 Vojo Deretic
Vojo Deretic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Tuberc. , 29 September 2023
Sec. Pathogen and Host Biology of Tuberculosis
Volume 1 - 2023 | https://doi.org/10.3389/ftubr.2023.1275882
Nearly two decades have passed since the first report on autophagy acting as a cell-autonomous defense against Mycobacterium tuberculosis. This helped usher a new area of research within the field of host-pathogen interactions and led to the recognition of autophagy as an immunological mechanism. Interest grew in the fundamental mechanisms of antimicrobial autophagy and in the prophylactic and therapeutic potential for tuberculosis. However, puzzling in vivo data have begun to emerge in murine models of M. tuberculosis infection. The control of infection in mice affirmed the effects of certain autophagy genes, specifically ATG5, but not of other ATGs. Recent studies with a more complete inactivation of ATG genes now show that multiple ATG genes are indeed necessary for protection against M. tuberculosis. These particular ATG genes are involved in the process of membrane atg8ylation. Atg8ylation in mammalian cells is a broad response to membrane stress, damage and remodeling of which canonical autophagy is one of the multiple downstream outputs. The current developments clarify the controversies and open new avenues for both fundamental and translational studies.
The most devastating infectious diseases of humans are often the result of novel encounters between the microbe and the human host, as reflected in the recent, past and ongoing pandemics caused by coronaviruses, HIV, infleunza, tuberculosis, and others. They often have history of initial maladaptation between the host and the pathogen causing high lethality, subsequently moderated by the evolution of lineages that balance transmission with pathogenicity. The association between tuberculosis and humans is estimated to had started ca., 50,000–96,000 years ago based on archeological estimates of the most recent common ancestor, with evidence of continuing co-evolution of the pathogen and the host still underway as reflected in different lineages of Mycobacterium tuberculosis (Mtb) (1) and high mortality of tuberculosis globally (2). The present-day host pathogen interactions in tuberculosis are complex at cell-autonomous and innate and adaptive immunity levels (3–6) underlying the infectious cycle in human populations, including transmission (7), primary active, latent and reactivation disease (8). The clinical presentation and disease outcomes are governed by predisposing conditions, comorbidities, age, coinfections, population and socioeconomic determinants, further complicated by the notorious emergence of antibiotic resistance (8). Among the repertoire of host cell-autonomous defenses and inflammation modulators engaged during Mtb infection (6) is the process of autophagy (9, 10), which represents one of the outputs of a broader membrane stress response, termed atg8ylation (11). The topic of this review article is the evolving understanding of membrane atg8ylation (Figure 1A), autophagy and other atg8ylation-dependent processes (12–15) (Figure 1B), with a focus on their anti-inflammatory and antimicrobial effects during Mtb infection (Figure 2) and tuberculosis pathogenesis.
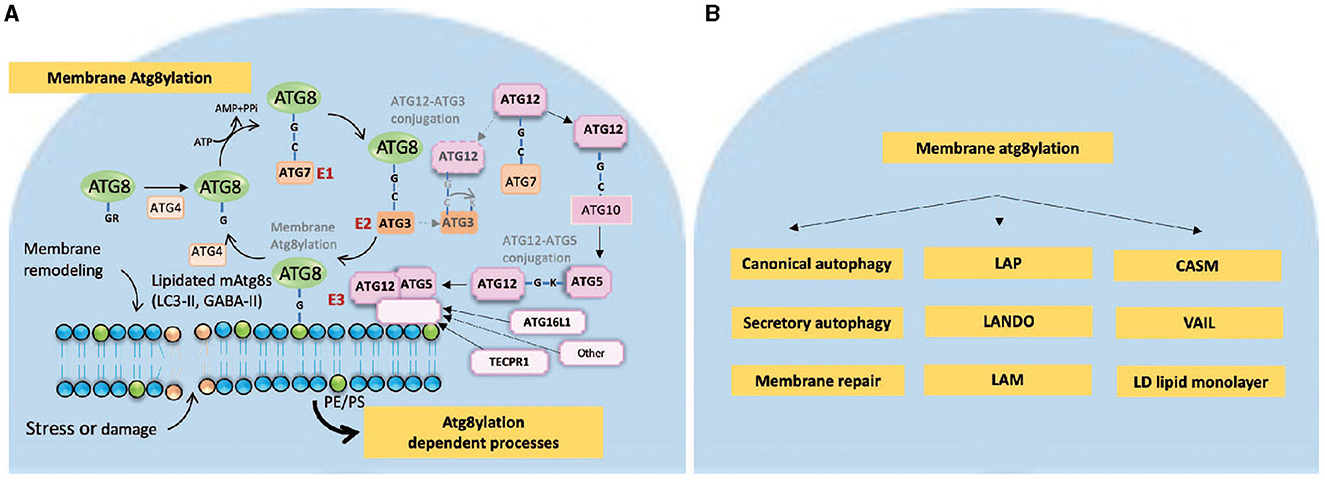
Figure 1. Membrane atg8ylation. (A) Membrane atg8ylation process. Membrane atg8ylation is a pathway of covalent membrane modification by ubiquitin-like proteins of the mammalian ATG8 family (LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2). Membrane atg8ylation occurs in response to membrane stress, damage and remodeling signals and participates in various atg8ylation-dependent processes including canonical autophagy as well as a number of non-canonical and non-autophagy phenomena [listed in (B)]. The cycle of membrane atg8ylation and de-atg8ylation resembles the process of protein ubiquitylation and deubiquitylation. It involves activation by ATP of the ATG8 C-terminal Gly residue (once it is exposed following proteolytic processing by ATG4 peptidases) to make a conjugate with E1 enzyme ATG7 via a Gly-Cys high energy thioester bond. ATG8s are then transferred to the E2 enzyme ATG3, which itself has intrinsic affinity for membranes. The final step is the transfer of ATG8s from the ATG8-ATG3 conjugates to form a covalent amide bond with the headgroups of phospholipids such as PE (phosphatidylethanolamine) and alternatively PS (phosphatidylserine). This final step is catalyzed by E3 ligases, which include as a catalytic component the ATG12-ATG5 covalent protein conjugate formed in a separate cycle. ATG12-ATG5 conjugation starts with activation of ATG12, which is another ubiquitin-like molecule and its sequential transfer to ATG7 and ATG10 before making an isopeptide bond between the C-terminal Gly of ATG12 and Lys (K130) of ATG5. The ATG12-ATG5 conjugate binds to a membrane binding component, such as ATG16L1 or TECPR1, to form the E3 holocomplexes (ATG12-ATG5/ATG16L1 or ATG12-ATG5/TECPR1). ATG16L1 or TACPR1 serve to bring to specific membrane sites the ATG12-ATG5 conjugate where it in a spatially restricted manner catalyzes the transfer of ATG8s from the ATG8-ATG3 intermediate to the phospholipids (PE or PS). ATG16L1 recruits the E3 holoenzyme ATG12-ATG5/ATG16L1 to phosphatidylinositol-3-P (PI3P)-marked membrane sites via its binding to WIPI2 protein which in turn recognizes PI3P (omitted from the drawing for clarity). TECPR1 performs identical function by recruiting the E3 holoenzyme ATG12-ATG5/TECPR1 to other membranes tagged by sphingomyelin marks, a situation that can occur on the cytofacial side of endomembranes upon stress or damage. It is postulated here that there are other membrane binding components equivalent to ATG16L1 or TECPR1 that can guide ATG12-ATG5 to other signals on membranes and perform atg8ylation in response to a variety of membrane damage, stress or remodeling cues. The downstream effector processes resulting from atg8ylation of membranes are listed in panel B, whereas the relationship to M. tuberculosis host-pathogen interactions including protection of the host are illustrated in the schematic in Figure 2. (B) Membrane atg8ylation outputs and downstream processes. Canonical autophagy is a process of formation of autophagosomes in the cytoplasm which can capture and remove by digestion, after their fusion with lysosomal organelles, exogenous or endogenous irritants ranging from microbes or their products to defunct organelles, innate immunity signaling platforms, protein aggregates and danger-associated molecular patterns. At times of starvation, canonical autophagy also plays nutritional role, for example by digesting cytosolic macromolecules and replenishing amino acids and nucleoside pools for biosynthetic needs. Secretory autophagy is an outcome of various canonical and non-canonical autophagy-related processes that lead to exocytosis and release of cargo to the extracellular milieu rather than digestion in lysosomal organelles. Atg8ylation also participates in membrane repair and other adjustments following damage. A wide range of atg8ylation processes on cellular endomembranes (delimiting membranes of various organelles) includes: LAP (LC3-associated phagocytosis), LANDO (LC3-associated endocytosis), LAM (LC3-associated micropinocytosis), CASM (conjugation of ATG8 to single membranes), and VAIL (V-ATPase-ATG16L1-induced LC3 lipidation) (12–14). Of note, atg8ylation requires only the cytofacial monolayer of phospholipids and does not need “single membrane” which by definition consists of a lipid bilayer. This is well-illustrated by atg8ylation of lipid droplets (LD), which are neutral lipid storage organelles delimited by a monolayer of phospholipids that separates the LD core consisting of neutral lipids such as triglycerides and choleteryl esters from the aqueous phase of the cytosol. Note that canonical autophagy, secretory autophagy, LAP, LD stores, V-ATPase, and membrane repair have all been implicated in host-pathogen interactions during M. tuberculosis phagocytosis, cytosol invasion, and infection of and replication in macrophages.
