 Sadiya Parveen
Sadiya Parveen William R. Bishai
William R. Bishai- Center for Tuberculosis Research, Department of Medicine, Johns Hopkins School of Medicine, Baltimore, MD, United States
Mycobacterium tuberculosis (Mtb) has remained one of the major infectious disease killers for generations and generations. In 2023 alone, this ancient disease was responsible for the death of 1.4 million individuals and has infected 10.6 million people. With the ever-evolving multi- and extremely resistant Mtb strains, the need for novel and effective drugs requiring shorter treatment regimens represents an urgent medical need for the development of new drugs. Over the last two decades, the field of host-directed therapy as a potential novel avenue for new approaches to TB treatment, either as a mono or adjuvant therapy, has garnered increasing attention. Among many host-directed targets, host immunometabolism has emerged as one of the most attractive targets for developing new host-directed therapies. As one of the most successful bacterial pathogens, Mtb has evolved several mechanisms to modulate numerous host metabolic pathways, including glycolysis, glutaminolysis, Kreb cycle, and oxidative phosphorylation. This mini review will focus on glutamine metabolism and its emergence as a potential target for treating tuberculosis (TB). In the last several decades, the role of glutamine metabolism in cancer and neurological disorders has been extensively studied. However, the association of glutamine metabolism with infectious disease has remained underappreciated. The aim of this review is to not only discuss the current knowledge in the field but also the existing knowledge gap that needs further exploration.
Introduction
Mycobacterium tuberculosis (Mtb) is one of the most successful infectious agents worldwide. In the year 2023 alone, Mtb infected 10.6 million individuals and killed 1.3 million individuals (1). Mtb, the ancient pathogen, is known to modulate several host pathways to aid its own establishment, progression, and dissemination. These host pathways include various metabolic pathways such as glycolysis, pentose phosphate pathway, glutamine metabolism, fatty acid oxidation, Kreb cycle, and oxidative phosphorylation (2–5). Over the past several decades, glutamine metabolism has garnered increasing attention as a therapeutic target in the field of cancer and neurological disorders. However, there is a fundamental gap in the knowledge of how glutamine metabolism contributes to tuberculosis pathogenesis. Over the last few years, several researchers have published data that strongly supports the notion that glutamine metabolism plays a critical role in TB pathogenesis. This review attempts to compile all the studies and present them in the context of TB pathogenesis. The mini-review also discusses the unanswered questions in the field and potential strategies (including pitfalls) to find the answers.
Glutamine is the most abundant amino acid in the human body
Glutamine is the most abundant amino acid of all 20 amino acids in the human body. The average human plasma contains ~500–800 μM/L glutamine (after 12 h fasting), which is 20% of the total amino acid pool in the plasma (6). In tissues like skeletal muscles and liver, glutamine concentration can account for as much as 40%−80% of the total amino acid pool, further reinforcing the status of glutamine as the most abundant amino acid in the body (7, 8). Despite this heavy abundance, glutamine supplementation remains non-essential under normal physiological conditions. As a versatile amino acid, glutamine participates in a variety of metabolic pathways, such as synthesis of amino acids (e.g., asparagine), purines, pyrimidines, amino sugars, nicotinamide adenine dinucleotide phosphate (NADPH), glutathione, in addition to providing substrates for tricarboxylic acid (TCA) cycle. In short, glutamine serves as a fuel and nitrogen donor via ammonia for the pathways critical for cellular growth, proliferation, differentiation, and maintenance.
Glutamine transport and its cellular fates
As shown in Figure 1, Glutamine enters the cells via one of the several glutamine transporters like SLC1A5 (ASCT2), SLC38A1, or SLC38A2 present in the plasma membrane (9). Once inside the cell, glutamine may either stay in the cytosol and participate in the synthesis of purines, pyrimidines, amino sugars, or non-essential amino acids. Alternatively, glutamine can be transported to the mitochondrial matrix via a mitochondrial glutamine transporter (a SLC1A5 variant), where it undergoes glutaminolysis. First, glutaminases (GS) convert glutamine to glutamate, and then glutamate dehydrogenase (and several other aminotransferases) convert glutamate into alpha-ketoglutarate (ɑ-KG), which then serves as a substrate for the Kreb's cycle, eventually fueling the oxidative phosphorylation and ATP generation. Under hypoxic conditions, SLC25A11-driven transport of ɑ-KG from mitochondria to cytosol plays a critical role in mTROC1 activation and epigenetic modification via ɑ-KG-dependent dioxygenases (10). In addition to ɑ-KG, mitochondria can also export glutamine-derived glutamate to the cytosol via SLC25A18/22, which drives the synthesis of glutathione and non-essential amino acid synthesis in addition to cystine import (11).
