 Baishuang Yin1†
Baishuang Yin1† Shanshan Qi2†
Shanshan Qi2† Wanli Sha1Hongyu Qin1Liming Liu1Jinyan Yun1Jinhai Zhu2Guojiang Li1*
Wanli Sha1Hongyu Qin1Liming Liu1Jinyan Yun1Jinhai Zhu2Guojiang Li1* Dongbo Sun1,2*
Dongbo Sun1,2*- 1College of Animal Science and Technology, Jilin Agricultural Science and Technology University, Jilin, China
- 2Laboratory for the Prevention and Control of Swine Infectious Diseases, College of Animal Science and Veterinary Medicine, Heilongjiang Bayi Agricultural University, Daqing, China
Porcine reproductive and respiratory syndrome virus (PRRSV) causes a highly contagious disease and brings huge economic losses to commercial pork production worldwide. PRRSV causes severe reproductive failure in sows and respiratory distress in piglets. To trace the evolution of PRRSV in pigs with respiratory diseases in some regions of China, 112 samples were collected from nine provinces in China during 2016–2018. All samples were detected by RT-PCR and analyzed by the Nsp2/ORF5 (ORF5a)-genes-phylogeny. Sequence analysis and recombination analysis were conducted on the Nsp2/ORF5 (ORF5a) genes of the identified strain in the study. The RT-PCR result shown that the positive rate of PRRSV was 50.89% (57/112). Phylogenetic analysis showed that the identified PRRSV strains were all NA genotype and belonged to lineage 1, 3, and 8. The Nsp2 gene of identified PRRSV strains exhibited nucleotide homologies of 53.0 ~ 99.8%, and amino acid homologies of 46.8 ~ 99.7%. The ORF5 gene of identified PRRSV strains exhibited nucleotide homologies of 82.4 ~ 100%, and amino acid homologies of 79.6 ~ 100%. Sequence analysis revealed that a discontinuous 30-amino-acid deletion (positions 481 and 533–561) and a 131-amino-acid discontinuity deletion (positions 323–433, 481, and 533–551) in Nsp2 of PPRSV isolates; all identified strains in this study may be wild strains, and most identified strains may be highly virulent strains. Sequence analysis of ORF5 and ORF5a revealed that the mutation sites of GP5 were mainly concentrated in the signal peptide and epitopes region, while the mutation sites of ORF5a were mainly concentrated in the transmembrane and the intramembrane region. The recombination analysis indicated that there may be multiple recombination regions in identified strains, and the recombination pattern was more complex. This study showed that the prevalent PRRSV strain in some regions of China was still HP-PRRSV, while NADC30 strain also occupied a certain proportion; different types of PRRSV strains showed different patterns and variation in China. This study suggested that the monitoring of PRRSV prevalence and genetic variation should be further strengthened.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is a major threat to the global swine industry, causing significant economic losses each year. The causative agent is PRRS virus (PRRSV), a member of the Arteriviridae family, order Nidoviridales. PRRSV is a single positive-strand RNA virus with a genome length of ~15.4 kb (1, 2). The PRRSV contains at least 10 open reading frames (ORFs), which are ORF1a, ORF1b, ORF2a, ORF2b, ORF5a, and ORF3 ~ 7 from the 5' to the 3' untranslated regions (UTR) (3, 4). ORF1a and ORF1b are cleaved into at least 13–16 non-structural proteins (Nsps) by a complex proteolytic cascade (3).
PRRSV was first reported in commercial pigs by the United States in 1987 (5), and the disease quickly spread worldwide with frequent break outs. PRRSV is still considered a highly contagious disease in the pig industry and creates huge economic losses (1, 6, 7). PRRSV was divided into two genotypes: the European genotype (type I) and North American genotype (type II) (3). There are three main subtypes of PRRSV (type II) isolates in Chinese pig populations: classical PRRSV (type II) including CH-1a, S1, and BJ-4; highly pathogenic PRRSV (HP-PRRSV) including JXA1, HuN4, and TJ; and NADC30-like PRRSV including JL580, CHsx1401, and HNjz15 (8). The genetic characteristic of HP-PRRSV isolates have a discontinuous 30-amino-acid deletion in Nsp2, and NADC30-like PRRSV isolates have a discontinuous 131-amino-acid deletion in Nsp2 (9, 10). PRRSV has mutated in the epidemic process to produce new strains due to the high frequency of gene mutation and recombination, in which new strains often have stronger environmental adaptations. The above factors have made the PRRSV epidemic more complicated, and it also brings great difficulties to disease prevention (11, 12). Nsp2 and ORF5 (ORF5a) are highly variable and ORF5 is associated with the neutralizing epitope (13, 14). They are usually used as target genes for PRRSV molecular epidemiological surveillance.
This study intends to reveal the prevalence and genetic evolution of PRRSV during 2016–2018 in different regions of China. The current study used the Nsp2 and ORF5 (ORF5a) genes to analyze the genetic evolution of the identified PRRSV strains. Our aim is to provide a theoretical basis for further monitoring of genetic variations of PRRSV in China.
Methods
Sampling
In total, 112 samples of the lung or lymph node tissues from pigs with respiratory diseases were collected between 2016 and 2018 in nine provinces or municipalities of China, including Heilongjiang, Jilin, Liaoning, Hubei, Jiangsu, Jiangxi, Zhejiang, Hebei, and the Inner Mongolia Autonomous Region. The lung or lymph node tissues were ground to powder with liquid nitrogen and diluted with three volumes of phosphate-buffered saline (PBS). The samples were centrifuged at 5,000 × g for 15 min at 4°C and the supernatants were transferred to a 1.5 mL tube. The genomic RNA was extracted from the supernatant using a commercial TIANamp Stool RNA Kit (Tiangen Biotech Co., Ltd, Beijing, China). The viral cDNA was synthesized using Moloney murine leukemia virus (RNaseH-) reverse transcriptase (Novoprotein Scientific Inc., Shanghai, China) in conjunction with six-random-nucleotide primers. The extracted genomic RNA and cDNA was stored at −80°C.