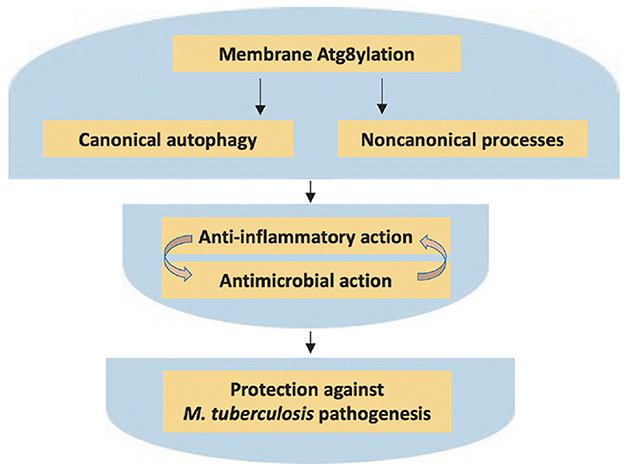
Figure 2. Atg8ylation in control of M. tuberculosis pathogenesis. The atg8ylation process and its outputs, as detailed in Figure 1 and this figure, and how it affects host-pathogen interactions in tuberculosis. Both canonical and non-canonical outputs of atg8ylation afford protection against extensive cell-autonomous and tissue inflammation, pro-inflammatory cell death, necrosis, and excessive tissue damage. These processes also contribute to control of intracellular bacteria; note that inflammation and cell death complicate interpretations of the in vivo effects on bacterial elimination vs. intracellular bacterial replication. The significance of these processes during tuberculosis infection is supported by the existence of a plethora of M. tuberculosis virulence factors that counter atg8ylation outputs, as reviewed elsewhere. All of the above processes, when functional and stimulated by appropriately sequenced immunological responses while avoiding excess or prolonged neutrophilic and other inflammation, contribute to protection of the host reducing tissue damage during M. tuberculosis infection.
Canonical autophagy (15) has been implicated in defense against Mtb (9, 16) contemporaneously with the initial recognition of the broader roles of autophagy in health and disease (17–19) which includes its immune functions (20–24). In principle, autophagy is a homeostatic process that cleanses host cell cytoplasm, turns over long lived proteins and other macromolecules, removes defunct or surplus organelles (15), adjusts cellular metabolic needs (10), and eliminates intracellular microbes and microbial or host products causing inflammation (24, 25). The simplest rendition of autophagy as immune mechanism is that autophagy acts as a cell-autonomous defense (20, 24), whereby it seeks out and eliminates pathogens that manage to penetrate cellular interiors. Autophagy also has a much broader role in innate and adaptive immunity (21, 23, 24). It is of particular significance as a versatile process that suppresses inflammation by removing microbial products collectively referred to as pathogen associated molecular patterns (PAMPs) and endogenous irritants collectively referred to as damage associated molecular patterns (DAMP) or acting upon the signaling platforms that detect PAMPs and DAMPs, the so-called PRRs or pattern recognition receptors (24, 25).
The mechanism of autophagosome generation in mammalian cells is far more complex than in yeast, the model organism that served to genetically unlock the initial aspects of the canonical autophagy pathway (26). Recently, major strides have been made toward understanding molecular mechanisms driving canonical autophagic membrane formation in mammalian cells via prophagophore and phagophore stages (27–32), phagophore reshaping (33, 34) and expansion via lipid transfer (35–37), cargo capture (38), closure of the nascent phagosomes (39–42), and their fusion with endolysosomal organelles where the cargo is degraded as a primary path to elimination or recycling (43) or alternatively secreted in a process termed secretory autophagy (44).
Membrane atg8ylation, also known as mATG8 lipidation, is the process of covalent modification of various membranes by mammalian ATG8 proteins (mATG8s) that takes place as a response to membrane stress, damage, and remodeling signals (Figure 1A) (11). This process is best understood by analogy to ubiquitylation: atg8ylation is to membranes what ubiquitylation is to proteins (11). The principal protagonists of atg8ylation and ubiqutylation, mATG8s and ubiquitin, are homologous, and the conjugation cascades that activate them and result in membrane atg8ylation or protein ubiquitylation are very similar involving ATP, E1, E2 and E3 ligases (45). The specific factors leading to atg8ylation include two enzymatic cascades with ATG12-ATG5 and mATG8-phosphatidylethanolamine (PE) conjugates as their end products. The former protein-protein conjugate combines with additional proteins to form E3 ligases (46–50) to guide the latter protein-lipid conjugate resulting in atg8ylation of specific membrane domains.
The conjugation cascade follows typical ubiqitylation-like steps (Figure 1A). After proteolytic exposure of the Gly residue at the C-termini, mATG8s are activated by ATP, and transferred to ATG7 (E1) to form mATG8-ATG7 conjugate, then transferred to ATG3 (E2) to form mATG8-ATG3 conjugate and finally to PE in membranes guided by the E3 enzymes. The known E3 enzymes consist of the ATG12-ATG5 conjugate associated with ATG16L1 (46) or (thus far) TECPR1 (47–50). The ATG12-ATG5 conjugate component of the atg8ylation E3 ligases is generated in its own cascade (46). Of note, ATG12 is, like mATG8s, a ubiquitin-like molecule. It is activated by ATP, conjugated to ATG7 (E1), then to ATG10 (E2) and finally to ATG5, forming the ATG12-ATG5 conjugate which then noncovalently associates with ATG16L1 (46) or TECPR1 (48–50) to form E3 ligases directing atg8ylation of different membranes, including autophagosomal and others (11). In preparation for the next step, an ATG12-ATG5 containing E3 ligase activates its substrate mATG8-ATG3 by exposing the thioester bond of the ATG8-G-C-ATG3 intermediate for transfer of the ATG8 to the membrane via formation of an amide bond with the ethanolamine headgroup of the PE phospholipid (46). There are additional branches of these conjugation cascades (Figure 1A), whereby ATG12 can make a non-canonical sidestep conjugate with ATG3 (ATG12-ATG3) (51, 52) which is enhanced in the absence of ATG5 (53).
The specialization of atg8ylation for membranes is ensured by the two extra (relative to ubiquitin) α-helices at the N-terminus of mATG8s with concealed affinities for membranes actuated during atg8ylation (54) and intrinsic membrane affinities of the atg8ylation ligase components ATG3 (55, 56) and ATG16L1 (57, 58). During canonical autophagy, WIPI2, an effector of phosphatidylinositol-3-phosphate (PI3P, a stress-signaling phosphoinositide phospholipid) and a known interactor of ATG16L1 (59–61), helps dock ATG12-ATG5/ATG16L1 and ATG3 to the PI3P-marked membranes to present activated mATG8s for conjugation to the phospholipid PE embedded within the target membrane (58, 62). It is likely that membrane atg8ylation processes other than canonical autophagy may employ yet to be identified functional equivalents of ATG16L1 and WIPI2, as already exemplified with TECPR1 (48–50), to guide atg8ylation on other non-autophagic membranes. Finally, there are exceptions to a strict separation between membrane atg8ylation and protein ubiquitylation as evidenced by crossovers such as atg8ylation of proteins (63–66) and ubiquitylation of membranes (67, 68).
