 Olivia K. Favor
Olivia K. Favor James J. Pestka
James J. Pestka Melissa A. Bates
Melissa A. Bates Kin Sing Stephen Lee
Kin Sing Stephen Lee- 1Department of Pharmacology and Toxicology, College of Osteopathic Medicine, Michigan State University, East Lansing, MI, United States
- 2Institute for Integrative Toxicology, Michigan State University, East Lansing, MI, United States
- 3Department of Food Science and Human Nutrition, Michigan State University, East Lansing, MI, United States
- 4Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, United States
- 5Department of Chemistry, Michigan State University, East Lansing, MI, United States
Exposure to exogenous particles found as airborne contaminants or endogenous particles that form by crystallization of certain nutrients can activate inflammatory pathways and potentially accelerate autoimmunity onset and progression in genetically predisposed individuals. The first line of innate immunological defense against particles are myeloid-lineage phagocytes, namely macrophages and neutrophils, which recognize/internalize the particles, release inflammatory mediators, undergo programmed/unprogrammed death, and recruit/activate other leukocytes to clear the particles and resolve inflammation. However, immunogenic cell death and release of damage-associated molecules, collectively referred to as “danger signals,” coupled with failure to efficiently clear dead/dying cells, can elicit unresolved inflammation, accumulation of self-antigens, and adaptive leukocyte recruitment/activation. Collectively, these events can promote loss of immunological self-tolerance and onset/progression of autoimmunity. This review discusses critical molecular mechanisms by which exogenous particles (i.e., silica, asbestos, carbon nanotubes, titanium dioxide, aluminum-containing salts) and endogenous particles (i.e., monosodium urate, cholesterol crystals, calcium-containing salts) may promote unresolved inflammation and autoimmunity by inducing toxic responses in myeloid-lineage phagocytes with emphases on inflammasome activation and necrotic and programmed cell death pathways. A prototypical example is occupational exposure to respirable crystalline silica, which is etiologically linked to systemic lupus erythematosus (SLE) and other human autoimmune diseases. Importantly, airway instillation of SLE-prone mice with crystalline silica elicits severe pulmonary pathology involving accumulation of particle-laden alveolar macrophages, dying and dead cells, nuclear and cytoplasmic debris, and neutrophilic inflammation that drive cytokine, chemokine, and interferon-regulated gene expression. Silica-induced immunogenic cell death and danger signal release triggers accumulation of T and B cells, along with IgG-secreting plasma cells, indicative of ectopic lymphoid tissue neogenesis, and broad-spectrum autoantibody production in the lung. These events drive early autoimmunity onset and accelerate end-stage autoimmune glomerulonephritis. Intriguingly, dietary supplementation with ω-3 fatty acids have been demonstrated to be an intervention against silica-triggered murine autoimmunity. Taken together, further insight into how particles drive immunogenic cell death and danger signaling in myeloid-lineage phagocytes and how these responses are influenced by the genome will be essential for identification of novel interventions for preventing and treating inflammatory and autoimmune diseases associated with these agents.
Introduction
Exogenous and endogenous particles have profound effects on human health. The concept of particle toxicology was first introduced in the 15th century when occupational exposure to dust was etiologically linked to lung disease [reviewed by Donaldson and Seaton (2012)]. Paracelsus, the toxicologist who famously quoted “The dose makes the poison,” documented in a 1567 book his observations of lung disease symptoms in smelters and miners. In 1700, these observations were expanded upon by Bernardino Ramazzini, also known as the father of occupational medicine, who recognized that human disease could be triggered by environmental factors in his work Diseases of Workers. Industrialization in the 19th century elicited a rise in occupationally related diseases such as silicosis, asbestosis, lung cancer, and pulmonary fibrosis, leading to a significant increase in both in vitro and in vivo particle toxicology studies in the 20th century (Perlman and Maier, 2019).
Over the past 50 years, the field of particle toxicology has expanded to encompass not only pathological impacts of environmental particles but also of endogenously formed crystals, hereafter referred to as endogenous particles (Donaldson and Seaton, 2012). Growing interest in endogenous particles is largely attributed to increased worldwide prevalence of genetic hyperuricemia and familial hypercholesterolemia, which are predispositions for crystallization of monosodium urate (MSU) and cholesterol, respectively (Beheshti et al., 2020; Butler et al., 2021). Hyperuricemia is a risk factor for gout, coronary heart disease, and neurodegenerative disorders (Jin et al., 2012; Rock et al., 2013), and hypercholesterolemia is a risk factor for coronary heart disease (Goldstein and Brown, 2015), atherosclerosis (Nidorf et al., 2020), non-alcoholic steatohepatitis (NASH) (Ioannou, 2016), and cholesterol gallstone disease (Di Ciaula et al., 2018). The observed pathological outcomes associated with MSU and cholesterol crystals have spurred ongoing in vitro and in vivo studies to determine the mechanisms by which these endogenous particles, as well as other types of endogenous particles (e.g., calcium-containing salts) elicit toxicity.
In parallel with the growing interest in particle toxicology, immunologist Polly Matzinger and her colleagues introduced the “danger model” to explain the development of autoimmune disease, which contrasts the classic “self/non-self model” (Matzinger, 1994; Gallucci and Matzinger, 2001; Matzinger, 2002). While the self/non-self model posits that autoreactivity occurs when the adaptive immunity mistakenly recognizes host “self” tissues as foreign “non-self” tissues, the danger model suggests that accumulation of dead cell corpses and released danger signals (e.g., cytokines, chemokines, alarmins, nucleic acids) contribute to heightened proinflammatory responses in innate immune cells, activation of antigen-presenting cells, and differentiation of autoreactive T and B cells, leading to loss of immunological self-tolerance and autoimmunity (Tveita, 2010). In the context of particle toxicology, Matzinger’s danger model provides a useful framework for understanding the mechanisms by which exogenous and endogenous particles induce inflammation and autoimmunity.
The purpose of this review is to provide an overview of critical molecular mechanisms by which exogenous particles (i.e., silica, asbestos, carbon nanotubes, titanium dioxide, aluminum-containing salts) and endogenous particles (i.e., MSU, cholesterol crystals, calcium-containing salts) promote unresolved inflammation and autoimmunity by inducing toxic responses in myeloid-lineage phagocytes with emphases on inflammasome activation and necrotic and programmed cell death pathways. Autoimmune diseases are defined by uncontrolled innate immunity leading to hyperactivation of adaptive immunity, the latter of which drives tissue damage and disease pathogenesis (Doria et al., 2012). The review will focus specifically on myeloid-lineage phagocytes (i.e., macrophages, neutrophils), as these cells comprise the first line of immunological defense against particles (Boraschi et al., 2017).
Exogenous Particles, Endogenous Particles, and Their Sources
Exogenous particles are defined herein as any particles originating from environmental or synthetic sources. These include silicon dioxide (SiO2), asbestos, carbon nanotubes (CNTs), titanium dioxide (TiO2), and aluminum-containing salts (alum). SiO2 is one of the most abundant compounds in the Earth’s crust (Mossman and Glenn, 2013) and is classified based on its level of crystallinity, with crystalline SiO2 (cSiO2) demonstrating a periodic order of atoms and amorphous SiO2 (aSiO2) having either an anarchic order of atoms or crystalline structures (Mulay and Anders, 2016). Asbestos refers to a broad group of fibrous, chain-like silicate minerals that have high tensile strength, large surface area, and resistance to abrasion and chemical corrosion—all characteristics that made it ideal for construction, mining, and other industrial applications such as pipefitting, shipyard work, insulation manufacturing, and textile production in the 20th century (Kusiorowski et al., 2012; Goswami et al., 2013). Like asbestos, CNTs are fibrous, carbon-containing materials that have high tensile strength and large surface area (Iijima 1991), rendering them useful in construction and electronics (Zhang and Lieber, 2016; Venkataraman et al., 2019). TiO2 can exist as either nanospheres or nanobelts (Porter et al., 2013), giving them versatile use in construction, agriculture, food additives, cosmetics, and biomedicine (Mohajerani et al., 2019; Baranowska-Wojcik et al., 2020; Musial et al., 2020). Alum was serendipitously discovered as a vaccine adjuvant nearly 100 years ago (Glenny et al., 1926) and is now the most utilized adjuvant in the world (Tomljenovic and Shaw, 2011). Another highly relevant exogenous particle is particulate matter (PM), which may consist of carbon, sulfate, nitrate, silicon, ammonium, and sodium emissions from both manmade and organic sources (Dominici et al., 2015). Due to the complex and heterogenous composition of PM, its toxic mechanisms are much more difficult to characterize than the previously mentioned particles. A detailed discussion of PM toxicity falls outside the scope of this review, but the reader is referred to several excellent reviews on this topic (Adams et al., 2015; Peixoto et al., 2017; Park et al., 2018; Zhao et al., 2019; Kelly and Fussell, 2020; Curtis, 2021).
Exposure to exogenous particles can occur by inhalation, ingestion, or injection. SiO2 was first identified as an inhalation hazard in the 1920s when it was etiologically linked to silicosis in miners (Elliott, 1923; Heffernan, 1929). Today, SiO2 remains an occupational inhalation hazard in construction, mining, ceramic manufacturing, dental mold production, and jewelry production (Fazen et al., 2020; Hall et al., 2020; Russ et al., 2020). Asbestos exposure primarily occurs by inhalation (Mossman et al., 2011), and despite decreased industrial use in the United States and Europe, industrial asbestos use is being deferred to Asian and Latin-American countries (Pira et al., 2018). CNTs can either pose as respirable toxicants similar to asbestos fibers in industrial settings (Donaldson et al., 2010) or function as carrier systems in targeted drug, vaccine, cancer, and gene therapies (Beg et al., 2011; Negri et al., 2020). TiO2 exposure can occur by inhalation in industrial environments or ingestion of commercial products, and it exhibits toxicity in the lungs, digestive tract, brain, and cardiovascular system (Baranowska-Wojcik et al., 2020). Exposure to alum occurs primarily by injection as a vaccine adjuvant (Gherardi and Authier, 2012) but can also occur by inhalation in foundry work and related occupations (Rifat et al., 1990; Polizzi et al., 2002). While the National Institute for Occupational Safety and Health (NIOSH) recommends using respirators in occupations with high, prolonged particle exposure (NIOSH, 2019), low compliance with such guidelines is associated with respirator discomfort, lack of training on health hazards, self-employment, and breathing problems that would be aggravated by respirator use (Fukakusa et al., 2011).
Endogenous particles are defined as any particles that form within biological systems. From an environmental perspective, many of these are formed by crystallization of nutrients, typically in individuals with corresponding genetic predispositions. Endogenous particles include MSU, cholesterol crystals (CCs), and calcium salts such as calcium phosphate (CaP) and calcium oxalate (CaOx). MSU originates from crystallized uric acid, a byproduct of purine nucleic acid catabolism released by dying cells (Gallo and Gallucci, 2013). Cholesterol is derived from dietary sources and biosynthesis in the liver (Pownall and Gotto, 2019). Dysregulated cholesterol metabolism can contribute to deposition of low-density lipoproteins (LDLs) and high-density lipoproteins (HDLs) in tissues, engulfment of LDLs and HDLs by recruited macrophages and dendritic cells (DCs), and intracellular CC formation (Schaftenaar et al., 2016; Defesche et al., 2017; Varsano et al., 2018). Like cholesterol, calcium occurs both in dietary and body sources, and it can crystallize as CaP and CaOx salts within renal tubules and blood vessels (Khan et al., 2016; Karasawa and Takahashi, 2017). While biomolecules and minerals found in endogenous particles can originate from diet and/or metabolism, crystal formation itself occurs in myeloid phagocytes and along tubular structures within the body.
Endogenous particles are thought to form by crystallization resulting from supersaturation of biological molecules (e.g., cholesterol, uric acid) and minerals (e.g., calcium) in the joints, arteries, and urinary tract (Mulay and Anders, 2016). Although the precise mechanisms for crystal formation have yet to be elucidated, genome-wide associated studies have identified loci that contribute to overproduction and insufficient metabolism of uric acid, LDL, HDL, and calcium-containing salts (Holdt and Teupser, 2013; Dron and Hegele, 2017; Sayer, 2017; Kawamura et al., 2019; Tai et al., 2019). Overabundance of these biomolecules in synovial fluid, serum, or urine creates conditions for supersaturation, increasing the likelihood of crystallization and disease development (Table 1).