PCR Detection and Sequencing of PRRSV Strains
The primer of ORF5 full-length gene (including complete ORF5a gene) can be found in the report by Cao et al. (15). A pair of primers of Nsp2 gene were designed based on the alignment of published PRRSV genome sequences obtained from the NCBI GenBank database. Primer information is shown in Table 1. The amplification reactions were carried out in a 25 μL reaction volume containing 12.5 μl of EmeraldAmp® PCR Master Mix (2×Premix) (TaKaRa Biotechnology Co., Ltd., Dalian, China), 0.5 μM of the forward primer, 0.5 μM of the reverse primer, 1 μL of cDNA, and an appropriate volume of double-distilled (dd) H2O. The cycling parameters of ORF5 gene were: 36 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, followed by a final extension at 72°C for 10 min. The cycling parameters of Nsp2 gene were: 35 cycles of 95°C for 30 s, 57.6°C for 30 s, and 72°C for 3 min, followed by a final extension at 72°C for 10 min. The PCR products were analyzed by electrophoresis in a 1% agarose gel under UV light, and the samples with positive results were recorded. After the amplification, products were purified using the AxyPrep DNA Gel Extraction kit (A Corning Brand, Suzhou, China), and cloned into pGM-T Vector (TaKaRa Biotechnology Co., Ltd., Dalian, China). Each fragment was sequenced at least three times. All nucleotide sequences generated in this study have been submitted to the GenBank database.

Table 1. Primers specific for ORF5 and Nsp2 genes of PRRSV.
Phylogenetic and Sequence Analysis of PRRSV Strains
For the phylogenetic analysis, the Nsp2 and ORF5 (ORF5a) genes of PRRSV reference strains were retrieved from the NCBI nucleotide database as reference sequences. Detailed information and the GenBank number of PRRSV reference strains is shown in Supplementary Table 1. To construct phylogenetic trees, nucleotide sequences of the target gene using the ClustalX alignment tool in the MEGA 6.06 software (16). Neighbor-joining phylogenetic trees were constructed with 1,000 bootstrap replicates and the remaining default parameters in the MEGA 6.06 software. The generated phylogenetic tree was annotated using the online software ITOL (https://itol.embl.de/) (17). The PRRSV-identified strains and reference strains were analyzed by MegAlign program in DNASTAR™ 5.06 software. Nucleotide/amino acid homology of Nsp2 and ORF5 (ORF5a) genes of PRRSV-identified strains, and reference strains were gained using the Pairwise/Multiple Align function in Geneious Prime software.
Recombination Analysis
When RDP, GENECONV, BootScan, MaxChi, Chimera, SiScan, and 3eq methods were used to detect potential recombinant events, five or more methods were identified as gene recombination and P < 0.05 in RDP4.0 software. The strains in this event were determined to be recombinant strains. In addition, the detected recombination events were further confirmed by SimPlot 3.5.1.
Results
Detection and Analysis of PRRSV
In this study, 112 samples from nine provinces or municipalities of China were detected for PRRSV by RT-PCR. Of these tested samples, 50.89% (57/112) were positive for PRRSV. The infection rate of PRRSV was the highest in East China (85.71%, 12/14), including Jiangsu, Jiangxi, and Zhejiang province, and the lowest in Central China (11.11%, 1/9), including Hubei province. The infection rate of PRRSV in Northeast China was 52.3% (34/65), including Heilongjiang, Jilin, and Liaoning province, while the infection rate of PRRSV in North China was 43.5% (10/23), including Hebei province and the Inner Mongolia Autonomous Region (Table 2). Although the sources of the samples in different regions were non-uniform, the results showed that the presence of PRRSV was severe in some parts of China.
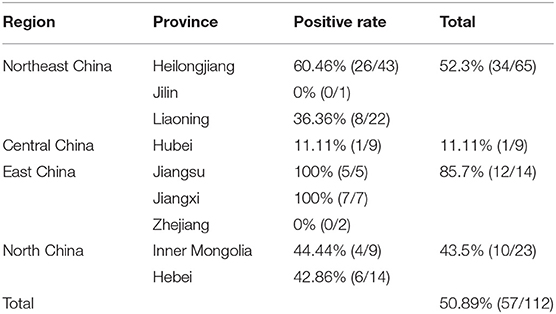
Table 2. The results of PRRSV sample positive rate in China between 2016 and 2018.
Phylogenetic Analysis of PRRSV
In 2010, Shi et al. conducted a systematic classification of type II PRRSVs based on ORF5 gene, and classified the PRRSVs into nine Lineage and 37 sublineage (18). Referring to the reported genotyping study by Shi et al., all the previously identified lineage reference sequences were also clustered in the same lineage in the phylogenetic tree of this study. The Nsp2 and ORF5-genes-based phylogenetic analysis revealed that all 56 PRRSV strains identified in this study belong to the North American genotype and were distributed in lineage 1, 3, and 8, among which the proportion of identified strains is the highest in sublineage 8.7 (73.2%, 41/56) and the lowest in lineage 3 (1.8%, 1/56) (Figures 1, 2). In addition, some identified strains were distributed differently in phylogenetic trees constructed with different genes. For instance, HeB/2016/1014a and HLJ/HEB/2016/1031(c, d) belong to the sublineage 8.7 while HLJ/HEB/2016/1031e and JX/FC/2017/914c belong to lineage 1 of the phylogenetic tree based on Nsp2 gene. However, the distribution of the identified strains was reversed in the phylogenetic tree constructed with the ORF5 gene. With the rapid growth of sequence deposition into the databases, it would be complicated for the diversity of PRRSV sequences.
