 Ahmad Nadzri Nur-Farahiyah1
Ahmad Nadzri Nur-Farahiyah1 Kiven Kumar2
Kiven Kumar2 Abd Rahaman Yasmin1,3*
Abd Rahaman Yasmin1,3* Abdul Rahman Omar3,4Siti Nazrina Camalxaman5
Abdul Rahman Omar3,4Siti Nazrina Camalxaman5- 1Department of Veterinary Laboratory Diagnosis, Faculty of Veterinary Medicine, Universiti Putra Malaysia, UPM Serdang, Selangor, Malaysia
- 2Department of Pathology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, UPM Serdang, Selangor, Malaysia
- 3Laboratory of Vaccines and Biomolecules, Institute of Bioscience, Universiti Putra Malaysia, UPM Serdang, Selangor, Malaysia
- 4Department of Veterinary Pathology and Microbiology, Faculty of Veterinary Medicine, Universiti Putra Malaysia, UPM Serdang, Selangor, Malaysia
- 5Department of Medical Laboratory Technology, Faculty of Health Sciences, Universiti Teknologi MARA (UiTM), Selangor, Malaysia
Naïve Felidae in the wild may harbor infectious viruses of importance due to cross-species transmission between the domesticated animals or human–wildlife contact. However, limited information is available on virus shedding or viremia in the captive wild felids, especially in Malaysia. Four infectious viruses of cat, feline herpesvirus (FHV), feline calicivirus (FCV), canine distemper virus (CDV), and canine parvovirus (CPV), were screened in leopards, feral cats, and tigers in Malaysia based on virus isolation in Crandell-Rees feline kidney (CRFK) cells, PCR/RT-PCR, and whole-genome sequencing analysis of the positive isolate. From a total of 36 sera collected, 11 samples showed three consecutive cytopathic effects in the cell culture and were subjected to PCR using specific primers for FHV, FCV, CDV, and CPV. Only one sample from a Malayan tiger was detected positive for CPV. The entire viral genome of CPV (UPM-CPV15/P. tigris jacksoni; GenBank Accession number MW380384) was amplified using the Sanger sequencing approach. Genome sequencing of the isolate revealed 99.13, 98.65, and 98.40% close similarity to CPV-31, CPV-d Cornell #320, and CPV-15 strains, respectively, and classified as CPV-2a. Time-scaled Bayesian Maximum Clade Credibility tree for the non-structural (NS) genes of CPV showed a close relationship to the isolates CPV-CN SD6_2014 and KSU7-SD_2004 from China and USA, respectively, while the capsid gene showed the same ancestor as the FPV-BJ04 strain from China. The higher evolution rate of the capsid protein (CP) (VP 1 and VP2) [1.649 × 10−5 (95% HPD: 7.626 × 10−3 to 7.440 × 10−3)] as compared to the NS gene [1.203 × 10−4 (95% HPD: 6.663 × 10−3 to 6.593 × 10−3)] was observed in the CPV from this study, and fairly higher than other parvovirus species from the Protoparvovirus genus. Genome sequencing of the isolated CPV from a Malayan tiger in the present study provides valuable information about the genomic characteristics of captive wild felids, which may add information on the presence of CPV in species other than dogs.
Introduction
Parvoviridae is a family of non-enveloped and single-stranded DNA virus with a 4–6 kb linear genome. It derives its name from the Latin word “parvus,” which means small and widely dispersed in nature (1). Parvoviridae family is divided into three subfamilies, namely, Parvovirinae (vertebrate viruses), Densovirinae (invertebrate viruses), and Hamaparvovirinae (found in insects). Members of Parvoviridae consist of parvovirus that can infect a diverse range of mammalian, avian, and reptilian hosts. The subfamily Parvovirinae are categorized into 10 recognized genera, namely, Amdoparvovirus, Artiparvovirus, Aveparvovirus, Bocaparvovirus, Copiparvovirus, Dependoparvovirus, Erythroparvovirus, Loriparvovirus, Protoparvovirus, and Tetraparvovirus (2). Among Protoparvovirus, examples of virus species are canine parvovirus (CPV) (known as carnivore protoparvovirus 1), feline panleukopenia virus (FPV) (known as carnivore protoparvovirus 1), porcine parvovirus (PPV) (known as ungulate parvovirus 1), rodent protoparvovirus, rodent protoparvovirus 2 (rat parvovirus 1), mink enteritis virus (MEV), and Kilham's rat virus (KRV) (3).
One such important veterinary pathogen known to infect carnivore species is the CPV, a pathogenic agent that causes severe haemorrhagic gastroenteritis in dogs. It emerged during the late 1970's in the canine population (4). Currently, there are three antigenic types of CPV-2 named CPV-2a,−2b, and−2c. The CPV-2 is considered to be derived from FPV of domestic cats (5). As a result of adaptation to feline transferrin receptor, CPV variants are able to infect not only canines but also felines (6, 7). Although, these antigenic variants occur negligibly, they remain important because they can infect cats (8–11), while remaining a threat to dogs. The CPV genome consists of two open reading frames (ORFs) that encode for non-structural proteins (NS1 and NS2) and structural proteins (VP1 and VP2) (12).
Currently, in Malaysia, abundant studies have been exploring the prevalence of common viral pathogens of domesticated cats and dogs, such as feline herpesvirus (FHV), feline calicivirus (FCV), canine distemper virus (CDV) and CPV (13, 14). However, there are limited data on the viremia status of those viruses in the captive wild felids. As felid species are also susceptible to viral infections, this study was performed to investigate the potentially important viruses in diverse species of felids, namely, leopards, feral cats, and tigers in Malaysia from archive sera samples. Consecutively, the study was also attempted to characterize the isolated virus via full-genome sequencing analysis.
Materials and Methods
Screening of Viruses
Sera from 36 unvaccinated various feline species (three Sunda clouded leopard, two Leopard cats, 27 feral cats, and four Malayan tiger) were obtained from archives of Veterinary Laboratory Service Unit (VLSU), Faculty of Veterinary Medicine, Universiti Putra Malaysia. Since archive samples were used in this study, animal ethical approval was not required. The samples were received between the year 2014 and 2015 from zoos located in Sabah, Selangor, and Pahang for diagnostic purposes and general screening, and a year later, the archived sera were used in this study. Briefly, 100 μl of each felid serum was seeded in the 75% confluent Crandell-Rees feline kidney (CRFK) (ATCC CCL94) monolayer cell line in 25-cm3 cell culture flasks (Nunc, Germany). Then, the samples were incubated at 37°C with 1 ml of growth medium for 1 h at 5% CO2. After an hour of incubation, the growth medium was decanted and replaced with PBS to wash the cells thrice. Following that, 5 ml of MEM (GibcoBRL, USA) supplemented with 1% FBS (GibcoBRL, USA) (maintenance media) was added. The inoculated cells were then incubated again in 37°C incubators supplemented with 5% CO2 and were monitored daily for any appearance of cytopathic effect (CPE) in three blind passages. Any serum inoculated cells with CPE were further harvested and subjected to viral genome extraction.
PCR and RT-PCR Analysis
Samples that showed consecutive CPE were subjected to viral DNA and RNA extraction using DNeasy Blood and Tissue Kit (Qiagen, Germany) and RNeasy Kit (Qiagen, Germany), respectively, according to the manufacturer's protocol. The extracted DNA was subjected to FHV and CPV PCR protocol, whereas, RNA was subjected to FCV and CDV RT-PCR protocol in a Mastercycler® gradient thermal cycler (Eppendorf, Germany). Information on the target genes, primer sequences used, positive control, PCR, and RT-PCR protocol, and expected amplicon size are shown in Supplementary Table 1 (15–18). Gel electrophoresis using 1.5% (w/v) agarose gel (Vivantis, USA) and 6× Loading Dye (Fermentas, USA) was conducted to view the amplification of positive reactions visualized by the presence of an amplicon fragment between target genes aligned with a positive control band using a UV transilluminator (Biorad, USA).