There are six principal mATG8s: LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2 (69–71). The yeast's sole Atg8 has served to delineate the posttranslational modification at the C-terminus of ATG8s with the lipid phosphatidylethanolamine (PE) (72, 73), which can also occur with phosphatidylserine (PS) (74). In mammalian cells, this process, referred to as “LC3 lipidation” and LC3 puncta formation (46, 75), has been widely considered as being synonymous with autophagy and thus has been somewhat indiscriminately used to report observations and measurements of canonical autophagy (76). Whereas LC3B continues to be used to monitor autophagy, it has become evident that it can be present on membranes other than autophagosomes (77). Thus far, mammalian membranes that have been reported to be atg8ylated (covalently modified by mATG8s, typically reported as LC3B but not limited to this mATG8) include: conventional phagosomes harboring pathogens or microbial products (78–81), various types of stressed or signaling endosomal compartments (12–14, 79, 82), lysosomes (48, 66, 83–86), exocytic compartments releasing exosomes (52, 87), ER during its piecemeal ESCRT-dependent lysosomal degradation (88), and lipid droplets (89). Of note, the delimiting membrane of lipid droplets modified by LC3B is not even a full lipid bilayer but a monolayer of phospholipids (89). Thus, the repertoire of cellular membranes that undergo atg8ylation is not limited to double or not even to single membranes. Apparently, all that is needed as a substrate is a PE- or PS-containing phospholipid hemilayer.
Preceding the unified model for atg8ylation (11, 45), the phenomena dependent on atg8ylation have been historically recognized as an assortment of ‘non-canonical autophagy' processes (77) reflected in various terms describing them (Figure 1B): LAP (LC3-associated phagocytosis) (78), LANDO (LC3-associated endocytosis) (90), LC3-associated micropinocytosis (LAM) (82), CASM (conjugation of ATG8 to single membranes) (86, 91), and “vATPase-ATG16L1 axis xenophagy” (80) that later received an acronym VAIL (V-ATPase-ATG16L1-induced LC3 lipidation) (12–14).
Despite a growing plethora of “non-canonical” processes and phenomena that have no autophagic functions and yet engage mATG8s (11, 77), atg8ylation remains an important aspect of canonical autophagy (Figure 2) (11). Although the initial stages of autophagy such as crescent phagophores can form and close in cells lacking all principal mATG8s (42, 65, 92) or in cells defective for mATG8 lipidation (93), their size (92) and contents (93) as well as quality (42) are significantly affected. Among the known functions of the atg8ylation of the autophagosomal membranes are membrane remodeling during autophagosome biogenesis (54, 56, 94–96), its kinetic acceleration (97), the enhancement of selective cargo sequestration into autophagosomes (38), as well as autophagosome-lysosome fusion (98). Recent studies have identified a new role of mATG8s in ESCRT-dependent sealing of autophagic membranes and their maintenance in an impervious state (42). In the absence of mATG8s, the autophagic membrane are permeable to solutes and small macromolecules, arrested at the stage termed amphisome (a hybrid organelles between autophagosomes and multivesicular body endosomes) and cannot progress to autolysosomes (42).
Macrophages are the key cell type where Mtb normally replicates and where many aspects of critical host-pathogen interactions occur (3, 99–104). The early seminal studies (9, 16, 105) have shown that induction of autophagy by starvation or IFN-γ, measured by methods available prior to the recent differentiation between atg8ylation and autophagy (11), can restrict virulent Mtb in human and mouse macrophages. A number of subsequent studies, carried out either in vitro with macrophages and neutrophils or in murine models of tuberculosis, are consistent with the notion that atg8ylation or autophagy (or both) are involved in control of Mtb pathogenesis (Figure 2) (53, 106–124). The in vivo effects of autophagy on bacterial burden in the mouse models of Mtb infection (110, 111) have been reported as negligible (120, 121) or fluctuating (122) depending on the experimental conditions. In vivo, any effects directly on bacterial growth (110) are overshadowed by the dominant anti-inflammatory effects of autophagy and atg8ylation as shown in one of the first in vivo reports (111) and re-affirmed in subsequent studies (120, 121) (Figure 2). Cell death pathways triggered in infected macrophages (100, 125), modulated by atg8ylation (111, 122, 123), can be proinflammatory or non-inflammatory and may contribute to the inflammation-driven outcomes in vivo.
The increased inflammation, persistent neutrophilic infiltration, excessive IL-17 production and Th17 polarization have been initially described in a mouse model of Mtb infection with murine Atg5 inactivated specifically in myeloid cells (Atg5fl/fl LysM-Cre mice) (111). A number of studies (126) including those in mice (127) and human populations (128, 129) have shown that neutrophils may contribute to protection early in infection with Mtb (130–132) but when they linger for prolonged periods of time, i.e., when Th17 does not mature into Th1 response, they can contribute to tuberculosis pathogenesis (133–138). A study with depletion of neutrophils (120) affirmed the prior findings (111) and additionally demonstrated their key role in causing high mortality of Mtb-infected Atg5fl/fl LysM-Cre mice (120).
The 2004 report that autophagy is a new cell-autonomous defense against Mtb (9) was initially validated in 2012 in vivo by two groups using mouse models of Mtb respiratory infection (110, 111). However, these studies used conditional inactivation of only one Atg gene, i.e., Atg5 (Atg5fl/fl LysM-Cre mice). A role for autophagy as a pathway was subsequently challenged in 2015 (120). Whereas, the same report confirmed that Atg5fl/fl LysM-Cre mice were more susceptible to Mtb, the authors reported that inactivating additional autophagy genes did not increase susceptibility to Mtb (120). Worth mentioning is that these were short term studies of up to 80 days post-infection and the efficacy of inactivation of other autophagy genes was not fully validated (120). Nevertheless, this report undercut the burgeoning interest in autophagy as a defense against Mtb (139). However, a newer well-designed study demonstrated that dismissal of autophagy was premature and that a more thorough inactivation of additional canonical autophagy genes did confer protection against tuberculosis (122). Shortly afterwards, the same group that initially dismissed autophagy published a revised conclusion (121) that autophagy does matter and that autophagy prevents excessive proinflammatory responses and neutrophil recruitment during Mtb infection.
The trajectory of this line of investigation by different groups shows that details do matter and that interpretations of the findings must be scrutinized before making emphatic conclusions. There were both nuanced and major differences in experimental setups of the above studies in murine models of Mtb infection deserving further dissection and discussion. The published studies used either low dose (110, 111, 120) or in addition high dose aerosol exposure (111), and the investigators tested chronic (110, 111) or acute (111, 120) disease. Typically, chronic infection initiated by low dose aerosol exposure in mice requires observations of up to 200 days post-infection, dependent on the effect size and penetrance of genetic alterations (53, 110, 122, 140). Acute effects are either observed very early in infection with a low dose (53, 110, 111, 120) or require higher doses of aerosol infection (140–142). The initial analysis (120) of multiple Atg genes with low initial lung deposition of Mtb was however limited to short-term, acute disease observations ending at 80 days post-infection. Whereas that study confirmed that Atg5fl/fl LysM-Cre mice were particularly susceptible to Mtb, i.e., displayed high mortality early on even during low dose infection due to the strength of the effect of Atg5, mice with conditional knockouts in other autophagy genes tested, including Atg14, Ulk1/2 (considered to be the mouse Atg1 orthologs), and LC3-lipidation/atg8ylation enzymatic components Atg3, Atg7, Atg12, and Atg16L1, did not die within the 80 days of reported observation following Mtb infection (120). A 2023 study (122) revisited this issue by monitoring mice over a longer, more typical time course, and reported that defects in other autophagy genes, albeit testing only those carrying out atg8ylation (124), rendered mice susceptible to Mtb. In this well-executed study, the authors also considered the efficacy of conditional inactivation of the floxed genes (122). They enhanced Cre-driven LoxP-excisions by increasing Cre recombinase gene dosage in mice by breading animals to be homozygous for LysM-Cre (LysM-Cre+/+) instead of carrying only one LysM-Cre allele (LysM-Cre+/−) used in previous studies (110, 111, 120). This resulted in better inactivation of Atg16L1 and Atg7 and in increased susceptibility of Atg16L1fl/fl LysM-Cre+/+ and Atg7fl/fl LysM-Cre+/+ to Mtb compared to their Cre− littermates. Even the mice carrying only one LysM-Cre allele (LysM-Cre+/− Atg16L1fl/fl) displayed increased mortality when infected with Mtb, illustrating the fact that these effects were missed in the previous study limited by shorter duration (120). The Golovkine et al. (122) study reported dynamic changes in bacterial burden in mice with properly Cre-excised floxed Atg7 and Atg16L1 genes, characterized by an early lung-specific increase. Regardless of the details on bacterial burden, which fluctuate, all observations agree on increased lesions and neutrophilic infiltration (110, 111, 120) and have been recapitulated in LysM-Cre+/+ Atg7fl/fl and LysM-Cre+/+ Atg16L1Fl/Fl mice (122).