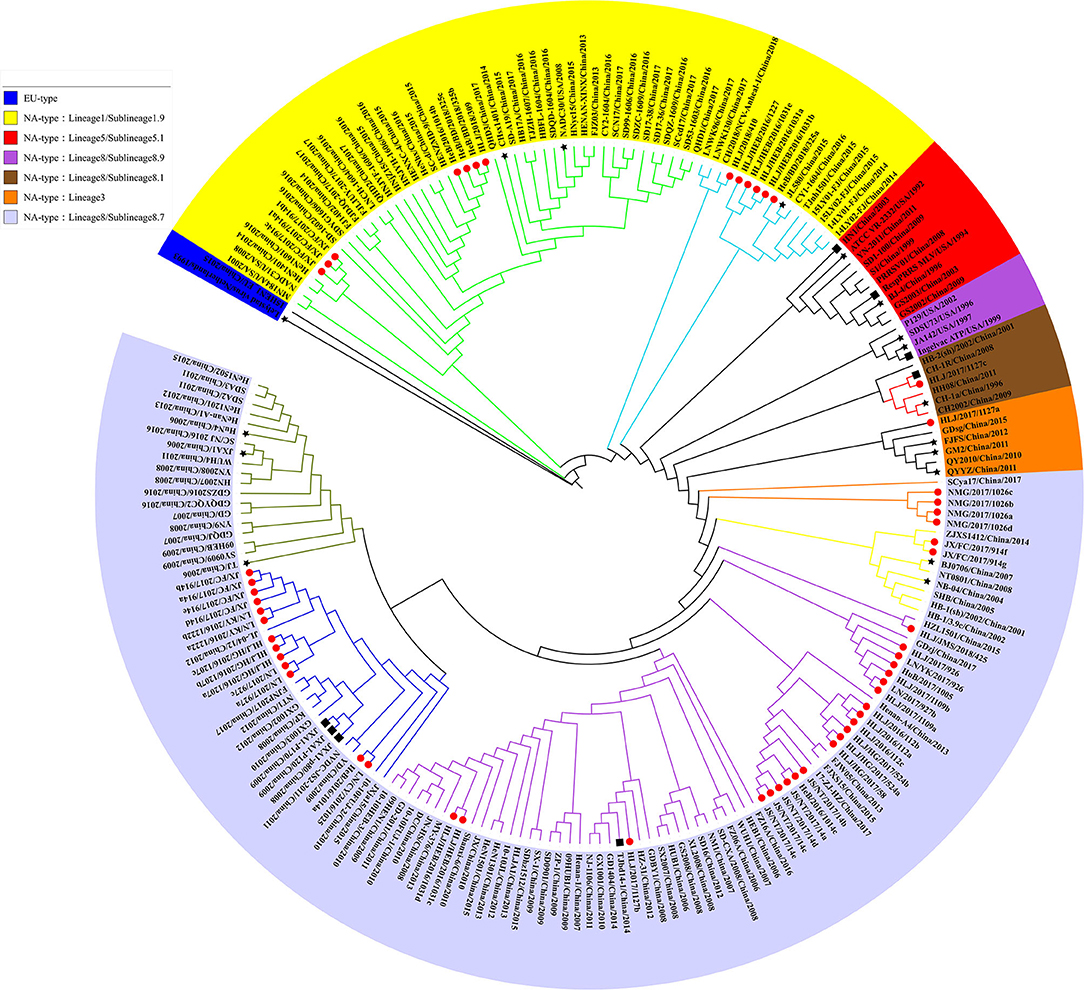
Figure 1. Phylogenetic analysis based on Nsp2 gene. The red circle diagram represents PRRSV strains identified in our study, black star diagram represents PRRSV reference strains, and the black square diagram represents PRRSV vaccine strains.
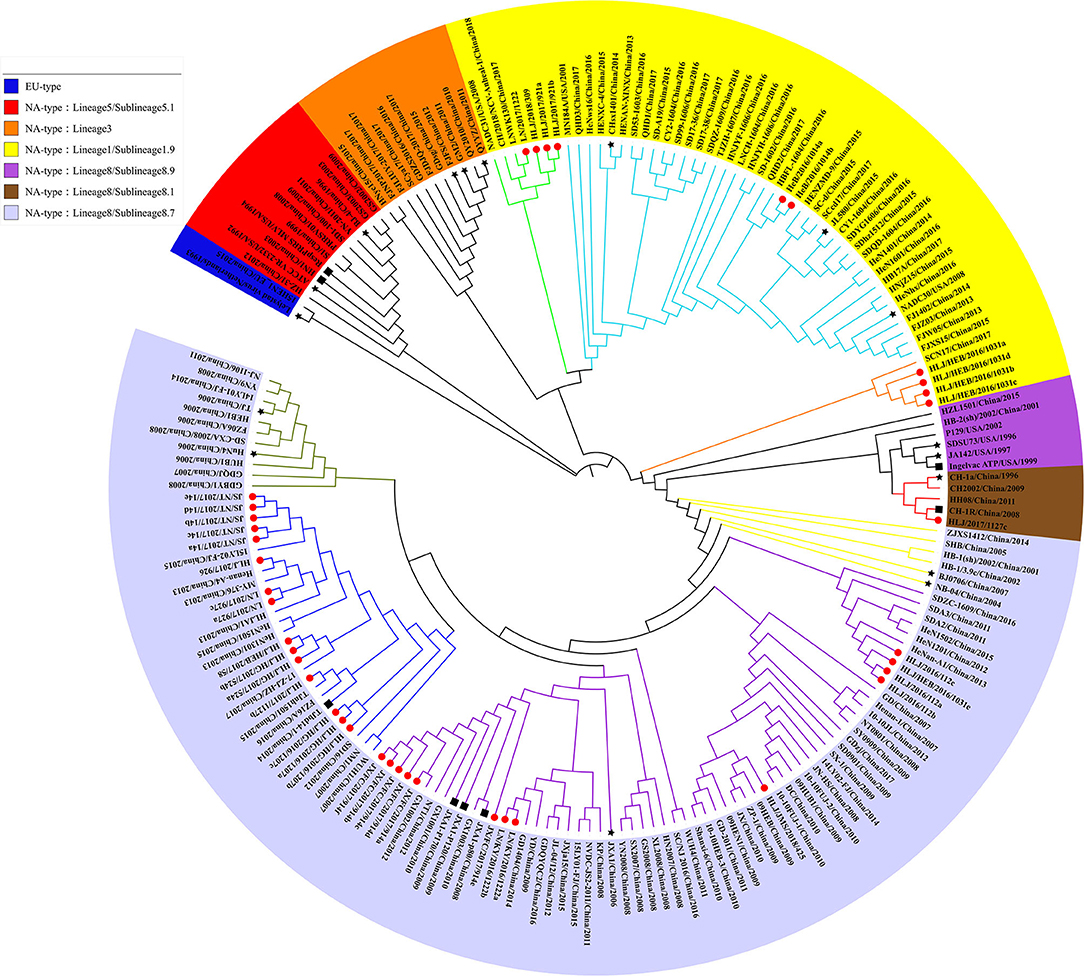
Figure 2. Phylogenetic analysis based on ORF5 gene. The red circle diagram represents PRRSV strains identified in our study, the black star diagram represents PRRSV reference strains, and the black square diagram represents PRRSV vaccine strains.
