules are housed. The sulfated GAGs attached to proteoglycans include heparan, chondroitin, and dermatan sulfate. Heparan sulfate is the most abundant GAG on syndecans and glypican-1, which is why proteoglycans are often referred to as heparan sulfate proteoglycans. An additional GAG, hyaluronan, is not typically associated with a proteoglycan; instead, it is attached to the endothelium via receptors such as CD44 or other GAG molecules (Figure 1) (14, 15). Hyaluronan is a long GAG of varying lengths that weaves its way through the tall “forest” of proteoglycans, with their sulfated GAG “branches”. These GAG chains contribute to the barrier function of the ESL of excluding large molecules (16). Glycoproteins reside on the luminal surface of the endothelium, hidden within the ESL, and include adhesion molecules such as integrins and selectins (17). Glycoproteins play an important role in leucocyte trafficking during states of inflammation (18); many of their functions are only initiated once the ESL has been shed or thinned. Finally, mobile components residing within the forest of the ESL include proteins, such as albumin, and anticoagulants, such as antithrombin and tissue factor pathway inhibitor (15, 19). The presence of plasma proteins are likely important for maintaining the normal structure and permeability of the ESL, as well as providing an anticoagulated surface at the blood interface.
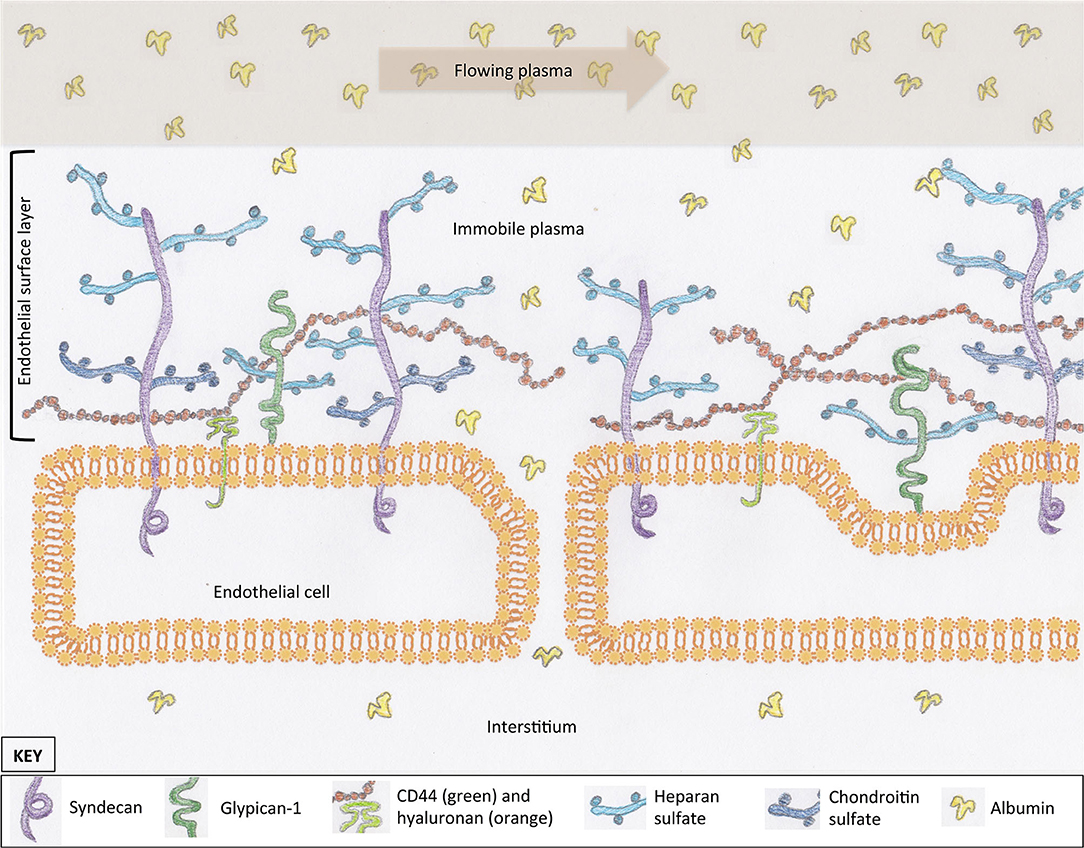
Figure 1. The scaffold of the endothelial glycocalyx, within the endothelial surface layer, is provided by proteoglycans, syndecan (four subtypes), and glypican-1. Glycosaminoglycans are attached to proteoglycans (e.g., heparan sulfate) or the endothelial surface (hyaluronan). Molecules suspended in the plasma of the endothelial surface layer include proteins such as albumin. These proteins create a protein-poor sub-glycocalyx area that is important for transvascular colloid osmotic pressure balance. For simplicity, structures within the interendothelial cleft are not represented.
The EG serves a range of functions, many of which are still being characterized. These functions include maintenance of a surface barrier that buffers circulating leucocytes, inhibits coagulation, regulates fluid flux, and communicates changes in vascular wall shear forces. The sulfated GAGs on the surface of the endothelium generate and maintain a net negative charge, which repels similarly charged cells from the endothelial surface (20). Although albumin has an overall negative charge, it is likely that the positively charged groups within the molecular structure are what allow for incorporation of albumin into the ESL (21). Maintenance of this negative charge is important for endothelial integrity; loss of negative charge on the luminal surface of the endothelium leads to extravasation of albumin (22–24). In addition to the importance of electrostatic charge, the ESL provides a barrier to fluid filtration, thus creating a protein-poor sub-glycocalyx layer and maintaining a colloid osmotic pressure (COP) gradient favoring fluid retention within the vasculature (20).
The traditional Starling hypothesis describes a relationship between the intravascular and the interstitial colloid osmotic pressure and implies the importance of this relationship in determining net transvascular fluid filtration. Many studies in the last four decades, however, have built a body of evidence stating that it is mainly the low COP in the sub-glycocalyx area that creates the pressure gradient opposing capillary hydrostatic pressure (25, 26). The theory is that the sub-glycocalyx fluid space, within the inter-endothelial cleft, has a very low concentration of macromolecules. This is due to the high impermeability of the ESL to macromolecules and the rapid flow of water and solutes through the sub-glycocalyx space (27). In the steady state with an intact ESL, net filtration of fluid occurs across the blood vessel wall, with only a small limitation from intravascular COP. Increasing intravascular COP does not, and cannot, serve to reverse net fluid transudation nor does increasing interstitial COP lead to an increase in fluid transudation (28, 29). A large drop in hydrostatic pressure, such as that may occur during circulatory shock, may reverse transvascular fluid flow to a resorptive state; however, this effect is transient (29). These hypotheses explain why fluid would not be “drawn” out of the interstitium by increasing intravascular COP, such as the use of synthetic colloid fluids. Rather, they can reduce extravasation compared to crystalloid fluid. The COP of the intravascular space, and even of the sub-glycocalyx space, is microanatomically and physiologically distant from the interstitial COP. However, the fluid dynamics across the endothelium in disease states with a denuded ESL, removing the influence of the sub-glycocalyx layer, is yet to be fully characterized.
The EG also plays an important role to changing pressures and flow within the intravascular space. Proteoglycans, especially those with heparan sulfate chains, play an important role in responding to changes in vascular wall shear stress or changes in intravascular pressure (10, 30, 31). Detection of these mechanical forces leads to morphological changes in endothelial cells and release of nitric oxide (32–35). Shear stress can also lead to a change in location of proteoglycans or upregulation of their cell surface expression (36–38). It is possible that change in location or upregulation of these molecules during microcirculatory disturbances may also affect shedding of the extracellular components. This becomes relevant when discussing biomarkers for EG shedding below.
Shedding, or modification, of the ESL is a vital step after tissue injury in order to facilitate leucocyte and platelet adherence (39). Shedding reproduced in cell culture or ex vivo models leads to increased expression of adhesion molecules, increased leucocyte adhesion, and increased cytokine production (32, 40–43). This process is likely more complex in vivo, whereby certain disease states may have varying effects, from selective removal of glycocalyx components to complete denudation. Although ESL shedding is a necessary step in localized inflammation, it is becoming clearer that systemic-wide shedding is associated with severity of illness and poor outcome. There has also been a growing concern in recent years that interventions that promote ESL shedding may worsen clinical outcomes for critically ill patients. Given that ESL shedding is associated with exacerbating inflammation and increasing vascular permeability in laboratory models, there is theoretical plausibility that limiting ESL shedding may improve clinical outcome.
Shedding alters capillary perfusion, causing a decrease in functional capillary density (44), in addition to increased endothelial permeability (45). A decrease in functional capillary density means that some vessels within a given area do not have red blood cells traversing their course, consistent with microcirculatory dysfunction. This can create regions of tissue hypoxia. A clear association between ESL shedding and impairment of microcirculatory blood flow is yet to be demonstrated in either an in vivo animal model or clinical study (46). However, given that ESL shedding via artificial means can reduce capillary blood flow (44), it is mechanistically plausible that shedding of the ESL in critical illness plays a role in altered microcirculation. It has been well-demonstrated that heterogeneity of capillary blood flow can persist in critical illness despite normalization of macrohemodynamic variables, such as blood pressure and cardiac output (47–51). Further, persistence of altered microcirculatory flow in critically ill people is associated with severity of illness and poor outcome (47, 51–53). Consequently, it appears important to identify causes of microcirculatory dysfunction that occurs independent of circulatory shock. More evidence is required to determine a causal link between ESL shedding and microcirculatory dysfunction, and whether protection of the ESL can prevent the latter.
There may be downstream consequences to the release of EG components into circulation. Shed components can stimulate inflammation by acting as danger-associated molecular patterns or “alarmins.” Soluble heparan sulfate molecules play a key role in modulating inflammation, including leukocyte activation, increasing production of cytokines, and endothelial activation (54, 55). Low molecular weight hyaluronan can also stimulate production of inflammatory mediators (56–59). In contrast, shed components such as syndecan-1 and−4 ectodomains can have indirect anti-inflammatory effects by facilitating neutrophil cytotoxicity (60, 61). The complexity of EG components acting as effector molecules in the systemic circulation may be analogous to the systemic inflammatory response, where some cytokine release is beneficial to the host response, whereas a “cytokine storm” creates pathological consequences.