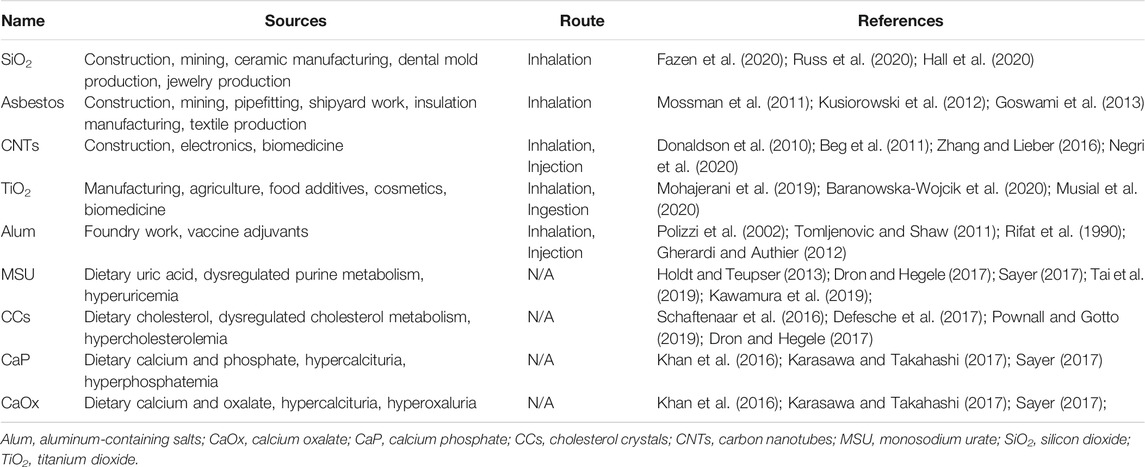
TABLE 1. Sources and common exposure routes of exogenous and endogenous particles.
Recognition of Exogenous and Endogenous Particles by Myeloid-Lineage Phagocytes
Particles can stimulate multiple types of surface receptors to promote incorporation into phagosomes, or intracellular vesicles that transport phagocytosed particles. Macrophages, neutrophils, and DCs can recognize particles through a diverse repertoire of surface receptors (Figure 1). For instance, SiO2 and TiO2 both bind to members of the class A scavenger receptor family including SR-A1 and macrophage receptor with collagenous structure (MARCO). However, SiO2 also binds the class B scavenger receptors SR-B1 and CD36/SR-B2, whereas TiO2 does not (Thakur et al., 2009; Tsugita et al., 2017b; Nishijima et al., 2017). In macrophages, stimulation of class A and class B scavenger receptors by their respective ligands has been associated with p38 MAPK and JNK activation and enhanced particle endocytosis (Zani et al., 2015). Alternatively, CNTs, which are more fibrous than SiO2 and TiO2 particles, are recognized by the phosphatidylserine receptor T cell immunoglobulin mucin 4 (Tim4) (Omori et al., 2021).
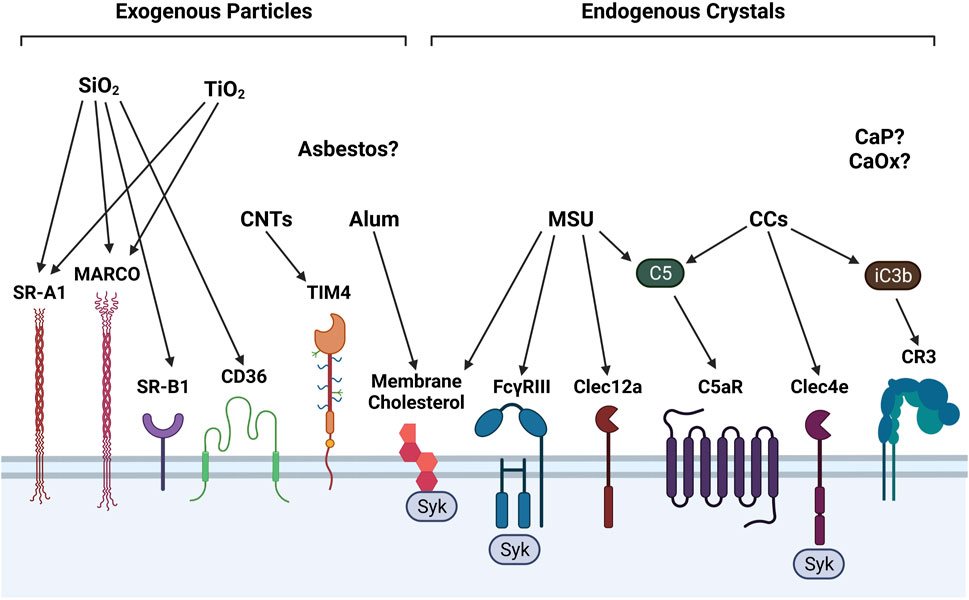
FIGURE 1. Surface receptors involved in detecting exogenous and endogenous particles. Phagocytes employ a diverse assortment of membrane receptors to recognize and ultimately phagocytose particles, some of which are depicted in this illustration. SiO2 is recognized by scavenger receptors SR-A1, MARCO, SR-B1, CD36. TiO2 is recognized only by SR-A1 and MARCO. CNTs are recognized by phosphatidylserine receptor Tim4. Alum and MSU interact directly with membrane cholesterol moieties to stimulate Syk signaling. MSU and CCs activate complement components C5 and iC3b, which stimulate C5aR and CR3, respectively. MSU also binds to FcγRIII/CD16 and C-type lectin (Clec)-12a. On human phagocytes only, CCs are recognized by Clec4e. Surface receptors for asbestos fibers and calcium-containing salts (e.g., CaP, CaOx) have not yet been identified.
Contrary to exogenous particles, endogenous particles are recognized by a more diverse set of surface receptors and elicit different intracellular signaling pathways. For example, MSU crystals interact with C-type lectin (Clec)-12a on macrophages and DCs (Neumann et al., 2014; Li et al., 2019a) and FcγRIII/CD16 on neutrophils (Barabe et al., 1998). FcγRIII is also expressed in murine macrophages and DCs (Nimmerjahn and Ravetch, 2008). On human macrophages, neutrophils, and DCs, CCs can bind to Clec4e to potentiate proinflammatory immune responses (Kiyotake et al., 2015). FcγRIII stimulation by MSU and Clec4e stimulation by CCs trigger downstream spleen tyrosine kinase (Syk) signaling (Desaulniers et al., 2001; Clement et al., 2016). Alternatively, both MSU and alum can directly interact with membrane cholesterol moieties and induce Syk signaling in DCs, potentially by lipid membrane sorting (Ng et al., 2008; Flach et al., 2011).
Surface receptors for asbestos, CaP, and CaOx have not yet been identified, but it is possible that phagocytes recognize these particles directly by membrane lipid binding or indirectly through complement receptor signaling. Accordingly, complement C5 binding to the C5a receptor (C5aR) can amplify MSU-driven toxicity (An et al., 2014). In addition, activation of C5aR by C5 and complement receptor 3 (CR3) by complement factor iC3b can augment CC-induced toxic responses (Niyonzima et al., 2017).
Differential Expression of Particle-Sensing Receptors in Myeloid-Lineage Phagocytes
Not only is it important to consider the types of surface receptors that can be stimulated by particles, but it is also crucial to further emphasize which myeloid-lineage phagocytes express which receptors, because different particles might activate different subsets of myeloid cells. For instance, SR-A1 is expressed by macrophages, monocytes, and DCs, while MARCO is primarily expressed by macrophages and DCs (Ingersoll et al., 2010; Stephen et al., 2010). CD36 is expressed by many cell types including macrophages, monocytes, DCs, and non-hematopoietic cells, whereas SR-B1 is predominantly expressed by macrophages and hepatocytes (Prabhudas et al., 2014; Li et al., 2020). Macrophages and DCs have been shown to express Tim4, but data pertaining to Tim4 expression in neutrophils is currently lacking (Wong et al., 2010; Caronni et al., 2021). On the other hand, macrophages, neutrophils, and DCs express Clec12a (Neumann et al., 2014; Li et al., 2019a; Vaillancourt et al., 2021), FcγRIII (Barabe et al., 1998; Nimmerjahn and Ravetch, 2008), and Clec4e (in humans only) (Kiyotake et al., 2015). Collectively, these observations suggest that myeloid-lineage phagocytes might be better prepared to respond to endogenous particles compared to exogenous particles. Nevertheless, additional research is required to confirm or reject such a hypothesis.
Several studies published over the past decade have shed additional light on differential expression patterns of particle-sensing receptors in tissue-resident macrophages that commonly interact with particles, including bone marrow-derived macrophages (BMDMΦs), alveolar macrophages (AMΦs), and hepatic Kupffer cells (KCs). A comprehensive gene expression review across different tissue-resident macrophage types found that SR-A1 expression is high in BMDMΦs and low in both AMΦs and KCs, whereas MARCO expression is low in BMDMΦs and high in both AMΦs and KCs (Ley et al., 2016). In the same analysis, notable observations were made in relation to the other receptors mentioned in the present review: 1) SR-B1 expression is higher in AMΦs and KCs compared to BMDMΦs; 2) CD36 expression is high in BMDMΦs but lower in AMΦs and KCs; 3) Tim4 expression is low in BMDMΦs and AMΦs but high in KCs; 4) Clec12a is highly expressed in BMDMΦs but not in AMΦs or KCs; 5) Clec4e expression is high in BMDMΦs and AMΦs but low in KCs; 6) FcγRIII is highly expressed in BMDMΦs, AMΦs, and KCs; and 7) C5aR expression is high only in BMDMΦs (Ley et al., 2016). In two different studies, MARCO and Tim4 expression were found to be lower in BMDMΦs compared to KCs (Beattie et al., 2016; A-Gonzalez et al., 2017). Two other studies also showed that Clec4e expression increases in macrophages localized to the kidneys during acute renal inflammation, suggesting Clec4e perpetuates proinflammatory cytokine signaling and cell death in the kidney (Lv et al., 2017; Tanaka et al., 2020).
Not only do tissue-resident macrophages demonstrate differential expression patterns for many particle-sensing surface receptors, but similar patterns can be detected in blood-derived monocytes. A single-cell gene expression analysis with human monocytes found that expression levels for SR-A1, MARCO, CD36, and Clec4e significantly differed between classical monocytes (CD14++CD16–), intermediate monocytes (CD14++CD16+), and non-classical monocytes (CD14+CD16++) (Gren et al., 2015). A different study comparing FcγRIII expression in classical and non-classical monocytes found that expression was higher in classical monocytes than non-classical monocytes in mice, but expression was lower in classical monocytes than non-classical monocytes in humans (Kerntke et al., 2020). Furthermore, FcγRIII expression in murine classical monocytes was similar to that in neutrophils, while expression in human neutrophils was remarkably higher than both classical and non-classical monocytes (Kerntke et al., 2020). Although surface receptor expression patterns were not compared between monocytes and macrophages in either of these studies, such distinctions might require a case-by-case basis approach. For instance, monocytes and BMDMΦs express similar levels of Clec12a (Lobato-Pascual et al., 2013), but CD36 expression increases in monocytes differentiating into BMDMΦs (Huh et al., 1996). Accordingly, future research in this area would provide valuable insight into specific myeloid-lineage phagocyte subsets that respond to different types of exogenous and endogenous particles. Future therapies for particle-induced inflammatory and autoimmune diseases may potentially include antagonists that prevent particle-receptor interactions and downstream toxicity.
Inflammasome Activation: A Central Mechanism of Particle-Induced Toxicity and Proinflammatory Immune Responses
Following phagocytosis, one central mechanism of toxicity initiated by exogenous and endogenous particles alike is inflammasome activation (Martinon et al., 2006; Dostert et al., 2008; Hornung et al., 2008; Tsugita et al., 2017a; Dominguez-Gutierrez et al., 2018; Karasawa and Takahashi, 2019; Luz et al., 2019). Inflammasomes are cytosolic multiprotein complexes that assemble upon sensing diverse stimuli—including microbial moieties, endogenous danger signals, and particles—to promote proinflammatory signaling (Strowig et al., 2012; Sayan and Mossman, 2016). Because of their importance in orchestrating innate immune responses, inflammasomes are primarily studied in innate immune cells, most notably macrophages; however, other investigators are beginning to investigate their roles in adaptive immune cells and nonhematopoietic cells (Gasteiger et al., 2017). Pattern recognition receptors (PRRs) from the nucleotide-binding oligomerization domain (NOD) leucine-rich region-containing receptor (NLR) family, including NLRP1, NLRP3, and NLRC4, as well as absent-in-melanoma 2 (AIM2) and pyrin, form well-defined inflammasome complexes (Haneklaus and O’Neill, 2015; Duncan and Canna, 2018; Heilig and Broz, 2018; Lugrin and Martinon, 2018; Mitchell et al., 2019). In addition, the NLRs NLRP2, NLRP6, NLRP7, NLRP12, and NLRC5, as well as interferon-inducible protein 16 (IFI16), form inflammasome complexes, albeit less well-characterized or atypical complexes (Elinav et al., 2011; Khare et al., 2012; Vladimer et al., 2012; Janowski and Sutterwala, 2016; Matsuoka et al., 2019).