Sequence Analysis of PRRSV
Sequence Analysis of Nsp2 Genes of PRRSV
The 56 Nsp2 genes and 39 ORF5 (ORF5a) of PRRSV were successfully sequenced. The detailed GenBank number of the 56 PRRSV strains is shown in Supplementary Table 2. A sequence comparison of the Nsp2 genes revealed nucleotide homologies of 53.0 ~ 99.8% and deduced amino acid homologies of 46.8 ~ 99.7% among the 56 PRRSV strains. Along with the reference strain, its nucleotide and amino acid homologies compared with HP-PRRSV JXA1 strain was the highest (63.5 ~ 98.8% and 57.3 ~ 97.9%), and its nucleotide and amino acid homologies compared with NADC30 strain was the lowest (54.9 ~ 93.9% and 50.1 ~ 92.0%) (Table 3). In addition, the nucleotide and amino acid homologies of the identified strain in sublineage 8.1 and lineage 3 were higher than that of some identified strains in sublineage 8.7 from the same lineage. For example, HLJ/2017/1127c and HLJ/2017/1127b exhibited nucleotide and amino acid homologies of 76.6 and 73.1%, respectively. However, HLJ/2017/1127c and HLJ/2017/1127a exhibited nucleotide and amino acid homologies of 82.5 and 78.0%. The nucleotide and amino acid homologies of HLJ/2017/1127a and the representative strain QYYZ of lineage 3 was lower than the representative strain JXA1 of the 8.7 sublineage. This further suggests HLJ/2017/1127a is likely to be generated by recombinant strains in lineage 3 and sublineage 8.7. The classical PRRSV (type II) ATCC VR-2332 was used as the reference standard; the identified strain HLJ/2017/1127c had the same mutation pattern with vaccine strain CH-1R, which lacked a V at position 630 aa in the Nsp2. The identified strains of sublineage 8.7 and lineage 3 all showed a discontinuous 30-amino-acid deletion (positions 481 and 533–561) that conforms to the classical deletion mutation pattern of the HP-PRRSV-like strain. Excluding HLJ/2018/410, all strains identified in lineage 1 showed 131-amino-acid discontinuity deletion (positions 323–433, 481, and 533–551), that conforms to the classical deletion mutation pattern of the NADC30-like strain (Figures 3–6). The prevalent PRRSV strain in some regions of China was still HP-PRRSV, while NADC30 strain also occupied a certain proportion.
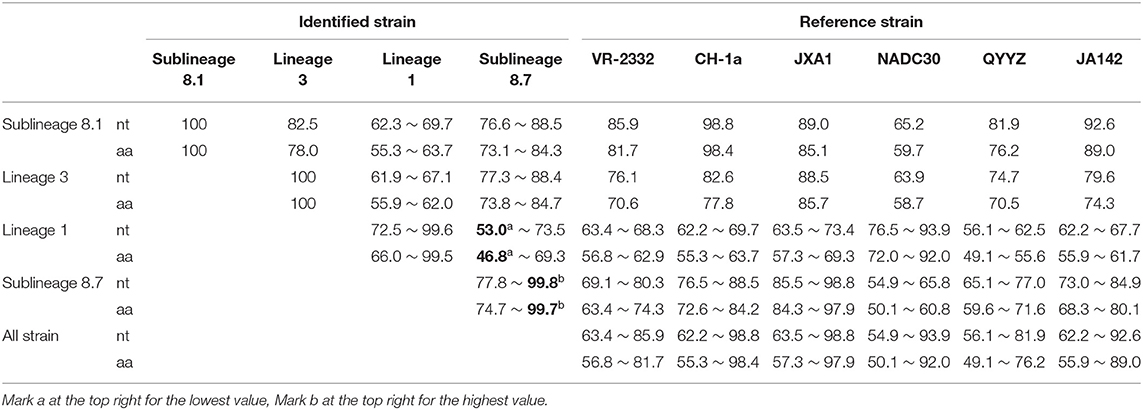
Table 3. Nucleotide and deduced amino acid homologies analysis based on Nsp2 gene (%).
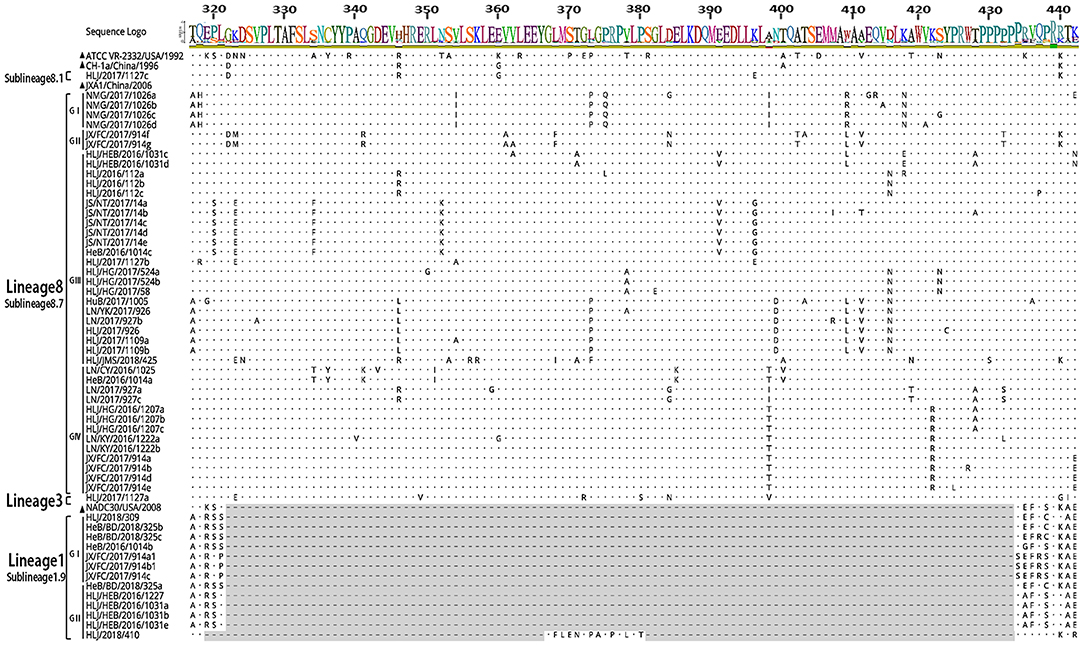
Figure 3. Amino acid sequence alignment of Nsp2 protein.

Figure 4. Amino acid sequence alignment of Nsp2 protein.

Figure 5. Amino acid sequence alignment of Nsp2 protein.

Figure 6. Amino acid sequence alignment of Nsp2 protein.
Sequence Analysis of ORF5 Genes of PRRSV
A sequence comparison of the ORF5 genes revealed nucleotide homologies of 82.4 ~ 100% and deduced amino acid homologies of 79.6 ~ 100% among the 39 PRRSV strains. The nucleotide and amino acid homologies compared with JXA1 strain was the highest (84.9 ~ 99.7%, 84.1 ~ 99.0%), and its nucleotide and amino acid homologies compared with QYYZ strain was the lowest (81.9 ~ 84.6%, 80.1 ~ 86.6%) (Table 4). The mutation sites of ORF5 were mainly concentrated in the signal peptide and epitopes region. GP5 virulence-related sites showed that nine of the 39 identified strains had mutated at the position 13th aa (R → Q). A total of 12 identified strains of sublineage 8.7 and lineage 1 had mutated at position 151 aa (R → K), The 137th aa of all identified strains was conservative and was S (Figure 7). It is observed that all identified strains may be wild strains, and most identified strains may be highly virulent strains in the study.