It is unclear how long it takes for ESL recovery to occur. Most evidence is based on data from in vitro cell culture or ex vivo vascular models, which are unable to fully replicate in vivo conditions. That being said, individual components can be regenerated within 24 h (37, 62), but restoration of the structure itself can take up to 7 days (32, 63). During critical illness, inflammation likely continues the shedding process, delaying the repair. Further in vivo research is required using real-time videomicroscopy or similar means in order to characterize the temporal changes during ESL recovery.
Shedding of the ESL can be detected by a number of means. Laboratory studies utilize several methods, including measurement of circulating components of the EG (proteoglycan ectodomains and GAGs), detection of EG components on the surface of the endothelium, direct visualization of the ESL via tissue fixation and microscopy, and indirect visualization via real-time videomicroscopy. Clinical studies usually rely on measurement of circulating components of the EG, or EG biomarkers, in serum or plasma samples to assess systemic shedding. The most frequently reported EG biomarkers are syndecan-1, heparan sulfate, and hyaluronan. There are several limitations to relying on this kind of assessment of the ESL. Firstly, studies often only measure a single component of the EG at a single point in time. That particular component may have other sources of shedding. For example, syndecan-1 is not only present on the surface of the endothelium but also on epithelial cells and leucocytes (64–68). Also, highly relevant to intravenous fluid therapy, hyaluronan is abundant throughout the interstitium and can be “flushed” through the lymphatics back into systemic circulation (69–71). It is unclear in critical illness to what degree other sources of glycocalyx shedding contribute to serum or plasma concentrations. Further, tissue injury and inflammation during critical illness may upregulate cell surface expression of these biomarkers, especially the proteoglycans (38, 72–83). Therefore, an increase in circulating concentration may reflect an increase in cell turnover rather than primarily cell surface shedding. Several recent studies have demonstrated temporal differences in the shedding of several EG components in people presenting to an emergency department with sepsis, whereby hyaluronan increased early in treatment whereas sydnecan-1 increased later (84–86). This raises questions as to why some biomarkers increase earlier than others. It is possible that the temporal differences relate to either alternative sources of the biomarker or differences in upregulation.
Ideally, it would be best to visualize shedding of the EG in vivo rather than interpreting circulating biomarker concentrations. These techniques are mostly reserved for use in laboratory models rather than clinical use. Specialized tissue fixation techniques can be used to directly visualize the ESL via electron microscopy, often employed in non-survival rodent models (28, 87–91). Given the fragility of the ESL, it is best that the tissue is preserved via perfusion with fixative prior to death, either perfusion of an isolated organ or the whole body. Tissue immersion techniques that avoid whole-body perfusion have also been recently described (92). Anecdotally, it is challenging to achieve quality images of intact ESL in tissue sections. Alternatively, components of the EG may be fluorescently labeled and visualized by confocal microscopy of cell culture or tissue models (93). Other laboratory techniques include dye exclusion with intravital microscopy to estimate ESL thickness (94–96). Finally, ESL thickness may be estimated in real-time by sidestream dark field microscopy in both large animal models and clinical research (46, 97–99). This technology approximates ESL thickness by measuring the perfused boundary region, which is the region within the blood vessel that is peripheral to the flow of red blood cells, or the immobile plasma layer. Although this technique shows promise for future use in veterinary clinical research (100), image acquisition can be technically difficult and image quality is currently limited for reliably estimating ESL thickness.
In the patient requiring bolus fluid therapy, it is likely that alteration of the ESL has already occurred due to the effects of inflammatory mediators. For example, tumor necrosis factor-α, a potent pro-inflammatory cytokine, causes reduction in thickness of the ESL, shedding of syndecan-1,-4, and GAGs from the endothelium, as well as upregulation of glycocalyx components on the endothelial surface (76, 81, 90, 101). Matrix metalloproteinases, one of the main perpetrators for glycocalyx shedding, are released by activated leucoytes (17). Tissue injury, such as ischemia and reperfusion, and production of reactive oxygen species also cause EG shedding (43, 102–104). Activation of coagulation may also affect the EG, as both plasmin and thrombin can cleave syndecan ectodomains (74, 105). Relevant to sepsis, bacterial components such as lipopolysaccharide and chemotactic peptides also cause shedding of the EG (43, 77, 81, 106–108). Therefore, it is likely that partial or complete denudation of the ESL has already occurred before fluid therapy is administered.
Shedding of the EG in states of critical illness, such as shock, has been demonstrated in animal models before any fluid therapy has commenced. In mice, hemorrhagic shock was associated with a thinner pulmonary ESL, as measured by electron microscopy, and downregulation of syndecan-1 on the endothelial surface, compared to a sham model (109, 110). In a canine hemorrhagic shock model comparing different fluid interventions (111), plasma hyaluronan concentration was significantly increased across treatment groups after atraumatic blood removal, but before fluid, compared to baseline (unpublished analysis). An increase in plasma hyaluronan concentration has also been demonstrated in a rodent sepsis model, 4 h after Escherichia coli lipopolysaccharide infusion, where blood pressure was maintained by norepinephrine administration (112). There are limited data on EG shedding in the clinical setting before fluid therapy; however, serum syndecan-1 concentrations above healthy control levels has been demonstrated pre-hospital in human trauma patients, though the temporal relationship to fluid administration by first responders is unknown (113).
Bearing in mind that alterations to the ESL may already exist in critically ill patients, there is growing evidence that administration of “clear” fluids may propagate ESL shedding. Several clinical studies in critically ill people have identified an association between volume of fluid administered and EG biomarker concentrations, including increased hyaluronan (84), syndecan (114), and heparan sulfate concentrations (115). Putting aside the confounders of inflammation and severity of illness, bolus fluid therapy may have a direct effect on the ESL via hemodilution and production of natriuretic peptides. Ex vivo vascular models provide important data in regards to this issue, as they exclude the effects of the varying pharmacodynamic properties of fluid types administered in vivo. These vascular models have shown that dilution of plasma with crystalloid reduces ESL thickness. Notably, a mathematical model derived from meta-analysis of several studies demonstrated that dilution of blood with fluid reduced vascular resistance, independent of hematocrit and COP (116). Also, the decrease in resistance caused by saline was reduced in magnitude when the vasculature was pre-treated with heparinase. Heparinase sheds heparan sulfate side-chains from proteoglycans, thus thinning the glycocalyx (117). From this, it may be inferred that the decrease in resistance caused by saline infusion is due to not only changes in hemorrheology but also loss of the ESL. Further, several studies have shown that perfusion of blood vessels with crystalloid solution increases vascular permeability. One such study showed an inverse linear relationship between albumin concentration in the perfusate and hydraulic conductivity, or permeability, of vasculature (118). Return to baseline permeability was not achieved by simply increasing the albumin concentration back to baseline levels; it required a much higher albumin concentration. This implies modification of the ESL and its affinity for albumin after crystalloid infusion. Further experiments showed that perfusion of blood vessels with plasma was more effective at restoring normal vascular permeability after crystalloid infusion (119), compared with an albumin solution, indicating that substances within plasma are important for maintaining normal transvascular fluid flux. A more recent study showed that infusion of guinea pig coronary vasculature with 0.9% saline significantly increased fluid extravasation, compared to low molecular weight (LMW) hydroxyethyl starch (HES) (Voluven®) and albumin solution (45). Pretreatment with heparinase combined with saline infusion did not yield higher fluid extravasation; stripping the EG before infusion was just as detrimental as saline infusion alone. Interestingly, no differences between fluid treatments in the appearance of the ESL were appreciated using electron microscopy. Finally, a murine hemorrhagic shock model compared 15 mL/kg of either fresh whole blood (FWB), packed red blood cells in lactated Ringer's solution (PRBC in LRS), or washed PRBC in LRS, or 75 mL/kg of plain LRS (120). Rats that received either washed PRBC or LRS had decreased ESL thickness compared to baseline, as measured by intravital microscopy, whereas those that received either FWB or PRBC did not show a significant difference to baseline. Results were similar for change in plasma heparan sulfate concentration. Therefore, it appeared that the presence of protein within the FWB and unwashed PRBC products was effective at mitigating ESL shedding.
Another mechanism of EG shedding during bolus fluid therapy is the action of natriuretic peptides on the endothelium. Atrial and brain natriuretic peptides (ANP and BNP, respectively) are released from the cardiac atria and ventricles during stretch of the myocardium (121). These peptides counteract the effects of hypervolemia by decreasing systemic vascular resistance and causing a natriuresis, among other mechanisms. Natriuretic peptides also increase vascular permeability (122, 123). A more recently discovered role of these peptides is shedding of the EG. All three major natriuretic peptides (ANP, BNP, and C-type NP) can shed the EG, as measured by increased syndecan-1 and heparan sulfate concentrations in the effluent of guinea pig coronary arteries (88, 124). These studies also demonstrated increased vascular permeability and visualized denudation of the ESL via electron microscopy.
Fast intravenous administration of isotonic crystalloid fluid may stimulate natriuretic peptide release during the bolus phase. The dose of crystalloid administered for treatment of shock far exceeds the volume remaining in circulation after redistribution. This rapid rise and fall of blood volume during a crystalloid bolus was demonstrated by Silverstein and others (7), whereby the blood volume rapidly rose by 76% during a large bolus of 0.9% saline before dissipating. Therefore, it is possible that atria may become “over-stretched” during crystalloid bolus fluid therapy and release natriuretic peptides, which may then contribute to systemic EG shedding. Two studies in human surgical patients have shown a rise in ANP in parallel with EG biomarker concentrations after fluid loading (125, 126). These studies showed a significant increase in serum syndecan-1 and hyaluronan concentrations but not heparan sulfate. One additional study showed an increase in ANP and all three EG biomarker concentrations in cardiac surgical patients; in both those undergoing cardiopulmonary bypass and those undergoing “off-pump” procedures (127). In contrast, one study in people undergoing hysterectomy (n = 26) showed no significant rise in BNP or biomarker concentrations (syndecan-1 and heparan sulfate) after 25 mL/kg of LRS given over 30 min during surgery (128). An additional study showed no significant rise in BNP or any EG biomarker concentrations (syndecan-1, heparan sulfate, hyaluronan) in human surgical patients or healthy volunteers during a modest fluid load of 3 mL/kg of 20% albumin, though sample size was small (n = 15 per group) (129). A recent canine hemorrhagic shock study did not observe a significant increase in plasma ANP concentration after 80 mL/kg of balanced isotonic crystalloid given over 20 min, however small sample size (n = 6) and baseline variability may have hampered the ability to detect a difference (111).