The NLRP3 inflammasome is the most studied inflammasome due to its putative roles in various pathologies including rheumatic disease (So et al., 2013), Alzheimer’s disease (Heneka et al., 2018), acute myocardial infarction (Toldo and Abbate, 2018), kidney disease (Komada and Muruve, 2019), type 2 diabetes (Wada and Makino, 2016), obesity (Rheinheimer et al., 2017), cancer (Moossavi et al., 2018), and COVID-19, which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (Freeman and Swartz, 2020). This inflammasome also plays a pertinent role in particle-driven diseases such as pulmonary fibrosis, asthma, chronic obstructive pulmonary disease (COPD), malignant mesothelioma, and other lung cancers (Sayan and Mossman, 2016). NLRP3 inflammasome oligomers consist of the NOD-like receptor NLRP3, the adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and pro-caspase-1 as an effector (Kelley et al., 2019). Three distinct pathways are implicated for NLRP3 inflammasome activation: 1) the canonical pathway, 2) the alternative pathway, and 3) the noncanonical pathway (He et al., 2016). The alternative and noncanonical pathways fall beyond the scope of this review, though readers are directed to other excellent discussions of these topics for further information (Gaidt and Hornung, 2017; Yi, 2017).
Step 1: Priming
Canonical inflammasome activation occurs in a two-step process that first requires a priming signal to promote transcriptional upregulation of inflammasome-related proteins and a subsequent activating signal to trigger inflammasome oligomerization and caspase-1 activation (Zheng et al., 2020). Priming can be accomplished upon recognition of damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), or cytokines by specific surface receptors. For example, the bacterial PAMP lipopolysaccharide (LPS) activates toll-like receptor (TLR)-4, the endogenous DAMP high group mobility group box 1 (HMGB1) activates TLR2/4/9, and tumor necrosis factor (TNF)-α and interleukin (IL)-1α activate the TNF and IL-1 receptors, respectively (Rai and Agrawal, 2017; McKee and Coll, 2020; Wang and Zhang, 2020). These binding events contribute to phosphorylation of the inhibitor of nuclear factor kappa-B kinase (IKK)-β subunit within the cytosolic IKK2 complex. IKKβ then phosphorylates IκBα and targets it for K48-ubiquitination and proteasomal degradation. Degradation of IκBα liberates the dimeric transcription factor nuclear factor-kappa B (NF-κB), allowing its translocation into the nucleus where it upregulates the inflammasome subunits NLRP3, ASC, and pro-caspase-1 as well as pro-IL-1β and pro-IL-18 (Dorrington and Fraser, 2019) (Figure 2). Under homeostatic conditions, DAMPs and proinflammatory cytokines are typically contained inside phagocytes; however, these danger signals can be released into the extracellular environment following particle-induced cell death (Zitvogel et al., 2010; Rabolli et al., 2016). If clearance of extracellular particles, DAMPs, and cytokines is hindered, perpetual stimulation of DAMP/cytokine receptors and particle-sensing receptors may ensue, leading to aberrant inflammasome priming and activation.
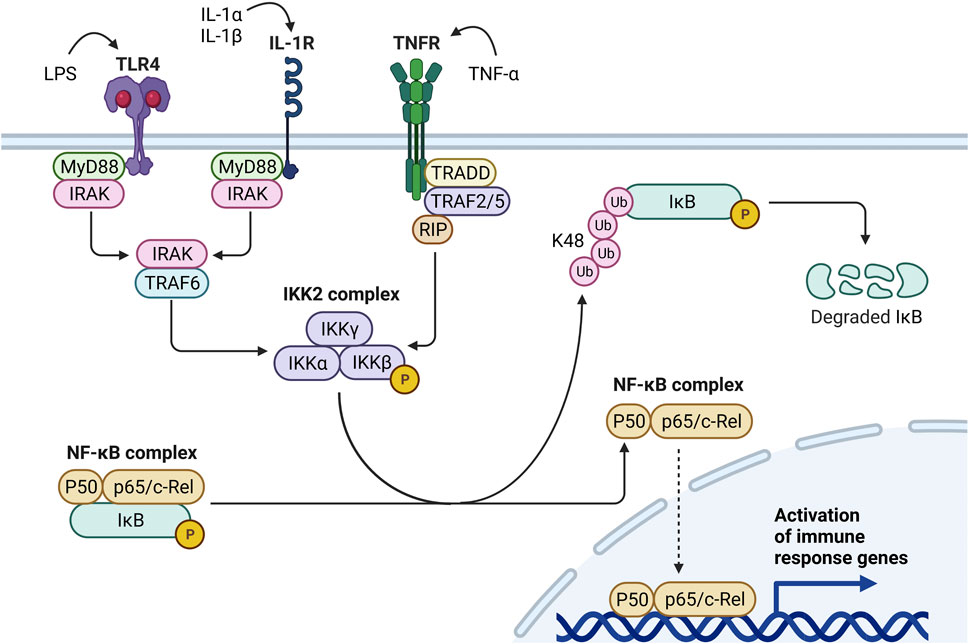
FIGURE 2. Mechanisms of Signal 1 inflammasome priming. Inflammasome priming can be triggered by diverse stimuli including bacterial molecules (e.g., LPS), alarmins (e.g., IL-1α), or proinflammatory cytokines (e.g., IL-1β, TNF-α). LPS binds to TLR4, activates the MyD88-IRAK-TRAF6 pathway, and induces IKKβ activity within the IKK2 complex. Likewise, by binding IL-1R, IL-1α and IL-1β promote IKKβ activity through the MyD88-IRAK-TRAF6 pathway. Conversely, when TNF-α binds TNFR, the TRADD-TRAF2/5-RIP pathway induces IKKβ activity. Once activated, IKKβ phosphorylates IκB within the NF-κB complex, targeting IκB for K48 polyubiquitination and proteasomal degradation. IκB degradation liberates the NF-κB complex (i.e., P50 and p65/c-Rel) and enables its translocation to the nucleus, where it upregulates proinflammatory cytokines, chemokines, and other immune response genes.
Step 2: Activation
Following the priming signal, a separate activating signal triggers inflammasome assembly and caspase-1 maturation. Contrary to the priming step, which is initiated by a select set of ligands, the activating step can be triggered by many different stimuli including ATP (Di Virgilio et al., 2017), mitochondrial reactive oxygen species (mtROS) (Zhou et al., 2011), mitochondrial DNA (mtDNA) (Shimada et al., 2012), ceramide (Scheiblich et al., 2017), bacterial toxins (Munoz-Planillo et al., 2013), and particles (Sayan and Mossman, 2016). The diverse nature of these stimuli suggests they do not directly act upon inflammasome subunits but rather induce a few common intracellular events that lead to inflammasome oligomerization. Lawlor and Vince propose that these signals may converge on lysosomal rupture, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress (Lawlor and Vince, 2014) (Figure 3).
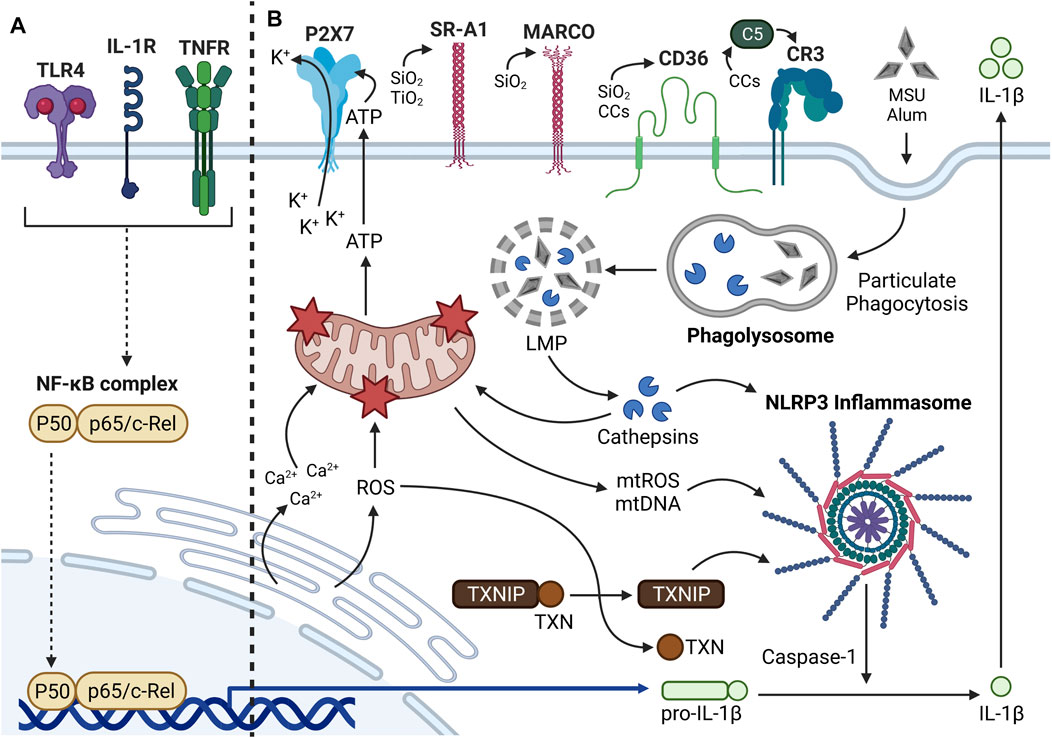
FIGURE 3. Mechanisms of Signal 2 inflammasome activation. (A) Summary of Signal 1 inflammasome priming. Translocation of NF-κB into the nucleus leads to upregulation of proinflammatory cytokines such as pro-IL-1β and inflammasome subunits (i.e., NLRP3, ASC, pro-caspase-1; not shown). (B) The NLRP3 inflammasome is a cytosolic multiprotein complex that promotes proinflammatory cytokine production in response to extracellular stimuli and intracellular stress. Many extracellular and intracellular components can be involved in particle-driven inflammasome oligomerization and activity. Some particles (e.g., SiO2, TiO2, CCs) bind transmembrane receptors prior to phagocytosis, whereas other particles (e.g., MSU, alum) interact directly with the plasma membrane. Following phagocytosis, the particle-containing phagosomes fuse with a lysosome to form a phagolysosome. Through undefined mechanisms, the particles aggravate the phagolysosomal membrane and induce lysosomal membrane permeabilization (LMP), which causes release of lysosomal proteases called cathepsins into the cytosol. Some cathepsins such as cathepsin B can directly trigger inflammasome oligomerization. Cathepsins can cause mitochondrial dysfunction and release of mtDAMPs (e.g., ATP, mtROS, mtDNA) into the cytosol. ATP released from dying phagocytes can interact with P2X7 receptors and trigger K+ efflux, which can contribute to inflammasome activation. mtROS and mtDNA can also contribute significantly to inflammasome oligomerization. Mitochondrial dysfunction can alternatively be elicited by CHOP-mediated Ca2+ release and ROS production from the ER. Cytosolic ROS contributes to dissociation of TXN from TXNIP, the latter of which can promote inflammasome activation. Once the inflammasome is assembled, pro-caspase-1 proteolytically activates adjacent pro-caspase-1 moieties. Activated caspase-1 then proteolytically processes pro-IL-1β to IL-1β, which is ultimately released from the cell to interact with IL-1R on neighboring phagocytes.
Lysosomal Membrane Permeabilization
Once particles or other danger signals are incorporated into a phagosome, the phagosome fuses with a lysosome to form an intracellular phagolysosome (Rosales and Uribe-Querol, 2017). The role of the phagolysosome is to digest internalized materials; however, many crystalline particles such as cSiO2, CCs, alum, and MSU disrupt the phagolysosomal membrane in a process called lysosomal membrane permeabilization (LMP) (Hornung et al., 2008; Campden and Zhang, 2019). LMP describes any process by which the lysosomal membrane is disrupted and lysosomal enzymes including cathepsins are released into the cytosol (Boya and Kroemer, 2008). Although the precise mechanisms by which particles induce LMP remain unknown, one critical study recently found that a subfamily of silanols, termed “nearly free silanols,” on the surface of cSiO2 and aSiO2 particles promote membranolysis by direct membrane interaction (Pavan et al., 2020). Once cathepsins are released from ruptured phagolysosomes, some cathepsins may directly activate the inflammasome (Niemi et al., 2011; Orlowski et al., 2015; Chevriaux et al., 2020) or elicit dysfunction of other intracellular organelles, including the mitochondria and ER, that can indirectly activate the inflammasome. Accordingly, exogenous and endogenous particles that are engulfed by phagocytes can directly elicit LMP and indirectly promote mitochondrial and ER stress.