Complete Genome Sequence of CPV
Viral DNA of CPV was extracted from the viral isolate, and the entire ORF (including NS1 and VP2 genes for CPV) was amplified using four pairs of primers (Supplementary Table 2). Primers for the virus genome sequence were designed using Primer-BLAST along with its specificity information (https://www.ncbi.nlm.nih.gov/tools/primer-blast/), according to the reference of a full complete sequence of a CPV isolate obtained through GenBank (NC_001539).
Positive isolate was run by PCR using designated primers targeted for each region of the virus genome. Primer target regions for PCRs for the different gene segments were selected from the conserved regions of the respective aligned gene sequences. Single intact band observed using agarose gel electrophoresis was sent for Sanger sequencing (Apical Scientific Sdn. Bhd, Malaysia).
Phylogenetic Analysis
Eighty complete genome sequences of the viruses from the subfamily of Parvovirinae that represents eight different genera were included in this study (Supplementary Table 3). The sequences were aligned by using the online tool MAFFT version 7. The Neighbor-Joining tree was constructed with 1,000 replications using Mega-7 with Jukes-Cantor as substitution model. The Newick Export file was generated using MEGA-7, and the tree was viewed and edited using Interactive Tree of Life (iTOL) V5.
Estimation of Divergence
About 176 and 181 of CP and NS genes from PPV, FPV, and CPV were retrieved from GenBank (Supplementary Table 4). The divergence times for parvoviruses were identified by the Bayesian Markov chain Monte Carlo (MCMC) method as applied in BEAST2 (Version 2.6.2) (19). The investigation was done under the GTR + I + G substitution model for C and NS gene sequence and used an unrelaxed log-normal-distributed (Ucld) relaxed molecular clock. The MCMC run was at 3 × 107 steps long, with sampling at every 3,000 steps. Convergence was measured using Tracer software version 1.7.1 (available from http://beast.bio.ed.ac.uk/Tracer) by examining the Effective Sample Sizes (ESS > 200), and the 95% HDP values provide the degree of uncertainty in each parameter estimate. To reconstruct plausible ancestral states, all nodes in the MCC tree were annotated with posterior probability values using TreeAnnotator v.2.6.2, and the tree was visualized with FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree). The demographical history of CPV NS and capsid was estimated using the Bayesian skyline plot method applied in Tracer V1.7.1.
Selection Pressure Analysis
The selection pressure of NS and CP genes from CPV was used to estimate the distinction between the non-silent site (dN) and silent site (dS) substitution rates by using MEGA version 7.0 software (20). The parameter was set as follows: dN/dS ratio >0 is positive selection; dN/dS ratio <0 is negative selection; and dN/dS ratio equal to 0 is neutral evolution (21). This ratio will measure the selection pressures by equating the rate of substitutions at silent sites (dS), which are assumed neutral, to the rate of substitutions at non-silent sites (dN), which are probably involved in selection (22). The entropy was used to calculate the genetic density and determined by using BioEdit vision 7.2.5. The amino acid sequences of Protoparvovirus were compared with CPV capsid and NS genes.
Results
CPE and Virus Detection by PCR
As observed in Figure 1, the CRFK cells from a Malayan tiger showed formation of CPE evidenced by cytoplasmic extension and clusters formation. Samples that tested positive with three consecutive CPEs were tested by PCR using virus-specific primers to detect FHV, FCV, CDV, and CPV. Only one sample of Malayan tiger species was positive for CPV. A single DNA band of the expected size (583 bp), corresponding to the partial amplification of VP2 gene of the CPV, was detected following gel electrophoresis.
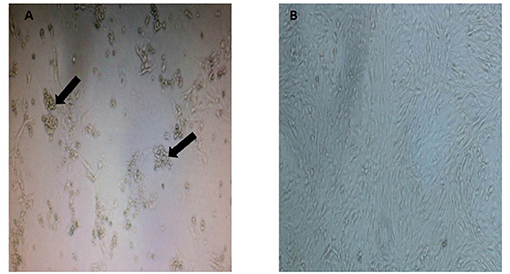
Figure 1. Appearance of cytopathic effect in CRFK cells in (A) a Malayan tiger (MT4), observed as clumping of cells with cluster formation and cytoplasmic extension (indicated by arrows at 8 d.p.i.) and (B) mock-infected control CRFK (magnification at 40×).
Full-Genome Sequencing of UPM-CPV15/P. Tigris Jacksoni
The positive isolate denoted as UPM-CPV15/P. tigris jacksoni was approximately 4,712 base pairs (bp) in size (GenBank Accession number MW380384). The complete genome sequence of isolate UPM-CPV15/P. tigris jacksoni was checked for specificity using BLAST (blast.ncbi.nlm.nih.gov/Blast.cgi). The specificity of isolate UPM-CPV15/P. tigris jacksoni was found to be highly similar to CPV-31 (GenBank accession no: M24000.1), CPV-d Cornell #320 (GenBank accession no: M23255.1), and CPV-15 (GenBank accession no: M24003.1) strains with 99.13, 98.65, and 98.40%, respectively, and classified as CPV-2a.
Bioinformatic Analysis of UPM-CPV15/P. Tigris Jacksoni
The phylogenetic tree was constructed for viruses from different genera of the subfamily Parvovirinae, including Amdoparvovirus, Aveparvovirus, Bocaparvovirus, Copiparvovirus, Dependoparvovirus, Erythroparvovirus, Protoparvovirus, and Tetraparvovirus, which were used to create a Neighbor-Joining phylogenetic tree. The complete genome of UPM-CPV15/P. tigris jacksoni shares the same branch with CPV, FPV, and MEV under the genus Protoparvovirus (Figure 2).