One of the challenges of interpreting studies that measure EG biomarker concentrations as the primary assessment of EG shedding is the variability in choice of biomarker. Across multiple studies investigating the effect of fluid loading on the EG, it appears that hyaluronan is consistently increased immediately after a large volume of crystalloid, or synthetic colloid fluid in the setting of hypervolemia (111, 125, 126). Syndecan-1 concentration significantly increases after fluid in humans and rats (110, 125, 126, 130–132), but not in all studies (128, 133). Heparan sulfate has not been shown to significantly increase in humans (125, 126, 128) but has been shown to increase in rats (120, 130, 133). Unpicking these consistencies is difficult due to differences across species, study designs, fluid doses, timing of intervention and blood sampling, and choice of comparator (other fluid vs. no fluid vs. sham). Studies may also vary in whether or not they adjust the biomarker concentration for the effects of hemodilution, such as indexing to albumin, hemoglobin, or other tracer concentration in the blood. Although this may help to account for the different pharmacodynamics of each fluid type, and their variable dilution of blood tracer components, indexation of biomarker concentrations can create a margin of error. On closer inspection of individual studies, inconsistency has been shown within the studies themselves, showing a significant change in one biomarker but not the other (125, 126, 133). As mentioned above, the source of circulating EG biomarkers may not be restricted to the endothelium and other sources, such as the interstitium or surface of other cells, may be contributing. Therefore, a healthy sceptism should be applied to any conclusion drawn from biomarker concentrations alone.
Much of the evidence concerning EG or ESL shedding after fluid therapy concerns the use of large volumes of crystalloid fluid. Administering other fluid types that have less redistribution to the interstitium may theoretically be associated with less EG shedding. This section assesses the evidence for the comparative effect of crystalloid, synthetic colloid, and hypertonic fluids on the EG or ESL.
Multiple rodent studies have shown that administration of large volumes of crystalloid fluid for hemorrhagic shock is associated with more EG shedding, compared to fluids containing protein (109, 110, 120, 130–133). However, it is difficult to compare interventions in regards to effects on the EG in many of these studies, as cardiovascular parameters are either not closely monitored or are different between treatment groups. Shock itself, without fluid resuscitation, can cause EG shedding (109, 110, 131, 134–136), which becomes a confounder when comparing resuscitation strategies that provide inequitable blood volume expansion. However, several key studies allow some assessment of this issue. A murine hemorrhagic shock model showed that administration of protein-poor fluids, either washed PRBCs or LRS, was associated with a thinner ESL, whereas protein-containing fluids (FWB and PRBCs) were not (120). Cardiovascular parameters after resuscitation appeared similar across these groups, based on the limited data available. Another murine hemorrhagic shock model compared seven different fluid strategies: 15 mL/kg of FWB, PRBC, fresh frozen plasma (FFP) or 5% albumin, 8 mL/kg of 3% saline, 45 mL/kg of 0.9% saline, or 75 mL/kg of LRS (130). Shock index, usually defined by heart rate divided by systolic blood pressure, was compared before hemorrhage and immediately after fluid resuscitation within each fluid group. Rats that received either 5% albumin, LRS, 3% saline, or 0.9% saline had a significantly increased shock index after fluid resuscitation, whereas rats that received FWB, PRBC, and FFP showed no change in shock index. When comparing only the fluid types that did not resolve shock, based on shock index, rats that received 5% albumin had higher ESL thickness and lower EG biomarker concentrations, compared to the crystalloid groups. Finally, a murine hemorrhagic shock model used a blood pressure-targeted resuscitation method for comparing crystalloid fluid with plasma (109). Administration of LRS was associated with a thinner ESL, visualized on electron microscopy, and decreased endothelial expression of syndecan-1, compared to administration of plasma.
There is a scarcity of “large animal” models, or those including pigs, sheep, and dogs, that have assessed ESL shedding after fluid resuscitation. This may be partially due to a limitation on validated assays available for measuring glycocalyx biomarkers across these species. Current commercially available validated options for assessing EG shedding are restricted to circulating hyaluronan concentration. A canine hemorrhagic shock model did not detect a significant difference in plasma hyaluronan concentrations when comparing dogs that received 20 mL/kg of FWB (n = 6) with those that received 80 mL/kg of Plasmalyte-148® (n = 6) (111). Other differences in parameters were seen between the crystalloid and colloid fluid groups in this study, which is discussed further below. In an ovine endotoxemia model, a pressure-targeted resuscitation method using vasopressor therapy was used to compare either norepinephrine alone or 0.9% saline (40 mL/kg) in combination with norepinephrine (137). Although there was no comparison to resuscitation with a protein-containing fluid, the comparison of crystalloid fluid to no fluid in the setting of sepsis provided interesting results. There was no significant difference in serum hyaluronan concentrations between groups over time, though sheep that received 0.9% saline showed a greater rate of increase in this biomarker. This greater increase was not observed directly after the fluid bolus, like in the aforementioned canine study, but beyond 6 h, which may have been related to worsening septic shock in this group. Atrial natriuretic peptide was significantly higher at the end of the fluid bolus in the sheep that received fluid (see previous discussion on natriuretic peptides). Detection of between-group differences was also likely affected by small sample size in this study (n = 8 per group), similar to the study in dogs.
Given that crystalloid fluids generally require a larger volume to expand circulating blood volume to the same extent as colloid fluids (6–9), crystalloids cause greater hemodilution during the fluid bolus and, potentially, greater natriuretic peptide release. This begs the question if administration of colloid fluids causes less EG shedding than crystalloid fluid. Two main types of synthetic colloid are currently in use: HES and gelatin products. The aforementioned canine hemorrhagic shock study included both of these fluid types at 20 mL/kg, as comparator fluids to 80 mL/kg of Plasmalyte-148® (111). Dogs administered HES had significantly lower plasma hyaluronan concentration immediately after the fluid bolus, compared to both FWB and crystalloid groups. In contrast, the group that received 4% succinylated gelatin had significantly increased plasma hyaluronan concentration 40 and 100 min after the end of the fluid bolus, compared to FWB. Given that inflammation may cause EG shedding, these differential effects observed between these two colloids may be due to release of inflammatory mediators; HES has been associated with mitigation of inflammation (138–141) whereas gelatin has been associated with pro-inflammatory effects (142). However, there were no differences in plasma concentrations of inflammatory mediators between the two colloid groups in this study (111). Therefore, the effect of gelatin on hyaluronan shedding may be a direct effect of the fluid, rather than pro-inflammatory effects, such as washout of the interstitium. Another hemorrhagic shock study in rats replaced the shed volume with crystalloid, at either shed volume, two times shed volume, or three times shed volume, or shed volume with HES (n = 6 per group) (141). This study did not identify any significant differences between groups in response of blood pressure to fluid resuscitation, organ syndecan-1 expression, or circulating syndecan-1 concentration. A hemorrhagic shock, stroke volume-targeted resuscitation model in pigs compared balanced crystalloid to HES and found no differences in post-fluid serum syndecan-1 or glypican concentrations (6). Blood was sampled 120 min after commencement of fluids, therefore any peaks in EG biomarker shedding may have been missed. Given the limited evidence for different effects of colloid fluids on the EG or ESL, compared to crystalloids, a conclusion cannot be currently drawn.
Hypertonic solutions exert immunomodulatory effects both in in vitro and animal model studies, including decreased leucocyte response to pro-inflammatory stimuli or circulatory shock states, when compared to other fluid types (143–156). Administration of hypertonic saline to human trauma patients also reduced inflammatory biomarker concentration, compared to isotonic saline (157, 158). Several studies have also shown a reduction in vascular leakage of macromolecules (147, 151, 155). Given these effects, it is possible that hypertonic solutions may have a beneficial effect in regards to reducing ESL shedding. In the rodent hemorrhagic shock model previously discussed comparing many different type of fluids, rats that received 8 mL/kg of 3% saline had a lower increase from baseline in plasma heparan sulfate concentration, compared to both isotonic crystalloid groups (45 mL/kg of 0.9% saline or 75 mL/kg of LRS), and a lower loss of ESL thickness (130). This difference was despite a persistence of an elevated shock index and hyperlactatemia after resuscitation with 3% saline. Therefore, although hypertonic saline may have had less impact on the EG, it may have also provided inadequate fluid resuscitation. A preliminary randomized clinical trial in people with suspected sepsis could not demonstrate a difference between groups in hyaluronan concentrations after treatment allocation of either 5 mL/kg of 3% saline or 10 mL/kg of 0.9% saline (86). Serum syndecan-1 concentration decreased after 0.9% saline, compared to baseline, whereas serum syndecan-1 concentration did not change after 3% saline. Though this significant difference between groups may reflect hemodilution, as twice the volume of 0.9% saline was administered, this was not reflected in differences in hemoglobin concentration. It is unclear what the clinical significance is of this subtle difference in change in serum syndecan-1 concentration, whether it reflects more syndecan-1 shedding in the hypertonic saline group or not. Limitations of this study included a low severity of illness and small sample size, and further studies are needed to clarify the effects of hypertonic solutions on the EG.
The clinical relevance of ESL shedding due to fast or large-volume fluid administration is yet to be determined. This type of fluid administration can cause shedding of the ESL, which may propagate microcirculatory dysfunction, inflammation, and procoagulation. Therefore, there has been much interest developing in “glycoprotective” fluid strategies or adjunctive therapies during the resuscitation phase.
Blood volume expansion is the cornerstone of managing circulatory shock, with some types of shock an exception such as cardiogenic shock. Conventionally, this is achieved by administering large (more than 20 mL/kg) volumes of crystalloid fluid intravenously as rapidly as possible, or as a bolus (159, 160). This serves to increase cardiac preload and improve cardiac output. The need for this intervention is intuitive for shock caused by hypovolemia; however, some types of shock have a component of vasodilation and microcirculatory dysfunction. This includes states of shock due to a systemic inflammatory response, such as sepsis and blunt trauma. For these types of shock, improvement of macrohemodynamic variables, such as cardiac output and blood pressure, may not improve microcirculatory blood flow in a linear way. This is particularly pertinent to septic shock, where the role of fluid bolus therapy has been called into question, with a movement toward early vasopressor therapy instead (161, 162). Further to the lack of improvement in microcirculatory flow, it has been suggested that fluid bolus therapy in sepsis can propagate vasodilation, due to endothelial shear stress, release of nitric oxide, and direct vasodilatory effects of natriuretic peptides. This may contribute to some patients becoming refractory to vasopressor therapy. In an ovine endotoxemia model, administration of 40 mL/kg of 0.9% saline was associated with a significantly higher dose of noradrenaline subsequently administered in order to maintain blood pressure, compared to sheep that received only noradrenaline (137). Given the concern that bolus fluids may cause harm in sepsis, a preliminary randomized clinical trial was completed demonstrating feasibility of restricting crystalloid fluid early in the treatment of sepsis in people (163). Following on from this, a large multi-center human randomized clinical trial is underway to compare a restrictive fluid strategy with a more liberal one in people with early sepsis (164).