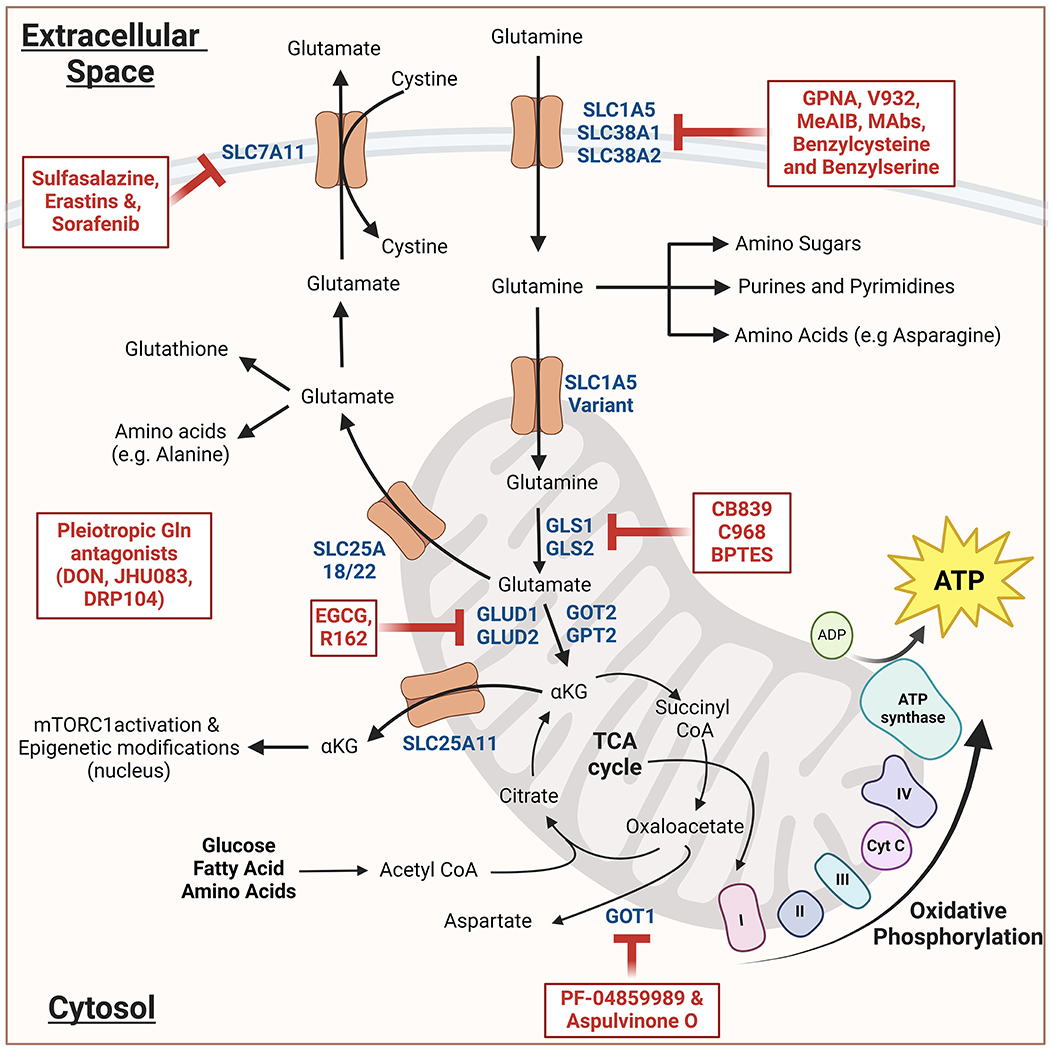
Figure 1. Schematic representation of glutamine metabolism and its inhibitors: Glutamine enters the cytosol and then either engages in various biosynthetic pathways or enters the mitochondria wherein it undergoes glutaminolysis. The resulting alpha-ketoglutarate (αKG) either enters Kreb's cycle leading to oxidative phosphorylation and ATP generation. Alternatively, it can lead to the mTORC1 activation and epigenetic modifications. The resulting glutamate when transported to the cytosol, generates glutathione and non-essential amino acids. All the inhibitors are indicated in red; dark blue indicates the important components of glutamine metabolism. The schematic was created using Biorender.com (Tornonto, Canada) by SP.
Glutamine distribution in the tissues
Glutamine concentration in various organs and tissues is driven by its synthesis, release, and uptake. Tissues like brain, skeletal muscles, lungs, adipose tissue, and liver exhibit high de novo glutamine synthesis activity. Glutamine synthesis is driven by an ATP-dependent glutamine synthetase (GS) that produces glutamine from glutamate and ammonia. De novo Glutamine synthesis serves two important functions; first, it provides glutamine required for rapid cell growth and proliferation. Second, it aids pH homeostasis by removing excess ammonia from the cells. Alterations in GLS activity have been associated with various pathologies like cancer, hyperammonemia, and neurological disorders, including behavior abnormalities, as well as cognitive and motor deficits (12). Tissues like intestinal mucosa, kidney, leukocytes, and vascular endothelial cells tend to heavily rely on glutamine consumption by upregulating the expression of glutaminase (GLS), the enzyme that catalyzes the conversion of glutamine to glutamate. There are two distinct GLS isoforms, (1) kidney-type glutaminase (GLS1) and (2) liver-type glutaminase (GLS2), encoded by genes located on chromosome 2 and 12 respectively (13–18). GLS1 has three additional isoforms; GLS or KGA (which corresponds to a longer transcript isoform), GLS C or GAC (which corresponds to a shorter transcript isoform), and GAM (with no catalytic activity). While GLS1 upregulation causes tumor progression in most cancers, the role of GLS2 seems more context specific. While GLS2 acts as a tumor-suppressor gene and prevents metastasis in many cancers (19), its upregulation has been shown to potentiate the occurrence and development of neuroblastoma tumors (20, 21).
Glutamine as a conditionally essential immunonutrient
Under normal physiological conditions, glutamine remains non-essential as the human body can endogenously produce from 40 to 80 g/L glutamine (22, 23). However, under catabolic conditions such as infections, glutamine becomes conditionally essential. Additionally, by virtue of working under these harsh and nutrient-deprived/restricted conditions, immune cells tend to heavily rely on glutamine as both the fuel and nitrogen source (24, 25). Paradoxically, under such highly catabolic conditions, several tissues increase their glutamine consumption while skeletal muscles tend to decrease their contribution to the serum glutamine concentration, further limiting glutamine availability for the immune cells. Such glutamine-depleted conditions often diminish the ability of the immune system to optimally combat the pathological agent/condition and lead to the worsening of the pathology. Hence, glutamine has long been considered a critical nutrient for the immune system, “an immunonutrient.” Depending upon the pathological condition, glutamine supplementation has been shown to improve clinical outcomes and prevent life-threatening conditions (26, 27). Almost all immune cells, including T-cells, macrophages, B-cells, and neutrophils, depend on glutamine uptake and utilization for their proliferation, differentiation, and activation (28). This heavy reliance of the immune system upon glutamine is a critical immune vulnerability. It is possible that pathogens may have evolved mechanisms to exploit this vulnerability to promote their own replication and dissemination.
Glutamine as one of the drivers of immunosuppression
During pathological conditions such as cancer and infections, the immune cells driving immunosuppression include myeloid-derived suppressor cells (MDSCs), regulatory T-cells (Tregs), and M2 macrophages (29–32). These immunosuppressive cells create an artificial scarcity of glutamine by ramping up the import and utilization of this amino acid. The artificial scarcity of glutamine hampers T-cells' ability to proliferate, activate, and differentiate into helper T-cells. Glutamine-deprived T-cells have also been shown to differentiate into FoxP3+ Tregs instead of Th1 helper cells, a defect that could be circumvented by simply feeding cells the cell-permeable ɑ-KG (33). L-glutamine also fuels the maturation process of MDSCs and is essential for their immunosuppressive activity (34). Additionally, glutamine favors the activation of M2 macrophages through the glutamine–UDP-N-acetylglucosamine pathway and glutamine-derived ɑ-KG (28, 35). Additionally, B-cells, especially the IL-10 expressing regulatory B-cells, which are critical for immune tolerance, have been shown to upregulate glutaminolysis (36, 37). Accordingly, in murine cancer models, blockade of glutamine metabolism has been shown to deplete MDSCs and Tregs populations and to metabolically reprogram M2 macrophages, alleviating immunosuppression and enhancing anti-tumor T-cell immunity (38–40).