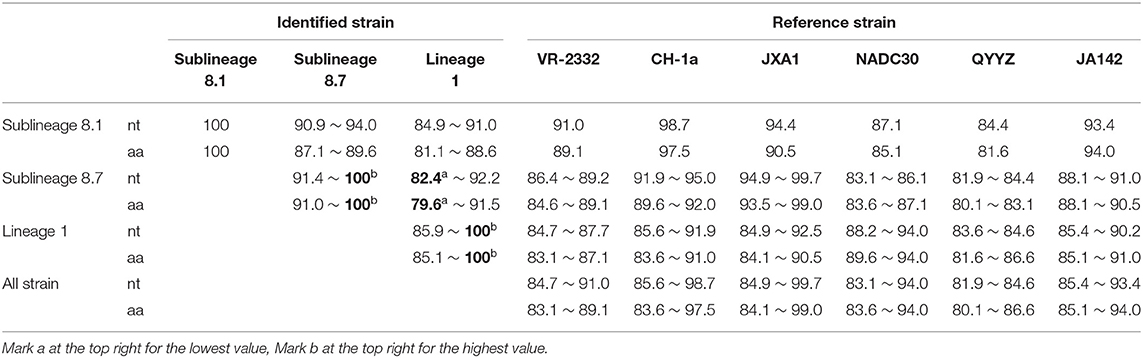
Table 4. Nucleotide and deduced amino acid homologies analysis based on ORF5 gene (%).
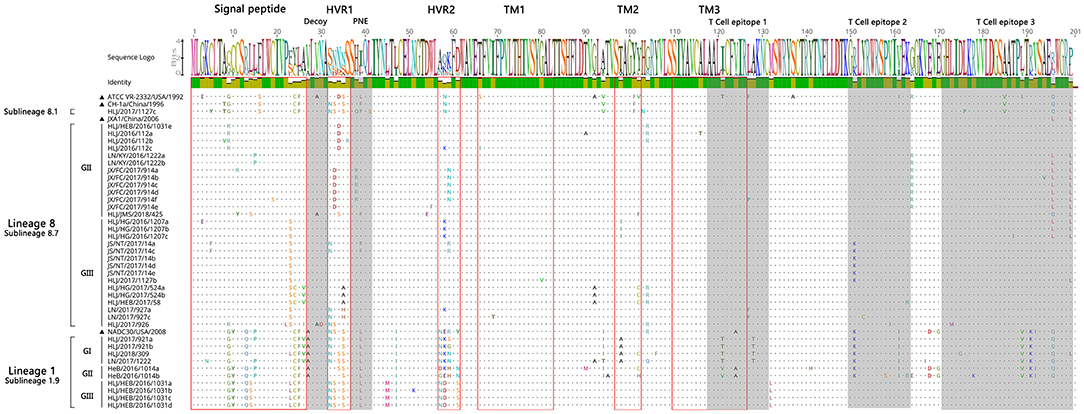
Figure 7. Amino acid sequence alignment of GP5 protein.
Sequence Analysis of ORF5a Genes of PRRSV
A sequence analysis of the ORF5a genes among the 39 PRRSV strains exhibited nucleotide homologies of 84.0 ~ 100% and deduced amino acid homologies of 80.8 ~ 100%. The nucleotide and amino acid homologies compared with JXA1 strain was the highest (87.2 ~ 99.4%, 84.6 ~ 100%), and its nucleotide and amino acid homologies compared with QYYZ strain was the lowest (70.5 ~ 79.5%, 67.3 ~ 78.8%) (Table 5). All the ORF5a proteins of the identified strains encoded 46 aa, and HLJ/2017/1127c exhibited high sequence similarity compared with CH-1a strain, with only a few point mutations, such as M8 → I8, F20 → L20, and S42/F42 → I42; A part of identified strains had two amino acid mutations in the transmembrane region, G12/V12 → A12, R24 → C24, and two amino acid mutations in the intramembrane region, Q36 → R36, Q38 → R38/P38. Compared with NADC30, some of the strains identified had one amino acid mutation in the transmembrane region, V26/I26 → T26, and one amino acid conformity mutation in the intramembrane region, S42 → F42/L42 (Figure 8). This shows that the mutation sites of ORF5a were mainly concentrated in the transmembrane region and the intramembrane region.
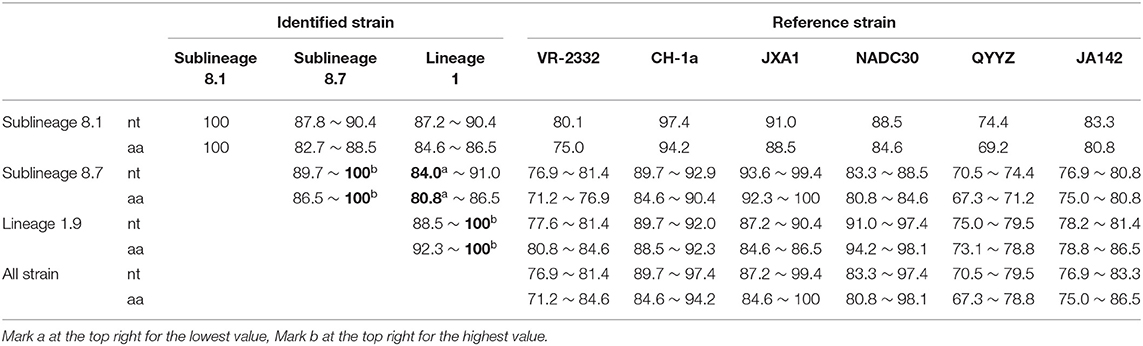
Table 5. Nucleotide and deduced amino acid homologies analysis based on ORF5a gene (%).

Figure 8. Amino acid sequence alignment of GP5a protein.