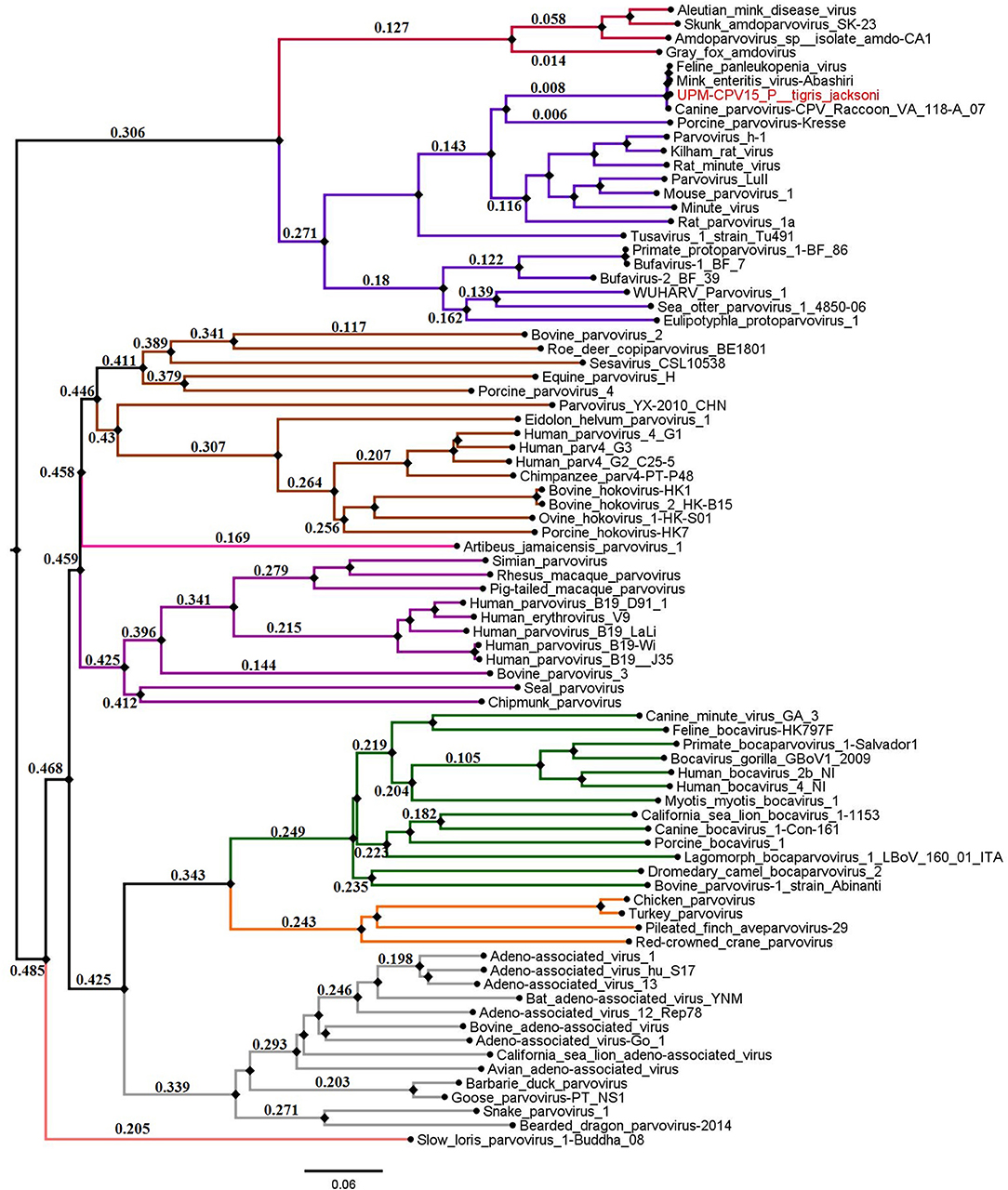
Figure 2. Neighbor joining tree of the subfamily Parvovirinae. The branches of the tree was colored according to the genera; Bocaparvovirus was colored forest green, Amdoparvovirus was colored carmine red, Protoparvovirus was colored electric violet, Aveparvovirus was colored orange, Dependoparvovirus was colored gray, unclassified was colored baby blue, Erythroparvovirus was colored Byzantium violet, and Tetraparvovirus was colored cinnamon brown. Local isolate strains UPM-CPV15/P. tigris jacksoni were highlighted in red.
Phylogenetic trees were constructed for CPV capsid protein CP (VP 1 and VP2) and NS gene using 223 and 201 sequences, respectively, from the 1970's to 2020, from countries in North and South America, Asia, and Europe (Figures 3, 4). The tree was scaled to time (years); the time to the most recent common ancestor (tMRCA) with its corresponding 95% highest posterior density (HPD) interval for CPV was estimated in the unit of years. The tMRCA tress of 11 and 25 major branches for NS and CP genes, respectively, as highlighted in different colors (Figures 3, 4). The NS gene of UPM-CPV15/P. tigris jacksoni shared the same branch with CPV8 Iran strain isolated in 2020 from Iran. Both UPM-CPV15/P. tigris jacksoni and CPV_8_Iran strains shared the same node with raccoon-related CPV, which is isolated in 2009 (Figure 3), while the tMRCA of NS and CP genes of CPV and FPV was predicted to originate in the late 1970's (95% HPD: 1973–1982) and (95% HPD: 1973–1979), respectively. The CP gene of UPM-CPV15/P. tigris jacksoni shared the same branch with NNGag isolated in 2012 from Argentina (Figure 4). Both NNGag and UPM-CPV15/P. tigris jacksoni shared the same node with CPV220, which was detected from dog in 1993 from USA. This clade consisted of the older clade (red star) of CPV before the year 2000.
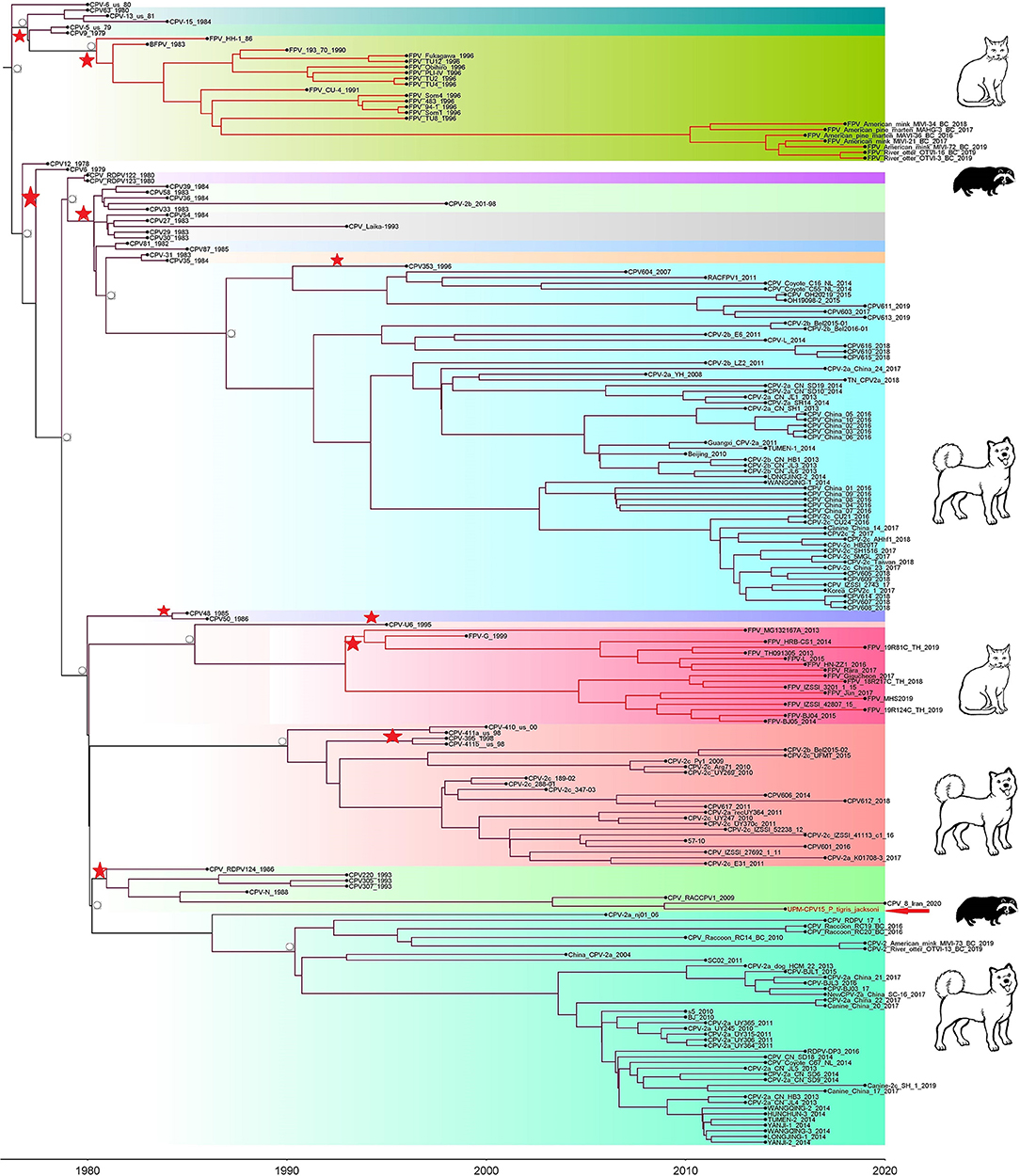
Figure 3. Time-scaled Bayesian Maximum Clade Credibility tree for the non-structure (NS) genes of porcine, canine, and feline panleukopenia viruses. The local isolate was highlighted in golden color. PPV, CPV, and FPV were highlighted in red, green, and blue color, respectively. Each branch was colored according to posterior probability (PP) value; blue bars at nodes indicate 95% highest probability density (HPD). Tree nodes with a posterior probability >0.8 was displayed at tree nodes with red color.