It is much less known if restricting crystalloid fluid in other types of shock may be beneficial. In the setting of human trauma, discussion concerning restrictive crystalloid fluid strategies focuses on delayed resuscitation or permissive hypotension in order to avoid clot disruption and dilutional coagulopathy before definitive hemostasis (4, 165). Although animal models have shown that fluid resuscitation is likely beneficial for severe uncontrolled hemorrhage, it increases the risk of mortality in lower grades of severity of bleeding, compared to not administering any fluid (166). Hypotensive resuscitation, in parallel with lower volumes of administered fluids, is also associated with a reduced risk of mortality in human trauma patients (167, 168), though the evidence is mixed (169). This benefit is likely related to improved coagulation and clot stability; it is unknown if there are also benefits associated with reducing endothelial dysfunction. Currently, the recommendation in human trauma medicine is for limitation of “clear” fluid and early, yet judicious, administration of blood products. This includes packed red blood cells, plasma, and platelets in parallel during resuscitation. The early administration of plasma may not only serve to limit dilutional coagulopathy but also assist with repairing the ESL. It is difficult to translate this practice to veterinary medicine, given the cost of transfusion, limited availability, and variability in blood banking practices. Given there are many factors entering into the cost to benefit assessment of administering plasma or albumin products to individuals, theoretical benefits of plasma for the ESL are of lesser importance, until more is known. Also, caution should be applied when restricting crystalloid fluid to any patient with shock, as the benefits of a restrictive strategy may be only relevant to certain patient populations, such as sepsis. This concept was highlighted by a recent randomized clinical trial in human surgical patients whereby participants randomized to receive a restrictive fluid strategy had a significantly higher rate of acute kidney injury and surgical site infection, compared to those randomized to a liberal fluid strategy (170).
Fast fluid administration serves to rapidly improve clinical signs and macrohemodynamic variables, and has been the bread-and-butter of emergency and critical care medicine for decades. However, normalization of macrohemodynamic variables in people does not always translate to improved microcirculatory flow (47–49, 51). This is termed a lack of hemodynamic coherence (171). Intravenous fluid delivered rapidly during the stabilization phase may be contributing to persistence of disturbed microcirculatory flow. When viewing the microcirculation using sidestream dark field microscopy during anesthesia in pigs, fast fluid administration (20 mL/kg/hr) for 3 h was associated with development of heterogeneity in capillary blood flow, compared to “standard” anesthesia fluid rates (5 mL/kg/hr) (172). Although a direct relationship between estimated ESL thickness and microcirculatory flow has not been established in the clinical setting (46), it is possible that amplification of ESL shedding caused by fast fluid administration may be contributing to microcirculatory dysfunction. It is possible that slower fluid administration may achieve the same resuscitation end-points while avoiding some of these deleterious effects on the endothelium. This concept has been explored in a handful of studies. In a rodent model where 40% of the blood volume was removed by atraumatic hemorrhage, rats were assigned to receive no fluid, rapid fluid (0.9% saline at three times shed volume over 30 min), or slow fluid (0.9% saline at three times shed volume over 12 h) (173). Rats in the fast fluid group showed higher inflammatory cytokine concentrations early in the study and some markers of worse outcome over the latter part of the study, including lower mean arterial blood pressure, higher blood glucose and lactate concentrations, and an increase in markers of lung injury, compared to rats in the slow fluid group. As the rats were euthanased after 24 h, it is unknown how their recovery progressed. In a human open randomized clinical trial, including 50 surgical and 16 septic patients, no significant difference was found in estimated ESL thickness between patients that received fast crystalloid administration (5–10 min) and slow crystalloid administration (20–30 min) (48). However, an overall decrease in estimated ESL thickness after fluid administration was observed. Given that there is some evidence that rapid fluid administration (i.e., within 10 min) may not provide any clinical benefit over slower fluid administration (i.e., within 20 to 60 min) (8, 174), larger clinical trials are justified to explore effects of fluid rate on the microcirculation and clinical outcomes.
Intense ongoing debate surrounds the choice of fluid for critically ill patients, including crystalloid, synthetic colloid, and protein-containing solutions. These conversations surround relative fluid effectiveness and adverse effects, many of which are still yet to be fully elucidated for clinical relevance in certain species or disease subsets. Adding in the potential benefit or detriment of certain fluid types to preservation of the ESL and optimizing microcirculation is still premature. Bearing that in mind, the current evidence supports mitigating hemodilution and avoiding hypervolemia in order to optimize ESL recovery. There is theoretical benefit for the ESL to the use of protein-containing solutions, such as plasma and albumin, rather than “clear” fluids, but this must be carefully considered against any detrimental effects of blood products, including financial cost. At this stage, no clear recommendation can be made for veterinary medicine, and more clinical research is required.
Many drugs have been explored as to their protective or resurrecting actions for the ESL and have been reviewed in detail elsewhere (175, 176). These therapies may become viable in the future for use during the optimization and stabilization phase of fluid resuscitation (177). Therapies that are in current use in veterinary medicine that have been shown to have beneficial effects on the EG or ESL include albumin solution, plasma, hydrocortisone, heparin, and N-acetylcysteine.
The coating of endothelium by the EG and its associated molecules serves many functions in the body, both intact and when denuded. Infusion of large volumes of fluid causes disruption of this layer, which may propagate interstitial edema and inflammation. There is some evidence that slow fluid administration or restricting the volume of crystalloid fluid for shock resuscitation may benefit the patient. Exactly how to do this and for which patient subset this strategy benefits in veterinary medicine remains to be identified. Clinical veterinary research assessing the effect of glycoprotective therapy or strategies is currently limited by a lack of validated EG biomarker assays and affordable, reliable technology for estimating ESL thickness in vivo. Further work is required on developing validated, reliable EG biomarker assays and determining the relationship between these biomarkers and shedding of the ESL in vivo, especially for dogs and cats. Despite these current limitations, future research directions should also focus on strategies limiting crystalloid fluid volumes, especially in the setting of sepsis and early vasopressor drug therapy.
LS: preparation of manuscript and final approval. DH: revision of manuscript and final approval. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Angus DC, Barnato AE, Bell D, Bellomo R, Chong CR, Coats TJ, et al. A systematic review and meta-analysis of early goal-directed therapy for septic shock: the ARISE, ProCESS and ProMISe Investigators. Intensive Care Med. (2015) 41:1549–60. doi: 10.1007/s00134-015-3822-1
2. Maitland K, Babiker A, Kiguli S, Molyneux E. The FEAST trial of fluid bolus in African children with severe infection. Lancet. (2012) 379:613. doi: 10.1016/S0140-6736(12)60260-8
3. Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, et al. Comparison of two fluid-management strategies in acute lung injury. New Eng J Med. (2006) 354:2564–75. doi: 10.1056/NEJMoa062200
4. Albreiki M, Voegeli D. Permissive hypotensive resuscitation in adult patients with traumatic haemorrhagic shock: a systematic review. Eur J Trauma Emerg Surg. (2018) 44:191–202. doi: 10.1007/s00068-017-0862-y
5. Yozova ID, Howard J, Sigrist NE, Adamik KN. Current trends in volume replacement therapy and the use of synthetic colloids in small animals-an internet-based survey 2016. Front Vet Sci. (2017) 4:140. doi: 10.3389/fvets.2017.00140
6. Laszlo I, Demeter G, Oveges N, Erces D, Kaszaki J, Tanczos K, et al. Volume-replacement ratio for crystalloids and colloids during bleeding and resuscitation: an animal experiment. Intensive Care Med Exp. (2017) 5:52. doi: 10.1186/s40635-017-0165-y
7. Silverstein DC, Aldrich J, Haskins SC, Drobatz KJ, Cowgill LD. Assessment of changes in blood volume in response to resuscitative fluid administration in dogs. J Vet Emerg Crit Care. (2005) 15:185–92. doi: 10.1111/j.1476-4431.2005.00138.x
8. Ho L, Lau L, Churilov L, Riedel B, McNicol L, Hahn RG, et al. Comparative evaluation of crystalloid resuscitation rate in a human model of compensated haemorrhagic shock. Shock. (2016) 46:149–57. doi: 10.1097/SHK.0000000000000610
9. Orbegozo Cortes D, Gamarano Barros T, Njimi H, Vincent JL. Crystalloids versus colloids: exploring differences in fluid requirements by systematic review and meta-regression. Anesth Analg. (2015) 120:389–402. doi: 10.1213/ANE.0000000000000564
10. Tarbell JM, Pahakis MY. Mechanotransduction and the glycocalyx. J Intern Med. (2006) 259:339–50. doi: 10.1111/j.1365-2796.2006.01620.x
11. Rosenberg RD, Shworak NW, Liu J, Schwartz JJ, Zhang L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J Clin Invest. (1997) 100(Suppl. 11):S67–75.