Collectively, the above studies indicate that multiple autophagy genes are needed for disease control in the mouse model of tuberculosis (Figure 2). They also confirm the well-established notion that in vivo autophagy primarily manifests as an anti-inflammatory and tissue-sparing process (21, 24, 25) as reported in one of the two initial studies with Mtb infection in mice (111). Ex vivo, autophagy can undoubtedly control multiple intracellular microbes in infected cells including Mtb and other pathogens (143–149). However, the in vivo data with Mtb by multiple groups indicate that changes in bacterial burden in infected murine lungs are minor relative to the anti-inflammatory power of autophagy (111), and are not detected at all (121, 150) or fluctuating (122). It is easy to reconcile these apparent discrepancies as fluctuations or minor changes in bacillary loads in the lungs given that autophagy and atg8ylation control inflammation whereas inflammation affects antimicrobial action (Figure 2; middle box) (24), thus confounding bacillary load measurements making data highly dependent on timing and phase of infection. Furthermore as a contributor to modulation of inflammatory responses, in the absence of autophagy (or atg8ylation), cell death occurs more readily thus eliminating cells in which Mtb would have an opportunity to replicate. This is further underscored by the findings that absence of autophagy or atg8ylation increases inflammatory responses to microbes (25) and that this paradoxically and artificially protects the host in certain mouse models of infection (151), e.g., with Listeria monocytogenes (152), herpesviruses (153), and influenza (154, 155).
Of further import is the fact that the field of autophagy has in the meantime evolved in major ways, and it is necessary to view old and new studies in the broader context of canonical and non-canonical processes. Whereas, these processes are still being linked to the catchall term “autophagy,” they do represent a collection of much wider and disparate membrane stress and remodeling responses (11, 77).
Mirroring the growth in appreciation of broader biological roles of atg8ylation, of which canonical autophagy is one of the outputs, are the more recent studies with Mtb, attempting to differentiate between canonical autophagy and atg8ylation in control of Mtb, both in vitro and in vivo. This is a difficult task, as atg8ylation and canonical autophagy are intertwined via the protein conjugation systems (Figure 1A). Furthermore, the full spectrum and ramifications of diverse processes that are autophagy-unrelated or only partially overlapping with autophagy (Figure 1B) are not completely known at this point in time (11, 77).
Interpretation of the available published information is confounded by the emerging relationships and it is difficult to deconvolute atg8ylation-dependent mechanisms capable of controlling Mtb. As discussed above, atg8ylation participates in a variety of membrane stress and remodeling responses (11), one of which is LAP (77). Using an Mtb mutant in a gene called CpsA (Rv3484) which is susceptible to LAP, an apparent role in control of Mtb for LAP has been unmasked (117). This was based on the effects of inactivating/downregulating in murine macrophages genes selectively implicated in LAP (Rubcn) or shared with canonical autophagy (atg8ylation factors Atg5, Atg7 and Atg16L1 and Beclin 1), but not in others that are considered specific for canonical autophagy (Ulk1 and Atg14). The susceptibility was observed only during innate immunity responses, as IFN-γ and adaptive immunity abrogated LAP effects both ex and in vivo (117). Of note IFN-γ induces classical autophagy (9, 105, 115), so it is possible that engagement of atg8ylation machinery in LAP is competing with canonical autophagy for the available atg8ylation resources, as in the case of CASM competing with other atg8ylation processes (86, 156). CpsA inhibits NOX2 (117) implicated in RUBCN (Rubicon)-dependent LAP (157). However, complicating the issues, CasA also binds NDP52 and TAX1BP1 (117, 158), two classical receptors involved in selective canonical autophagy of mitochondria and microbes (149). It is also not known how CpsA enters the host cytosol and could this be dependent on permeabilization of phagosomal membranes by ESX-1 (159), a process that induces canonical autophagy (110). In a SCID mouse model of aerosol infection the animals were susceptible to Mtb H37Rv but not to its cpsA mutant in experiments carried out up to 120 days post-infection (117). It is important to note that while these studies reveal a hidden role for LAP, this role remains masked unless cpsA is inactivated and is thus probably not dominant during infection with wild type virulent Mtb.
Another recent study addressed the effects of LAP specifically on neutrophilic inflammation in the murine model of tuberculosis (121). Whereas Atg16L1fl/fl LysM-Cre mice infected with Mtb Erdman showed neutrophilic infiltration akin to the Atg5fl/fl LysM-Cre mice, Rubcn−/− mice did not. Since Rubcn (Rubicon) is key to the atg8ylation process known as LAP (77), based on the above study and its chosen experimental parameters this specific manifestation (LAP) of atg8ylation was deemed not to be involved (121). However, the study (121) did not test cpsA mutant, and thus the role of LAP might have remained masked in these experiments. Regardless, the identification of CasA as an inhibitor of LAP and the unmasking of the role of LAP (117) (or perhaps other atg8ylation processes) in Mtb pathogenesis represent a major step in deconvoluting different atg8ylation outputs in the context of Mtb infection.
In yet another study with human iPSC-derived macrophages (123), the authors aimed to assess the relative contributions of LAP and canonical autophagy in control of Mtb. For this, they (123) compared a strain of Mtb (cpsA mutant) that cannot inhibit LAP with an Mtb strain (esx mutant) that is assumed not to elicit canonical autophagy. The latter assumption is based on the notion that esx mutant cannot permeabilize the membrane of phagosomes harboring Mtb, an event necessary to induce canonical autophagy to counter Mtb's access to the host cell cytosol (110). The loss of ATG7 (which blocks atg8ylation affecting both canonical autophagy and non-canonical outputs including LAP) permitted growth of wild type H37Rv but not of its esx and cpsA mutants. This affirmed the notion that atg8ylation matters for control of Mtb ex vivo but could not discern between canonical autophagy and other non-canonical manifestations of atg8ylation. A loss in iPSC-derived macrophages of ATG14, believed to be specific for canonical autophagy, permitted growth of wild type H37Rv Mtb (123), which can be interpreted as an affirmation of canonical autophagy's role in control of Mtb in human macrophages (9). Loss of ATG14 also promoted replication of the esx mutant (123), contrary to the expectation that M. tuberculosis incapable of escaping the phagosome would not be able to induce canonical autophagy (110). This led investigators to propose a further branching of processes controlled by ATGs, whereby ATG14 may affect phagosomal maturation (123) in addition to the conventional role in canonical autophagy.
The mystery of the uniquely penetrant effects of Atg5 on susceptibility to Mtb in vivo has recently been partially solved (53). As a reminder, even the latest analyses with a more complete excision of floxed Atg alleles in mice and standard long-term post-infection observations (53, 122) confirmed the uniqueness of the Atg5 phenotype (110, 111, 120). The Atg5fl/fl LysM-Cre+/+ mice and even Atg5fl/fl LysM-Cre+/− mice infected with Mtb died much faster compared to Atg16L1fl/fl LysM-Cre+/+ and Atg7fl/fl LysM-Cre+/+ mice (122). In another independent study, Atg5fl/fl LysM-Cre+/− mice succumbed much faster to Mtb than Atg7fl/fl LysM-Cre+/− mice (53). Hence, Atg5 has additional roles, past the canonical autophagy and even past the atg8ylation processes (53, 124).