Glutamine metabolism in infectious diseases (bacterial, viral, and fungal)
The speculation that glutamine plays a substantial role in infectious diseases dates back to 1975 when depletion of skeletal muscle Glutamine pool was detected under stress conditions like surgery, trauma, and inflammatory conditions. Several studies have shown that glutamine supplementation improved clinical outcomes and decreased mortality of severely ill patients with sepsis. In prolonged abdominal sepsis patients (n = 14), muscle glutamine concentration was identified as the single most reliable factor that effectively discriminated survivors from non-survivors (6, 8). The survivors exhibited higher glutamine and lower branched-chain amino acid levels in their muscles. In another study, low plasma glutamine concentration in critically ill patients was directly correlated with higher mortality rates (41). However, this initial enthusiasm was modestly dampened by the studies that found no correlation between plasma glutamine concentration and favorable outcomes in critically ill patients (41, 42). Despite these contradictory findings, several studies have observed substantial derangements in the glutamine distribution, transport, and synthesis in the tissues of patients carrying severe infections (43). During infections, skeletal muscles release two-fold as much glutamine, indicating a substantial increase in the de novo GS-mediated glutamine synthesis.
Alterations in glutamine metabolism have also been observed in several bacterial, viral, and fungal infections, including Human Immunodeficiency Virus (HIV), Mycobacterium tuberculosis (Mtb), COVID-19 and Aspergillus fumigatus. In 2017, Djoko et al. (44) demonstrated that Escherichia coli alters its glutamine metabolism and may utilize glutamine to overcome host-imposed metal toxicity. More recently, Turner et al. (45) showed that glutamine supplementation can protect host tissues from cholesterol-dependent cytotoxin secreted by pathogenic bacteria. HIV infection elevates glutamine levels and alters several key glutamine metabolism enzymes in human T-cells (46). In addition, the blockade of Glutamine metabolism was also shown to reverse a cognition deficit in the mouse model of HIV-associated neurocognitive disorders (47). In COVID-19 patients, a decline in circulatory glutamine levels directly correlates with the disease severity (48–54). In another meta-analysis study performed with West African cohorts, high plasma glutamine concentration was associated with a lower risk of COVID-19 infections and disease severity (55). Blocking glutamine metabolism was also reported to check the progression of Aspergillus fumigatum in macrophages and in the experimental model of Aspergillosis (56). These studies strongly support that alterations in the host glutamine metabolism may be a unique metabolic signature of several bacterial, viral, and fungal infections.
Glutamine metabolism alterations as the potential driver of TB pathogenesis
In the majority of patients who present with TB, the lung is the primary organ affected. The lung is also the organ with the second most abundant glutamine levels, with hitherto de novo glutamine synthesis activity. Alterations in lung glutamine levels and metabolism have been observed in several critical illnesses. For example, the lungs of septic surgical patients had 850% higher glutamine levels compared to the preoperative controls. Exogenous glutamine supplementation is still used in the Intensive Care Unit to prevent post-operative complications. Glutamine supplementation has also exerted beneficial effects on several respiratory and pulmonary ailments, such as asthma and acute respiratory distress syndrome. However, the impact of exogenous glutamine supplementation on TB progression has not been investigated so far.
Nonetheless, several studies have linked glutamine metabolism to TB pathogenesis. Glutamine is the primary nitrogen donor in Mtb-infected macrophages (57). With one exception (58), several studies have identified decreased levels of glutamine in serum/blood as one of the prominent diagnostic markers that distinguish active TB patients from latent TB patients and healthy controls (59–62). Mtb infection has also been shown to affect the expression of several genes associated with glutamine metabolism in both human macrophages and TB patients (63). The same study also demonstrated that perturbing glutamine pathway either via glutamine depletion or by using specific chemical inhibitors or due to genetic polymorphism (e.g., GLS2, SLC7A5, and SLC1A5), decreased T-cell associated cytokine production by the macrophages (PMID: 30541099).
Similarly, Vreiling et al. (64) demonstrated substantial alterations in the expression of multiple glutamine metabolism pathway transcripts in Mtb-infected M2 macrophages. More recently, Jiang et al. (65) using stable isotope labeling of glutamine and glucose followed by metabolomics, reported that M1 macrophages preferentially utilize glutamine, and the chemical or genetic ablation of glutamine synthase perturbs M1 polarization of the macrophages. Parveen et al. (66, 67) have reported that inhibiting glutamine metabolism in a murine model of TB has dual immunomodulatory and antibacterial activities. The study demonstrated that glutamine metabolism inhibition causes early recruitment of activated T-cells, reduced frequency of immunosuppressive myeloid cells, and enhanced antimycobacterial activity of macrophages concomitant with upregulation of host-protective metabolic pathways. Eventually, glutamine metabolism inhibition was also shown to reduce lung bacillary burden and improve lung histopathology and survival in murine hyperacute models of TB (66, 67).
Overall, these studies demonstrate that Mtb infection alters glutamine metabolism in infected macrophages, mice, and TB patients, and hence modulation of the glutamine levels and/or metabolism has the potential as an effective host-directed therapy.
Glutamine supplementation as an adjunctive therapy for TB
Several studies performed with patient cohorts from Africa, Indonesia, and Georgia have identified lower circulatory glutamine levels as a potential diagnostic signature of TB patients. The lower glutamine levels could be due to either decreased GLS-mediated synthesis or increased glutamine utilization/uptake. Further studies need to be performed to dissect the mechanism driving the lower glutamine levels in TB patients. Regardless of the mechanism, however, glutamine supplementation to restore circulatory glutamine levels should be tested as an adjunctive therapeutic approach. It is also clear that glutamine facilitates host-protective M1 polarization of the infected mouse macrophages, enhancing their antimycobacterial and inflammatory properties (65). In HIV patients, glutamine supplementation (20 g/day for 7 days) has improved serum glutamine levels (68).
While glutamine supplementation may represent a promising approach, the ex-vivo macrophage infection model is unsuitable for these studies as the macrophage culture media already contains milli-molar concentrations of glutamine. Testing glutamine supplementation in murine models of TB as an adjunctive therapy, despite being non-trivial, holds promise.