12. Couchman JR. Syndecans: proteoglycan regulators of cell-surface microdomains? Nat Rev Mol Cell Biol. (2003) 4:926–37. doi: 10.1038/nrm1257
13. Bame KJ. Heparanases: endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology. (2001) 11:91–8r. doi: 10.1093/glycob/11.6.91R
14. Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med. (1997) 242:27–33. doi: 10.1046/j.1365-2796.1997.00170.x
15. Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. (2007) 454:345–59. doi: 10.1007/s00424-007-0212-8
16. Henry CB, Duling BR. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol. (1999) 277:H508–14. doi: 10.1152/ajpheart.1999.277.2.H508
17. Kolarova H, Ambruzova B, Svihalkova Sindlerova L, Klinke A, Kubala L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediators Inflamm. (2014) 2014:694312. doi: 10.1155/2014/694312
18. Paulus P, Jennewein C, Zacharowski K. Biomarkers of endothelial dysfunction: can they help us deciphering systemic inflammation and sepsis? Biomarker. (2011) 16(Suppl. 1):S11–21. doi: 10.3109/1354750X.2011.587893
19. Bashandy GM. Implications of recent accumulating knowledge about endothelial glycocalyx on anesthetic management. J Anesth. (2015) 29:269–78. doi: 10.1007/s00540-014-1887-6
20. Becker BF, Chappell D, Jacob M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol. (2010) 105:687–701. doi: 10.1007/s00395-010-0118-z
21. Schnitzer JE, Carley WW, Palade GE. Specific albumin binding to microvascular endothelium in culture. Am J Physiol. (1988) 254(Pt. 2):H425–37. doi: 10.1152/ajpheart.1988.254.3.H425
22. Chang SW, Westcott JY, Henson JE, Voelkel NF. Pulmonary vascular injury by polycations in perfused rat lungs. J Appl Physiol. (1987) 62:1932–43. doi: 10.1152/jappl.1987.62.5.1932
23. Antunes E, Mariano M, Cirino G, Levi S, de Nucci G. Pharmacological characterization of polycation-induced rat hind-paw oedema. Br J Pharmacol. (1990) 101:986–90. doi: 10.1111/j.1476-5381.1990.tb14193.x
24. Vehaskari VM, Chang CT, Stevens JK, Robson AM. The effects of polycations on vascular permeability in the rat. A proposed role for charge sites. J Clin Invest. (1984) 73:1053–61. doi: 10.1172/JCI111290
25. Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res. (2010) 87:198–210. doi: 10.1093/cvr/cvq062
26. Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth. (2012) 108:384–94. doi: 10.1093/bja/aer515
27. Adamson RH, Michel CC. Pathways through the intercellular clefts of frog mesenteric capillaries. J Physiol. (1993) 466:303–27.
28. Jacob M, Bruegger D, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, et al. The endothelial glycocalyx affords compatibility of Starling's principle and high cardiac interstitial albumin levels. Cardiovasc Res. (2007) 73:575–86. doi: 10.1016/j.cardiores.2006.11.021
29. Hu X, Adamson RH, Liu B, Curry FE, Weinbaum S. Starling forces that oppose filtration after tissue oncotic pressure is increased. J Physiol Heart Circ Physiol. (2000) 279:H1724–36. doi: 10.1152/ajpheart.2000.279.4.H1724
30. Cruz-Chu ER, Malafeev A, Pajarskas T, Pivkin IV, Koumoutsakos P. Structure and response to flow of the glycocalyx layer. Biophys J. (2014) 106:232–43. doi: 10.1016/j.bpj.2013.09.060
31. Dull RO, Mecham I, McJames S. Heparan sulfates mediate pressure-induced increase in lung endothelial hydraulic conductivity via nitric oxide/reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L1452–8. doi: 10.1152/ajplung.00376.2006
32. McDonald KK, Cooper S, Danielzak L, Leask RL. Glycocalyx degradation induces a proinflammatory phenotype and increased leukocyte adhesion in cultured endothelial cells under flow. PLoS ONE. (2016) 11:e0167576. doi: 10.1371/journal.pone.0167576
33. Yao Y, Rabodzey A, Dewey CF Jr. Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol. (2007) 293:H1023–30. doi: 10.1152/ajpheart.00162.2007
34. Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. (2003) 93:e136–42. doi: 10.1161/01.RES.0000101744.47866.D5
35. Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol. (2014) 6:338–47. doi: 10.1039/C3IB40199E
36. Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong EE, Rizzo V, et al. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am J Physiol Heart Circ Physiol. (2013) 305:H811–20. doi: 10.1152/ajpheart.00764.2012
37. Zeng Y, Tarbell JM. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS ONE. (2014) 9:e86249. doi: 10.1371/journal.pone.0086249
38. Liu JX, Yan ZP, Zhang YY, Wu J, Liu XH, Zeng Y. Hemodynamic shear stress regulates the transcriptional expression of heparan sulfate proteoglycans in human umbilical vein endothelial cell. Cell Mol Biol. (2016) 62:28–34. doi: 10.14715/cmb/2016.62.8.5
39. Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng. (2012) 40:840–8. doi: 10.1007/s10439-011-0427-x
40. Lygizos MI, Yang Y, Altmann CJ, Okamura K, Hernando AA, Perez MJ, et al. Heparanase mediates renal dysfunction during early sepsis in mice. Physiol Rep. (2013) 1:e00153. doi: 10.1002/phy2.153
41. Bitan M, Weiss L, Zeira M, Zcharia E, Slavin S, Nagler A, et al. Heparanase promotes engraftment and prevents graft versus host disease in stem cell transplantation. PLoS ONE. (2010) 5:e10135. doi: 10.1371/journal.pone.0010135
42. Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. (2003) 23:1541–7. doi: 10.1161/01.ATV.0000085630.24353.3D
43. Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. (2004) 286:H1672–80. doi: 10.1152/ajpheart.00832.2003
44. Cabrales P, Vazquez BY, Tsai AG, Intaglietta M. Microvascular and capillary perfusion following glycocalyx degradation. J Appl Physiol. (2007) 102:2251–9. doi: 10.1152/japplphysiol.01155.2006
45. Jacob M, Bruegger D, Rehm M, Welsch U, Conzen P, Becker BF. Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiol. (2006) 104:1223–31. doi: 10.1097/00000542-200606000-00018
46. Rovas A, Seidel LM, Vink H, Pohlkötter T, Pavenstädt H, Ertmer C, et al. Association of sublingual microcirculation parameters and endothelial glycocalyx dimensions in resuscitated sepsis. Crit Care. (2019) 23:260. doi: 10.1186/s13054-019-2542-2
47. De Backer D, Donadello K, Sakr Y, Ospina-Tascon G, Salgado D, Scolletta S, et al. Microcirculatory alterations in patients with severe sepsis: impact of time of assessment and relationship with outcome. Crit Care Med. (2013) 41:791–9. doi: 10.1097/CCM.0b013e3182742e8b
48. Pouska J, Tegl V, Astapenko D, Cerny V, Lehmann C, Benes J. Impact of intravenous fluid challenge infusion time on macrocirculation and endothelial glycocalyx in surgical and critically ill patients. Biomed Res Int. (2018) 2018:1–11. doi: 10.1155/2018/8925345
49. Jhanji S, Vivian-Smith A, Lucena-Amaro S, Watson D, Hinds CJ, Pearse RM. Haemodynamic optimisation improves tissue microvascular flow and oxygenation after major surgery: a randomised controlled trial. Crit Care. (2010) 14:R151. doi: 10.1186/cc9220
50. Trieu M, van Meurs M, van Leeuwen ALI, Van Slyke P, Hoang V, Geeraedts LMG, et al. Vasculotide, an angiopoietin-1 mimetic, restores microcirculatory perfusion and microvascular leakage and decreases fluid resuscitation requirements in hemorrhagic shock. Anesthesiology. (2018) 128:361–74. doi: 10.1097/ALN.0000000000001907
51. Tachon G, Harrois A, Tanaka S, Kato H, Huet O, Pottecher J, et al. Microcirculatory alterations in traumatic hemorrhagic shock. Crit Care Med. (2014) 42:1433–41. doi: 10.1097/CCM.0000000000000223
52. van Genderen ME, Lima A, Akkerhuis M, Bakker J, van Bommel J. Persistent peripheral and microcirculatory perfusion alterations after out-of-hospital cardiac arrest are associated with poor survival. Crit Care Med. (2012) 40:2287–94. doi: 10.1097/CCM.0b013e31825333b2
53. den Uil CA, Lagrand WK, van der Ent M, Jewbali LS, Cheng JM, Spronk PE, et al. Impaired microcirculation predicts poor outcome of patients with acute myocardial infarction complicated by cardiogenic shock. Eur Heart J. (2010) 31:3032–9. doi: 10.1093/eurheartj/ehq324
54. Wrenshall LE, Stevens RB, Cerra FB, Platt JL. Modulation of macrophage and B cell function by glycosaminoglycans. J Leukoc Biol. (1999) 66:391–400. doi: 10.1002/jlb.66.3.391
55. Akbarshahi H, Axelsson JB, Said K, Malmstrom A, Fischer H, Andersson R. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J Transl Med. (2011) 9:219. doi: 10.1186/1479-5876-9-219
56. Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. (2006) 177:1272–81. doi: 10.4049/jimmunol.177.2.1272
57. McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest. (1996) 98:2403–13. doi: 10.1172/JCI119054
58. Lenart M, Rutkowska-Zapala M, Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Smallie T, et al. Hyaluronan carried by tumor-derived microvesicles induces IL-10 production in classical (CD14++CD16-) monocytes via PI3K/Akt/mTOR-dependent signalling pathway. Immunobiology. (2017) 222:1–10. doi: 10.1016/j.imbio.2015.06.019
59. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. The J Exp Med. (2002) 195:99–111. doi: 10.1084/jem.20001858
60. Webb LM, Ehrengruber MU, Clark-Lewis I, Baggiolini M, Rot A. Binding to heparan sulfate or heparin enhances neutrophil responses to interleukin 8. Proc Natl Acad Sci USA. (1993) 90:7158–62. doi: 10.1073/pnas.90.15.7158
61. Kainulainen V, Wang H, Schick C, Bernfield M. Syndecans, heparan sulfate proteoglycans, maintain the proteolytic balance of acute wound fluids. J Biol Chem. (1998) 273:11563–9. doi: 10.1074/jbc.273.19.11563
62. Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M, et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem. (1999) 274:25085–92. doi: 10.1074/jbc.274.35.25085
63. Potter DR, Jiang J, Damiano ER. The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ Res. (2009) 104:1318–25. doi: 10.1161/CIRCRESAHA.108.191585
64. Wang JB, Zhang YJ, Guan J, Zhou L, Sheng Y, Zhang Y, et al. Enhanced syndecan-1 expression on neutrophils in patients with type 2 diabetes mellitus. Acta Diabetol. (2012) 49:41–6. doi: 10.1007/s00592-011-0265-1
65. Dhodapkar MV, Sanderson RD. Syndecan-1 (CD 138) in myeloma and lymphoid malignancies: a multifunctional regulator of cell behavior within the tumor microenvironment. Leuk Lymph. (1999) 34:35–43. doi: 10.3109/10428199909083378
66. Yeaman C, Rapraeger AC. Post-transcriptional regulation of syndecan-1 expression by cAMP in peritoneal macrophages. J Cell Biol. (1993) 122:941–50. doi: 10.1083/jcb.122.4.941
67. Bartlett AH, Hayashida K, Park PW. Molecular and cellular mechanisms of syndecans in tissue injury and inflammation. Mol Cells. (2007) 24:153–66.
68. Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. (2005) 96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8
69. Townsley MI, Reed RK, Ishibashi M, Parker JC, Laurent TC, Taylor AE. Hyaluronan efflux from canine lung with increased hydrostatic pressure and saline loading. Am J Respir Crit Care Med. (1994) 150(Pt. 1):1605–11. doi: 10.1164/ajrccm.150.6.7952622
70. Reed RK, Townsley MI, Zhao Z, Ishibashi M, Laurent TC, Taylor AE. Lymphatic hyaluronan flux from skin increases during increased lymph flow induced by intravenous saline loading. Int J Microcirc Clin Exp. (1994) 14:56–61. doi: 10.1159/000178207
71. Ostgaard G, Reed RK. Intravenous saline infusion in rat increases hyaluronan efflux in intestinal lymph by increasing lymph flow. Acta Physiol Scand. (1993) 147:329–35. doi: 10.1111/j.1748-1716.1993.tb09506.x
72. Gallo R, Kim C, Kokenyesi R, Scott Adzick N, Bernfield M. Syndecan-1 and−4 are induced during wound repair of neonatal but not fetal skin. J Invest Dermatol. (1996) 107:676–83.
73. Rops AL, van den Hoven MJ, Baselmans MM, Lensen JF, Wijnhoven TJ, van den Heuvel LP, et al. Heparan sulfate domains on cultured activated glomerular endothelial cells mediate leukocyte trafficking. Kidney Int. (2008) 73:52–62. doi: 10.1038/sj.ki.5002573
74. Subramanian SV, Fitzgerald ML, Bernfield M. Regulated shedding of syndecan-1 and−4 ectodomains by thrombin and growth factor receptor activation. J Biol Chem. (1997) 272:14713–20. doi: 10.1074/jbc.272.23.14713
75. Okuyama E, Suzuki A, Murata M, Ando Y, Kato I, Takagi Y, et al. Molecular mechanisms of syndecan-4 upregulation by TNF-alpha in the endothelium-like EAhy926 cells. J Biochem. (2013) 154:41–50. doi: 10.1093/jb/mvt024
76. Ramnath R, Foster RR, Qiu Y, Cope G, Butler MJ, Salmon AH, et al. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor alpha: a contributor to endothelial cell glycocalyx dysfunction. FASEB J. (2014) 28:4686–99. doi: 10.1096/fj.14-252221
77. Strand ME, Herum KM, Rana ZA, Skrbic B, Askevold ET, Dahl CP, et al. Innate immune signaling induces expression and shedding of the heparan sulfate proteoglycan syndecan-4 in cardiac fibroblasts and myocytes, affecting inflammation in the pressure-overloaded heart. FEBS J. (2013) 280:2228–47. doi: 10.1111/febs.12161
78. Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, et al. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest. (2001) 107:R9–14. doi: 10.1172/JCI10559
79. Fitzgerald ML, Wang Z, Park PW, Murphy G, Bernfield M. Shedding of syndecan-1 and−4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J Cell Biol. (2000) 148:811–24. doi: 10.1083/jcb.148.4.811
80. Zhang Y, Pasparakis M, Kollias G, Simons M. Myocyte-dependent regulation of endothelial cell syndecan-4 expression. Role of TNF-alpha. J Biol Chem. (1999) 274:14786–90. doi: 10.1074/jbc.274.21.14786
81. Klein NJ, Shennan GI, Heyderman RS, Levin M. Alteration in glycosaminoglycan metabolism and surface charge on human umbilical vein endothelial cells induced by cytokines, endotoxin and neutrophils. J Cell Sci. (1992) 102 (Pt. 4):821–32.
82. Mohamadzadeh M, DeGrendele H, Arizpe H, Estess P, Siegelman M. Proinflammatory stimuli regulate endothelial hyaluronan expression and CD44/HA-dependent primary adhesion. J Clin Invest. (1998) 101:97–108. doi: 10.1172/JCI1604
83. Rouschop KM, Roelofs JJ, Claessen N, da Costa Martins P, Zwaginga JJ, Pals ST, et al. Protection against renal ischemia reperfusion injury by CD44 disruption. J Am Soc Nephrol. (2005) 16:2034–43. doi: 10.1681/ASN.2005010054
84. Smart L, Macdonald SPJ, Burrows S, Bosio E, Arendts G, Fatovich DM. Endothelial glcyocalyx biomarkers increase in patients with infection during Emergency Department treatment. J Crit Care. (2017) 42:304–9. doi: 10.1016/j.jcrc.2017.07.001
85. Smart L, Bosio E, Macdonald SPJ, Dull R, Fatovich DM, Neil C, et al. Glycocalyx biomarker syndecan-1 is a stronger predictor of respiratory failure in patients with sepsis due to pneumonia, compared to endocan. J Crit Care. (2018) 47:93–8. doi: 10.1016/j.jcrc.2018.06.015
86. Smart L, Macdonald SPJ, Bosio E, Fatovich D, Neil C, Arendts G. Bolus therapy with 3% hypertonic saline or 0.9% saline in emergency department patients with suspected sepsis: a pilot randomised controlled trial. J Crit Care. (2019) 52:33–9. doi: 10.1016/j.jcrc.2019.03.009
87. Ebong EE, Macaluso FP, Spray DC, Tarbell JM. Imaging the endothelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy. Arterioscler Thromb Vasc Biol. (2011) 31:1908–15. doi: 10.1161/ATVBAHA.111.225268
88. Bruegger D, Jacob M, Rehm M, Loetsch M, Welsch U, Conzen P, et al. Atrial natriuretic peptide induces shedding of endothelial glycocalyx in coronary vascular bed of guinea pig hearts. Am J Physiol Heart Circ Physiol. (2005) 289:H1993–9. doi: 10.1152/ajpheart.00218.2005
89. Chappell D, Jacob M, Paul O, Rehm M, Welsch U, Stoeckelhuber M, et al. The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture. Circ Res. (2009) 104:1313–7. doi: 10.1161/CIRCRESAHA.108.187831
90. Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, et al. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol. (2009) 104:78–89. doi: 10.1007/s00395-008-0749-5
91. Xia Y, Fu BM. Investigation of endothelial surface glycocalyx components and ultrastructure by single molecule localization microscopy: stochastic optical reconstruction microscopy (STORM). Yale J Biol Med. (2018) 91:257–66.
92. Mukai S, Takaki T, Nagumo T, Sano M, Kang D, Takimoto M, et al. Three-dimensional electron microscopy for endothelial glycocalyx observation using Alcian blue with silver enhancement. Med Mol Morphol. (2020). doi: 10.1007/s00795-020-00267-1. [Epub ahead of print].
93. van den Berg BM, Spaan JA, Vink H. Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflugers Archiv. (2009) 457:1199–206. doi: 10.1007/s00424-008-0590-6
94. Long DS, Smith ML, Pries AR, Ley K, Damiano ER. Microviscometry reveals reduced blood viscosity and altered shear rate and shear stress profiles in microvessels after hemodilution. Proc Natl Acad Sci USA. (2004) 101:10060–5. doi: 10.1073/pnas.0402937101
95. Vink H, Duling BR. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ Res. (1996) 79:581–9. doi: 10.1161/01.RES.79.3.581
96. Kataoka H, Ushiyama A, Kawakami H, Akimoto Y, Matsubara S, Iijima T. Fluorescent imaging of endothelial glycocalyx layer with wheat germ agglutinin using intravital microscopy. Microsc Res Tech. (2016) 79:31–7. doi: 10.1002/jemt.22602
97. Donati A, Damiani E, Domizi R, Romano R, Adrario E, Pelaia P, et al. Alteration of the sublingual microvascular glycocalyx in critically ill patients. Microvasc Res. (2013) 90:86–9. doi: 10.1016/j.mvr.2013.08.007
98. Rovas A, Lukasz AH, Vink H, Urban M, Sackarnd J, Pavenstädt H, et al. Bedside analysis of the sublingual microvascular glycocalyx in the emergency room and intensive care unit - the GlycoNurse study. Scand J Trauma Resusc Emerg Med. (2018) 26:16. doi: 10.1186/s13049-018-0483-4
99. Cerny V, Astapenko D, Burkovskiy I, Hyspler R, Ticha A, Trevors MA, et al. Glycocalyx in vivo measurement. Clin Hemorheol Microcirc. (2017) 67:499–503. doi: 10.3233/CH-179235
100. Gommeren K, Allerton FJ, Morin E, Reynaud A, Peeters D, Silverstein DC. Evaluation of a rapid bedside scoring system for microcirculation videos acquired from dogs. J Vet Emerg Crit Care. (2014) 24:554–61. doi: 10.1111/vec.12212
101. Henry CB, Duling BR. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. (2000) 279:H2815–23. doi: 10.1152/ajpheart.2000.279.6.H2815
102. Kurzelewski M, Czarnowska E, Beresewicz A. Superoxide- and nitric oxide-derived species mediate endothelial dysfunction, endothelial glycocalyx disruption, and enhanced neutrophil adhesion in the post-ischemic guinea-pig heart. J Physiol Pharmacol. (2005) 56:163–78.