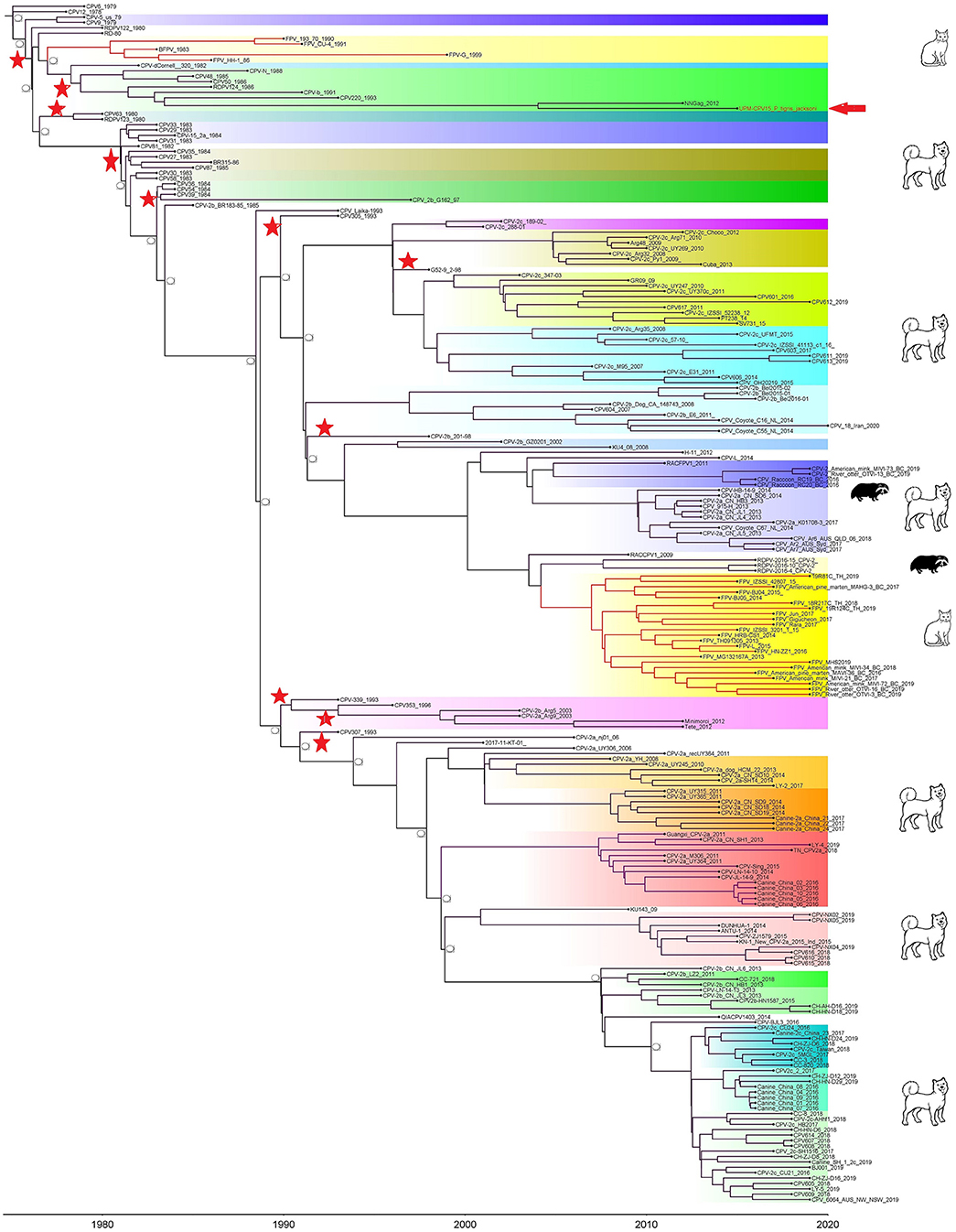
Figure 4. Time-scaled Bayesian Maximum Clade Credibility tree for the capsid genes (VP) of porcine, canine, and feline panleukopenia viruses. The local isolate was highlighted in golden color. PPV, CPV, and FPV were highlighted in red, green, and blue color, respectively. Each branch was colored according to posterior probability (PP) value; blue bars at nodes indicate 95% highest probability density (HPD). Tree nodes with a posterior probability >0.8 was displayed at tree nodes with red color.
Figure 5 demonstrates the demographic history of CPV NS and CP genes. An S-shape was observed over the 41 years (1978 to 2019). Both NS (a) and capsid (b) genes of CPV populations have had a stable size until the first decline inferred range between the years 2000 and 2010. A steady population size followed the decrease until the current date. Table 1 shows the evolution rate of the CP (VP 1 and VP2) and NS gene of Protoparvovirus. Overall, the capsid of PPV, FPV, and MPV shows higher nucleotide substitutions/site/year. While the NS gene showed lower nucleotide substitutions/site/year as compared to PPV, RPV, and FPV.
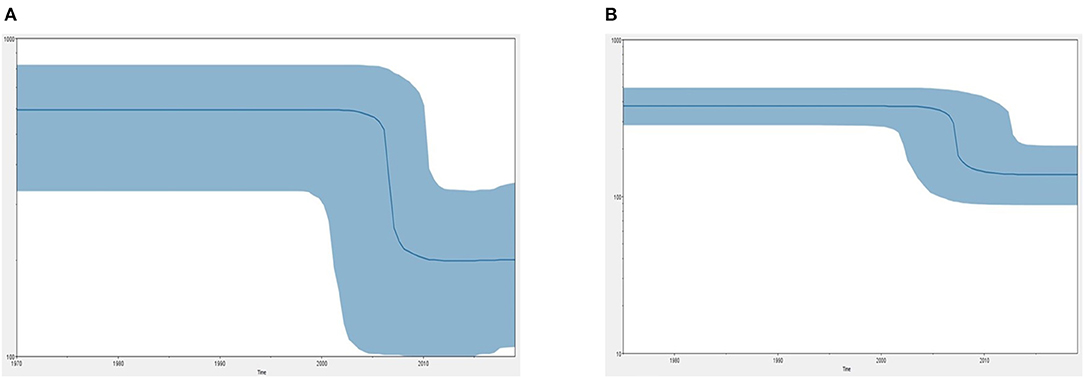
Figure 5. Bayesian skyline plot (BSP) shows the changes in effective population size over time of canine parvovirus. (A) The demographical history of the canine parvovirus NS gene. (B) The demographical history of the canine parvovirus capsid gene. The blue line denotes the median posterior value, and the blue area represents the 95% HPD intervals. The x-axis is labeled years (time) and the y-axis is labeled effective population size.