Mitochondrial Dysfunction
As mentioned in the previous section, cathepsins released by particle-triggered LMP may promote downstream mitochondrial dysfunction (Thibodeau et al., 2004; Joshi and Knecht, 2013). Mitochondrial dysfunction has been linked to inflammasome activation specifically by the release of mitochondrial DAMPs (mtDAMPs) such as ATP, oxidized mtDNA, and mtROS (Zhou et al., 2011; Mills et al., 2017). A large body of evidence suggests ATP can trigger inflammasome assembly and caspase-1 activation in macrophages, specifically by promoting K+ ion efflux through either the P2X7 surface receptor or the TWIK2 K+ channel (Luna-Gomes et al., 2015; Di Virgilio et al., 2017; Martinez-Garcia et al., 2019). In phagocytes, particle exposure also can trigger apoptosis, a process that can begin in the mitochondria (Hamilton et al., 1996; Iyer et al., 1996; Lim et al., 1999; Thibodeau et al., 2003; Hu et al., 2006; Hirano et al., 2017). It is possible that opening of the mitochondrial permeability transition pore (MPTP) during apoptosis allows oxidized mtDNA and mtROS to exit depolarized mitochondria and activate the inflammasome, but this requires additional investigation. Once in the cytosol, oxidized mtDNA can directly bind NLRP3 to promote caspase-1 activation and resultant IL-1β maturation (Shimada et al., 2012; Zhong et al., 2018).
Conversely, the requirement of mtROS in inflammasome activation is debatable, with some investigators arguing that mtROS are indispensable for inflammasome activation and others suggesting that mtROS only partially contribute to inflammasome activity (Munoz-Planillo et al., 2013; Harijith et al., 2014; Gross et al., 2016). Of interest, activation of the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), which mediates transcription of antioxidant genes, has been shown to inhibit inflammasome-driven IL-1β maturation, supporting a clear role for total cellular ROS in promoting inflammasome assembly (Tsai et al., 2011; Ka et al., 2015; Hennig et al., 2018). It is currently unclear how much of this response is driven by mtROS specifically; however, it is reasonable to expect mtROS play a fairly large role because mitochondria are major drivers of ROS production (Brieger et al., 2012). Evidence suggests mtROS can further disrupt lysosomal compartments (Heid et al., 2013). On the other hand, lysosomal leakage has been previously shown to occur upstream from perturbations in mitochondrial membrane potential following cSiO2 exposure in AMΦs (Thibodeau et al., 2004; Joshi and Knecht, 2013). Taken together, these findings suggest that lysosomal and mitochondrial dysfunction might reciprocally influence one another in the context of particle-induced toxicity. Such a notion requires additional study, as the mechanisms driving cyclical lysosomal and mitochondrial dysfunction remain unclear.
Endoplasmic Reticulum Stress
Similar to mitochondrial dysfunction, ER stress has also been linked to inflammasome activation. Extracellular ATP, a mtDAMP released from dying cells, stimulates the transcription factor CCAAT enhancer binding protein homologous protein (CHOP) in LPS-primed BMDMΦs to induce Ca2+ signaling, which promotes Ca2+ efflux from the ER, downstream mitochondrial damage, and resultant caspase-1 activation (Murakami et al., 2012). Additionally, ER stress promotes NF-κB-dependent transcription of pro-IL-1β and activation of the oxidative protein folding pathway to induce ROS production (Kim et al., 2014). Elevated ROS levels initiate the dissociation of thioredoxin-interacting protein (TXNIP) from thioredoxin (TXN) and its subsequent association with the LRR of NLRP3, which promotes inflammasome oligomerization and caspase-1 activation (Kim et al., 2014). Furthermore, ER stress can activate inositol-requiring enzyme 1 alpha (IRE1α), which promotes translocation of TXNIP to the mitochondria and the release of mtDAMPs including mtROS and mtDNA (Zhou et al., 2020). In previous studies, it has been demonstrated in macrophages that SiO2 upregulates CHOP (Chen et al., 2019), asbestos increases CHOP expression and cytosolic Ca2+ (Ryan et al., 2014), and MWCNTs promote intracellular lipid accumulation, CHOP phosphorylation, and CD36 expression (Long et al., 2019). Additional research is needed to determine the specific steps that occur between particle phagocytosis and downstream ER stress.
Taken together, inflammasome-activating exogenous (Table 2) and endogenous (Table 3) particles have multifaceted impacts on intracellular lysosomal, mitochondrial, and ER-related functionality, and these pathways can feed into each other to mount robust inflammatory responses that drive rheumatic and autoimmune disease.
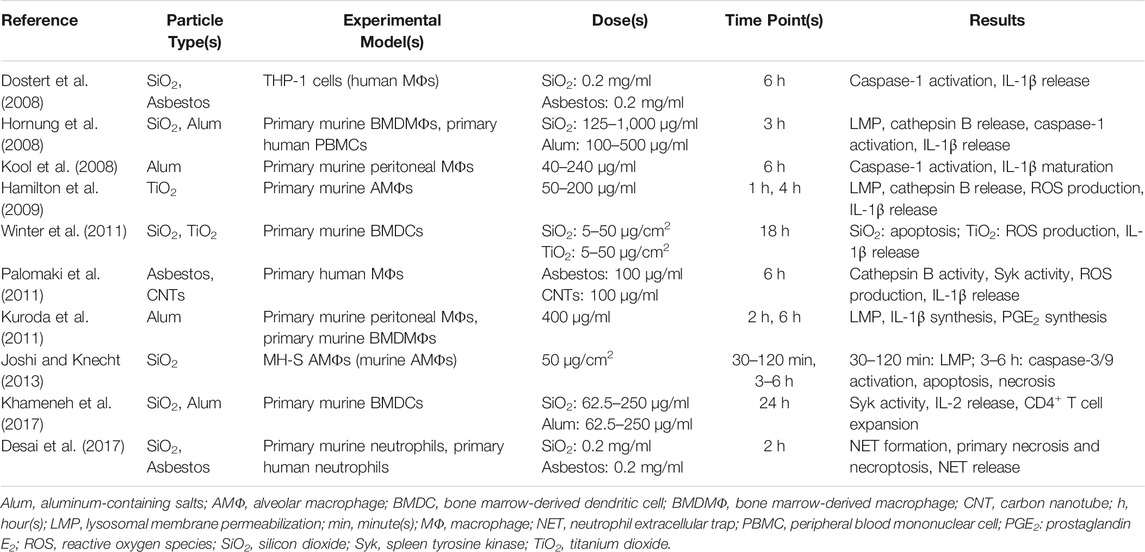
TABLE 2. Examples of studies demonstrating toxic responses of exogenous particles.
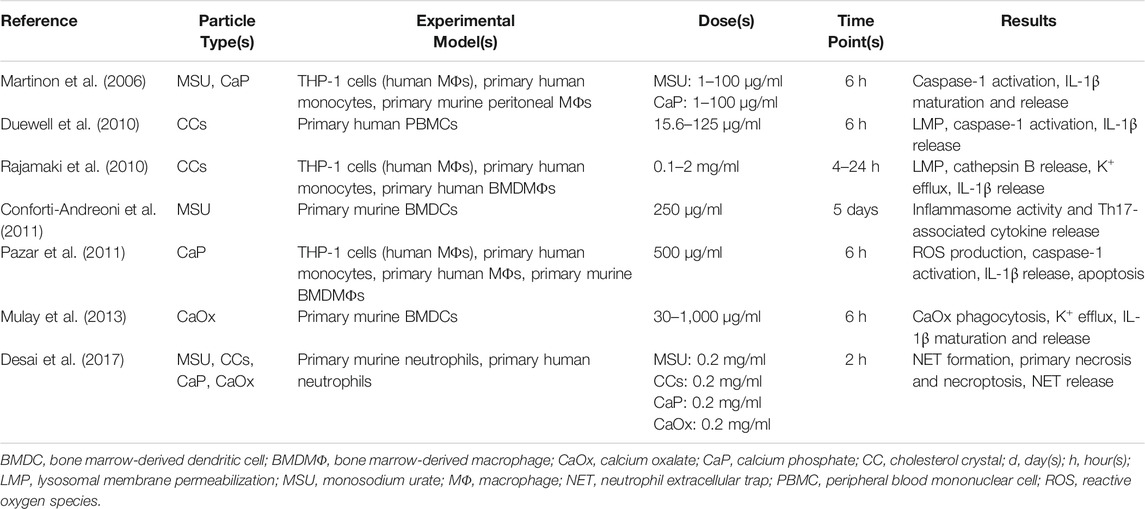
TABLE 3. Examples of studies demonstrating toxic responses of endogenous particles.
Particle-Induced Cell Death Pathways That Contribute to Innate and Adaptive Immune Responses
Consistent with Matzinger’s danger model (Gallucci and Matzinger, 2001), exposure to exogenous (Table 2) and endogenous (Table 3) particles can trigger inflammasome-dependent and -independent cell death pathways in phagocytes, resulting in the release of DAMPs and autoantigens that can activate innate and adaptive immunity. Of note for the present review are necrosis, pyroptosis, apoptosis, necroptosis, and NETosis. In addition, we provide a brief perspective on PANoptosis, a recently proposed unified cell death pathway involving pyroptosis, apoptosis, and necroptosis.
Necrosis
Necrosis is an unprogrammed cell death pathway characterized by organellar disorganization, cellular swelling, plasma membrane rupture, and DAMP release (Rello et al., 2005). No specific signaling pathway is associated with necrosis, but it is usually preceded by lysosomal rupture, mitochondrial swelling, and ROS production (Green and Llambi, 2015; Niemann and Rohrbach, 2016). Necrosis is generally considered a proinflammatory mode of cell death, as DAMPs released from dying cells provoke inflammatory gene expression and signaling in neighboring innate and adaptive immune cells (Davidovich et al., 2014) (Figure 4A). Both exogenous and endogenous particles have been shown to provoke necrotic cell death in a variety of cell types including AMΦs, fibroblasts, mesothelial cells, and kidney epithelial cells. Primary mechanisms by which particles induce necrosis include upstream LMP, mitochondrial depolarization, and ROS production (Joshi and Knecht, 2013; Mulay et al., 2019b; Ito et al., 2020), though it might also be possible that particles directly disrupt the plasma membrane (Pavan et al., 2020; Reus et al., 2020) or promote necrosis through other unelucidated mechanisms. Infectious agents, mechanical stress, hypoxia, and chemical and radiation exposure can also compromise the integrity of the cell membrane, leading to necrosis (Nirmala and Lopus, 2020). When particles induce necrosis, the dying cell releases particles and DAMPs, which can perpetuate unresolved inflammation if not efficiently cleared.