Mechanistically, ATG5 affects exocytosis through multiple mechanisms underlying excessive neutrophil activation during Mtb infection (53). In the absence of ATG5, instead of the canonical ATG12-ATG5 conjugate, ATG12 engages in the formation of the non-canonical ATG12-ATG3 conjugate (51) (Figure 1A) which in turn binds the ESCRT protein ALIX (160). This has multiple effects unrelated to autophagy or atg8ylation and is based on ALIX's involvement in maintaining lysosomal membranes intact thus keeping lysosomes in good repair (142, 161, 162). Stress in endolysosomal compartments (66, 140, 142, 159, 163–166) and other membranes (167, 168), which occurs during Mtb infection, is exacerbated for lysosomes in ATG5 knockout cells since ALIX is sequestered away by ATG12-ATG3 for the exocytic processes instead of being available for lysosomal repair (53). The pools of ALIX, redirected in the absence of ATG5, augment exocytic events leading to excessive neutrophilic activation and degranulation (53). Thus, the excessive Mtb pathogenesis encountered in Atg5fl/fl LysM-Cre+/− mice (111) is caused by hyperactive exocytosis and degranulation by neutrophils further compounded by the inability to maintain functional lysosomes (53).
The focus on canonical autophagy as a potential anti-Mtb mechanism has now evolved into a new stage following the improved understanding of the processes controlled by the ATG genes. It has become evident that canonical autophagy is merely a subset of a much broader cellular stress response termed membrane atg8ylation. Atg8ylation pathways that do not engage all parts of the canonical autophagy pathway have many manifestations affecting membranes of various intracellular compartments. This includes variations known under the acronyms LAP, LANDO, LAM, CASM, and VAIL. There is also a ‘sidestep' atypical ATG12-ATG3 conjugation which is enhanced in the absence of ATG5. This atypical ATG conjugate promotes exocytic events, excessive activation of neutrophils and their degranulation, as well as ties up the pools of available ESCRT proteins diverting them away from repair of cellular endomembranes. Several of these processes have already been shown to protect the host against Mtb under certain conditions. The improved understanding of atg8ylation and its branching outputs, of which autophagy is one of the best studied but not the only one, offers a new conceptual framework and beckons for cogent repositioning of the field while opening prospects for reformulated fundamental, preclinical, and clinical studies.
VD: Conceptualization, Writing—original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH grants R37AI042999 and R01AI111935, and center grant P20GM121176 to VD.
The author thanks his present and past collaborators, associates, and trainees, who have made key contributions to linking autophagy and tuberculosis pathogenesis as well as contributed to the development of fundamental aspects of autophagy and atg8ylation as immune mechanisms.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Brites D, Gagneux S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev. (2015) 264:6–24. doi: 10.1111/imr.12264
3. Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. (2004) 20:367–94. doi: 10.1146/annurev.cellbio.20.010403.114015
4. Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. (2012) 12:581–91. doi: 10.1038/nri3259
5. Flynn JL, Chan J. Immune cell interactions in tuberculosis. Cell. (2022) 185:4682–702. doi: 10.1016/j.cell.2022.10.025
6. Rahlwes KC, Dias BRS, Campos PC, Alvarez-Arguedas S, Shiloh MU. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence. (2023) 14:2150449. doi: 10.1080/21505594.2022.2150449
7. Ruhl CR, Pasko BL, Khan HS, Kindt LM, Stamm CE, Franco LH, et al. Mycobacterium tuberculosis sulfolipid-1 activates nociceptive neurons and induces cough. Cell. (2020) 181:293–305. doi: 10.1016/j.cell.2020.02.026
8. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers. (2016) 2:16076. doi: 10.1038/nrdp.2016.76
9. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
10. Deretic V, Kroemer G. Autophagy in metabolism and quality control: opposing, complementary or interlinked functions. Autophagy. (2021) 21:1–10. doi: 10.1080/15548627.2021.1933742
11. Deretic V, Lazarou M, A. guide to membrane atg8ylation and autophagy with reflections on immunity. J Cell Biol. (2022) 221:83. doi: 10.1083/jcb.202203083
12. Fischer TD, Wang C, Padman BS, Lazarou M, Youle RJ. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J Cell Biol. (2020) 21:128. doi: 10.1083/jcb.202009128
13. Xu Y, Cheng S, Zeng H, Zhou P, Ma Y, Li L, et al. ARF GTPases activate Salmonella effector SopF to ADP-ribosylate host V-ATPase and inhibit endomembrane damage-induced autophagy. Nat Struct Mol Biol. (2022) 29:67–77. doi: 10.1038/s41594-021-00710-6
14. Xu Y, Ding J. Endomembrane damage sensing by V-ATPase recruits ATG16L1 for LC3 lipidation in situ. Autophagy. (2022) 18:2751–3. doi: 10.1080/15548627.2022.2062889
15. Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol. (2019) 35:453–75. doi: 10.1146/annurev-cellbio-100818-125300
16. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. (2006) 313:1438–41. doi: 10.1126/science.1129577
17. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
18. Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. (2020) 383:1564–76. doi: 10.1056/NEJMra2022774
19. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. (2021) 40:e108863. doi: 10.15252/embj.2021108863
20. Deretic V. Autophagy in innate and adaptive immunity. Trends Immunol. (2005) 26:523–8. doi: 10.1016/j.it.2005.08.003
21. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammatio,n and immunity. Nat Rev Immunol. (2013) 13:722–37. doi: 10.1038/nri3532
22. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. (2013) 39:211–27. doi: 10.1016/j.immuni.2013.07.017
23. Clarke AJ, Simon AK. Autophagy in the renewal, differentiatio,n and homeostasis of immune cells. Nat Rev Immunol. (2019) 19:170–83. doi: 10.1038/s41577-018-0095-2
24. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. (2021) 54:437–53. doi: 10.1016/j.immuni.2021.01.018
25. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. (2018) 8:1–9. doi: 10.1080/15548627.2017.1402992
26. Yamamoto H, Zhang S, Mizushima N. Autophagy genes in biology and disease. Nat Rev Genet. (2023) 24:382–400. doi: 10.1038/s41576-022-00562-w
27. Kumar S, Javed R, Mudd M, Pallikkuth S, Lidke K, Jain A, et al. Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell. (2021) 184:5950–69. doi: 10.1016/j.cell.2021.10.017
28. Nguyen A, Lugarini F, David C, Hosnani P, Alagoz C, Friedrich A, et al. Metamorphic proteins at the basis of human autophagy initiation and lipid transfer. Mol Cell. (2023) 83:2077–90. doi: 10.1016/j.molcel.2023.04.026
29. Olivas TJ, Wu Y, Yu S, Luan L, Choi P, Guinn ED, et al. ATG9 vesicles comprise the seed membrane of mammalian autophagosomes. J Cell Biol. (2023) 222:7. doi: 10.1083/jcb.202208088
30. Kannangara AR, Poole DM, McEwan CM, Youngs JC, Weerasekara VK, Thornock AM, et al. BioID reveals an ATG9A interaction with ATG13-ATG101 in the degradation of p62/SQSTM1-ubiquitin clusters. EMBO Rep. (2021) 22:e51136. doi: 10.15252/embr.202051136
31. Ren X, Nguyen TN, Lam WK, Buffalo CZ, Lazarou M, Yokom AL, et al. Structural basis for ATG9A recruitment to the ULK1 complex in mitophagy initiation. Sci Adv. (2023) 9:eadg2997. doi: 10.1126/sciadv.adg2997