Glutamine metabolism inhibition as a host-directed therapy for TB
Several strategies have been employed to target glutamine metabolism in cancer models. These strategies can be majorly classified into four categories; (1) glutaminase inhibitors, (2) glutamine transporter inhibitors, (3) glutamine synthetases inhibitors, and (4) pleiotropic glutamine antagonists (Table 1, Figure 1) (98–100). While numerous studies have tested these approaches in various cancer models, only a handful of such studies have been performed in experimental models of TB. In a study by Koekan et al. (63) pharmacological inhibitors such as GPNA, BPTES, C968, and DON were shown to adversely impact the cytokine production by Mtb-stimulated peripheral blood mononuclear cells, particularly T-cell cytokines such as IFNy, IL-22, and IL-17. However, the impact of these inhibitors on the bacillary burden was not explored. More recently, Roca et al. (69) have demonstrated that TNF-treated infected macrophages upregulate glutaminolysis to induce pathogenic mitochondrial reactive oxygen species production (mROS). The pathogenic mROS production in macrophages could be successfully perturbed by BPTES and CB-839 (GLS1 inhibitors) and R162 (GLUD1 inhibitor) (69). Interestingly, the effect of GLS1 inhibitors was limited to the infected macrophages with excess TNF. A more recent report has shown that the glutamine metabolism antagonist prodrug, JHU083, significantly reduced the lung bacillary burden, improving lung histopathology and survival of Mtb-infected mice. The authors also noted significant immunological and metabolic reprogramming in the infected lungs, potentially driving the therapeutic benefit. As the drug was administered early after infection, future studies in acute and chronic models of TB infections will be required to assess the true translational potential of the drug (66, 67). All these early studies suggest that glutamine metabolism inhibition has the potential to be developed as an effective host-directed therapy option for TB.
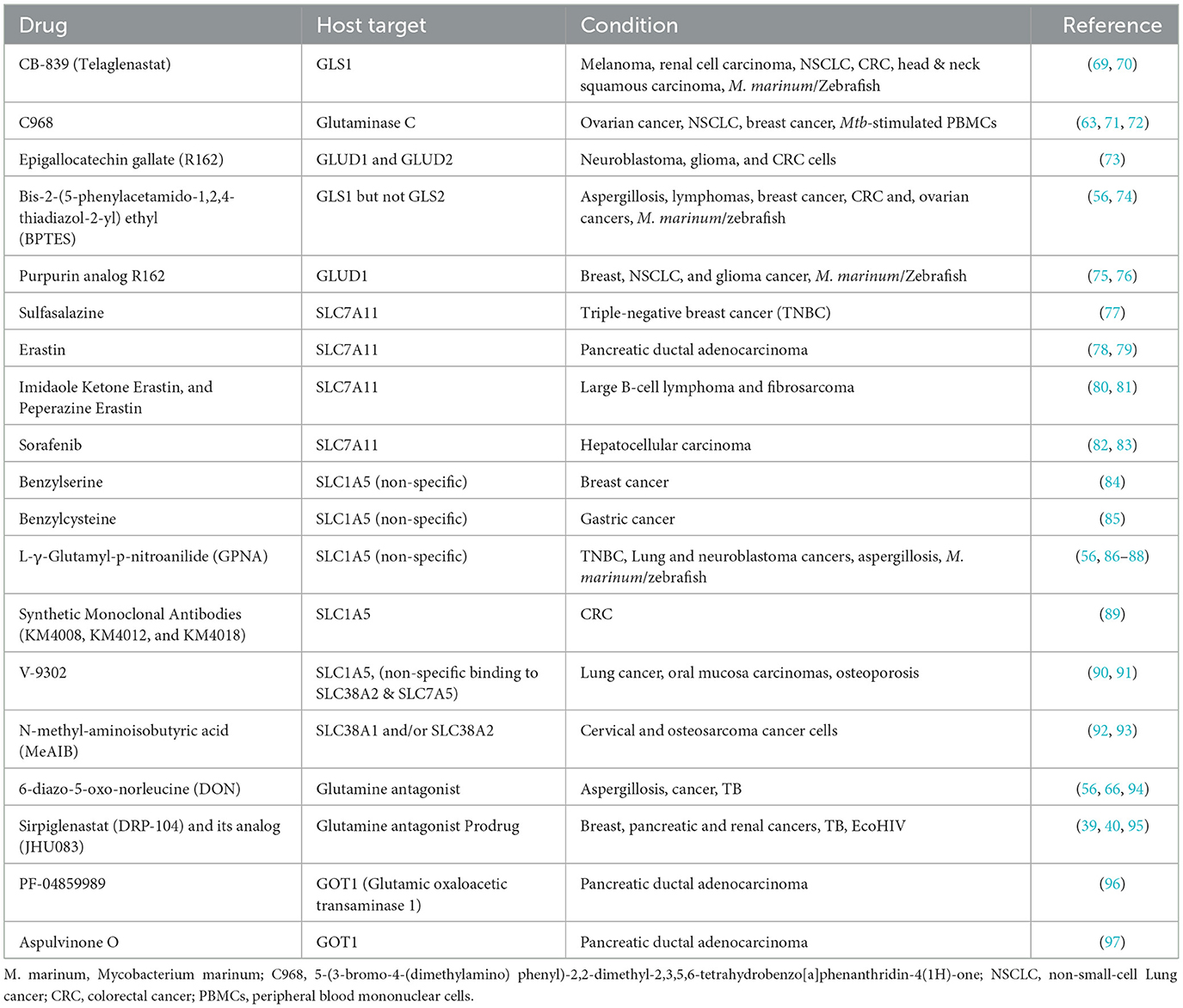
Table 1. List of glutamine metabolism inhibitors used in various pathological conditions.
Future of glutamine metabolism in TB pathogenesis
More studies are required to demonstrate the precise link between glutamine metabolism and TB pathogenesis. Several research avenues need further investigation. First and foremost, what signals/factors trigger the upregulation of glutamine metabolism during Mtb infection? Are these signals/triggers host-derived or secreted by Mtb? Is glutamine metabolism upregulation an early event during Mtb infection or a sustained phenotype of infected lungs? Does glutamine metabolism upregulation contribute to granuloma formation? And if yes, how does glutamine metabolism upregulation contribute to granuloma formation? How, why, and when do granulomas undergo metabolic reprogramming during different phases of infection? What are the differences in the metabolic profile of granuloma vs. non-granulomatous regions of Mtb-infected lungs? Do all lung cells undergo similar metabolic reprogramming, or is the effect driven by a handful of cell types? Do non-immune lung cells also undergo metabolic reprogramming upon Mtb infection? While numerous questions can be asked in the context of glutamine metabolism and TB pathogenesis, a solid first step could be investigating the impact of Mtb upon the metabolic profiles of the immune cells and using the knowledge to block glutamine metabolism mindfully and selectively. One of the important considerations will be to strike a delicate balance of selective and targeted inhibition of the problematic immunosuppressive cell populations while maintaining the wellbeing of host-protective cell types (38).