Recombination Analysis
The recombination analysis of Nsp2 gene showed that there were five potential recombination events (Table 6). The recombination analysis of Nsp2 gene showed that the recombinant strains in event 1 and 2 were produced by recombination of lineage 1 and sublineage 8.7 (Supplementary Figure 1). The high frequency mutation and recombination make the virus gain more genetically diverse (19). This recombination pattern is the most common in PRRSV recombinant strains in China, and animal tests have confirmed that the virulence of some recombinant strains is higher than the prototype strain NADC30 (9). The identified strain HLJ/2017/1127a in recombinant event 3 was produced by recombination of lineage 3 and sublineage 8.7 wild strain. The main parental strain was FZ06A, while the minor parental strain was QYYZ. Previous studies have shown that the low virulence prototype strain QYYZ even became highly virulent after recombination with the vaccine strain derived from HP-PRRSV (20). The identified strain JS/NT/2017/14b in recombinant event four showed that the main parental strain HLJ/2017/1127b belongs to subline 8.7, while the minor parental strain JS/NT/2017/14a belongs to subline 8.7. Three recombinant strains in recombinant event five were from the same origin as JS/NT/2017/14b, but the recombinant sites are different. The recombination analysis of ORF5 genes showed that the recombinant event included four recombinant strains of lineage 1 (Table 7). The main parental strain CY1-1604 belongs to lineage 1, while the minor parental strain GS2008 belongs to sublineage 8.7 (Supplementary Figure 2). Combined with the recombination analysis of Nsp2 gene, the identified strain HLJ/HEB/2016/1031 (a, b) was also recombined in ORF5 gene, which indicated that there may be multiple recombination regions in identified strains, and the recombination pattern was more complex.
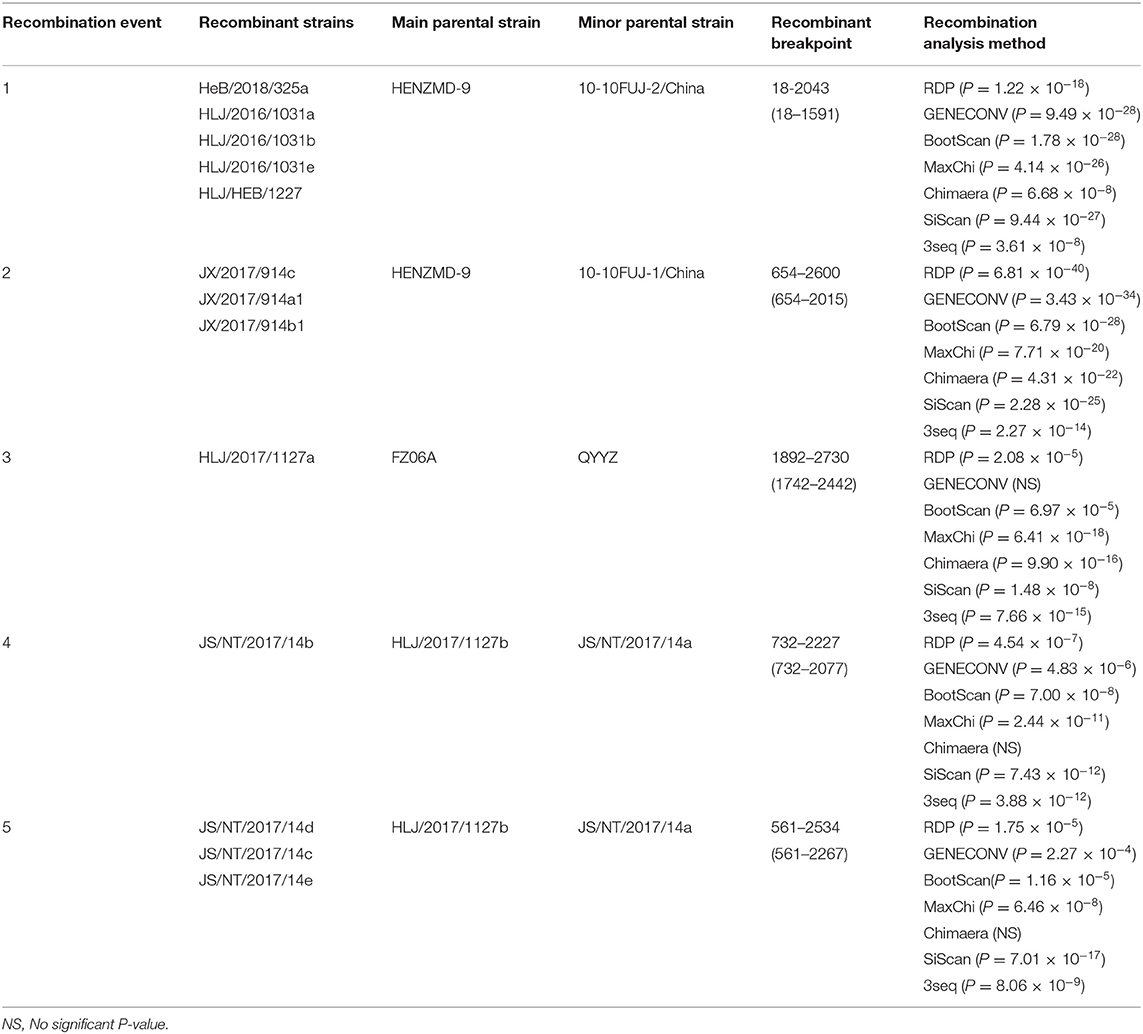
Table 6. Recombination analysis of Nsp2 gene.

Table 7. Recombination analysis of ORF5 gene.
Discussion
Since the outbreak of HP-PRRSV in 2006, PRRSV has been widely spread across the world. In previous studies, the positive rate of PRRSV was shown to be 55.21% (7,490/11,3567) in 29 provinces of China in 2012–2015 (21). In Central and Southern China, there was a positive rate of 50.62% (530/1,047) of PRRSV among 257 pig farms (22). In our study, the total positive rate of PRRSV was 50.89% (57/112) in nine provinces of China from 2016 to 2018, which was in accordance with the above scholars. PRRS is one of the most prevalent and threatening infectious diseases in Chinese pig farms.
Nsp2 and ORF5 (ORF5a) genes have the highest variability in PRRSV genome and are used as main target genes for PRRSV genetic variation. Phylogenetic tree analysis showed that all the 56 PRRSV strains identified in this study belong to the North American genotype and were distributed in lineage 1, 3, and 8 according to Shi et al. (18). In this study, the HP-PRRSV strain accounted for the highest proportion of epidemic strains in China; the NADC30-like strain had increased gradually, which was in accordance with the results of Gao et al. (23). However, some studies have shown that the NADC30-like strain in some regions of China have replaced the HP-PRRSV strain, and has become a new dominant strain (22). The rising infection rate of HP-PRRSV and NADC30-like strains may lead to a significant decrease in the effective protection rate of vaccines on pig farms.