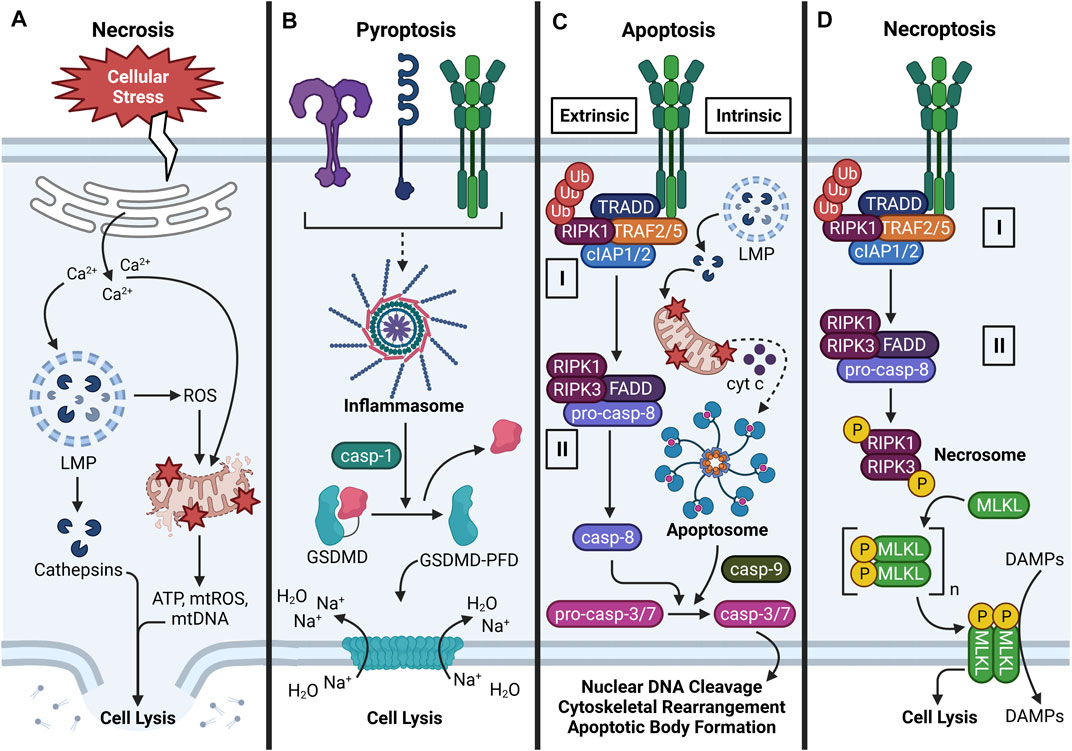
FIGURE 4. Major cell death pathways induced by particles. (A) Overview of necrosis. Necrosis can be triggered by various stimuli that provoke cellular stress. Common hallmarks of necrosis include Ca2+ efflux from the ER, Ca2+-induced LMP and cathepsin release, ROS-driven mitochondrial dysfunction, cellular swelling, plasma membrane rupture, and DAMP release. (B) Overview of pyroptosis. Following NLRP3 inflammasome oligomerization and activation, caspase-1 proteolytically processes GSDMD to expose its N-terminal pore-forming domain (PFD). GSDMD-PFD polymerizes into a pore in the plasma membrane, which allows Na+ to move along its electrochemical gradient into the cell. By osmosis, water enters the cell, causes cellular swelling, and cell lysis. (C) Overview of apoptosis (extrinsic and intrinsic). Extrinsic apoptosis is triggered by activation of a death receptor (e.g., TNFR), which promotes assembly of Complex I. Complex I consists of TRADD, TRAF2/5, cIAP1/2, and ubiquitinated RIPK1. Inhibition of cIAP1/2 and/or deubiquitylation of RIPK1 by CYLD (not shown) induces formation of cytosolic Complex II, which consists of RIPK1, RIPK3, FADD, and pro-caspase-8 oligomers that proteolytically activate themselves. Intrinsic apoptosis is defined by release of cytochrome c (cyt c) from perturbed mitochondria, formation of a multiprotein apoptosome, and activation of caspase-9. In certain cases, LMP-driven cathepsin release may contribute to mitochondrial dysfunction. Caspase-8/9 proteolytically activates caspase-3/7, which promotes nuclear DNA cleavage, cytoskeletal rearrangement, and apoptotic body formation. (D) Overview of necroptosis. Necroptosis is characterized by activation of a death receptor (e.g., TNFR), Complex I formation, and Complex II formation as in extrinsic apoptosis. Inhibition of pro-caspase-8 activation allows formation of the RIPK1-RIPK3 necrosome, which phosphorylates MLKL. Phospho-MLKL monomers polymerize into a pore-shaped complex at phosphatidylinositol 3-phosphate sites in the inner leaflet of the plasma membrane. Consequently, cell lysis occurs and DAMPs are released from the cell. Some steps in the depicted cell death pathways are omitted for clarity.
Pyroptosis
Pyroptosis is a programmed lytic cell death pathway that is dependent on inflammasome activation (Lamkanfi and Dixit, 2014; Abe and Morrell, 2016). As previously discussed, many different types of exogenous and endogenous particles can activate the inflammasome (Martinon et al., 2006; Dostert et al., 2008; Hornung et al., 2008; Tsugita et al., 2017a; Dominguez-Gutierrez et al., 2018; Karasawa and Takahashi, 2019; Luz et al., 2019). When the NLRP3 inflammasome assembles and activates caspase-1 following particle exposure, caspase-1 not only converts pro-IL-1β and pro-IL-18 to their mature forms but also cleaves the N-terminal pore-forming domain (PFD) of gasdermin D (GSDMD). PFD monomers oligomerize and insert into the plasma membrane, which destabilizes plasma membrane potential and leads to an osmotic movement of water into the cell that mediates cellular swelling and lysis (Ros et al., 2020) (Figure 4B). Like necrosis, pyroptosis is considered a proinflammatory cell death pathway because the GSDMD pore and resultant lysis caused by its insertion into the plasma membrane allows passage of DAMPs from intracellular to extracellular environments (Davidovich et al., 2014).
Apoptosis
Exposure to exogenous and exogenous particles such as SiO2, asbestos, CCs, MSU, and CaP can induce apoptosis in macrophages (Hamilton et al., 1996; Geng et al., 2003; Pazar et al., 2011; Joshi and Knecht, 2013; Kim et al., 2016). Unlike necrosis, apoptosis is morphologically defined by nuclear DNA cleavage, cytoskeletal rearrangement, cellular shrinkage, and plasma membrane blebbing (Rello et al., 2005) (Figure 4C). In apoptosis, the plasma membrane does not rupture but rather invaginates organelles and DAMPs in apoptotic bodies that are engulfed by phagocytes (Santavanond et al., 2021). Accordingly, apoptosis is a quiescent mode of cell death; however, if apoptotic bodies are insufficiently removed, they undergo secondary necrosis, which releases DAMPs into the extracellular space (Nagata, 2018). Apoptosis can be induced by death receptor (DR) signaling (extrinsic pathway), mitochondrial signaling (intrinsic pathway), or perforin/granzyme signaling (Elmore, 2007; Nirmala and Lopus, 2020). The perforin/granzyme pathway falls outside the scope of the present review, but readers are encouraged to consult other excellent reviews on this topic (Trapani and Smyth, 2002; Voskoboinik et al., 2010; Voskoboinik et al., 2015). While particles have not been shown to bind DRs and particle-sensing receptors are not known to activate signaling components downstream from DRs (Thibodeau et al., 2003; Hu et al., 2006), an overview of extrinsic apoptosis is warranted because particle exposure can induce expression and secretion of DR ligands such as TNF-α (Dubois et al., 1989; Perkins et al., 1993; Gozal et al., 2002; Brown et al., 2007; Chen et al., 2018; Dominguez-Gutierrez et al., 2018). In the context of particle-triggered apoptosis, however, the intrinsic pathway is most relevant because particles can indirectly elicit mitochondrial stress (Thibodeau et al., 2003; Hu et al., 2006; Joshi and Knecht, 2013).
In the extrinsic pathway, the initiation phase is triggered by activation of a DR in the TNF receptor superfamily [e.g., TNF receptor (TNFR)-1 or Fas receptor (FasR)] by its corresponding ligand [e.g., TNF-α or Fas ligand (FasL)], which triggers association of an adapter protein to the intracellular domain of the DR (Nirmala and Lopus, 2020). The recruited adapter protein differs depending on the DR activated: FasL recruits Fas-associated protein with death domain (FADD) to FasR, and TNF-α recruits TNFR1-associated death domain protein (TRADD) to TNFR1 (Elmore, 2007). Specific to TNFR1, TNF receptor associated factor (TRAF)-2/5, receptor-interacting serine/threonine-protein kinase (RIPK)-1, and cellular inhibitor of apoptosis protein (cIAP)-1/2 are subsequently recruited to the intracellular receptor domain of TNFR1 and associate with TRADD (i.e., Complex I). Cylindromatosis tumor suppressor protein (CYLD) then deubiquitylates RIPK1 which allows this protein to leave Complex I and leads to association of FADD and RIPK3 (i.e., Complex II). Following these events, FADD associates with multiple pro-caspase-8 proteins to form a death-inducing signaling complex (DISC) that cleaves pro-caspase-8 to caspase-8 (Tummers and Green, 2017). Caspase-8 then proteolytically activates caspase-3 and -7 and triggers the execution phase of apoptosis (Elmore, 2007). During the execution phase, mature caspases-3 and -7 cleave nuclear DNA and intracellular proteins, which are encapsulated in apoptotic bodies (Walsh et al., 2008). Apoptotic cells express phosphatidylserine (PS) in the outer leaflet of the plasma membrane, which serves as an “eat me” signal for phagocytes to engulf the dying cells. This process, termed efferocytosis, functions to remove apoptotic bodies, thus preventing secondary necrosis and DAMP release (Segawa and Nagata, 2015).
In the intrinsic pathway, particle-driven organellar dysfunction leads to MPTP opening, as described in the previous section. This releases cytochrome c (cyt c) into the cytosol, where it binds with apoptotic protease activating factor 1 (Apaf-1) and pro-caspase-9 to form a multiprotein apoptosome complex that is structurally and functionally analogous to the inflammasome. During this process, mitochondrial second mitochondria-derived activator of caspases (SMAC) and high temperature requirement protein A2 (HtrA2) block the activity of inhibitors of apoptosis proteins (IAPs) to promote apoptosis (D’Arcy, 2019). Pro-caspase-9 moieties proteolytically activate each other within the apoptosome in a manner that resembles caspase-1 activation in the inflammasome. Activated caspase-9 then activates caspase-3, which activates caspase-activated DNase (CAD). Subsequently, caspase-3 cleaves nuclear DNA, triggers cytoskeletal rearrangement, and induces formation of apoptotic bodies, which are cleared by phagocytes under normal conditions (Elmore, 2007). However, phagocytotic capacity might be exhausted under conditions of persistent particle exposure, which raises the likelihood of secondary necrosis, DAMP release, and ongoing inflammatory signaling.
Necroptosis
Exogenous particles (e.g., SiO2, TiO2) and endogenous particles (e.g., CCs, MSU, CaP, CaOx) have been demonstrated to induce necroptosis in neutrophils, with less well-defined effects in macrophages (Desai et al., 2017; Honarpisheh et al., 2017). Necroptosis is a programmed cell death pathway that morphologically resembles necrosis but shares cellular machinery with the extrinsic apoptotic pathway. Accordingly, the early steps of necroptosis involve DR activation and recruitment of signaling proteins to the intracellular domain of the DR (e.g., TNFR1) to form Complex I as previously described (Frank and Vince, 2019). TNFR1 endocytosis, cIAP1/2 inhibition, and RIPK1 deubiquitylation by CYLD triggers formation of cytosolic Complex II, which involves dissociation of TRAF2/5 and cIAP1/2 and association of FADD and pro-caspase-8 as previously described (Newton and Manning, 2016; Su et al., 2016). Under normal conditions, Complex II can induce extrinsic apoptosis. However, impairment of pro-caspase-8 activity allows formation of a RIPK1- and RIPK3-containing complex called the necrosome (Galluzzi et al., 2017). The necrosome facilitates activation of the pseudokinase mixed lineage kinase domain-like (MLKL) via phosphorylation, and MLKL monomers forms oligomers at phosphatidylinositol 3-phosphate sites on the inner leaflet of the plasma membrane. Consequently, the MLKL oligomers elicit plasma membrane permeabilization by currently undefined mechanisms, leading to destabilization of membrane potential and cell lysis (Figure 4D). As with necrosis and pyroptosis, necroptosis allows DAMP release from the cell, and these DAMPs can induce downstream inflammatory responses (Newton and Manning, 2016). While the exact mechanisms of particle-induced necroptosis have yet to be fully elucidated, it is possible that cathepsins released from disrupted phagolysosomes promote assembly of the RIPK1-RIPK3 necrosome, which promotes MLKL polymerization (Honarpisheh et al., 2017). Another possibility is that TNF-α released from dying cells interacts with TNFR1 on viable nearby cells, promoting either extrinsic apoptosis or necroptosis depending on pro-caspase-8 activity.
PANoptosis
PANoptosis is a recently coined term that unifies inflammatory cell death involving simultaneous activation of pyroptosis, apoptosis, and necroptosis (Malireddi et al., 2019). Currently, two models have been proposed for PANoptosis-induced cell death. In the first model, an inflammatory stimulus simultaneously activates the inflammasome, apoptosome, and necrosome, which execute their respective forms of cell death. In the second model, PANoptosis is induced through inflammatory stimuli that trigger formation of a multiprotein complex called the PANoptosome, which triggers pyroptosis, apoptosis, and necroptosis at the same time. In myeloid-lineage phagocytes (e.g., neutrophils and macrophages exposed to LPS), caspase-8 (apoptosis), FADD (apoptosis and necroptosis), RIPK1 (necroptosis), NLRP3 (pyroptosis), ASC (pyroptosis), and caspase-1 (pyroptosis) can assemble into the PANoptosome. Accordingly, the PANoptosome can trigger apoptosis by caspase-8-dependent activation of caspase-3/7, pyroptosis by caspase-1-dependent cleavage of GSDMD, and necroptosis by RIPK3-dependent phosphorylation of MLKL (Samir et al., 2020) (Figure 5). The result is a detrimental cell death pathway that permits release of inflammatory DAMPs into the extracellular space. While it is still unclear which factors dictate execution of PANoptosis versus individual activation of pyroptosis, apoptosis, or necroptosis, inhibition of TGF-β-activated kinase 1 (TAK1) has previously been associated with PANoptosome formation in macrophages (Malireddi et al., 2020).