Challenges of the new field and potential solutions
Investigating the impact of glutamine metabolism on TB pathogenesis will require extensive deployment of metabolomics tools. There are a few unique challenges specific to TB research: first and foremost, as single-cell metabolomics is still in the initial phases of development, metabolomics of individual cell types remains challenging. Worldwide, most TB researchers (including those in developed countries) do not have access to a flow sorter in a BSL3-containment facility, making it harder to work with individual cell types. While immortalized immune cell lines are an option, magnetic bead-based enrichment of the primary lung-derived cells may be helpful in several cases. Over the years, there has been tremendous development and expansion of bead-based kits. However, these kits need extensive standardization and optimization when working with complex tissue samples. The second challenge will be distinguishing the host vs. bacterial-derived metabolites in the infected samples. Novel isotope labeling methods may need to be developed to circumvent this issue. Third, metabolite fluxes tend to be extremely sensitive to their milieu and are potentially affected during tissue collection, processing, sorting, and metabolite extraction, causing extensive batch-to-batch variations and processing artifacts. Fast, efficient, and homogenous processing of the infected samples, while not impossible to achieve, all these factors can be challenging in a BSL3 containment facility. Such processing artifacts can be avoided using stable isotope-labeled nutrients (e.g., [13C] glucose or [13C] glutamine) to probe the metabolic fluxes within the intact lungs in vivo. This method was recently developed by Faubert et al. (101) and was used to investigate the metabolic activity of tumors in vivo using the [13C] glucose label. Despite the unique challenges faced by TB researchers, the glutamine metabolism and TB pathogenesis field holds tremendous promise and needs our immediate attention.
Conclusion
In conclusion, sufficient data supports the claim that glutamine metabolism is a promising host-directed mechanism that can be targeted to perturb TB pathogenesis. Several inhibitors, originally identified by cancer researchers, can be tested as potential host-directed therapy. However, several missing pieces in the puzzles must be placed to successfully predict and implement these therapies and evaluate their therapeutic potential.
Author contributions
SP: Conceptualization, Funding acquisition, Investigation, Software, Supervision, Visualization, Writing—original draft, Writing—review & editing. WRB: Conceptualization, Funding acquisition, Software, Supervision, Visualization, Writing—review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors gratefully acknowledge the support of the JHU-TRAC development award grant for covering the publishing cost of the minireview.
Acknowledgments
We are grateful to John R. Murphy, Jonathan Powell, and Barbara Slusher for their advice and insights. SP acknowledges the support of the JHU-TRAC development award grant from NIH grant P30AI18436. WRB acknowledges the grant support from NIH grants P30AI18436 and R01AI155602.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, et al. (1)H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res. (2011) 10:2238–47. doi: 10.1021/pr101054m
3. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML, et al. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. (2015) 5:18176. doi: 10.1038/srep18176
4. Kim JS, Kim YR, Yang CS. Host-directed therapy in tuberculosis: targeting host metabolism. Front Immunol. (2020) 11:1790. doi: 10.3389/fimmu.2020.01790
5. Collins JM, Jones DP, Sharma A, Khadka M, Liu KH, Kempker RR, et al. TCA cycle remodeling drives proinflammatory signaling in humans with pulmonary tuberculosis. PLoS Pathog. (2021) 17:e1009941. doi: 10.1371/journal.ppat.1009941
6. Roth E. Nonnutritive effects of glutamine. J Nutr. (2008) 138:2025s−31s. doi: 10.1093/jn/138.10.2025S
7. Labow BI, Souba WW, Abcouwer SF. Mechanisms governing the expression of the enzymes of glutamine metabolism–glutaminase and glutamine synthetase. J Nutr. (2001) 131(9 Suppl):2467S−74S; discussion 2486S−7S. doi: 10.1093/jn/131.9.2467S
8. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. (2018) 10:1564. doi: 10.3390/nu10111564
9. Scalise M, Pochini L, Galluccio M, Console L, Indiveri C. Glutamine transport and mitochondrial metabolism in cancer cell growth. Front Oncol. (2017) 7:306. doi: 10.3389/fonc.2017.00306
10. de la Cruz López KG, Toledo Guzmán ME, Sánchez EO, García Carrancá A. mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front Oncol. (2019) 9:1373. doi: 10.3389/fonc.2019.01373
11. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. (2020) 52:1496–516. doi: 10.1038/s12276-020-00504-8
12. He Y, Hakvoort TBM, Köhler SE, Vermeulen JLM, de Waart DR, de Theije C, et al. Glutamine synthetase in muscle is required for glutamine production during fasting and extrahepatic ammonia detoxification. J Biol Chem. (2010) 285:9516–24. doi: 10.1074/jbc.M109.092429
13. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. (2016) 126:3313–35. doi: 10.1172/JCI86387
14. Guo Y, Deng Y, Li X, Ning Y, Lin X, Guo S, et al. Glutaminolysis was induced by TGF-β1 through PP2Ac regulated Raf-MEK-ERK signaling in endothelial cells. PLoS ONE. (2016) 11:e0162658. doi: 10.1371/journal.pone.0162658
15. Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML, et al. The role of glutaminase in cancer. Histopathology. (2020) 76:498–508. doi: 10.1111/his.14014
16. Matés JM, Campos-Sandoval JA, de Los Santos-Jiménez J, Segura JA, Alonso FJ, Márquez J, et al. Metabolic reprogramming of cancer by chemicals that target glutaminase isoenzymes. Curr Med Chem. (2020) 27:5317–39. doi: 10.2174/0929867326666190416165004
17. Yu W, Yang X, Zhang Q, Sun L, Yuan S, Xin Y, et al. Targeting GLS1 to cancer therapy through glutamine metabolism. Clin Transl Oncol. (2021) 23:2253–68. doi: 10.1007/s12094-021-02645-2
18. Wang S, Yan Y, Xu WJ, Gong SG, Zhong XJ, An QY, et al. The role of glutamine and glutaminase in pulmonary hypertension. Front Cardiovasc Med. (2022) 9:838657. doi: 10.3389/fcvm.2022.838657
19. Zhang C, Liu J, Zhao Y, Yue X, Zhu Y, Wang X, et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife. (2016) 5:e10727. doi: 10.7554/eLife.10727
20. Katt WP, Lukey MJ, Cerione RA. A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future Med Chem. (2017) 9:223–43. doi: 10.4155/fmc-2016-0190
21. Dias MM, Adamoski D, Dos Reis LM, Ascenção CFR, de Oliveira KRS, Mafra ACP, et al. GLS2 is protumorigenic in breast cancers. Oncogene. (2020) 39:690–702. doi: 10.1038/s41388-019-1007-z
22. Berg A, Norberg A, Martling CR, Gamrin L, Rooyackers O, Wernerman J, et al. Glutamine kinetics during intravenous glutamine supplementation in ICU patients on continuous renal replacement therapy. Intensive Care Med. (2007) 33:660–6. doi: 10.1007/s00134-007-0547-9
23. Wernerman J. Clinical use of glutamine supplementation. J Nutr. (2008) 138:2040s−4s. doi: 10.1093/jn/138.10.2040S
24. Curi R, Newsholme P, Newsholme EA. Intracellular distribution of some enzymes of the glutamine utilisation pathway in rat lymphocytes. Biochem Biophys Res Commun. (1986) 138:318–22. doi: 10.1016/0006-291X(86)90282-2
25. Newsholme EA, Newsholme P, Curi R. The role of the citric acid cycle in cells of the immune system and its importance in sepsis, trauma and burns. Biochem Soc Symp. (1987) 54:145–62.