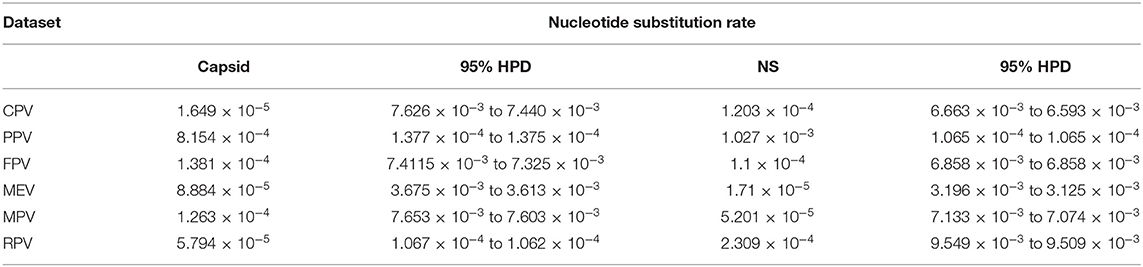
Table 1. Nucleotide substitution rates of capsid and NS gene of protoparvoviruses.
Figure 6 shows the three-dimensional structure comparison of CPV capsid and NS gene with other Protoparvovirus. The dN/dS ratio in this study showed that most codons fell under neutral or negative selection for both CP and NS genes (Figures 7A,B). High levels of variability were spotted at CP amino acid sequences compared to NS. The CP protein amino acid sequence was at the 4, 80–87, 93–103, 232, 267, 297, 300–305, 323–324, 370–375, 426, 440, and 564–568 position. Comparing the amino acid sequence of CPV capsid and NS genes with other Protoparvovirus shows that only FPV and MEV show high similarity, whereas, PPV, MPV, KRV, and RPV show less similarity with CPV (Table 2).
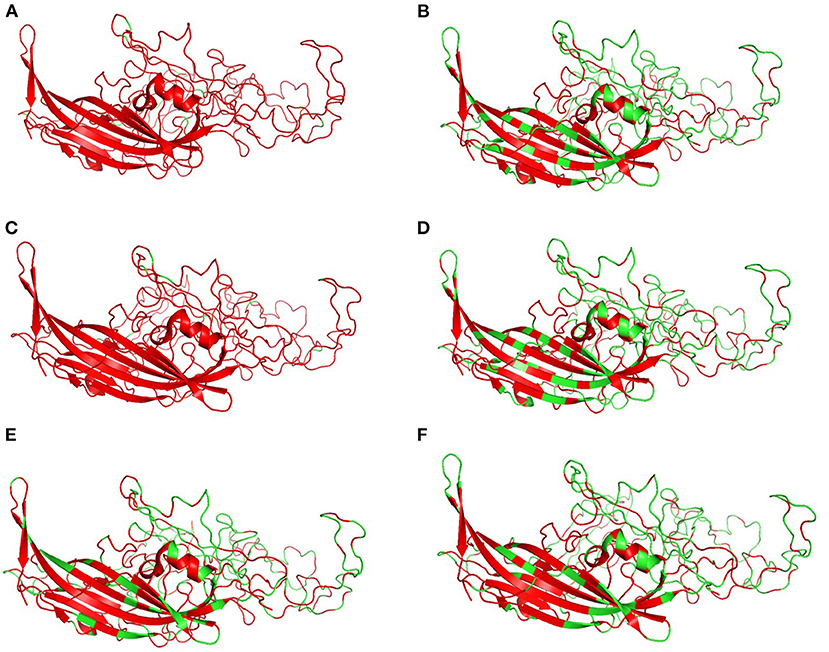
Figure 6. Changes in capsid: Three-dimensional structure of canine parvovirus capsid comparison with other Protoparvovirus. Red indicates the portion of similar amino acids shared, whereas, green is dissimilar amino acids. Comparison of CPV capsid protein with (A) FPV, (B) KRV, (C) MEV, (D) MPV, (E) PPV, and (F) RPV.
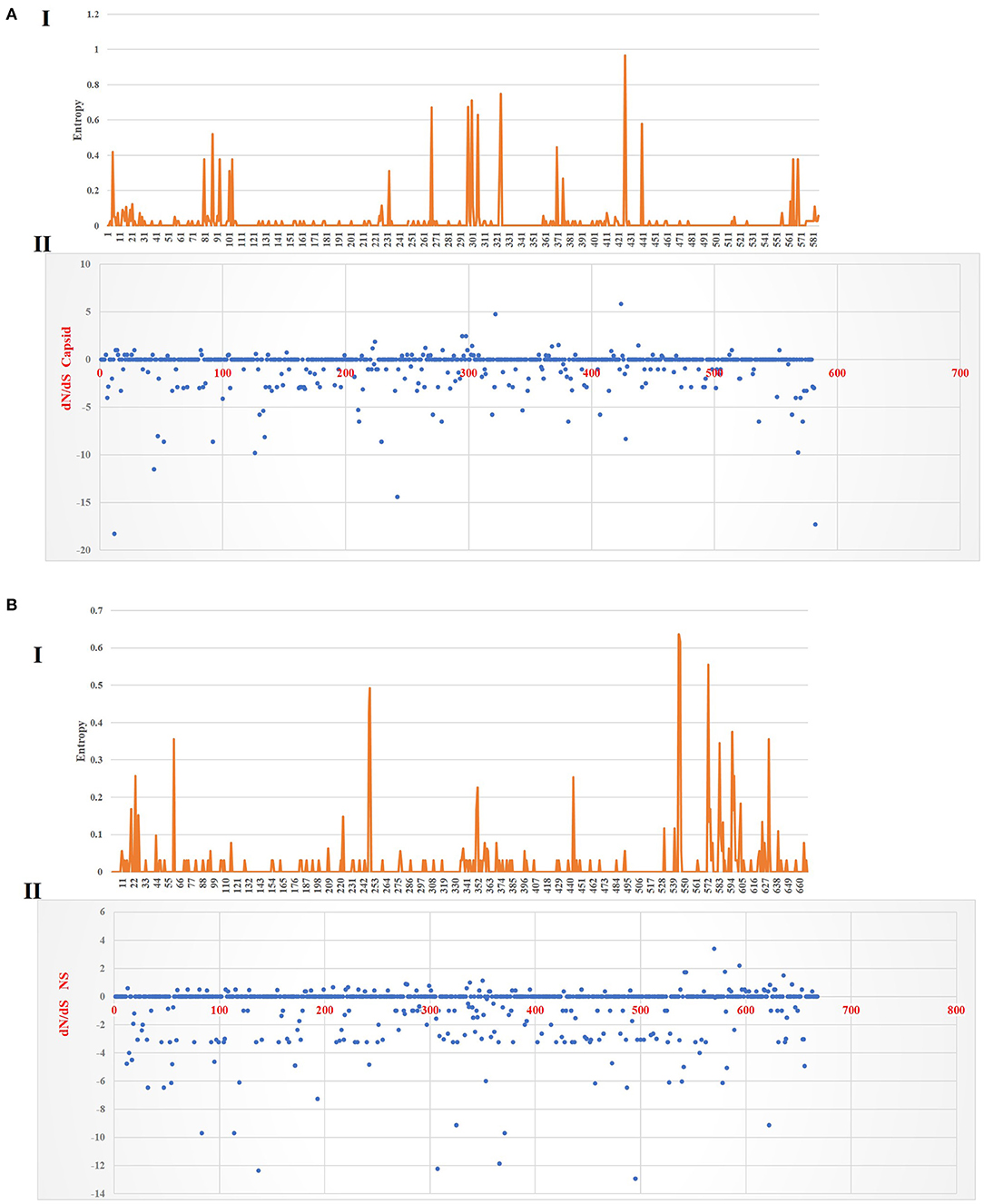
Figure 7. Plot for the distinction between non-synonymous and synonymous rates (dN–dS) and amino acid entropy rate for the CP and NS genes. The entropy for CP amino acid sequences shows exhibited high-level entropy [(A) I] and the dN–dS ratio falls under purified selection [(A) II]. The entropy for NS amino acid sequences shows exhibited low-level entropy [(B) I] and the dN–dS ratio falls under purified selection [(B) II].