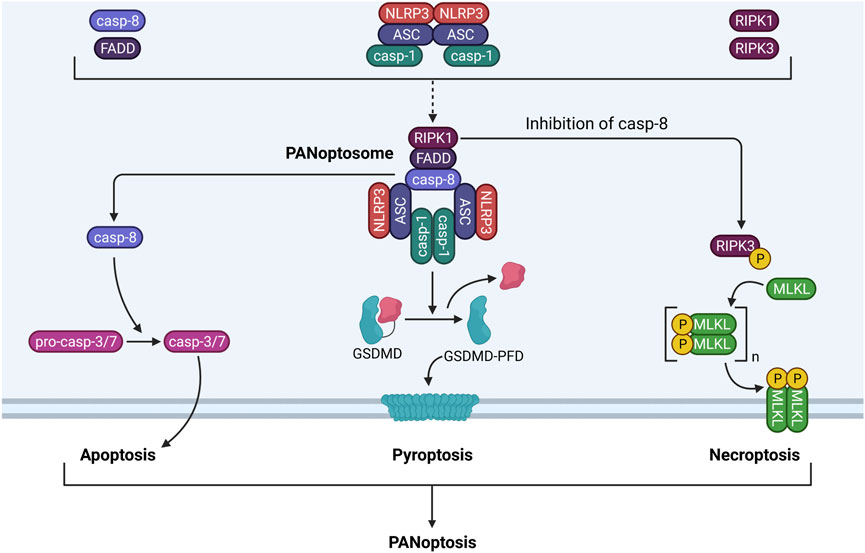
FIGURE 5. PANoptosome components and functionality. The PANoptosome is a multiprotein complex consisting of molecules from the apoptotic, pyroptotic, and necroptotic cell death pathways. Exposure to a proinflammatory stimulus such as LPS causes upregulation and activation of apoptotic proteins (i.e., caspase-8, FADD), pyroptotic proteins (i.e., NLRP3, ASC, caspase-1), and necroptotic proteins (i.e., RIPK1, RIPK3). These proteins associate with one another to form the PANoptosome. Following assembly, the PANoptosome can execute apoptosis, pyroptosis, and necroptosis simultaneously by driving caspase-3/7 activation by caspase-8, GSDMD processing by caspase-1, and MLKL phosphorylation and pore formation by RIPK1 and RIPK3. Cell death by concurrent apoptosis, pyroptosis, and necroptosis is termed PANoptosis.
Currently, there is no evidence linking particle exposure to PANoptosis in myeloid-lineage phagocytes, yet the current evidence supports such a possibility. Multiple particles have been previously reported to induce pyroptosis, apoptosis, and necroptosis in phagocytes [summarized by Mulay et al. (2019a)], but whether these multiple forms of cell death occur simultaneously in the same model is yet to be determined. Intriguingly, components of these three pathways can regulate one another. Not only can caspase-8 promote pyroptosis by cleaving GSDMD, but it can also prevent necroptosis by degrading RIPK. Necroptotic MLKL pore formation also can trigger NLRP3 inflammasome activity by K+ efflux (Schwarzer et al., 2020).
NETosis
In addition to pyroptosis, apoptosis, and necroptosis, exogenous particles (e.g., SiO2, alum) and endogenous particles (e.g., MSU, CCs, CaP) can induce NETosis [reviewed by (Rada, 2017) and (Li et al., 2018)]. NETosis describes the process by which neutrophil extracellular traps (NETs) are formed within and released from neutrophils (Sollberger et al., 2018). NETs are web-like structures composed of decondensed chromatin decorated with cytosolic myeloperoxidase (MPO) and neutrophil elastase (NE) (Papayannopoulos, 2018). NETs can be released from neutrophils by two mutually exclusive pathways: 1) suicidal NETosis and 2) vital NETosis (Yipp and Kubes, 2013) (Figure 6).
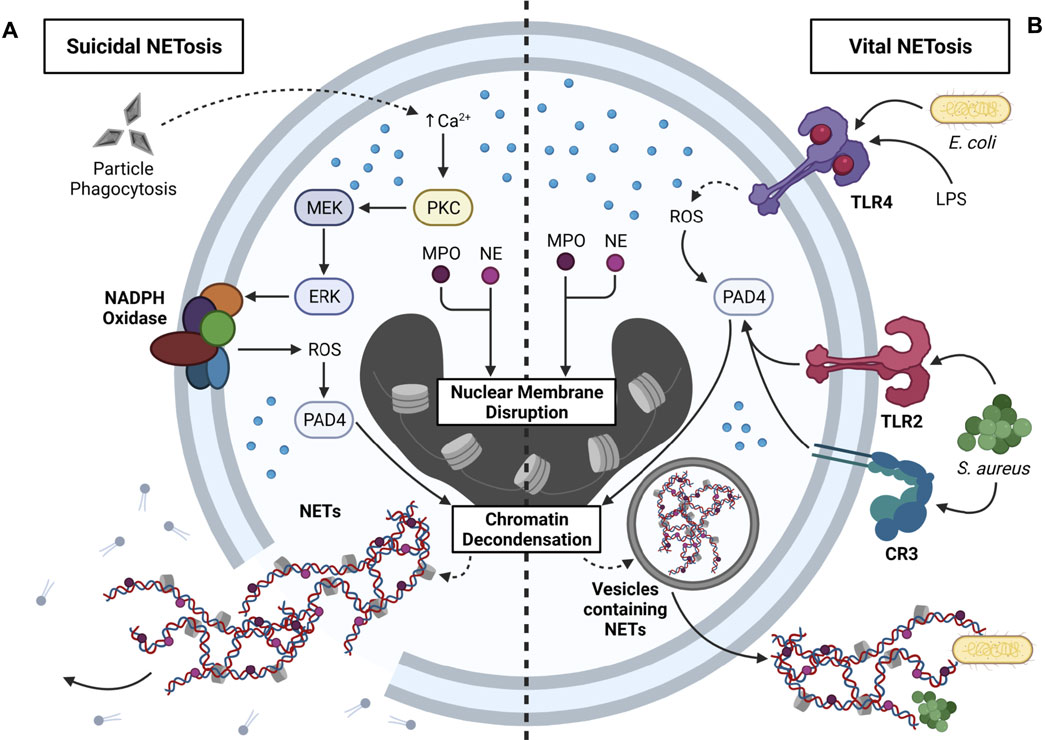
FIGURE 6. Mechanisms that contribute to NETosis. NETosis is the process by which neutrophil extracellular traps (NETs) are formed and released from neutrophils. Two primary forms of NETosis exist: suicidal and vital NETosis. (A) Overview of suicidal NETosis. Neutrophils phagocytose exogenous particles (e.g., SiO2) and endogenous particles (e.g., CCs), which triggers Ca2+ efflux from the ER. Intracellular Ca2+ efflux activates protein kinase C (PKC), PKC activates MEK, and MEK activates ERK. ERK stimulates NADPH oxidase via gp91phox phosphorylation, and NADPH oxidase produces ROS. ROS activates peptidyl arginine deiminase 4 (PAD4), which contributes to chromatin decondensation. Translocation of myeloperoxidase (MPO) and neutrophil elastase (NE) into the nucleus leads to nuclear membrane disruption and additional chromatin decondensation. Resultant NETs are directly released into the cytosol, and rupture of the plasma membrane contributes to extracellular NET release and neutrophil death. Suicidal NETosis occurs within a 2–4 h timeframe. (B) Overview of vital NETosis. Activation of TLR4 by LPS or Gram-negative bacteria (e.g., E. coli) contributes to ROS production, which is required for PAD4 activity. Alternatively, activation of TLR2 or CR3 by Gram-positive bacteria (e.g., S. aureus) leads to downstream PAD4 activation. As with suicidal NETosis, PAD4 triggers chromatin decondensation, and nuclear translocation of MPO and NE contributes to disruptions in the nuclear membrane. NETs are encased in nuclear vesicles, and NETs are released from viable neutrophils via exocytosis. Vital NETosis occurs within a 5–60 min timeframe, and released NETs can ensnare bacteria in the extracellular environment.
In suicidal NETosis, phagocytosis of particles elicits Ca2+ efflux from the ER, which triggers activation of protein kinase C (PKC). PKC activates the MEK/ERK pathway, ERK phosphorylates the gp91phox subunit of NADPH oxidase to induce ROS production, and increased cytosolic ROS activate peptidyl arginine deiminase 4 (PAD4). Together with MPO and NE, which translocate to the nucleus, PAD4 promotes chromatin decondensation and nuclear membrane disruption. Consequently, NETs are released from the nucleus into the cytosol, where they are further decorated with cytosolic proteins, and ultimately released into the extracellular environment upon cell lysis (Delgado-Rizo et al., 2017; Jorch and Kubes, 2017). Unlike suicidal NETosis, in vital NETosis, NETs are packaged into vesicles and released by exocytosis, and thus, the neutrophil remains viable. Stimulation of TLR2/4 or CR3 by Gram-positive bacteria (e.g., S. aureus) or Gram-negative bacteria (e.g., E. coli) activates PAD4, which partners with nuclear MPO and NE to induce nuclear membrane disruption and chromatin decondensation. (Delgado-Rizo et al., 2017; Jorch and Kubes, 2017). While released NETs can immobilize bacteria and viruses, they can also potentiate inflammation (Papayannopoulos, 2018). This raises a few questions pertaining to NETosis and particle toxicology. First, can released NETs capture extracellular particles and prevent their interactions with other phagocytes? Second, can NETs in particle-exposed neutrophils be decorated with particles prior to their release? Answering these questions could provide further insight into the protective and/or pathologic roles of NETs in particle-driven diseases.
Physicochemical Attributes That Influence Particle-Induced Toxicity and Proinflammatory Responses
Although many published studies suggest that different particles elicit similar toxic mechanisms in myeloid-lineage phagocytes, these responses depend greatly on the physicochemical attributes of the particle. Such attributes may include, but are not limited to, particle length (Hamilton et al., 2009; Murphy et al., 2011; Boyles et al., 2015), size (Fenoglio et al., 2012; Kusaka et al., 2014; Mischler et al., 2016), shape (Turci et al., 2016; Cohignac et al., 2018; Nahle et al., 2019), surface area (Sager and Castranova, 2009; Rabolli et al., 2010), and surface charge (Morishige et al., 2010; Hamilton et al., 2012; Hamilton et al., 2018; Pavan et al., 2020). An in-depth discussion of these attributes goes beyond the scope of this review, but the reader is encouraged to consult other previously published reviews on this topic (Aust et al., 2011; Rabolli et al., 2016; Sukhanova et al., 2018; Baranov et al., 2020). While current research has focused on characterizing relationships between particle attributes and toxic responses exhibited by exogenous particles, these relationships have not yet been characterized in relation to endogenous particles.
From Particle Exposure to Loss of Immunological Self-Tolerance
As discussed in previous sections, inflammasome activity and cell death induced by exogenous particles (Table 2) and endogenous particles (Table 3) permit DAMP release into the extracellular environment, where they can stimulate innate and adaptive immune cells. Released DAMPs include proinflammatory cytokines, nucleic acids, uric acid, cholesterol, heat shock proteins, HMGB1, type I interferons (IFNs), NETs, and mtDAMPs including mtDNA, ATP, cardiolipin, and cyt c [reviewed by Gallo and Gallucci (2013) and Grazioli and Pugin (2018)]. DCs, which are commonly referenced as bridges between innate and adaptive immunity (Balan et al., 2019), may also be bridges between particle exposure and loss of immunological self-tolerance because they interact with both particles and released DAMPs (Gallo and Gallucci, 2013). For example, DCs secrete cytokines involved in Th1 and Th17 differentiation (i.e., IL-1α, IL-1β, IL-2, IL-6, IL-17, IL-23) in response to MSU (Conforti-Andreoni et al., 2011), CCs (Westerterp et al., 2017), or alum (Khameneh et al., 2017). SiO2 and TiO2 induce caspase-1-dependent IL-1β maturation and apoptotic cell death in DCs (Winter et al., 2011), and extracellular IL-1β plays critical roles in promoting Th17 polarization (Sutton et al., 2006). In addition, HMGB1, ATP, TNF-α, and NETs can stimulate DC maturation, proinflammatory cytokine production (i.e., IL-6, CXCL8, IL-12, TNF-α), and subsequent T cell activation (Schnurr et al., 2000; Yang et al., 2007; Parackova et al., 2020). Furthermore, specific DC subsets secrete type I IFN and B-cell activating factor (BAFF), which regulate B cell differentiation into antibody-secreting plasma cells (Jego et al., 2005). Intriguingly, DCs also can promote and maintain immunological tolerance by inducing regulatory T cell (Treg) differentiation through cell-to-cell contact or secreted cytokines such as TGF-β and IL-10 (Raker et al., 2015; Hilpert et al., 2019). Consequently, activated Tregs can suppress proliferation and differentiation of naïve T cells into effector T cells, as well as the functions of activated CD4+ and CD8+ T cells, B cells, macrophages, and DCs. Treg depletion has been associated with exacerbated immune responses to self- and non-self antigens and development of autoimmunity (Sakaguchi et al., 2008; Ma, 2020). Nonetheless, the impacts of Treg function on particle-driven inflammation remain unclear. For instance, imbalances in the Treg/Th17 ratio significantly aggravate SiO2- and MSU-induced inflammation in the lungs and joints of mice, respectively (Dai et al., 2016; Dai et al., 2018), but inhaled SiO2 and asbestos elicit recruitment of Tregs to the lungs, which secrete TGF-β and IL-10 and contribute to resultant development of pulmonary fibrosis (Liu et al., 2010; Lo Re et al., 2011; Maeda et al., 2017). Accordingly, DCs play crucial roles in regulating T cell differentiation, interacting with proximal particles and DAMPs, and maintaining immunological self-tolerance. Dysregulated DC activation by particles and DAMPs, on the other hand, represents one major bridge connecting particle-induced innate immunity to irregular adaptive immunity.