26. Stehle P, Ellger B, Kojic D, Feuersenger A, Schneid C, Stover J, et al. Glutamine dipeptide-supplemented parenteral nutrition improves the clinical outcomes of critically ill patients: a systematic evaluation of randomised controlled trials. Clin Nutr ESPEN. (2017) 17:75–85. doi: 10.1016/j.clnesp.2016.09.007
27. Smedberg M, Helleberg J, Norberg Å, Tjäder I, Rooyackers O, Wernerman J, et al. Plasma glutamine status at intensive care unit admission: an independent risk factor for mortality in critical illness. Crit Care. (2021) 25:240. doi: 10.1186/s13054-021-03640-3
28. Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal. (2022) 20:114. doi: 10.1186/s12964-022-00909-0
29. Kumar P, Kumar A, Parveen S, Murphy JR, Bishai W. Recent advances with Treg depleting fusion protein toxins for cancer immunotherapy. Immunotherapy. (2019) 11:1117–28. doi: 10.2217/imt-2019-0060
30. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
31. Parveen S, Murphy JR, Bishai WR. Targeting inhibitory cells such as Tregs and MDSCs in the tuberculous granuloma. In: Karakousis PC, Hafner R, Gennaro ML, , editors. Advances in Host-Directed Therapies against Tuberculosis. Cham: Springer International Publishing (2021), p. 169–203. doi: 10.1007/978-3-030-56905-1_11
32. Lasser SA, Ozbay Kurt FG, Arkhypov I, Utikal J, Umansky V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol. (2024) 21:147–64. doi: 10.1038/s41571-023-00846-y
33. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. (2015) 8:ra97. doi: 10.1126/scisignal.aab2610
34. Hammami I, Chen J, Bronte V, DeCrescenzo G, Jolicoeur M. L-glutamine is a key parameter in the immunosuppression phenomenon. Biochem Biophys Res Commun. (2012) 425:724–9. doi: 10.1016/j.bbrc.2012.07.139
35. Ren W, Xia Y, Chen S, Wu G, Bazer FW, Zhou B, et al. Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv Nutr. (2019) 10:321–30. doi: 10.1093/advances/nmy084
36. Choi SC, Li W, Zhang X, Kanda N, Zeumer-Spataro L, Teng X, et al. Pharmacologically inferred glycolysis and glutaminolysis requirement of B cells in lupus-prone mice. J Immunol. (2022) 208:2098–108. doi: 10.4049/jimmunol.2100356
37. Liu JQ, Geng XR, Hu TY, Mo LH, Luo XQ, Qiu SY, et al. Glutaminolysis is required in maintaining immune regulatory functions in B cells. Mucosal Immunol. (2022) 15:268–78. doi: 10.1038/s41385-021-00481-9
38. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175:1780–95.e1719. doi: 10.1016/j.cell.2018.10.001
39. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. (2019) 366:1013–21. doi: 10.1126/science.aav2588
40. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. (2020) 130:3865–84. doi: 10.1172/JCI131859
41. Rodas PC, Rooyackers O, Hebert C, Norberg Å, Wernerman J. Glutamine and glutathione at ICU admission in relation to outcome. Clin Sci. (2012) 122:591–7. doi: 10.1042/CS20110520
42. Heyland D, Muscedere J, Wischmeyer PE, Cook D, Jones G, Albert M, et al. A randomized trial of glutamine and antioxidants in critically ill patients. N Engl J Med. (2013) 368:1489–97. doi: 10.1056/NEJMoa1212722
43. Karinch AM, Pan M, Lin CM, Strange R, Souba WW. Glutamine metabolism in sepsis and infection. J Nutr. (2001) 131(9 Suppl):2535S−8S; discussion 2550S−31S. doi: 10.1093/jn/131.9.2535S
44. Djoko KY, Phan MD, Peters KM, Walker MJ, Schembri MA, McEwan AG, et al. Interplay between tolerance mechanisms to copper and acid stress in Escherichia coli. Proc Natl Acad Sci U S A. (2017) 114:6818–23. doi: 10.1073/pnas.1620232114
45. Turner CT, Zeglinski MR, Richardson KC, Zhao H, Shen Y, Papp A, et al. Granzyme K Expressed by classically activated macrophages contributes to inflammation and impaired remodeling. J Invest Dermatol. (2019) 139:930–9. doi: 10.1016/j.jid.2018.09.031
46. Hegedus A, Kavanagh Williamson M, Khan MB, Dias Zeidler J, Poian ATD, El-Bacha T, et al. Evidence for altered glutamine metabolism in human immunodeficiency virus type 1 infected primary human CD4(+) T cells. AIDS Res Hum Retroviruses. (2017) 33:1236–47. doi: 10.1089/aid.2017.0165
47. Nedelcovych MT, Kim BH, Zhu X, Lovell LE, Manning AA, Kelschenbach J, et al. Glutamine antagonist JHU083 normalizes aberrant glutamate production and cognitive deficits in the ecoHIV murine model of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. (2019) 14:391–400. doi: 10.1007/s11481-019-09859-w
48. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. (2020) 182:59–72.e15. doi: 10.1016/j.cell.2020.05.032
49. Ansone L, Briviba M, Silamikelis I, Terentjeva A, Perkons I, Birzniece L, et al. Amino acid metabolism is significantly altered at the time of admission in hospital for severe COVID-19 patients: findings from longitudinal targeted metabolomics analysis. Microbiol Spectr. (2021) 9:e0033821. doi: 10.1128/spectrum.00338-21
50. Atila A, Alay H, Yaman ME, Akman TC, Cadirci E, Bayrak B, et al. The serum amino acid profile in COVID-19. Amino Acids. (2021) 53:1569–88. doi: 10.1007/s00726-021-03081-w