103. Chappell D, Jacob M, Hofmann-Kiefer K, Rehm M, Welsch U, Conzen P, et al. Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc Res. (2009) 83:388–96. doi: 10.1093/cvr/cvp097
104. Singh A, Ramnath RD, Foster RR, Wylie EC, Friden V, Dasgupta I, et al. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS ONE. (2013) 8:e55852. doi: 10.1371/journal.pone.0055852
105. Schmidt A, Echtermeyer F, Alozie A, Brands K, Buddecke E. Plasmin- and thrombin-accelerated shedding of syndecan-4 ectodomain generates cleavage sites at Lys(114)-Arg(115) and Lys(129)-Val(130) bonds. J Biol Chem. (2005) 280:34441–6. doi: 10.1074/jbc.M501903200
106. Strand ME, Aronsen JM, Braathen B, Sjaastad I, Kvaloy H, Tonnessen T, et al. Shedding of syndecan-4 promotes immune cell recruitment and mitigates cardiac dysfunction after lipopolysaccharide challenge in mice. J Mol Cell Cardiol. (2015) 88:133–44. doi: 10.1016/j.yjmcc.2015.10.003
107. Colburn P, Kobayashi E, Buonassisi V. Depleted level of heparan sulfate proteoglycan in the extracellular matrix of endothelial cell cultures exposed to endotoxin. J Cell Physiol. (1994) 159:121–30. doi: 10.1002/jcp.1041590116
108. Mulivor AW, Lipowsky HH. Inhibition of glycan shedding and leukocyte-endothelial adhesion in postcapillary venules by suppression of matrixmetalloprotease activity with doxycycline. Microcirculation. (2009) 16:657–66. doi: 10.3109/10739680903133714
109. Kozar RA, Peng Z, Zhang R, Holcomb JB, Pati S, Park P, et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. (2011) 112:1289–95. doi: 10.1213/ANE.0b013e318210385c
110. Peng Z, Pati S, Potter D, Brown R, Holcomb JB, Grill R, et al. Fresh frozen plasma lessens pulmonary endothelial inflammation and hyperpermeability after hemorrhagic shock and is associated with loss of syndecan 1. Shock. (2013) 40:195–202. doi: 10.1097/SHK.0b013e31829f91fc
111. Smart L, Boyd CJ, Claus MA, Bosio E, Hosgood G, Raisis A. Large-volume crystalloid fluid is associated with increased hyaluronan shedding and inflammation in a canine hemorrhagic shock model. Inflammation. (2018) 41:1515–23. doi: 10.1007/s10753-018-0797-4
112. Marechal X, Favory R, Joulin O, Montaigne D, Hassoun S, Decoster B, et al. Endothelial glycocalyx damage during endotoxemia coincides with microcirculatory dysfunction and vascular oxidative stress. Shock. (2008) 29:572–6. doi: 10.1097/SHK.0b013e318157e926
113. Naumann DN, Hazeldine J, Davies DJ, Bishop J, Midwinter MJ, Belli A, et al. Endotheliopathy of trauma is an on-scene phenomenon, and is associated with multiple organ dysfunction syndrome: a prospective observational study. Shock. (2018) 49:420–8. doi: 10.1097/SHK.0000000000000999
114. Tapking C, Hernekamp JF, Horter J, Kneser U, Haug V, Vogelpohl J, et al. Influence of burn severity on endothelial glycocalyx shedding following thermal trauma: a prospective observational study. Burns. (2020). doi: 10.1016/j.burns.2020.07.021. [Epub ahead of print].
115. Hippensteel JA, Uchimido R, Tyler PD, Burke RC, Han X, Zhang F, et al. Intravenous fluid resuscitation is associated with septic endothelial glycocalyx degradation. Crit Care. (2019) 23:259. doi: 10.1186/s13054-019-2534-2
116. Pries AR, Secomb TW, Sperandio M, Gaehtgens P. Blood flow resistance during hemodilution: effect of plasma composition. Cardiovasc Res. (1998) 37:225–35. doi: 10.1016/S0008-6363(97)00226-5
117. Chappell D, Jacob M, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, et al. Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol Chem. (2008) 389:79–82. doi: 10.1515/BC.2008.005
118. Huxley VH, Curry FE. Albumin modulation of capillary permeability: test of an adsorption mechanism. Am J Physiol. (1985) 248(Pt. 2):H264–73. doi: 10.1152/ajpheart.1985.248.2.H264
119. Huxley VH, Curry FE. Differential actions of albumin and plasma on capillary solute permeability. Am J Physiol. (1991) 260(Pt. 2):H1645–54. doi: 10.1152/ajpheart.1991.260.5.H1645
120. Torres LN, Sondeen JL, Dubick MA, Filho IT. Systemic and microvascular effects of resuscitation with blood products after severe hemorrhage in rats. J Trauma Acute Care Surg. (2014) 77:716–23. doi: 10.1097/TA.0000000000000448
121. D'Souza SP, Davis M, Baxter GF. Autocrine and paracrine actions of natriuretic peptides in the heart. Pharmacol Ther. (2004) 101:113–29. doi: 10.1016/j.pharmthera.2003.11.001
122. Chen W, Gassner B, Borner S, Nikolaev VO, Schlegel N, Waschke J, et al. Atrial natriuretic peptide enhances microvascular albumin permeability by the caveolae-mediated transcellular pathway. Cardiovasc Res. (2012) 93:141–51. doi: 10.1093/cvr/cvr279
123. Zimmerman RS, Trippodo NC, MacPhee AA, Martinez AJ, Barbee RW. High-dose atrial natriuretic factor enhances albumin escape from the systemic but not the pulmonary circulation. Circ Res. (1990) 67:461–8. doi: 10.1161/01.RES.67.2.461
124. Jacob M, Saller T, Chappell D, Rehm M, Welsch U, Becker BF. Physiological levels of A-, B- and C-type natriuretic peptide shed the endothelial glycocalyx and enhance vascular permeability. Basic Res Cardiol. (2013) 108:347. doi: 10.1007/s00395-013-0347-z
125. Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, et al. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care. (2014) 18:538. doi: 10.1186/s13054-014-0538-5
126. Belavić M, Sotošek TokmadŽić V, Fišić E, Brozović Krijan A, Strikić N, Lončarić Katušin M, et al. The effect of various doses of infusion solutions on the endothelial glycocalyx layer in laparoscopic cholecystectomy patients. Minerva Anestesiol. (2018) 84:1032–43. doi: 10.23736/S0375-9393.18.12150-X
127. Bruegger D, Schwartz L, Chappell D, Jacob M, Rehm M, Vogeser M, et al. Release of atrial natriuretic peptide precedes shedding of the endothelial glycocalyx equally in patients undergoing on- and off-pump coronary artery bypass surgery. Basic Res Cardiol. (2011) 106:1111–21. doi: 10.1007/s00395-011-0203-y
128. Nemme J, Krizhanovskii C, Ntika S, Sabelnikovs O, Vanags I, Hahn RG. Hypervolemia does not cause degradation of the endothelial glycocalyx layer during open hysterectomy performed under sevoflurane or propofol anesthesia. Acta Anaesthesiol Scand. (2020) 64:538–45. doi: 10.1111/aas.13511
129. Hasselgren E, Zdolsek M, Zdolsek JH, Björne H, Krizhanovskii C, Ntika S, et al. Long intravascular persistence of 20% albumin in postoperative patients. Anesth Analg. (2019) 129:1232–9. doi: 10.1213/ANE.0000000000004047
130. Torres Filho IP, Torres LN, Salgado C, Dubick MA. Plasma syndecan-1 and heparan sulfate correlate with microvascular glycocalyx degradation in hemorrhaged rats after different resuscitation fluids. Am J Physiol Heart Circ Physiol. (2016) 310:H1468–78. doi: 10.1152/ajpheart.00006.2016
131. Torres LN, Sondeen JL, Ji L, Dubick MA, Torres Filho I. Evaluation of resuscitation fluids on endothelial glycocalyx, venular blood flow, and coagulation function after hemorrhagic shock in rats. J Trauma Acute Care Surg. (2013) 75:759–66. doi: 10.1097/TA.0b013e3182a92514
132. Torres LN, Chung KK, Salgado CL, Dubick MA, Torres Filho IP. Low-volume resuscitation with normal saline is associated with microvascular endothelial dysfunction after hemorrhage in rats, compared to colloids and balanced crystalloids. Crit Care. (2017) 21:160. doi: 10.1186/s13054-017-1745-7
133. Nelson A, Statkevicius S, Schott U, Johansson PI, Bentzer P. Effects of fresh frozen plasma, Ringer's acetate and albumin on plasma volume and on circulating glycocalyx components following haemorrhagic shock in rats. Intensive Care Med Exp. (2016) 4:6. doi: 10.1186/s40635-016-0080-7
134. Aksu U, Bezemer R, Yavuz B, Kandil A, Demirci C, Ince C. Balanced vs unbalanced crystalloid resuscitation in a near-fatal model of hemorrhagic shock and the effects on renal oxygenation, oxidative stress, and inflammation. Resuscitation. (2012) 83:767–73. doi: 10.1016/j.resuscitation.2011.11.022
135. Sillesen M, Rasmussen LS, Jin G, Jepsen CH, Imam A, Hwabejire JO, et al. Assessment of coagulopathy, endothelial injury, and inflammation after traumatic brain injury and hemorrhage in a porcine model. J Trauma Acute Care Surg. (2014) 76:12–9; discussion 9–20. doi: 10.1097/TA.0b013e3182aaa675
136. Annecke T, Rehm M, Bruegger D, Kubitz JC, Kemming GI, Stoeckelhuber M, et al. Ischemia-reperfusion-induced unmeasured anion generation and glycocalyx shedding: sevoflurane versus propofol anesthesia. J Invest Surg. (2012) 25:162–8. doi: 10.3109/08941939.2011.618524
137. Byrne L, Obonyo NG, Diab SD, Dunster KR, Passmore MR, Boon AC, et al. Unintended consequences: fluid resuscitation worsens shock in an ovine model of endotoxemia. Am J Respir Crit Care Med. (2018) 198:1043–54. doi: 10.1164/rccm.201801-0064OC
138. Hofbauer R, Moser D, Hornykewycz S, Frass M, Kapiotis S. Hydroxyethyl starch reduces the chemotaxis of white cells through endothelial cell monolayers. Transfusion. (1999) 39:289–94. doi: 10.1046/j.1537-2995.1999.39399219286.x
139. Wang P, Li Y, Li J. Hydroxyethyl starch 130/0.4 prevents the early pulmonary inflammatory response and oxidative stress after hemorrhagic shock and resuscitation in rats. Int Immunopharmacol. (2009) 9:347–53. doi: 10.1016/j.intimp.2008.12.014
140. Pascual JL, Ferri LE, Chaudhury P, Seely AJ, Campisi G, Giannias B, et al. Hemorrhagic shock resuscitation with a low molecular weight starch reduces neutrophil-endothelial interactions and vessel leakage in vivo. Surg Infect. (2001) 2:275–87; discussion 87–8. doi: 10.1089/10962960152813313
141. Seo EH, Park HJ, Piao LY, Lee JY, Oh CS, Kim SH. Immune response in fluid therapy with crystalloids of different ratios or colloid for rats in haemorrhagic shock. Sci Rep. (2020) 10:8067. doi: 10.1038/s41598-020-65063-4
142. Ozturk T, Onur E, Cerrahoglu M, Calgan M, Nizamoglu F, Civi M. Immune and inflammatory role of hydroxyethyl starch 130/0.4 and fluid gelatin in patients undergoing coronary surgery. Cytokine. (2015) 74:69–75. doi: 10.1016/j.cyto.2014.10.002
143. Thiel M, Buessecker F, Eberhardt K, Chouker A, Setzer F, Kreimeier U, et al. Effects of hypertonic saline on expression of human polymorphonuclear leukocyte adhesion molecules. J Leuk Biol. (2001) 70:261–73. doi: 10.1189/jlb.70.2.261
144. Ciesla DJ, Moore EE, Musters RJ, Biffl WL, Silliman CA. Hypertonic saline alteration of the PMN cytoskeleton: implications for signal transduction and the cytotoxic response. J Trauma. (2001) 50:206–12. doi: 10.1097/00005373-200102000-00004
145. Banerjee A, Moore EE, McLaughlin NJ, Lee L, Jones WL, Johnson JL, et al. Hyperosmolarity attenuates TNF-alpha-mediated proinflammatory activation of human pulmonary microvascular endothelial cells. Shock. (2013) 39:366–72. doi: 10.1097/SHK.0b013e3182894016
146. Junger WG, Hoyt DB, Davis RE, Herdon-Remelius C, Namiki S, Junger H, et al. Hypertonicity regulates the function of human neutrophils by modulating chemoattractant receptor signaling and activating mitogen-activated protein kinase p38. J Clin Invest. (1998) 101:2768–79. doi: 10.1172/JCI1354
147. Rizoli SB, Kapus A, Fan J, Li YH, Marshall JC, Rotstein OD. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol. (1998) 161:6288–96.