32. Broadbent DG, Barnaba C, Perez GI, Schmidt JC. Quantitative analysis of autophagy reveals the role of ATG9 and ATG2 in autophagosome formation. J Cell Biol. (2023) 222:7. doi: 10.1083/jcb.202210078
33. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. (2008) 182:685–701. doi: 10.1083/jcb.200803137
34. Nahse V, Raiborg C, Tan KW, Mork S, Torgersen ML, Wenzel EM, et al. ATPase activity of DFCP1 controls selective autophagy. Nat Commun. (2023) 14:4051. doi: 10.1038/s41467-023-39641-9
35. Valverde DP Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol. (2019) 218:1787–98. doi: 10.1083/jcb.201811139
36. Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife. (2019) 8:777. doi: 10.7554/eLife.45777
37. Dabrowski R, Tulli S, Graef M. Parallel phospholipid transfer by Vps13 and Atg2 determines autophagosome biogenesis dynamics. J Cell Biol. (2023) 222:7. doi: 10.1083/jcb.202211039
38. Lamark T, Johansen T. Mechanisms of selective autophagy. Annu Rev Cell Dev Biol. (2021) 37:143–69. doi: 10.1146/annurev-cellbio-120219-035530
39. Flower TG, Takahashi Y, Hudait A, Rose K, Tjahjono N, Pak AJ, et al. A helical assembly of human ESCRT-I scaffolds reverse-topology membrane scission. Nat Struct Mol Biol. (2020) 27:570–80. doi: 10.1038/s41594-020-0426-4
40. Takahashi Y, Liang X, Hattori T, Tang Z, He H, Chen H, et al. VPS37A directs ESCRT recruitment for phagophore closure. J Cell Biol. (2019) 218:3336–54. doi: 10.1083/jcb.201902170
41. Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun. (2018) 9:2855. doi: 10.1038/s41467-018-05254-w
42. Javed R, Jain A, Duque T, Hendrix E, Paddar MA, Khan S, et al. Mammalian ATG8 proteins maintain autophagosomal membrane integrity through ESCRTs. EMBO J. (2023) 4:e112845. doi: 10.15252/embj.2022112845
43. Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol. (2019) 218:757–70. doi: 10.1083/jcb.201810099
44. Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V, et al. Secretory autophagy. Curr Opin Cell Biol. (2015) 35:106–16. doi: 10.1016/j.ceb.2015.04.016
45. Kumar S, Jia J, Deretic V. Atg8ylation as a general membrane stress and remodeling response. Cell Stress. (2021) 5:128–42. doi: 10.15698/cst2021.09.255
46. Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. (2020) 63:1–10. doi: 10.1016/j.ceb.2019.12.001
47. Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q, et al. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell. (2012) 45:629–41. doi: 10.1016/j.molcel.2011.12.036
48. Corkery DP, Castro-Gonzalez S, Knyazeva A, Herzog LK. An ATG12-ATG5-TECPR1 E3-like complex regulates unconventional LC3 lipidation at damaged lysosomes. EMBO Rep. (2023) 12:e56841. doi: 10.15252/embr.202356841
49. Kaur NLR, de la Ballina HS, Haukaas ML, Torgersen M, Radulovic MJ. TECPR1 is activated by damage-induced sphingomyelin exposure to mediate noncanonical autophagy. EMBO J. (2023) 5:e113105. doi: 10.15252/embj.2022113105
50. Boyle KB, Ellison CJ, Elliott PR, Schuschnig M, Grimes K, Dionne MS, et al. TECPR1 conjugates LC3 to damaged endomembranes upon detection of sphingomyelin exposure. EMBO J. (2023) 24:e113012. doi: 10.15252/embj.2022113012
51. Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. (2010) 142:590–600. doi: 10.1016/j.cell.2010.07.018
52. Leidal AM, Huang HH, Marsh T, Solvik T, Zhang D, Ye J, et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol. (2020) 22:187–99. doi: 10.1038/s41556-019-0450-y
53. Wang F, Peters R, Jia J, Mudd M, Salemi M, Allers L, et al. ATG5 provides host protection acting as a switch in the atg8ylation cascade between autophagy and secretion. Dev Cell. (2023) 58:866–84. doi: 10.1016/j.devcel.2023.03.014
54. Zhang W, Nishimura T, Gahlot D, Saito C, Davis C, Jefferies HBJ, et al. Autophagosome membrane expansion is mediated by the N-terminus and cis-membrane association of human ATG8s. Elife. (2023) 12:185. doi: 10.7554/eLife.89185
55. Ye Y, Tyndall ER, Bui V, Tang Z, Shen Y, Jiang X, et al. J: An N-terminal conserved region in human Atg3 couples membrane curvature sensitivity to conjugase activity during autophagy. Nat Commun. (2021) 12:374. doi: 10.1038/s41467-020-20607-0
56. Nishimura T, Lazzeri G, Mizushima N, Covino R, Tooze SA. Unique amphipathic alpha helix drives membrane insertion and enzymatic activity of ATG3. Sci Adv. (2023) 9:eadh1281. doi: 10.1126/sciadv.adh1281
57. Lystad AH, Carlsson SR, de la Ballina LR, Kauffman KJ, Nag S, Yoshimori T. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. (2019) 21:372–83. doi: 10.1038/s41556-019-0274-9
58. Jensen LE, Rao S, Schuschnig M, Cada AK, Martens S, Hummer G, et al. Membrane curvature sensing and stabilization by the autophagic LC3 lipidation machinery. Sci Adv. (2022) 8:eadd1436. doi: 10.1126/sciadv.add1436
59. Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA, et al. WIPI2 Links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. (2014) 55:238–52. doi: 10.1016/j.molcel.2014.05.021
60. Strong LM, Chang C, Riley JF, Boecker CA, Flower TG, Buffalo CZ, et al. Structural basis for membrane recruitment of ATG16L1 by WIPI2 in autophagy. Elife. (2021) 10:70372. doi: 10.7554/eLife.70372
61. Gong X, Wang Y, Tang Y, Wang Y, Zhang M, Li M, et al. ATG16L1 adopts a dual-binding site mode to interact with WIPI2b in autophagy. Sci Adv. (2023) 9:eadf0824. doi: 10.1126/sciadv.adf0824
62. Rao S, Strong LM, Ren X, Skulsuppaisarn M, Lazarou M, Hurley JH, et al. Three-step docking by WIPI2, ATG16L1 and ATG3 delivers LC3 to the phagophore. bioRxiv. (2023). doi: 10.1101/2023.07.17.549391
63. Agrotis A, von Chamier L, Oliver H, Kiso K, Singh T, Ketteler R, et al. Human ATG4 autophagy proteases counteract attachment of ubiquitin-like LC3/GABARAP proteins to other cellular proteins. J Biol Chem. (2019) 294:12610–21. doi: 10.1074/jbc.AC119.009977
64. Carosi JM, Nguyen TN. Lazarou M, Kumar S, Sargeant TJ. ATG8ylation of proteins: a way to cope with cell stress? J Cell Biol. (2021) 220:11. doi: 10.1083/jcb.202108120
65. Nguyen TN, Padman BS, Zellner S, Khuu G, Uoselis L, Lam WK, et al. ATG4 family proteins drive phagophore growth independently of the LC3/GABARAP lipidation system. Mol Cell. (2021) 81:2013–30. doi: 10.1016/j.molcel.2021.03.001
66. Jia J, Wang F, Bhujabal Z, Peters R, Mudd M, Duque T, et al. Stress granules and mTOR are regulated by membrane atg8ylation during lysosomal damage. J Cell Biol. (2022) 221:11. doi: 10.1083/jcb.202207091
67. Sakamaki JI Ode KL, Kurikawa Y, Ueda HR, Yamamoto H, Mizushima N, et al. Ubiquitination of phosphatidylethanolamine in organellar membranes. Mol Cell. (2022) 82:3677–92. doi: 10.1016/j.molcel.2022.08.008
68. Sakamaki JI, Mizushima N. Ubiquitination of non-protein substrates. Trends Cell Biol. (2023) 27:S0962-8924(23)00071-5. doi: 10.1016/j.tcb.2023.03.014
69. Xin Y, Yu L, Chen Z, Zheng L, Fu Q, Jiang J, et al. Cloning, expression patterns, and chromosome localization of three human and two mouse homologues of GABA(A) receptor-associated protein. Genomics. (2001) 74:408–13. doi: 10.1006/geno.2001.6555
70. He H, Dang Y, Dai F, Guo Z, Wu J, She X, et al. Y: Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem. (2003) 278:29278–87. doi: 10.1074/jbc.M303800200
71. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. (2010) 29:1792–802. doi: 10.1038/emboj.2010.74
72. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. (2000) 408:488–92. doi: 10.1038/35044114
73. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. (1998) 395:395–8. doi: 10.1038/26506
74. Durgan J, Lystad AH, Sloan K, Carlsson SR, Wilson MI, Marcassa E, et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol Cell. (2021) 81:2031–40. doi: 10.1016/j.molcel.2021.03.020
75. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
76. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. (2000) 19:5720–8. doi: 10.1093/emboj/19.21.5720
77. Galluzzi L, Green DR. Autophagy-Independent Functions of the Autophagy Machinery. Cell. (2019) 177:1682–99. doi: 10.1016/j.cell.2019.05.026
78. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. (2007) 450:1253–7. doi: 10.1038/nature06421
79. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. (2008) 27:1110–21. doi: 10.1038/emboj.2008.31
80. Xu Y, Zhou P, Cheng S, Lu Q, Nowak K, Hopp AK, et al. A Bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell. (2019) 178:552–66. doi: 10.1016/j.cell.2019.06.007
81. Ulferts R, Marcassa E, Timimi L, Lee LC, Daley A, Montaner B, et al. Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Rep. (2021) 37:109899. doi: 10.1016/j.celrep.2021.109899
82. Sonder SL, Hager SC, Heitmann ASB, Frankel LB, Dias C, Simonsen AC, et al. Restructuring of the plasma membrane upon damage by LC3-associated macropinocytosis. Sci Adv. (2021) 7:969. doi: 10.1126/sciadv.abg1969
83. Kumar S, Jain A, Choi SW, Silva GPD, Allers L, Mudd MH, et al. Mammalian Atg8 proteins and the autophagy factor IRGM control mTOR and TFEB at a regulatory node critical for responses to pathogens. Nat Cell Biol. (2020) 22:973–85. doi: 10.1038/s41556-020-0549-1
84. Nakamura S, Shigeyama S, Minami S, Shima T, Akayama S, Matsuda T, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol. (2020) 22:1252–63. doi: 10.1038/s41556-020-00583-9
85. Lee C, Lamech L, Johns E, Overholtzer M. Selective Lysosome Membrane Turnover Is Induced by Nutrient Starvation. Dev Cell. (2020) 55:289–97. doi: 10.1016/j.devcel.2020.08.008
86. Goodwin JM, Walkup WG, Hooper K, Li T, Kishi-Itakura C, NG A, et al. GABARAP sequesters the FLCN-FNIP tumor suppressor complex to couple autophagy with lysosomal biogenesis. Sci Adv. (2021) 7:eabj2485. doi: 10.1126/sciadv.abj2485
87. Guo H, Chitiprolu M, Roncevic L, Javalet C, Hemming FJ, Trung MT, et al. Atg5 disassociates the V1V0-ATPase to promote exosome production and tumor metastasis independent of canonical macroautophagy. Dev Cell. (2017) 43:716–30. doi: 10.1016/j.devcel.2017.11.018
88. Loi M, Raimondi A, Morone D, Molinari M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat Commun. (2019) 10:5058. doi: 10.1038/s41467-019-12991-z
89. Omrane M, Ben M'Barek K, Santinho A, Nguyen N, Nag S, Melia TJ, et al. LC3B is lipidated to large lipid droplets during prolonged starvation for noncanonical autophagy. Dev Cell. (2023) 58:1266–81. doi: 10.1016/j.devcel.2023.05.009
90. Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine Alzheimer's disease. Cell. (2019) 178:536–51. doi: 10.1016/j.cell.2019.05.056
91. Hooper KM, Jacquin E, Li T, Goodwin JM, Brumell JH, Durgan J, et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J Cell Biol. (2022) 221:112. doi: 10.1083/jcb.202105112
92. Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. (2016) 215:857–74. doi: 10.1083/jcb.201607039
93. Ohnstad AE, Delgado JM, North BJ, Nasa I, Kettenbach AN, Schultz SW, et al. Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. EMBO J. (2020) 39:e104948. doi: 10.15252/embj.2020104948
94. Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell. (2011) 20:444–54. doi: 10.1016/j.devcel.2011.02.006
95. Maruyama T, Alam JM, Fukuda T, Kageyama S, Kirisako H, Ishii Y, et al. Membrane perturbation by lipidated Atg8 underlies autophagosome biogenesis. Nat Struct Mol Biol. (2021) 28:583–93. doi: 10.1038/s41594-021-00614-5
96. Tooze SA, Zhang W, Lazzeri G, Gahlot D, Thukral L, Covino R, et al. Membrane association of the ATG8 conjugation machinery emerges as a key regulatory feature for autophagosome biogenesis. FEBS Lett. (2023). doi: 10.1002/1873-3468.14676. [Epub ahead of print].
97. Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. (2016) 354:1036–41. doi: 10.1126/science.aaf6136
98. McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. (2015) 57:39–54. doi: 10.1016/j.molcel.2014.11.006
99. Armstrong JA, Hart PDA. Response of Cultured Macrophages to Mycobacterium tuberculosis, with Observations on Fusion of Lysosomes with Phagosomes. J Exp Med. (1971) 134:713–40. doi: 10.1084/jem.134.3.713
100. Stutz MD, Allison CC, Ojaimi S, Preston SP, Doerflinger M, Arandjelovic P, et al. Macrophage and neutrophil death programs differentially confer resistance to tuberculosis. Immunity. (2021) 54:1758–71. doi: 10.1016/j.immuni.2021.06.009
101. Bade P, Simonetti F, Sans S, Laboudie P, Kissane K, Chappat N, et al. Integrative analysis of human macrophage inflammatory response related to Mycobacterium tuberculosis virulence. Front Immunol. (2021) 12:668060. doi: 10.3389/fimmu.2021.668060
102. Pisu D, Huang L, Narang V, Theriault M, Le-Bury G, Lee B, et al. Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J Exp Med. (2021) 218:9. doi: 10.1084/jem.20210615
103. Zhang L, Jiang X, Pfau D, Ling Y, Nathan CF. Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J Exp Med. (2021) 218:87. doi: 10.1084/jem.20200887
104. Chandra P, Coullon H, Agarwal M, Goss CW, Philips JA. Macrophage global metabolomics identifies cholestenone as host/pathogen cometabolite present in human Mycobacterium tuberculosis infection. J Clin Invest. (2022) 132:3. doi: 10.1172/JCI152509
105. Harris J, Haro SA, Master SS, Keane J, Roberts EA, Delgado M, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. (2007) 27:505–17. doi: 10.1016/j.immuni.2007.07.022
106. Alonso S, Pethe K, Russell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci U S A. (2007) 104:6031–6. doi: 10.1073/pnas.0700036104
107. Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. (2009) 6:231–43. doi: 10.1016/j.chom.2009.08.004
108. Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. (2010) 32:329–41. doi: 10.1016/j.immuni.2010.02.009
109. Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. (2012) 11:457–68. doi: 10.1016/j.chom.2012.03.008
110. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
111. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. (2012) 109:E3168–76. doi: 10.1073/pnas.1210500109
112. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. (2012) 37:223–34. doi: 10.1016/j.immuni.2012.04.015
113. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. (2014) 209:754–63. doi: 10.1093/infdis/jit550
114. van der Vaart M, Korbee CJ, Lamers GE, Tengeler AC, Hosseini R, Haks MC, et al. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense. Cell Host Microbe. (2014) 15:753–67. doi: 10.1016/j.chom.2014.05.005
115. Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, et al. Ubiquilin 1 promotes IFN-gamma-induced xenophagy of mycobacterium tuberculosis. PLoS Pathog. (2015) 11:e1005076. doi: 10.1371/journal.ppat.1005076
116. Horne DJ, Graustein AD, Shah JA, Peterson G, Savlov M, Steele S, et al. Human ULK1 variation and susceptibility to Mycobacterium tuberculosis infection. J Infect Dis. (2016) 214:1260–7. doi: 10.1093/infdis/jiw347
117. Koster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci U S A. (2017) 114:E8711–20. doi: 10.1073/pnas.1707792114
118. Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, et al. The ubiquitin ligase SMURF1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. (2017) 21:59–72. doi: 10.1016/j.chom.2016.11.002
119. Zhang S, Zhou X, Ou M, Fu X, Lin Q, Tao X, et al. Berbamine promotes macrophage autophagy to clear Mycobacterium tuberculosis by regulating the ROS/Ca(2+) axis. MBio. (2023) 5:e0027223.
120. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. (2015) 528:565–9. doi: 10.1038/nature16451
121. Kinsella RL, Kimmey JM, Smirnov A, Woodson R, Gaggioli MR. Autophagy prevents early proinflammatory responses and neutrophil recruitment during Mycobacterium tuberculosis infection without affecting pathogen burden in macrophages. PLoS Biol. (2023) 21:e3002159. doi: 10.1371/journal.pbio.3002159
122. Golovkine GR, Roberts AW, Morrison HM, Rivera-Lugo R, McCall RM, Nilsson H, et al. Autophagy restricts Mycobacterium tuberculosis during acute infection in mice. Nat Microbiol. (2023) 8:819–32. doi: 10.1038/s41564-023-01354-6
123. Aylan B, Bernard EM, Pellegrino E, Botella L, Fearns A, Athanasiadi N, et al. ATG7 and ATG14 restrict cytosolic and phagosomal Mycobacterium tuberculosis replication in human macrophages. Nat Microbiol. (2023) 8:803–18. doi: 10.1038/s41564-023-01335-9
124. Deretic V, Wang F. Autophagy is part of the answer to tuberculosis. Nat Microbiol. (2023) 8:762–3. doi: 10.1038/s41564-023-01373-3
125. Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage affects immunity to tuberculosis. Adv Exp Med Biol. (2013) 783:103–20. doi: 10.1007/978-1-4614-6111-1_6
126. Mayer-Barber KD. Granulocytes subsets and their divergent functions in host resistance to Mycobacterium tuberculosis- a 'tipping-point' model of disease exacerbation. Curr Opin Immunol. (2023) 84:102365. doi: 10.1016/j.coi.2023.102365
127. Torrado E, Cooper AM. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. (2010) 21:455–62. doi: 10.1016/j.cytogfr.2010.10.004
128. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. (2010) 466:973–7. doi: 10.1038/nature09247
129. Muefong CN, Owolabi O, Donkor S, Charalambous S, Mendy J, Sey ICM, et al. Major neutrophil-derived soluble mediators associate with baseline lung pathology and post-treatment recovery in tuberculosis patients. Front Immunol. (2021) 12:740933. doi: 10.3389/fimmu.2021.740933
130. Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. (2007) 8:369–77. doi: 10.1038/ni1449
131. Seiler P, Aichele P, Bandermann S, Hauser AE, Lu B. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur J Immunol. (2003) 33:2676–86. doi: 10.1002/eji.200323956
132. Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM, et al. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun. (2000) 68:577–83. doi: 10.1128/IAI.68.2.577-583.2000
133. Cruz A, Fraga AG, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med. (2010) 207:1609–16. doi: 10.1084/jem.20100265
134. Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. (2009) 31:974–85. doi: 10.1016/j.immuni.2009.10.007
135. Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. (2011) 208:2251–62. doi: 10.1084/jem.20110919
136. Lowe DM, Bandara AK, Packe GE, Barker RD, Wilkinson RJ, Griffiths CJ, et al. Neutrophilia independently predicts death in tuberculosis. Eur Respir J. (2013) 42:1752–7. doi: 10.1183/09031936.00140913
137. Han Y, Kim SJ, Lee SH, Sim YS Ryu YJ, Chang JH, et al. High blood neutrophil-lymphocyte ratio associated with poor outcomes in miliary tuberculosis. J Thorac Dis. (2018) 10:339–46. doi: 10.21037/jtd.2017.12.65
138. Carvalho ACC, Amorim G, Melo MGM, Silveira AKA, Vargas PHL, Moreira ASR, et al. Pre-treatment neutrophil count as a predictor of antituberculosis therapy outcomes: a multicenter prospective cohort study. Front Immunol. (2021) 12:661934. doi: 10.3389/fimmu.2021.661934
139. Behar SM, Baehrecke EH. Tuberculosis: Autophagy is not the answer. Nature. (2015) 528:482–3. doi: 10.1038/nature16324
140. Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. TRIMs and galectins globally cooperate and TRIM16 and Galectin-3 Co-direct autophagy in endomembrane damage homeostasis. Dev Cell. (2016) 39:13–27. doi: 10.1016/j.devcel.2016.08.003
141. Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, et al. Galectins control mTOR in response to endomembrane damage. Mol Cell. (2018) 70:120–35. doi: 10.1016/j.molcel.2018.03.009
142. Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, et al. Galectin-3 Coordinates a cellular system for lysosomal repair and removal. Dev Cell. (2020) 52:69–87. doi: 10.1016/j.devcel.2019.10.025
143. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science. (2004) 306:1037–40. doi: 10.1126/science.1103966
144. Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C, et al. Escape of intracellular Shigella from autophagy. Science. (2005) 307:727–31. doi: 10.1126/science.1106036
145. Levine B. Eating oneself and uninvited guests; autophagy-related pathways in cellular defense. Cell. (2005) 120:159–62. doi: 10.1016/S0092-8674(05)00043-7
146. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. (2009) 5:527–49. doi: 10.1016/j.chom.2009.05.016
147. Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. (2013) 340:701–6. doi: 10.1126/science.1233028
148. Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. (2014) 54:224–33. doi: 10.1016/j.molcel.2014.03.009
149. Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. (2014) 15:403–11. doi: 10.1016/j.chom.2014.03.012
150. Feng S, Nehls EM, Kinsella RL, Chavez SM, Naik SK, McKee SR, et al. Autophagy protects against high-dose Mycobacterium tuberculosis infection. bioRxiv. (2022) doi: 10.1101/2022.11.04.515158
151. Orvedahl A, McAllaster MR, Sansone A, Dunlap BF, Desai C. Autophagy genes in myeloid cells counteract IFNgamma-induced TNF-mediated cell death and fatal TNF-induced shock. Proc Natl Acad Sci U S A. (2019) 116:16497–506. doi: 10.1073/pnas.1822157116
152. Wang YT, Zaitsev K, Lu Q, Li S, Schaiff WT, Kim KW, et al. Select autophagy genes maintain quiescence of tissue-resident macrophages and increase susceptibility to Listeria monocytogenes. Nat Microbiol. (2020) 5:272–81. doi: 10.1038/s41564-019-0633-0
153. Park S, Buck MD, Desai C, Zhang X, Loginicheva E, Martinez J, et al. Autophagy genes enhance murine gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe. (2016) 19:91–101. doi: 10.1016/j.chom.2015.12.010
154. Lu Q, Yokoyama CC, Williams JW, Baldridge MT, Jin X. Homeostatic control of innate lung inflammation by vici syndrome gene epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe. (2016) 19:102–13. doi: 10.1016/j.chom.2015.12.011
155. Wang Y, Sharma P, Jefferson M, Zhang W, Bone B, Kipar A, et al. Non-canonical autophagy functions of ATG16L1 in epithelial cells limit lethal infection by influenza A virus. EMBO J. (2021) 40:e105543. doi: 10.15252/embj.2020105543
156. Durgan J, Florey O. Many roads lead to CASM: Diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci Adv. (2022) 8:eabo1274. doi: 10.1126/sciadv.abo1274
157. Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. (2015) 17:893–906. doi: 10.1038/ncb3192
158. Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, Porto M, et al. Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog. (2013) 9:e1003734. doi: 10.1371/journal.ppat.1003734
159. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. (2012) 11:469–80. doi: 10.1016/j.chom.2012.03.007
160. Murrow L, Malhotra R, Debnath J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat Cell Biol. (2015) 17:300–10. doi: 10.1038/ncb3112
161. Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. (2018) 360:6384. doi: 10.1126/science.aar5078
162. Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. (2018) 37:e753. doi: 10.15252/embj.201899753
163. Beatty WL, Rhoades ER, Hsu DK, Liu FT, Russell DG. Association of a macrophage galactoside-binding protein with Mycobacterium-containing phagosomes. Cell Microbiol. (2002) 4:167–76. doi: 10.1046/j.1462-5822.2002.00183.x
164. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. (2013) 501:512–6. doi: 10.1038/nature12566
165. Bell SL, Lopez KL, Cox JS, Patrick KL, Watson RO. Galectin-8 senses phagosomal damage and recruits selective autophagy adapter TAX1BP1 To control Mycobacterium tuberculosis infection in macrophages. MBio. (2021) 12:e0187120. doi: 10.1128/mBio.01871-20
166. Morrison HM, Craft J, Rivera-Lugo R, Johnson JR, Golovkine GR, Bell SL, et al. Deficiency in Galectin-3,−8, and−9 impairs immunity to chronic Mycobacterium tuberculosis infection but not acute infection with multiple intracellular pathogens. PLoS Pathog. (2023) 19:e1011088. doi: 10.1371/journal.ppat.1011088
167. Beckwith KS, Beckwith MS, Ullmann S, Saetra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun. (2020) 11:2270. doi: 10.1038/s41467-020-16143-6
Keywords: tuberculosis, autophagy, atg8ylation, macrophage, immunity
Citation: Deretic V (2023) Atg8ylation as a host-protective mechanism against Mycobacterium tuberculosis. Front. Tuberc. 1:1275882. doi: 10.3389/ftubr.2023.1275882
Received: 10 August 2023; Accepted: 05 September 2023;
Published: 29 September 2023.
Edited by:
Trude Helen Flo, Norwegian University of Science and Technology, NorwayReviewed by:
Oliver Florey, Babraham Institute (BBSRC), United KingdomCopyright © 2023 Deretic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vojo Deretic, dmRlcmV0aWNAc2FsdWQudW5tLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.