51. Rees CA, Rostad CA, Mantus G, Anderson EJ, Chahroudi A, Jaggi P, et al. Altered amino acid profile in patients with SARS-CoV-2 infection. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2101708118
52. Wu P, Chen D, Ding W, Wu P, Hou H, Bai Y, et al. The trans-omics landscape of COVID-19. Nat Commun. (2021) 12:4543. doi: 10.1038/s41467-021-24482-1
53. Lee JW, Su Y, Baloni P, Chen D, Pavlovitch-Bedzyk AJ, Yuan D, et al. Integrated analysis of plasma and single immune cells uncovers metabolic changes in individuals with COVID-19. Nat Biotechnol. (2022) 40:110–20. doi: 10.1038/s41587-021-01020-4
54. Masoodi M, Peschka M, Schmiedel S, Haddad M, Frye M, Maas C, et al. Disturbed lipid and amino acid metabolisms in COVID-19 patients. J Mol Med. (2022) 100:555–68. doi: 10.1007/s00109-022-02177-4
55. Li XK, Tu B, Zhang XA, Xu W, Chen JH, Zhao GY, et al. Dysregulation of glutamine/glutamate metabolism in COVID-19 patients: a metabolism study in African population and mini meta-analysis. J Med Virol. (2023) 95:e28150. doi: 10.1002/jmv.28150
56. Antunes D, Gonçalves SM, Matzaraki V, Rodrigues CS, Gonçales RA, Rocha J, et al. Glutamine metabolism supports the functional activity of immune cells against aspergillus fumigatus. Microbiol Spectr. (2023) 11:e0225622. doi: 10.1128/spectrum.02256-22
57. Borah K, Beyß M, Theorell A, Wu H, Basu P, Mendum TA, et al. Intracellular Mycobacterium tuberculosis exploits multiple host nitrogen sources during growth in human macrophages. Cell Rep. (2019) 29:3580–91.e3584. doi: 10.1016/j.celrep.2019.11.037
58. Zhou A, Ni J, Xu Z, Wang Y, Lu S, Sha W, et al. Application of (1)h NMR spectroscopy-based metabolomics to sera of tuberculosis patients. J Proteome Res. (2013) 12:4642–9. doi: 10.1021/pr4007359
59. Weiner J 3rd, Parida SK, Maertzdorf J, Black GF, Repsilber D, Telaar A, et al. Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS ONE. (2012) 7:e40221. doi: 10.1371/annotation/b7f554bc-ad78-4745-9cd6-e14954d6a01d
60. Albors-Vaquer A, Rizvi A, Matzapetakis M, Lamosa P, Coelho AV, Patel AB, et al. Active and prospective latent tuberculosis are associated with different metabolomic profiles: clinical potential for the identification of rapid and non-invasive biomarkers. Emerg Microbes Infect. (2020) 9:1131–9. doi: 10.1080/22221751.2020.1760734
61. Cho Y, Park Y, Sim B, Kim J, Lee H, Cho SN, et al. Identification of serum biomarkers for active pulmonary tuberculosis using a targeted metabolomics approach. Sci Rep. (2020) 10:3825. doi: 10.1038/s41598-020-60669-0
62. Magdalena D, Michal S, Marta S, Magdalena KP, Anna P, Magdalena G, et al. Targeted metabolomics analysis of serum and Mycobacterium tuberculosis antigen-stimulated blood cultures of pediatric patients with active and latent tuberculosis. Sci Rep. (2022) 12:4131. doi: 10.1038/s41598-022-08201-4
63. Koeken V, Lachmandas E, Riza A, Matzaraki V, Li Y, Kumar V, et al. Role of glutamine metabolism in host defense against Mycobacterium tuberculosis infection. J Infect Dis. (2019) 219:1662–70. doi: 10.1093/infdis/jiy709
64. Vrieling F, Kostidis S, Spaink HP, Haks MC, Mayboroda OA, Ottenhoff THM, et al. Analyzing the impact of Mycobacterium tuberculosis infection on primary human macrophages by combined exploratory and targeted metabolomics. Sci Rep. (2020) 10:7085. doi: 10.1038/s41598-020-62911-1
65. Jiang Q, Qiu Y, Kurland IJ, Drlica K, Subbian S, Tyagi S, et al. Glutamine is required for M1-like polarization of macrophages in response to Mycobacterium tuberculosis infection. MBio. (2022) 13:e0127422. doi: 10.1128/mbio.01274-22
66. Parveen S, Shen J, Lun S, Zhao L, Alt J, Koleske B, et al. Glutamine metabolism inhibition has dual immunomodulatory and antibacterial activities against Mycobacterium tuberculosis. Nat Commun. (2023) 14:7427. doi: 10.1038/s41467-023-43304-0
67. Parveen S, Shen J, Lun S, Zhao L, Koleske B, Leone RD, et al. Glutamine metabolism inhibition has dual immunomodulatory and antibacterial activities against Mycobacterium tuberculosis. bioRxiv. (2023). doi: 10.1101/2023.02.23.529704
68. Borges-Santos MD, Moreto F, Pereira PC, Ming-Yu Y, Burini RC. Plasma glutathione of HIV? patients responded positively and differently to dietary supplementation with cysteine or glutamine. Nutrition. (2012) 28:753–6. doi: 10.1016/j.nut.2011.10.014
69. Roca FJ, Whitworth LJ, Prag HA, Murphy MP, Ramakrishnan L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science. (2022)376: eabh. doi: 10.1126/science.abh2841
70. Biancur DE, Paulo JA, Małachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. (2017) 8:15965. doi: 10.1038/ncomms15965
71. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. (2010) 18:207–19. doi: 10.1016/j.ccr.2010.08.009
72. Yuan L, Sheng X, Clark LH, Zhang L, Guo H, Jones HM, et al. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am J Transl Res. (2016) 8:4265–77.