148. Ciesla DJ, Moore EE, Zallen G, Biffl WL, Silliman CC. Hypertonic saline attenuation of polymorphonuclear neutrophil cytotoxicity: timing is everything. J Trauma. (2000) 48:388–95. doi: 10.1097/00005373-200003000-00004
149. Angle N, Hoyt DB, Cabello-Passini R, Herdon-Remelius C, Loomis W, Junger WG. Hypertonic saline resuscitation reduces neutrophil margination by suppressing neutrophil L selectin expression. J Trauma. (1998) 45:7–12.
150. Gonzalez RJ, Moore EE, Ciesla DJ, Neto JR, Biffl WL, Silliman CC. Hyperosmolarity abrogates neutrophil cytotoxicity provoked by post-shock mesenteric lymph. Shock. (2002) 18:29–32. doi: 10.1097/00024382-200207000-00006
151. Pascual JL, Ferri LE, Seely AJ, Campisi G, Chaudhury P, Giannias B, et al. Hypertonic saline resuscitation of hemorrhagic shock diminishes neutrophil rolling and adherence to endothelium and reduces in vivo vascular leakage. Ann Surg. (2002) 236:634–42. doi: 10.1097/00000658-200211000-00014
152. Theobaldo MC, Barbeiro HV, Barbeiro DF, Petroni R, Soriano FG. Hypertonic saline solution reduces the inflammatory response in endotoxemic rats. Clinics. (2012) 67:1463–8. doi: 10.6061/clinics/2012(12)18
153. Petroni RC, Biselli PJ, de Lima TM, Theobaldo MC, Caldini ET, Pimentel RN, et al. Hypertonic saline (NaCl 7.5%) reduces LPS-induced acute lung injury in rats. Inflammation. (2015) 38:2026–35. doi: 10.1007/s10753-015-0183-4
154. Yu G, Chi X, Hei Z, Shen N, Chen J, Zhang W, et al. Small volume resuscitation with 7.5% hypertonic saline, hydroxyethyl starch 130/0.4 solution and hypertonic sodium chloride hydroxyethyl starch 40 injection reduced lung injury in endotoxin shock rats: comparison with saline. Pulm Pharmacol Ther. (2012) 25:27–32. doi: 10.1016/j.pupt.2011.10.003
155. Tan GK, Ng JK, Tan KW, Angeli V, Moochhala S, Ooi EE, et al. Hypertonic saline reduces vascular leakage in a mouse model of severe dengue. PloS ONE. (2013) 8:e61621. doi: 10.1371/journal.pone.0061621
156. Sun YX, Wu XS, Gao Z, Wang F, Liu S, Chen XL. Effect of 200 mEq/L Na+ hypertonic saline resuscitation on systemic inflammatory response and oxidative stress in severely burned rats. J Surg Res. (2013) 185:477–84. doi: 10.1016/j.jss.2013.06.039
157. Junger WG, Rhind SG, Rizoli SB, Cuschieri J, Shiu MY, Baker AJ, et al. Resuscitation of traumatic hemorrhagic shock patients with hypertonic saline-without dextran-inhibits neutrophil and endothelial cell activation. Shock. (2012) 38:341–50. doi: 10.1097/SHK.0b013e3182635aca
158. Rhind SG, Crnko NT, Baker AJ, Morrison LJ, Shek PN, Scarpelini S, et al. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J Neuroinflammation. (2010) 7:5. doi: 10.1186/1742-2094-7-5
159. de Laforcade A, Silverstein DC. Shock. In: Silverstein DC, Hopper K, editors. Small Animal Critical Care Medicine. 2nd ed. St. Louis, MI: Elsevier Saunders (2015). p. 26–30.
160. Boyd CJ, Smart L. Hypovolemic shock. In: Drobatz KJ, Hopper K, Rozanski E, Silverstein DC, editors. Textbook of Small Animal Emergency Medicine. 2nd ed. Hoboken, NJ: John Wiley & Sons (2018). p. 986–92.
161. Marik PE, Byrne L, van Haren F. Fluid resuscitation in sepsis: the great 30 mL per kg hoax. J Thor Dis. (2020) 12(Suppl. 1):S37–47. doi: 10.21037/jtd.2019.12.84
162. Macdonald SPJ, Taylor DM, Keijzers G, Arendts G, Fatovich DM, Kinnear FB, et al. REstricted Fluid REsuscitation in Sepsis-associated Hypotension (REFRESH): study protocol for a pilot randomised controlled trial. Trials. (2017) 18:399. doi: 10.1186/s13063-017-2137-7
163. Macdonald SPJ, Keijzers G, Taylor DM, Kinnear F, Arendts G, Fatovich DM, et al. Restricted fluid resuscitation in suspected sepsis associated hypotension (REFRESH): a pilot randomised controlled trial. Intensive Care Med. (2018) 44:2070–8. doi: 10.1007/s00134-018-5433-0
164. Self WH, Semler MW, Bellomo R, Brown SM, deBoisblanc BP, Exline MC, et al. Liberal versus restrictive intravenous fluid therapy for early septic shock: rationale for a randomized trial. Ann Emerg Med. (2018) 72:457–66. doi: 10.1016/j.annemergmed.2018.03.039
165. Bickell WH, Wall MJ Jr., Pepe PE, Martin RR, Ginger VF, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. New Eng J Med. (1994) 331:1105–9. doi: 10.1056/NEJM199410273311701
166. Mapstone J, Roberts I, Evans P. Fluid resuscitation strategies: a systematic review of animal trials. J Trauma. (2003) 55:571–89. doi: 10.1097/01.TA.0000062968.69867.6F
167. Safiejko K, Smereka J, Filipiak KJ, Szarpak A, Dabrowski M, Ladny JR, et al. Effectiveness and safety of hypotension fluid resuscitation in traumatic hemorrhagic shock: a systematic review and meta-analysis of randomized controlled trials. Cardiol J. (2020). doi: 10.5603/CJ.a2020.0096. [Epub ahead of print].
168. Wang CH, Hsieh WH, Chou HC, Huang YS, Shen JH, Yeo YH, et al. Liberal versus restricted fluid resuscitation strategies in trauma patients: a systematic review and meta-analysis of randomized controlled trials and observational studies*. Crit Care Med. (2014) 42:954–61. doi: 10.1097/CCM.0000000000000050
169. Kwan I, Bunn F, Chinnock P, Roberts I. Timing and volume of fluid administration for patients with bleeding. Cochrane Database Syst Rev. (2014) 2014:Cd002245. doi: 10.1002/14651858.CD002245.pub2
170. Myles PS, Bellomo R, Corcoran T, Forbes A, Peyton P, Story D, et al. Restrictive versus liberal fluid therapy for major abdominal surgery. New Eng J Med. (2018) 378:2263–74. doi: 10.1056/NEJMoa1801601
171. Chow RS. Terms, definitions, nomenclature and routes of fluid administration. Front Vet Sci. (2021) 7:591218. doi: 10.3389/fvets.2020.591218
172. Turek Z, Cerny V, Parizkova R, Samek J, Oberreiter M. Prolonged hypervolemic hemodilution decreases functional capillary density of ileal mucosa in pigs revealed by sidestream dark-field imaging. J Zhejiang Univ Sci B. (2011) 12:867–74. doi: 10.1631/jzus.B1000385
173. Yu TC, Yang FL, Hsu BG, Wu WT, Chen SC, Lee RP, et al. Deleterious effects of aggressive rapid crystalloid resuscitation on treatment of hyperinflammatory response and lung injury induced by hemorrhage in aging rats. J Surg Res. (2014) 187:587–95. doi: 10.1016/j.jss.2013.10.061
174. Iro MA, Sell T, Brown N, Maitland K. Rapid intravenous rehydration of children with acute gastroenteritis and dehydration: a systematic review and meta-analysis. BMC Pediatr. (2018) 18:44. doi: 10.1186/s12887-018-1006-1
175. Gaudette S, Hughes D, Boller M. The endothelial glycocalyx: structure and function in health and critical illness. J Vet Emerg Crit Care. (2020) 30:117–34. doi: 10.1111/vec.12925
176. Weinbaum S, Cancel LM, Fu BM, Tarbell JM. The glycocalyx and its role in vascular physiology and vascular related diseases. Cardiovasc Eng Technol. (2020) 12:37–71. doi: 10.1007/s13239-020-00485-9
Keywords: endothelium, glycocalyx, shock, fluid therapy, crystalloid, colloid, endothelial surface layer
Citation: Smart L and Hughes D (2021) The Effects of Resuscitative Fluid Therapy on the Endothelial Surface Layer. Front. Vet. Sci. 8:661660. doi: 10.3389/fvets.2021.661660
Received: 31 January 2021; Accepted: 16 March 2021;
Published: 07 May 2021.
Edited by:
Pablo Martín-Vasallo, University of La Laguna, SpainReviewed by:
Ivo P. Torres Filho, United States Army Institute of Surgical Research, United StatesCopyright © 2021 Smart and Hughes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Smart, bC5zbWFydEBtdXJkb2NoLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.