The sequence alignments of Nsp2 gene revealed that the identified strain HLJ/2017/1127c in subline 8.1 had a high similarity with vaccine strain CH-1R, and existed a V deletion in the 630aa, suggesting that the identified strain may be a vaccine strain or a recombinant strain of a vaccine strain. Sequence alignments identified a discontinuous 30-amino-acid deletion (positions 481 and 533–561) and a 131-amino-acid discontinuity deletion (positions 323–433, 481, and 533–551) in Nsp2 of PPRSV isolates. JX/FC/2017/914(c, a1, b1) had the same deletion pattern as PRRSV strains HeN1401 and HeN1601, isolated by Zhang et al. (24). The recombinant analysis of the two epidemic strains revealed that HeN1401 and HeN1601 strains were generated by the recombinant weak vaccine strains TJbd14-1 and NADC30 (24). This further suggests that the identified strain JX/FC/2017/914(c, a1, b1) may also be a recombinant strain.
The GP5 protein sequences of different subline strains showed high similarity with the representative strains of the subline. The mutation sites of GP5 were mainly concentrated in the signal peptide and epitopes region. But some identified strains also have some amino acid consistent mutations in immune-related regions. Allende et al. found that nine amino acid site mutations may be closely related to the virulence of the PRRSV and that two sites (13 and 151 aa) were located in GP5 protein. The GP5 protein of high virulence strains generally were shown as R13 and R151 (25). Wesley et al. showed that the 137aa of GP5 protein can distinguish the attenuated vaccine strain (A137) and the wild strain (S137) (26). Therefore, the above three amino acid sites are often used to predict the virulence of PRRSV strains. The sequence analysis of GP5 protein showed that only one mutation pattern (R13 → Q13 and R151 → K151) existed in this study. Nine identified strains had mutated at position 13 aa, and 12 identified strains mutated at position 151 aa. In addition, the 137 aa of all identified strains is S. The results suggest that all identified strains may be wild strains, and most identified strains may be highly virulent strains in nine provinces of China during 2016–2018.
Studies have shown that ORF5a protein is essential for viral viability and infectivity (27, 28). There is fairly limited information available on current genetic variations of PRRSV ORF5a gene (29). Therefore, this study explored the genetic variation of ORF5a gene of PRRSV epidemic strains in China by molecular biological methods. The ORF5a protein generally encoded 46–51 amino acids of which ORF5a protein of PRRSV strains encoded 46 amino acids in lineage 1 and 8, and 51 amino acids in lineage 3 and 5 (30). All the ORF5a proteins identified in this study encoded 46 amino acids. Compared with the reference strain, the identified strains also showed high sequence similarity, and the mutation sites of ORF5a were mainly concentrated in the transmembrane region and the intramembrane region, while the other region was highly conserved. Our study demonstrated the existence of multiple different strains in the same region and extensive genetic mutation of PRRSV in China from 2016 to 2018.
The recombination analysis indicated that there may be multiple recombination regions in identified strains, and the recombination pattern was more complex. At present, many studies have shown that PRRSV strain in lineage 1 is prone to recombinant mutation, and some of the recombinant strains are more virulent than others (10, 19). Although the recombination pattern of the virus identified in this study is in accordance with that reported by some previous scholars, the change of pathogenicity of PRRSV by gene recombination is not absolute. This study only carried out partial gene (Nsp2 and ORF5) recombination analysis without the virus isolation and whole genome sequences of PRRSV, so the recombination of whole genome sequences is more complicated and different.
Conclusion
This study showed that PRRSV infection was prevalent in nine provinces of China from 2016 to 2018, and the prevalent PRRSV strain in most regions was still HP-PRRSV, while the NADC30 strain also occupied a certain proportion. There was a discontinuous 30-amino-acid deletion (positions 481 and 533–561) and a 131-amino-acid discontinuity deletion (positions 323–433, 481, and 533–551) in Nsp2 of PPRSV isolates. All identified strains in this study may be wild strains, and most identified strains may be highly virulent strains. This study identified highly homologous HP-PRRSV variants with distinct genetic mutation, which contributes to further analyzing the epidemics and evolution of PRRSV in the field.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
DS and GL conceived the study. BY, SQ, and JZ analyzed the data. BY and SQ wrote the manuscript for submission. WS, HQ, LL, and JY participated in the design of the study, performed the data collection and analysis, and commented on the manuscript. All authors approved the final manuscript.
Funding
This work was supported by the Science and Technology Development Project of Jilin Province (20190301007NY), the Heilongjiang Province Postdoctoral Science Foundation of China (LBH-Q16188), and Heilongjiang Bayi Agricultural University Support Program for San Heng San Zong (TDJH201804).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2021.605832/full#supplementary-material
References
1. Baron T, Albina E, Leforban Y, Madec F, Guilmoto H, Plana Duran J, et al. Report on the first outbreaks of the porcine reproductive and respiratory syndrome (PRRS) in France. Diagnosis and viral isolation. Ann Rech Vet. (1992) 23:161–6.