Cells undergoing particle-induced death not only release DAMPs into the extracellular space but also autoantigens that can be recognized by T and B cells and consequently trigger autoimmunity. Autoantigens are self-proteins that are erroneously recognized as foreign proteins by the host’s immune system (Burbelo et al., 2021). When presented by DCs or other antigen-presenting cells, autoantigens promote activation of autoreactive T cells, which evade elimination in individuals with genetic predispositions to autoimmune disease and specifically target the presented self-proteins (Stranges et al., 2007). In addition, autoreactive T cells promote differentiation of autoreactive B cells into plasma cells, which secrete autoantibodies specific to the presented self-proteins (Riedhammer and Weissert, 2015). Autoantigens involved in systemic autoimmune diseases such as systemic lupus erythematosus (SLE) include dsDNA, small nuclear ribonucleoprotein (snRNP), cardiolipin, and histone proteins (i.e., H2B, H3, H4) (Doyle et al., 2014; Rosen and Casciola-Rosen, 2016). In some cases, autoantigens with post-translational modifications (PTMs), but not native self-proteins, are recognized by autoreactive T and B cells (Doyle et al., 2014). These PTMs include phosphorylation/dephosphorylation (Terzoglou et al., 2006; Nagai et al., 2012), methylation (Brahms et al., 2000), acetylation (van Bavel et al., 2009), citrullination (Lande et al., 2021), oxidation (Chang et al., 2004), and isomerization (Doyle et al., 2013). Since cytotoxic processes can contribute to modification of autoantigen structure and immunogenicity, it is tempting to speculate that intracellular mechanisms involved in inflammasome activation may also contribute to formation of PTM autoantigens and novel autoantigens. For example, cathepsins released from particle-containing phagolysosomes may non-specifically cleave mitochondrial and cytosolic proteins to create novel self-proteins that elicit immunological autoreactivity when released from dying cells. Caspase-1 may cleave mitochondrial and cytosolic proteins other than its identified substrates (i.e., pro-IL-1β, pro-IL-18, GSDMD) at specific sites, though this possibility seems less likely.
In addition to the roles that released DAMPs, autoantigens, and other danger signals play in aberrant activation of the immune system, genetics constitute a major determinant in the loss of immunological self-tolerance and resultant development of autoimmunity. Although some autoimmune diseases are monogenic, the majority are polygenic by nature (Doria et al., 2012). Genetic polymorphisms leading to increased expression and activation of inflammasome proteins (e.g., NLRP3), TLRs (e.g., TLR7, TLR9), transcription factors (e.g., STAT4), and IFN signaling proteins (e.g., IRF5) have been associated with increased susceptibility and severity of several autoimmune diseases including SLE, rheumatoid arthritis (RA), and multiple sclerosis (Cho and Gregersen, 2011; Yang and Chiang, 2015). In addition, loss-of-function mutations in efferocytosis receptors (e.g., MerTK), which leads to decreased engulfment of cytotoxic cell debris, have been associated with systemic autoimmunity (Lemke, 2013). Unique to autoimmune diseases are genetic polymorphisms in the major histocompatibility complex (MHC), or human leukocyte antigen (HLA) region in humans (Caso et al., 2018), which is crucial for presenting antigens to CD4+ helper T cells (Gough and Simmonds, 2007). Taken together, these genetic aberrations set the stage for increased inflammasome priming and activation, elevated proinflammatory cytokine and IFN production, and hindered cell debris clearance contributing to inflammatory tissue damage. In individuals susceptible to autoimmunity, these genetic variants may also contribute to enhanced autoantigen presentation to T and B cells, tissue damage by autoreactive T cells, and autoantibody production by autoreactive plasma cells, leading to development of autoimmunity.
Particle-Triggered Autoimmune and Autoinflammatory Diseases
Consistent with evoking inflammatory responses and cell death in phagocytes, exogenous and endogenous particles can trigger development of both chronic inflammatory and autoimmune diseases. Workplace inhalation of asbestos fibers has a long-recorded history of potentiating asbestosis and malignant mesothelioma (Westerfield, 1992; Brody, 1993; Bartrip, 2004). In rodents, CNT inhalation has been associated with proinflammatory AMΦ polarization and pulmonary fibrosis (Dong and Ma, 2018), (Kobayashi et al., 2017). TiO2 exposure has been connected to malabsorption, neuroinflammation, and cardiopulmonary inflammation in rodents and humans (Czajka et al., 2015; Zhao et al., 2018; Baranowska-Wojcik et al., 2020). MSU deposition in joints and blood vessels can promote gouty arthritis (Rock et al., 2013), coronary heart disease, and neurodegeneration (Jin et al., 2012). CCs can contribute to coronary heart disease (Goldstein and Brown, 2015), atherosclerosis (Nidorf et al., 2020), NASH (Ioannou, 2016), and cholesterol gallstone disease (Di Ciaula et al., 2018) if deposited in blood vessels, liver, or gallbladder, respectively. Furthermore, CaP and CaOx deposition can lead to pseudogout, nephropathy, and atherosclerosis (Lorenz et al., 2013; Kalampogias et al., 2016; Rosenthal and Ryan, 2016). Although different particles share similar mechanisms of promoting persistent inflammation, they elicit different pathologies depending on their routes of exposure and distribution in the body.
In addition to genetic predispositions, other factors that may modulate autoimmune susceptibility include particle exposure level, aging, and biological sex. Dose-response impacts of particle exposure on autoimmune pathogenesis remain largely uninvestigated. However, according to Paracelsus’s paradigm statement “The dose makes the poison,” it can be assumed that chronic exposures to many particles are more likely to induce aberrant inflammation and autoimmunity compared to acute exposures to few particles (Lison et al., 2014). This trend has been noted with respirable cSiO2 exposure in both mice (Bates et al., 2015; Mayeux et al., 2018) and humans (De Klerk et al., 2002; Boudigaard et al., 2021). Conversely, aging seems to have unclear impacts on the development of autoimmune disease. Older adults (>60 years) have higher prevalence of non-organ-specific autoantibodies than younger adults (20–60 years), but older adults are less likely than younger adults to develop autoimmune disease (Vadasz et al., 2013). Accordingly, aging contributes to restructuring of the immune system, leading to impaired immune responses, increased inflammation and oxidative stress, and increased autoantibody production (Watad et al., 2017). This suggests that the immune system is much more sensitive and reactive to autoantigens in younger adults compared to older adults, as many systemic autoimmune diseases manifest between 30 and 50 years of age (Amador-Patarroyo et al., 2012). A third factor that influences autoimmunity yet remains an enigma is biological sex. In general, autoimmune disease is more prevalent in women compared to men (Ngo et al., 2014). Postulated reasons for this observation include pregnancy and hormonal changes during puberty and menopause (Angum et al., 2020). While particle-induced inflammation and autoimmunity might be more biased toward men working in dusty occupations, more women are beginning to enter similar occupations, with emphases on making dental molds and using scouring powders in custodial work (Finckh et al., 2006; Pollard, 2012).
While exposure to exogenous and endogenous particles has been linked to inflammatory and autoimmune diseases, much less is known about their roles in initiating and exacerbating autoinflammatory disease. Briefly, autoinflammatory diseases are defined by uncontrolled innate immunity contributing to direct tissue damage and disease pathogenesis, whereas autoimmune diseases are potentiated by unresolved innate immunity leading to hyperactivation of adaptive immunity, the latter of which primarily drives tissue damage and disease pathogenesis (Doria et al., 2012). Most autoinflammatory diseases are caused by genetic mutations contributing to aberrant inflammasome activity, IL-1β activation, protein folding, IFN signaling, complement activation, and proinflammatory cytokine signaling (Krainer et al., 2020). Considering these mechanisms, it is not unreasonable to speculate that particles can worsen, or even trigger, autoinflammatory disease, beginning with myeloid-lineage phagocytes. Research in this area is crucial for verifying an etiological link between particle exposure and autoinflammatory disease and would provide additional rationale for regulating workplace particle exposure and fine-tuning dietary constituents for individuals predisposed to either autoinflammatory or autoimmune disease.
Linking Particle-Induced Inflammation to Autoimmune disease—Crystalline Silica as a Prototypical Example
Both preclinical and clinical studies have established that exposure to respirable cSiO2 contributes to SLE and other human autoimmune diseases (Parks et al., 2002; Pollard, 2016; Morotti et al., 2021). Patients with SLE typically have recurrent cycles of flaring and remission that eventually can cause cumulative damage to kidney, lung, heart, skin, and/or brain (Moulton et al., 2017). Intriguingly, both autoimmune flaring and disease progression can be induced by instilling cSiO2 to airways of mouse models of SLE (Brown et al., 2003; Brown et al., 2004; Brown et al., 2005; Bates et al., 2015; Clark et al., 2017; Bates et al., 2018; Foster et al., 2019). This is perhaps best exemplified in SLE-prone female New Zealand Black White (F1) (NZBWF1) mice which show autoantibody-driven glomerulonephritis with proteinuria by age 34 weeks resulting in death by age 52 weeks (Borchers et al., 2000). Our laboratory has demonstrated in this model that four weekly intranasal cSiO2 instillations of 1 mg trigger glomerulonephritis 12 weeks earlier than the conventional genome-driven model (Bates et al., 2015; Bates et al., 2018). Before glomerulonephritis onset in these mice, cSiO2 elicits severe pulmonary pathology involving continual accumulation of particle-laden AMsΦ, dying and dead cells resulting from PANoptosis, nuclear and cytoplasmic debris, and neutrophilic inflammation. Furthermore, there is buildup of large numbers of T and B cells, along with IgG-secreting plasma cells, suggestive of ectopic lymphoid tissue (ELT). Consistent with prolonged particle-induced pulmonary inflammation and ELT formation, lung fluid and blood from cSiO2-instilled mice have elevated proinflammatory cytokines, chemokines, and autoantibodies. As illustrated in Figure 7, these observations support the lung playing an essential role as the nexus for cSiO2-induced systemic autoimmune flaring and glomerulonephritis in the NZBWF1 mouse.
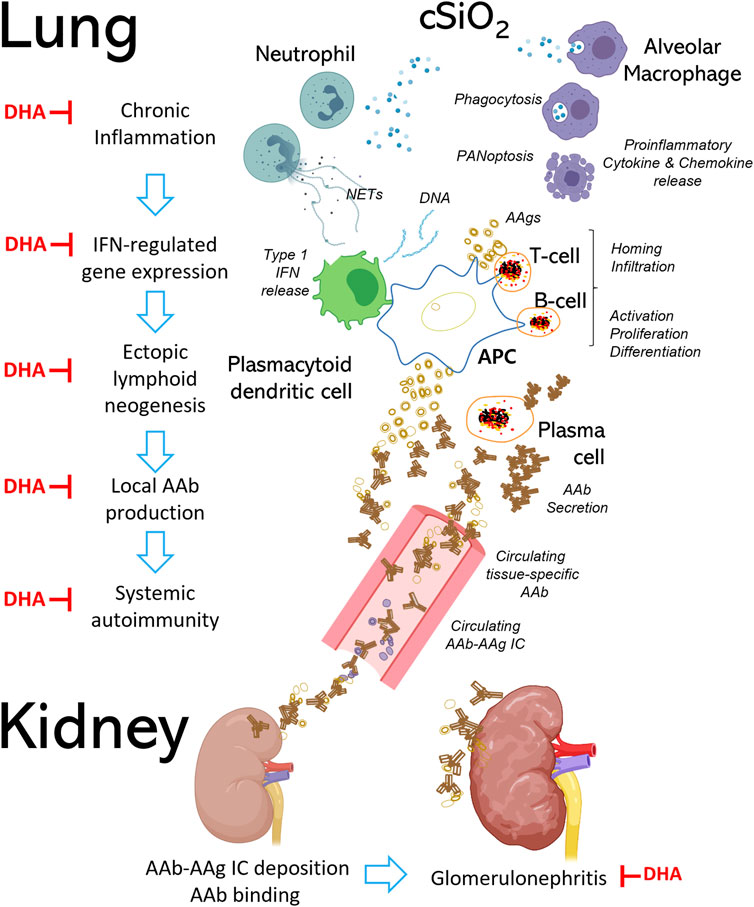
FIGURE 7. Respirable cSiO2 triggers autoimmune flaring and progression in the SLE-prone female NZBWF1 mouse. Chronic exposure to respirable cSiO2 particles contributes to irresolvable lung inflammation and systemic autoimmunity, resulting in end-stage glomerulonephritis and shortened lifespan in female NZBWF1 mice. Alveolar macrophages (AMΦs), which serve as one of the first lines of immunological defense in the lung, detect and phagocytose inhaled cSiO2. Resultantly, cSiO2 particles engulfed by AMΦs induce immunogenic cell death (i.e., pyroptosis, apoptosis, necrosis), proinflammatory cytokine and chemokine release, and NETosis in neighboring neutrophils. Aberrant accumulation of dead cell corpses, proinflammatory mediators, and host nucleic acids promotes recruitment of autoreactive T and B cells into the lung and type I interferon (IFN) release from plasmacytoid dendritic cells, leading to formation of ectopic lymphoid tissue (ELT). Type I IFN triggers maturation of B cells into plasma cells, which secrete IgG autoantibodies (AAb) that target local and systemic autoantigens (AAg). Binding of AAbs to their corresponding AAgs can lead to formation of immune complexes (ICs) that circulate in the body via blood vessels and deposit in other organs such as the kidneys. Once deposited, ICs recruit additional proinflammatory cells to the tissue, ultimately resulting in irreversible kidney damage and failure. Steps at which DHA has been shown to interfere with these pathways are indicated by red ┴ symbols.