73. Peeters TH, Lenting K, Breukels V, van Lith SAM, van den Heuvel CNAM, Molenaar R, et al. Isocitrate dehydrogenase 1-mutated cancers are sensitive to the green tea polyphenol epigallocatechin-3-gallate. Cancer Metab. (2019) 7:4. doi: 10.1186/s40170-019-0198-7
74. Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. (2012) 55:10551–63. doi: 10.1021/jm301191p
75. Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. (2015) 27:257–70. doi: 10.1016/j.ccell.2014.12.006
76. Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. (2018) 69:87–99.e7. doi: 10.1016/j.molcel.2017.11.025
77. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. (2013) 24:450–65. doi: 10.1016/j.ccr.2013.08.020
78. Jyotsana N, Ta KT, DelGiorno KE. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front Oncol. (2022) 12:858462. doi: 10.3389/fonc.2022.858462
79. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. (2020) 368:85–9. doi: 10.1126/science.aaw9872
80. Yan R, Xie E, Li Y, Li J, Zhang Y, Chi X, et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. (2022) 32:687–90. doi: 10.1038/s41422-022-00642-w
81. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. (2019) 26:623–33.e9. doi: 10.1016/j.chembiol.2019.01.008
82. Zhang L, Li XM, Shi XH, Ye K, Fu XL, Wang X, et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol Sin. (2023) 44:622–34. doi: 10.1038/s41401-022-00981-9
83. Zhang Y, Li Y, Qiu Q, Chen Z, Du Y, Liu X. MITD1 Deficiency suppresses clear cell renal cell carcinoma growth and migration by inducing ferroptosis through the TAZ/SLC7A11 pathway. Oxid Med Cell Longev. (2022) 2022:7560569. doi: 10.1155/2022/7560569
84. van Geldermalsen M, Quek LE, Turner N, Freidman N, Pang A, Guan YF, et al. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer. (2018) 18:689. doi: 10.1186/s12885-018-4599-8
85. Sun HJ, Meng LY, Shen Y, Zhu YZ, Liu HR. S-benzyl-cysteine-mediated cell cycle arrest and apoptosis involving activation of mitochondrial-dependent caspase cascade through the p53 pathway in human gastric cancer SGC-7901 cells. Asian Pac J Cancer Prev. (2013) 14:6379–84. doi: 10.7314/apjcp.2013.14.11.6379
86. van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. (2016) 35:3201–8. doi: 10.1038/onc.2015.381
87. Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. (2013) 19:560–70. doi: 10.1158/1078-0432.CCR-12-2334
88. Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H, et al. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol. (2015). 235:90–100. doi: 10.1002/path.4429
89. Suzuki M, Toki H, Furuya A, Ando H. Establishment of monoclonal antibodies against cell surface domains of ASCT2/SLC1A5 and their inhibition of glutamine-dependent tumor cell growth. Biochem Biophys Res Commun. (2017) 482:651–7. doi: 10.1016/j.bbrc.2016.11.089
90. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. (2018) 24:194–202. doi: 10.1038/nm.4464
91. Bröer A, Fairweather S, Bröer S. Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets. Front Pharmacol. (2018) 9:785. doi: 10.3389/fphar.2018.00785
92. Bröer A, Rahimi F, Bröer S. Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J Biol Chem. (2016) 291:13194–205. doi: 10.1074/jbc.M115.700534
93. Tang F, Zheng P. Tumor cells versus host immune cells: whose PD-L1 contributes to PD-1/PD-L1 blockade mediated cancer immunotherapy? Cell Biosci. (2018) 8:34. doi: 10.1186/s13578-018-0232-4
94. Lemberg KM, Vornov JJ, Rais R, Slusher BS. We're Not “DON” yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol Cancer Ther. (2018) 17:1824–32. doi: 10.1158/1535-7163.MCT-17-1148
95. Rais R, Lemberg KM, Tenora L, Arwood ML, Pal A, Alt J, et al. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci Adv. (2022) 8:eabq5925. doi: 10.1126/sciadv.abq5925
96. Yoshida T, Yamasaki S, Kaneko O, Taoka N, Tomimoto Y, Namatame I, et al. A covalent small molecule inhibitor of glutamate-oxaloacetate transaminase 1 impairs pancreatic cancer growth. Biochem Biophys Res Commun. (2020) 522:633–8. doi: 10.1016/j.bbrc.2019.11.130
97. Sun W, Luan S, Qi C, Tong Q, Yan S, Li H, et al. Aspulvinone O, a natural inhibitor of GOT1 suppresses pancreatic ductal adenocarcinoma cells growth by interfering glutamine metabolism. Cell Commun Signal. (2019) 17:111. doi: 10.1186/s12964-019-0425-4
98. Nachef M, Ali AK, Almutairi SM, Lee SH. Targeting SLC1A5 and SLC3A2/SLC7A5 as a potential strategy to strengthen anti-tumor immunity in the tumor microenvironment. Front Immunol. (2021) 12:624324. doi: 10.3389/fimmu.2021.624324
99. Halama A, Suhre K. Advancing cancer treatment by targeting glutamine metabolism-a roadmap. Cancers. (2022) 14:553. doi: 10.3390/cancers14030553
100. Jin J, Byun JK, Choi YK, Park KG. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. (2023) 55:706–15. doi: 10.1038/s12276-023-00971-9
Keywords: tuberculosis, host-directed therapies, glutamine metabolism, immunometabolism, infectious diseases
Citation: Parveen S and Bishai WR (2024) Role of glutamine metabolism in tuberculosis pathogenesis: a mini review. Front. Tuberc. 2:1432880. doi: 10.3389/ftubr.2024.1432880
Received: 14 May 2024; Accepted: 26 June 2024;
Published: 10 July 2024.
Edited by:
Alissa Rothchild, University of Massachusetts Amherst, United StatesReviewed by:
Bibhuti Bhusan Mishra, Albany Medical College, United StatesCopyright © 2024 Parveen and Bishai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sadiya Parveen, c3BhcnZlZTImI3gwMDA0MDtqaG1pLmVkdQ==; William R. Bishai, d2Jpc2hhaTEmI3gwMDA0MDtqaG1pLmVkdQ==