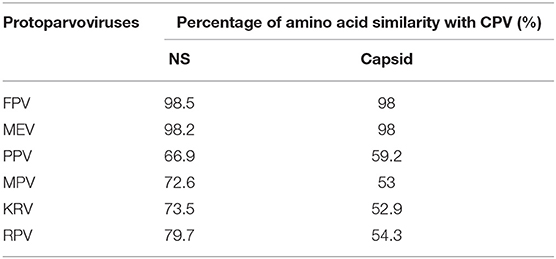
Table 2. Percentage of amino acid similarity with CPV non-structural and capsid proteins.
Discussion
Wild felids are susceptible to virus infection due to cross-species transmission potentially by domestic cats or dogs (23). Early detection of viral shedding of asymptomatic or symptomatic felids may help in elucidating the health status of wild felids in captive or nature. Since the status of virus infections among captivated wild felids in Malaysia is lacking, we aimed in this study to screen the presence of FHV, FCV, CDV, and CPV from serum samples in the leopard, feral cat, and tiger from zoo. Isolation based on cell cultures and detection of the viruses by gene-specific PCR/RT-PCR and full genome sequence analysis of the CPV isolate were carried out.
In general, parvovirus, especially CPV, has higher tropism in rapidly dividing cells such as the crypt of lieberkuhn in the intestine and is associated with clinical manifestation of diarrhea and gastroenteritis (1). In this study, serum sample was used instead of fecal sample primarily because the study was aimed to screen the presence of virus shedding from healthy captive felids. Several studies have included serum or plasma other than fecal or intestine tissue to diagnose presence of parvovirus DNA in human (24, 25), dog (26), and feline (27). Elucidating the status of virus shedding in the captive wild felids is part of a surveillance program that essentially will help in preventing occurrence of infectious disease outbreaks in the captivated wildlife.
Isolation of virus from various sample sources from either human, animal, or environment based on cell culture technique has been used extensively. However, in most cases, further, confirmatory analysis based on molecular tests is required to identify the virus since the detection of CPEs is a general indicator of viral-induced cellular changes and often offers inconclusive diagnosis (28). Isolation in CRFK cells was chosen as it is the most commonly used cell line for virus isolation in mostly feline samples, which include feline foamy virus (29), feline picornaviruses (30), FPV (31), feline leukemia virus (FeLV) (32), FHV (33), and CPV. Of the 36 serums inoculated in CRFK, 11 samples showed positive CPE up to three consecutive passages, whereas, only one sample was positive for PCR detection based on virus-specific primer combinations that detect CPV from a Malayan tiger. The other 10 isolates that were negative for PCR detection in the tested viruses were probably associated with unknown viruses or other types of viruses that were not included in this study, such as the feline immunodeficiency virus (FIV), FeLV, or feline coronavirus (FCoV).
In this study, UPM-CPV15/P. tigris jacksoni isolate was obtained through direct sequencing protocol based on Sanger sequencing. This isolate's complete genome was highly similar to CPV-31, CPV-d Cornell #320, and CPV-15 with 99.13, 98.65, and 98.40%, respectively. These strains are classified as CPV-2a and originated from USA. CPV-2a, however, is a very common CPV variant isolated from Asia (34). Although, the current strain was isolated in 2015, CPV-31, CPV-d Cornell #320, and CPV-15 were isolated in the early 1980's. Nevertheless, the CPV-31 strain shows high similarity with strains isolated from Beijing and Italy, namely, CPV-L (99.78%) (GenBank accession: MG763189.1) in 2014 and CPV_IZSSI_2323_11 (99.56%) (GenBank accession: KX434458.1) in 2011, indicating that the virus is still circulating, although, those strains were discovered three decades ago. Meanwhile, Wang et al. (35) and Jiao et al. (36) isolated parvovirus from the captive Siberian tigers, which was closely related to racoon parvovirus, indicating the susceptibility of felids to parvovirus. In regard to variant, CPV-2a and CPV-2b have been isolated from cheetah and tiger while CPV-2c was isolated from leopard (37, 38).
The Neighbor-Joining method is a suitable model to study evolution and has a well-chosen maximum likelihood model parameter that can often produce a reasonably good phylogenetic tree (39). Because of that, the Neighbor-Joining analysis method was chosen in order to better understand how divergent was isolate from this study within the species in the same genera and to verify that it clustered together with CPV. In genome diversity, mutation rate is an important parameter, which is the per-nucleotide or amino acid site mutation rate and the genome size. This mutation rate will determine the average number of mutations each offspring will have compared to the parental (or ancestral) genome. In this study, the phylogeny of the full length of the CP and NS gene was constructed separately based on the availability of sequences in GenBank. Comparing both phylogenetic tree, CP shows more branches as compared to NS. Variation in the CPV capsid from this study was also observable with FPV, a common variant of CPV. The structural protein CP (VP2 protein) produces multiple mutations during the evolution, causing more amino acid variation; thus, CP is denoted as the main antigenic determinant of CPV as it plays a key role in the mutation and changes in the capsid's amino acid sequences for adaptation to the host cellular receptors (40). Similarly, this study demonstrated higher nucleotide and amino acid sequence mutation in CP sequences as compared to the NS gene when compared with other species in Protoparvoviruses (Tables 1, 2).
Multiple statistical tests have been established to measure selection pressures that play a role in protein-coding regions. The dN/dS ratio is commonly used, owing in part to its simplicity and robustness. The dN/dS ratio in this study showed that most codons fell under neutral or less positive selection for both CP and NS genes. These findings are expected since positive selection and genetic recombination occur frequently in the parvovirus (41–43). High levels of variability were spotted at CP amino acid sequences as compared to NS. The CP protein amino acid sequence at position 4, 80–87, 93–103, 232, 267, 297, 300–305, 323–324, 370–375, 426, 440, and 564–568 demonstrated high complexity. This is comparable with the value based on the previous study by Zhou et al. (34), which is expecting 10−4 substitution of nucleotides per site per year, and amino acid mutation at position 267, 324, and 440. More variations observed in the phylogeny and amino acid sequences of the capsid of CPV strain from this study further articulate its crucial role in interacting with specific host cellular receptor for successive entry and penetration during replication. Mutation could be the main influence that changes the capsid's amino acid sequences for adaptation to the host cellular receptors (41). Meanwhile, the high similarity of current CPV NS amino acid sequences and phylogeny might be due to their definite role in the DNA replication mechanism (44).
Phylo-dynamics of CPV is important in an effort to shed light on how the transmission dynamics impact viral genetic differences and deliver important insights into epidemiology and virus evolution. To discover the demographic history of the CPV in the tested population over the time period, the relative actual population size over time was inferred by investigating the genetic diversity using the Bayesian skyline model. The demographic pattern of CPV NS and capsid dropped after the 2000's to 2010's (Figure 5). This could be explained by the reduction in the infection rate of the infected population due to immunization of inactivated and live vaccines that are used widely in dogs (12). However, vaccination against CPV could be responsible for the occurrence of antigenic variants through antigenic drift events to evade the host immune system (41, 45–49). Postulating exactly which animals specifically are the susceptible hosts for CPV and FPV particularly and how the genetic composition of the viruses will change in the different hosts are still unclear. Nevertheless, diverse extension of pathways and emergence of novel strain(s) might be contributed by several dynamics such as animal movement and the presence of the carrier or intermediate host that spread the virus causing cross-species transmission at large spatial scales worldwide (50). Thus far, a relatively low mutation has been observed in CPV over 40 years even though, rapid host adaptation was common, suggesting that continuous high diversities of genetic sequences of host between same species or interspecies are not necessarily required for virus host adaptation in CPV (51).