2. Li Y, Treffers EE, Napthine S, Tas A, Zhu L, Sun Z, et al. Transactivation of programmed ribosomal frameshifting by a viral protein. Proc Natl Acad Sci USA. (2014) 111:E2172–81. doi: 10.1073/pnas.1321930111
3. Fang Y, Treffers EE, Li Y, Tas A, Sun Z, van der Meer Y, et al. Efficient−2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc Natl Acad Sci USA. (2012) 109:E2920–8. doi: 10.1073/pnas.1211145109
4. Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, et al. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci. (2016) 4:129–54. doi: 10.1146/annurev-animal-022114-111025
6. Neumann EJ, Kliebenstein JB, Johnson CD, Mabry JW, Bush EJ, Seitzinger AH, et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc. (2005) 227:385–92. doi: 10.2460/javma.2005.227.385
7. Kuwahara H, Nunoya T, Tajima M, Kato A, Samejima T. An outbreak of porcine reproductive and respiratory syndrome in Japan. J Vet Med Sci. (1994) 56:901–9. doi: 10.1292/jvms.56.901
8. Chen N, Ye M, Huang Y, Li S, Xiao YZ, Li XS, et al. Identification of two porcine reproductive and respiratory syndrome virus variants sharing high genomic homology but with distinct virulence. Viruses. (2019) 11:875. doi: 10.3390/v11090875
9. Zhao K, Ye C, Chang XB, Jiang CG, Wang SJ, Cai XH, et al. Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J Virol. (2015) 89:10712–6. doi: 10.1128/JVI.01446-15
10. Zhou L, Wang Z, Ding Y, Ge X, Guo X, Yang H. NADC30-like strain of porcine reproductive and respiratory syndrome virus, China. Emerg Infect Dis. (2015) 21:2256–7. doi: 10.3201/eid2112.150360
11. Lu WH, Tun HM, Sun BL, Mo J, Zhou QF, Deng YX, et al. Re-emerging of porcine respiratory and reproductive syndrome virus (lineage 3) and increased pathogenicity after genomic recombination with vaccine variant. Vet Microbiol. (2015) 175:332–40. doi: 10.1016/j.vetmic.2014.11.016
12. Lu WH, Wei ZY, Zhang GQ, Li ZL, Ma JY, Xie QM, et al. Complete genome sequence of a novel variant porcine reproductive and respiratory syndrome virus (PRRSV) strain: evidence for recombination between vaccine and wild-type PRRSV strains. J Virol. (2012) 86:9543. doi: 10.1128/JVI.01341-12
13. Wang PP, Dong JG, Zhang LY, Liang PS, Liu YL, Wang L, et al. Sequence and phylogenetic analyses of the Nsp2 and ORF5 genes of porcine reproductive and respiratory syndrome virus in Boars from South China in 2015. Transbound Emerg Dis. (2017) 64:1953–64. doi: 10.1111/tbed.12594
14. Nilubol D, Tripipat T, Hoonsuwan T, Tipsombatboon P, Piriyapongsa J. Genetic diversity of the ORF5 gene of porcine reproductive and respiratory syndrome virus (PRRSV) genotypes I and II in Thailand. Arch Virol. (2013) 158:943–53. doi: 10.1007/s00705-012-1573-7
15. Cao ZX, Jiao PR, Huang YM, Qin HY, Kong LW, Pan QH, et al. Genetic diversity analysis of the ORF5 gene in porcine reproductive and respiratory syndrome virus samples from South China. Acta Vet Hung. (2012) 60:157–64. doi: 10.1556/avet.2012.013
16. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. (2013) 30:2725–9. doi: 10.1093/molbev/mst197
17. Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. (2007) 23:127–8. doi: 10.1093/bioinformatics/btl529
18. Shi M, Lam TT, Hon CC, Murtaugh MP, Davies PR, Hui RK, et al. Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol. (2010) 84:8700–11. doi: 10.1128/JVI.02551-09
19. Murtaugh MP, Stadejek T, Abrahante JE, Tommy TL, Frederick CL. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Research. (2010) 154:18–30. doi: 10.1016/j.virusres.2010.08.015
20. Dong J, Wang Y, Yu L, Zhang P, Liu X, Zhang L, et al. Pathogenicity of a newly emerged recombined porcine reproductive and respiratory syndrome virus strain (subgenotype III) in China. Vet Microbiol. (2017) 210:162–6. doi: 10.1016/j.vetmic.2017.09.018
21. Peng Z, Zhao T, Liang W, Song W, Gao Z, Tang X, et al. RT-PCR detection of porcine reproductive and respiratory syndrome virus based on the ORF5 gene in mainland China, 2012–2015. Acta Virol. (2017) 61:336–40. doi: 10.4149/av_2017_312
22. Liang W, Zhao T, Peng Z, Sun Y, Stratton CW, Zhou D, et al. Epidemiological and genetic characteristics of porcine reproductive and respiratory syndrome virus circulating in central and South China in 2016. Acta Trop. (2019) 190:83–91. doi: 10.1016/j.actatropica.2018.11.004
23. Gao JC, Xiong JY, Ye C, Chang XB, Guo JC, Jiang CG, et al. Genotypic and geographical distribution of porcine reproductive and respiratory syndrome viruses in mainland China in 1996–2016. Vet Microbiol. (2017) 208:164–72. doi: 10.1016/j.vetmic.2017.08.003
24. Zhang Z, Zhou L, Ge X, Guo X, Han J, Yang H. Evolutionary analysis of six isolates of porcine reproductive and respiratory syndrome virus from a single pig farm: MLV-evolved and recombinant viruses. Infect Genet Evol. (2018) 66:111–9. doi: 10.1016/j.meegid.2018.09.024
25. Allende R, Kutish GF, Laegreid W, Lu Z, Lewis TL, Rock DL, et al. Mutations in the genome of porcine reproductive and respiratory syndrome virus responsible for the attenuation phenotype. Arch Virol. (2000) 145:1149–61. doi: 10.1007/s007050070115
26. Wesley RD, Mengeling WL, Lager KM, Clouser DF, Landgraf JG, et al. Differentiation of a porcine reproductive and respiratory syndrome virus vaccine strain from North American field strains by restriction fragment length polymorphism analysis of ORF5. J Vet Diagn Invest. (1998) 10:140–4. doi: 10.1177/104063879801000204
27. van Dinten LC, den Boon JA, Wassenaar AL, Spaan WJ, Snijder EJ. An infectious arterivirus cDNA clone: identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc Natl Acad Sci USA. (1997) 94:991–6. doi: 10.1073/pnas.94.3.991
28. Firth AE, Brown CM. Detecting overlapping coding sequences in virus genomes. BMC Bioinformatics. (2006) 7:75. doi: 10.1186/1471-2105-7-75
29. Firth AE, Zevenhoven-Dobbe JC, Wills NM, Go YY, Balasuriya UB, Atkins JF, et al. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J Gen Virol. (2011) 92:1097–106. doi: 10.1099/vir.0.029264-0
Keywords: porcine reproductive and respiratory syndrome virus, Nsp2 gene, ORF5 gene, ORF5a gene, genetic evolution
Citation: Yin B, Qi S, Sha W, Qin H, Liu L, Yun J, Zhu J, Li G and Sun D (2021) Molecular Characterization of the Nsp2 and ORF5 (ORF5a) Genes of PRRSV Strains in Nine Provinces of China During 2016–2018. Front. Vet. Sci. 8:605832. doi: 10.3389/fvets.2021.605832
Received: 13 September 2020; Accepted: 09 February 2021;
Published: 04 March 2021.
Edited by:
Anuwat Wiratsudakul, Mahidol University, ThailandReviewed by:
Mohammed AbdelHameed AboElkhair, University of Sadat City, EgyptFrancisco Rivera-Benítez, Centro Nacional de Investigación Disciplinaria en Salud Animal e Inocuidad, Mexico
Copyright © 2021 Yin, Qi, Sha, Qin, Liu, Yun, Zhu, Li and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guojiang Li, amxsaWd1b2ppYW5nQDEyNi5jb20=; Dongbo Sun, ZG9uZ2Jvc3VuQDEyNi5jb20=
†These authors have contributed equally to this work