Conclusion
In conclusion, this study reports the presence of CPV (CPV-2a) in a Malayan tiger. Full-genome analysis denotes that the present isolate size is ~4.7 kbp and encodes for two major genes, the NS and the capsid gene. Genome sequencing provides valuable information on the genomic characteristics of the CPV from this study and other viruses from the Protoparvovirus genus.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the animal study because the samples used were archived and received for diagnostic screening.
Author Contributions
AN-F and KK were involved in writing the original draft preparation and conducted methodology by using software. AY, AO, and SC were involved in supervision, conceptualization, and writing in terms of reviewing and editing. All the authors have read and approved the final manuscript.
Funding
This research was supported by Universiti Putra Malaysia (UPM) under research grants UPM 700-2/1/GP-IPM/2016/9510500 and UPM 700-2/1/GP-IPS/2017/9547800.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank members of the Virology Laboratory of Faculty of Veterinary Medicine and the Institute of Bioscience from Universiti Putra Malaysia for the technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2021.660046/full#supplementary-material
References
1. Carter J, Saunders V. Virology: Principles and Applications. 1st edn. England: John Wiley & Sons Ltd (2007).
2. Pénzes J, Söderlund-Venermo M, Canuti M, Eis-Hübinger A, Hughes J, Cotmore S, et al. Reorganizing the family Parvoviridae: a revised taxonomy independent of the canonical approach based on host association. Arch Virol. (2020) 165:2133–46. doi: 10.1007/s00705-020-04632-4
3. Ros C, Bayat N, Wolfisberg R, Almendral JM. Protoparvovirus cell entry. Viruses. (2017) 9:313. doi: 10.3390/v9110313
4. Nandi S, Kumar M. Canine parvovirus: current perspective. Indian J Virol. (2010) 21:31–44. doi: 10.1007/s13337-010-0007-y
5. Appel MJ, Cooper BJ, Greisen H, Scott F, Carmichael LE. Canine viral enteritis. I. Status report on corona and parvo-like viral enteritides. Cornell Vet. (1979) 69:123–33.
6. Parrish CR. Mapping specific functions in the capsid structure of canine parvovirus and feline panleukopenia virus using infectious plasmid clones. Virology. (1991) 183:195–205. doi: 10.1016/0042-6822(91)90132-U
7. Van-Brussel K, Carrai M, Lin C, Kelman M, Setyo L, Aberdein D, et al. Distinct lineages of feline parvovirus associated with epizootic outbreaks in Australia, New Zealand and the United Arab Emirates. Viruses. (2019) 11:1155. doi: 10.3390/v11121155
8. Ikeda Y, Mochizuki M, Naito R, Nakamura K, Miyazawa T, Mikami T, et al. Predominance of canine parvovirus (CPV) in unvaccinated cat populations and emergence of new antigenic types of CPVs in cats. Virology. (2000) 278:13–9. doi: 10.1006/viro.2000.0653
9. Wang X, Li T, Liu H, Du J, Zhou F, Dong Y, et al. Recombinant feline parvovirus infection of immunized tigers in central China. Emerg Microbes Infect. (2017) 6:e42. doi: 10.1038/emi.2017.25
10. Awad RA, Khalil WKB, Attallah AG. Epidemiology and diagnosis of feline panleukopenia virus in Egypt: clinical and molecular diagnosis in cats. Vet World. (2018) 11:578–84. doi: 10.14202/vetworld.2018.578-584
11. Bergmann M, Schwertler S, Reese S, Speck S, Truyen U, Hartmann K. Antibody response to feline panleukopenia virus vaccination in healthy adult cats. J Feline Med Surg. (2018) 20:1087–93. doi: 10.1177/1098612X17747740
12. Franzo G, Tucciarone CM, Cecchinato M, Drigo M. Canine parvovirus type 2 (CPV-2) and Feline panleukopenia virus (FPV) codon bias analysis reveals a progressive adaptation to the new niche after the host jump. Mol Phylogenet Evol. (2017) 114:82–92. doi: 10.1016/j.ympev.2017.05.019
13. Ain-Fatin R, Nur-Fazila SH, Yasmin AR, Sifa-Shaida, AH, Ayuni WN, et al. Canine parvovirus (CPV) infection in a Great Dane puppy: a case report. Adv Anim Vet Sci. (2020) 8:1075–8. doi: 10.17582/journal.aavs/2020/8.10.1075.1078
14. Ling CH, Watanabe M, Zeenathul ZA, Megat-Rani PA. Detection of feline herpesvirus feline calicivirus in cats with feline upper respiratory tract disease presented to University Veterinary Hospital, Universiti Putra Malaysia. In: 9th Proceedings of the Seminar on Veterinary Sciences. UPM Serdang: Faculty of Veterinary Medicine, UPM (2014). p. 178.
15. Sykes JE, Allen JL, Studdert VP, Browning GF. Detection of feline calicivirus, feline herpesvirus 1 and Chlamydia Psittaci mucosal swabs by multiplex RT-PCR/PCR. Vet Microbiol. (2001) 81:95–108. doi: 10.1016/S0378-1135(01)00340-6
16. Marsilio F, Di Martino B, Decaro N, Buonavoglia C. A novel nested PCR for the diagnosis of calicivirus infections in the cat. Vet Microbiol. (2005) 105:1–7. doi: 10.1016/j.vetmic.2004.09.017
17. Elia G, Decaro N, Martella V, Cirone F, Lucente MS, Lorusso E, et al. Detection of canine distemper virus in dogs by real-time RT-PCR. J Virol Methods. (2006) 136:171–6. doi: 10.1016/j.jviromet.2006.05.004
18. Buonavoglia C, Martella V, Pratelli A, Tempesta M, Cavalli A, Buonavoglia D, et al. Evidence for evolution of canine parvovirus type 2 in Italy. J Gen Virol. (2001) 82:3021–5. doi: 10.1099/0022-1317-82-12-3021
19. Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol. (2012) 86:3995–4008. doi: 10.1128/JVI.06540-11
20. Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. (2005) 21:676–9. doi: 10.1093/bioinformatics/bti079
21. Hughes AL, Nei M. Nucleotide substitution at major histocompatibility complex class II loci: evidence for over dominant selection. PNAS. (1989) 86:958–62. doi: 10.1073/pnas.86.3.958
22. Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. (2008) 4:e1000304. doi: 10.1371/journal.pgen.1000304
23. O'Brien SJ, Troyer JL, Brown MA, Johnson WE, Antunes A, Roelke ME, et al. Emerging viruses in the Felidae: shifting paradigms. Viruses. (2012) 4:236–57. doi: 10.3390/v4020236
24. Nikkari S, Lappalainen H, Saario R, Lammintausta K, Kotilainen P. Detection of parvovirus B19 in skin biopsy, serum, and bone marrow of a patient with fever, rash, and polyarthritis followed by pneumonia, pericardial effusion, and hepatitis. Eur J Clin Microbiol Infect Dis. (1996) 15:954–7. doi: 10.1007/BF01690517
25. Pinho JRR, Alves VAF, Vieira AF, Moralez MOS, Fonseca LEP, Guz B, et al. Detection of human parvovirus B19 in a patient with hepatitis. Brazil J Med Biol Res. (2001) 34:1131–8. doi: 10.1590/S0100-879X2001000900005
26. Navarro C. Detection of canine parvovirus in dogs by means polymerase chain reaction. Am J Biomed Sci Res. (2020) 7. doi: 10.34297/AJBSR.2020.07.001219
27. Streck AF, Rüster D, Truyen U, Homeier T. An updated TaqMan real-time PCR for canine and feline parvoviruses. J Virol Methods. (2013) 193:6–8. doi: 10.1016/j.jviromet.2013.04.025
28. Freshney RI. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications. NJ: Wiley-Blackwell (2016).
29. Ledesma-Feliciano C, Troyer RM, Zheng X, Miller C, Cianciolo R, Bordicchia M, et al. Feline foamy virus infection: characterization of experimental infection and prevalence of natural infection in domestic cats with and without chronic kidney disease. Viruses. (2019) 11:662. doi: 10.3390/v11070662
30. Lau SKP, Woo PCY, Yip CCY, Choi GKY, Wu Y, Bai R, et al. Identification of a novel feline picornavirus from the domestic cat. J. Virol. (2012) 86:395–405. doi: 10.1128/JVI.06253-11
31. Liu C, Liu Y, Liu D, Qiu Z, Tian J, Guo D, et al. Complete genome sequence of feline panleukopenia virus strain hrb-cs1, isolated from a domestic cat in Northeastern China. Genome Announc. (2015) 3:e01556–14. doi: 10.1128/genomeA.01556-14
32. Powers JA, Chiu ES, Kraberger SJ, Roelke-Parker M, Lowery I, Erbeck K, et al. Feline Leukemia Virus (FeLV) disease outcomes in a domestic cat breeding colony: relationship to endogenous FeLV and Other chronic viral infections. J. Virol. (2018) 92:e00649–18. doi: 10.1128/JVI.00649-18
33. Tian J, Liu Y, Liu X, Sun X, Zhang J, Qu L. Feline Herpesvirus 1 US3 blocks the type I interferon signal pathway by targeting interferon regulatory factor 3 dimerization in a kinase-independent manner. J Virol. (2018) 92:e00047–18. doi: 10.1128/JVI.00047-18
34. Zhou P, Zeng W, Zhang X, Li S. The genetic evolution of canine parvovirus - A new perspective. PLoS ONE. (2017) 12:e0175035. doi: 10.1371/journal.pone.0175035
35. Wang K, Du S, Wang Y, Wang S, Luo X, Zhang Y, et al. Isolation and identification of tiger parvovirus in captive Siberian tigers and phylogenetic analysis of VP2 gene. Infect Genet Evol. (2019) 75:103957. doi: 10.1016/j.meegid.2019.103957
36. Jiao C, Zhang H, Liu W, Jin H, Liu D, Zhao J, et al. Construction and immunogenicity of virus-like particles of feline parvovirus from the tiger. Viruses. (2020) 12:315. doi: 10.3390/v12030315
37. Steinel A, Munson L, van Vuuren M, Truyen U. Genetic characterization of feline parvovirus sequences from various carnivores. J Gen Virol. (2000) 81(Pt. 2):345–50. doi: 10.1099/0022-1317-81-2-345
38. Ikeda Y, Nakamura K, Miyazawa T, Takahashi E, Mochizuki M. Feline host range of canine parvovirus: recent emergence of new antigenic types in cats. Emerg Infect Dis. (2002) 8:341–6. doi: 10.3201/eid0804.010228
39. Davidson, R, Martín Del Campo A. Combinatorial and computational investigations of neighbor-joining bias. Front Genet. (2020) 11:584785. doi: 10.3389/fgene.2020.584785
40. Allison AB, Kohler DJ, Ortega A, Hoover EA, Grove DM, Holmes EC, et al. Host-specific parvovirus evolution in nature is recapitulated by in vitro adaptation to different carnivore species. PLoS Pathog. (2014) 10:e1004475. doi: 10.1371/journal.ppat.1004475
41. Hoelzer K, Shackelton LA, Parrish CR, Holmes EC. Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. J Virol. (2008) 89:2280–9. doi: 10.1099/vir.0.2008/002055-0
42. Ohshima T, Mochizuki M. Evidence for recombination between feline panleukopenia virus and canine parvovirus type 2. J Vet Med Sci. (2009) 71:403–8. doi: 10.1292/jvms.71.403
43. Wang J, Cheng S, Yi L, Cheng Y, Yang S, Xu H, et al. Evidence for natural recombination between mink enteritis virus and canine parvovirus. Virol J. (2012) 9:252. doi: 10.1186/1743-422X-9-252
44. Li G, Ji S, Zhai X, Zhang Y, Liu J, Zhu M, et al. Evolutionary and genetic analysis of the VP2 gene of canine parvovirus. BMC Genom. (2017) 18:534. doi: 10.1186/s12864-017-3935-8
45. Decaro N, Buonavoglia C. Canine parvovirus a review of epidemiological and diagnostic aspects, with emphasis on type 2c. Vet Microbiol. (2012) 155:1–12. doi: 10.1016/j.vetmic.2011.09.007
46. Kapil S, Cooper E, Lamm C, Murray B, Rezabek G, Johnston, et al. Canine parvovirus types 2c and 2b circulating in North American dogs in 2006 and 2007. J Clin Microbiol. (2007) 45:4044–7. doi: 10.1128/JCM.01300-07
47. Geng Y, Guo D, Li C, Wang E, Wei S, Wang Z, et al. Co-circulation of the rare CPV-2c with unique Gln370Arg substitution, new CPV-2b with unique Thr440Ala substitution, and New CPV-2a with high prevalence and variation in Heilongjiang Province, Northeast China. PLoS ONE. (2015) 10:e0137288. doi: 10.1371/journal.pone.0137288
48. Decaro N, Desario C, Elia G, Martella V, Mari V, Lavazza A, et al. Evidence for immunisation failure in vaccinated adult dogs infected with canine parvovirus type 2c. New Microbiol. (2008) 31:125–130.
49. Calderón MG, Romanutti C, Antuono AD', Keller L, Mattion N, La Torre J. Evolution of canine parvovirus in Argentina between years 2003 and 2010: CPV2c has become the predominant variant affecting the domestic dog population. Virus Res. (2011) 157:106–10. doi: 10.1016/j.virusres.2011.02.015
50. Stucker KM, Pagan I, Cifuente JO, Kaelber JT, Lillie TD, Hafenstein S, et al. The role of evolutionary intermediates in the host adaptation of canine parvovirus. J Virol. (2012) 86:1514–21. doi: 10.1128/JVI.06222-11
Keywords: canine parvovirus, felids, Malayan tiger, full genome, capsid protein, non structural protein, evolution rate
Citation: Nur-Farahiyah AN, Kumar K, Yasmin AR, Omar AR and Camalxaman SN (2021) Isolation and Genetic Characterization of Canine Parvovirus in a Malayan Tiger. Front. Vet. Sci. 8:660046. doi: 10.3389/fvets.2021.660046
Received: 28 January 2021; Accepted: 23 June 2021;
Published: 03 August 2021.
Edited by:
Jesus Hernandez, Consejo Nacional de Ciencia y Tecnología (CONACYT), MexicoReviewed by:
Stefania Lauzi, University of Milan, ItalyJulian Ruiz-Saenz, Cooperative University of Colombia, Colombia
Copyright © 2021 Nur-Farahiyah, Kumar, Yasmin, Omar and Camalxaman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abd Rahaman Yasmin, bm9yeWFzbWluQHVwbS5lZHUubXk=