A potential promising intervention against cSiO2-induced chronic lung inflammation and resultant autoimmunity is increasing dietary intake of the marine polyunsaturated fatty acids (PUFAs) docosahexaenoic acid (C22:6 ω-3; DHA) and eicosapentaenoic acid (C20:5 ω-3; EPA) (Wierenga et al., 2019). Modes of action for ω-3 PUFAs’ ameliorative effects include 1) moderating membrane and lipid raft function, 2) up- and down-regulating gene expression, 3) competing with ω-6 PUFAs and their downstream proinflammatory eicosanoids, and 4) pro-resolving actions of their downstream metabolites [reviewed by Akbar et al. (2017), Calder (2017), Ferreira et al. (2019), and Wierenga et al. (2020)]. Preclinical (Halade et al., 2010; Halade et al., 2013; Pestka et al., 2014) and clinical investigations (Akbar et al., 2017; Li et al., 2019b; Charoenwoodhipong et al., 2020; Duarte-Garcia et al., 2020) indicate that ω-3 PUFAs can counter onset and progression of lupus symptoms, including nephritis. We have found that dietary DHA supplementation reflecting realistic human consumption (i.e., 2 and 5 g/d) can be employed as a prophylactic approach against cSiO2-triggered autoimmune flaring in NZBWF1 mice (Bates et al., 2018). DHA consumption specifically inhibited cSiO2-triggered pulmonary accumulation of B and T cells, follicular DCs, and IgG+ plasma cells. Importantly, DHA dose-dependently inhibited cSiO2-triggered lung mRNA signatures indicative of inflammation-, chemokine-, and interferon (IFN)-related gene pathways (Benninghoff et al., 2019). Additionally, DHA supplementation suppresses both cSiO2-induced autoantibody responses against a large number of SLE-associated autoantigens (Rajasinghe et al., 2020) and cSiO2-triggered glomerulonephritis (Bates et al., 2018). Lastly, we have recently demonstrated that DHA supplementation has value as a therapeutic intervention in this model (Pestka et al., 2021). The demonstration that DHA acts at many stages of cSiO2-induced autoimmunity (Figure 7) raises the possibility that ω-3 PUFA supplementation could be used as an intervention against other diseases associated with particle-triggered inflammation and autoimmunity.
Conclusions and Future Directions
Particle toxicology is a longstanding research field with origins in the 15th century. While this field primarily focused on toxic impacts of inhaled particles in the lung and their connections to occupational disease, it now encompasses a much broader arena that includes seeking to understand how exogenous and endogenous particles influence development of inflammatory and autoimmune diseases in diverse organs. Interestingly, the mechanisms by which particles trigger autoimmunity align with Polly Matzinger’s danger model, which argues that ongoing production and insufficient clearance of danger signals contributes to autoreactivity. Some outstanding knowledge gaps in the field of particle toxicology include understanding how genetics influence the immunotoxic potential of particles, how particles impact other immune cell populations (e.g., innate lymphoid cells, natural killer cells), and how particle toxicology studies can be performed in silico to assess risks associated with an individual’s environment and lifestyle. Answering these questions will lead to new understanding of the mechanisms by which particles elicit toxicity in the context of the genome, and will provide valuable insight into new interventions that can be used to prevent or treat particle-associated inflammatory and autoimmune diseases.
Author Contributions
OF: literature review, manuscript/figure preparation, manuscript submission. JP: manuscript/figure preparation, oversight, project funding. MB: manuscript preparation. KL: manuscript preparation, oversight.
Funding
This work was funded by NIH T32GM142521 (OF), NIH ES027353 (JP), Lupus Foundation of America (JP and MB), the Robert and Carol Deibel Family Endowment (JP), NIH ES024806 (KL), and NSF DMS-1761320 (KL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
alum, aluminum-containing salts; AMΦ, alveolar macrophage; BMDMΦ, bone marrow-derived macrophage; CaOx, calcium oxalate; CaP, calcium phosphate; CC, cholesterol crystal; CNT, carbon nanotube; DAMP, damage-associated molecular pattern; DC, dendritic cell; KC, Kupffer cell; MSU, monosodium urate; NET, neutrophil extracellular trap; PAMP, pattern-associated molecular pattern; ROS, reactive oxygen species; SiO2, silicon dioxide; TiO2, titanium dioxide; TLR, toll-like receptor.
References
A-Gonzalez, N., Quintana, J. A., García-Silva, S., Mazariegos, M., González de la Aleja, A., Nicolás-Ávila, J. A., et al. (2017). Phagocytosis Imprints Heterogeneity in Tissue-Resident Macrophages. J. Exp. Med. 214 (5), 1281–1296. doi:10.1084/jem.20161375
Abe, J.-i., and Morrell, C. (2016). Pyroptosis as a Regulated Form of Necrosis. Circ. Res. 118 (10), 1457–1460. doi:10.1161/CIRCRESAHA.116.308699
Adams, K., Greenbaum, D. S., Shaikh, R., van Erp, A. M., and Russell, A. G. (2015). Particulate Matter Components, Sources, and Health: Systematic Approaches to Testing Effects. J. Air Waste Manage. Assoc. 65 (5), 544–558. doi:10.1080/10962247.2014.1001884
Akbar, U., Yang, M., Kurian, D., and Mohan, C. (2017). Omega-3 Fatty Acids in Rheumatic Diseases. JCR: J. Clin. Rheumatol. 23 (6), 330–339. doi:10.1097/RHU.0000000000000563
Amador-Patarroyo, M. J., Rodriguez-Rodriguez, A., and Montoya-Ortiz, G. (2012). How Does Age at Onset Influence the Outcome of Autoimmune Diseases? Autoimmune Dis. 2012, 1–7. doi:10.1155/2012/251730
An, L.-L., Mehta, P., Xu, L., Turman, S., Reimer, T., Naiman, B., et al. (2014). Complement C5a Potentiates Uric Acid crystal-induced IL-1β Production. Eur. J. Immunol. 44 (12), 3669–3679. doi:10.1002/eji.201444560
Angum, F., Khan, T., Kaler, J., Siddiqui, L., and Hussain, A. (2020). The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 12 (5), e8094. doi:10.7759/cureus.8094
Aust, A. E., Cook, P. M., and Dodson, R. F. (2011). Morphological and Chemical Mechanisms of Elongated mineral Particle Toxicities. J. Toxicol. Environ. Health B 14 (1-4), 40–75. doi:10.1080/10937404.2011.556046
Balan, S., Saxena, M., and Bhardwaj, N. (2019). Dendritic Cell Subsets and Locations. Int. Rev. Cel Mol Biol 348, 1–68. doi:10.1016/bs.ircmb.2019.07.004
Barabé, F., Gilbert, C., Liao, N., Bourgoin, S. G., and Naccache, P. H. (1998). Crystal‐induced Neutrophil Activation VI. Involvement of FcγRIIIB (CD16) and CD11b in Response to Inflammatory Microcrystals. FASEB j. 12 (2), 209–220. doi:10.1096/fasebj.12.2.209
Baranov, M. V., Kumar, M., Sacanna, S., Thutupalli, S., and van den Bogaart, G. (2020). Modulation of Immune Responses by Particle Size and Shape. Front. Immunol. 11 (3854), 607945. doi:10.3389/fimmu.2020.607945
Baranowska-Wójcik, E., Szwajgier, D., Oleszczuk, P., and Winiarska-Mieczan, A. (2020). Effects of Titanium Dioxide Nanoparticles Exposure on Human Health-A Review. Biol. Trace Elem. Res. 193 (1), 118–129. doi:10.1007/s12011-019-01706-6
Bartrip, P. W. J. (2004). History of Asbestos Related Disease. Postgrad. Med. J. 80 (940), 72–76. doi:10.1136/pmj.2003.012526
Bates, M. A., Akbari, P., Gilley, K. N., Wagner, J. G., Li, N., Kopec, A. K., et al. (2018). Dietary Docosahexaenoic Acid Prevents Silica-Induced Development of Pulmonary Ectopic Germinal Centers and Glomerulonephritis in the Lupus-Prone NZBWF1 Mouse. Front. Immunol. 9 (September), 2002. doi:10.3389/fimmu.2018.02002
Bates, M. A., Brandenberger, C., Langohr, I., Kumagai, K., Harkema, J. R., Holian, A., et al. (2015). Silica Triggers Inflammation and Ectopic Lymphoid Neogenesis in the Lungs in Parallel with Accelerated Onset of Systemic Autoimmunity and Glomerulonephritis in the Lupus-Prone NZBWF1 Mouse. PLoS One 10 (5), e0125481. doi:10.1371/journal.pone.0125481
Beattie, L., Sawtell, A., Mann, J., Frame, T. C. M., Teal, B., de Labastida Rivera, F., et al. (2016). Bone Marrow-Derived and Resident Liver Macrophages Display Unique Transcriptomic Signatures but Similar Biological Functions. J. Hepatol. 65 (4), 758–768. doi:10.1016/j.jhep.2016.05.037
Beg, S., Rizwan, M., Sheikh, A. M., Hasnain, M. S., Anwer, K., and Kohli, K. (2011). Advancement in Carbon Nanotubes: Basics, Biomedical Applications and Toxicity. J. Pharm. Pharmacol. 63 (2), 141–163. doi:10.1111/j.2042-7158.2010.01167.x
Beheshti, S. O., Madsen, C. M., Varbo, A., and Nordestgaard, B. G. (2020). Worldwide Prevalence of Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 75 (20), 2553–2566. doi:10.1016/j.jacc.2020.03.057
Benninghoff, A. D., Bates, M. A., Chauhan, P. S., Wierenga, K. A., Gilley, K. N., Holian, A., et al. (2019). Docosahexaenoic Acid Consumption Impedes Early Interferon- and Chemokine-Related Gene Expression while Suppressing Silica-Triggered Flaring of Murine Lupus. Front. Immunol. 10 (2851), 2851. doi:10.3389/fimmu.2019.02851
Boraschi, D., Italiani, P., Palomba, R., Decuzzi, P., Duschl, A., Fadeel, B., et al. (2017). Nanoparticles and Innate Immunity: New Perspectives on Host Defence. Semin. Immunol. 34, 33–51. doi:10.1016/j.smim.2017.08.013
Borchers, A., Ansari, A. A., Hsu, T., Kono, D. H., and Gershwin, M. E. (2000). The Pathogenesis of Autoimmunity in New Zealand. Semin. Arthritis Rheum. 29 (6), 385–399. doi:10.1053/sarh.2000.7173
Boudigaard, S. H., Schlünssen, V., Vestergaard, J. M., Søndergaard, K., Torén, K., Peters, S., et al. (2021). Occupational Exposure to Respirable Crystalline Silica and Risk of Autoimmune Rheumatic Diseases: a Nationwide Cohort Study. Int. J. Epidemiol. 50 (4), 1213–1226. doi:10.1093/ije/dyaa287
Boya, P., and Kroemer, G. (2008). Lysosomal Membrane Permeabilization in Cell Death. Oncogene 27 (50), 6434–6451. doi:10.1038/onc.2008.310
Boyles, M. S. P., Young, L., Brown, D. M., MacCalman, L., Cowie, H., Moisala, A., et al. (2015). Multi-walled Carbon Nanotube Induced Frustrated Phagocytosis, Cytotoxicity and Pro-inflammatory Conditions in Macrophages Are Length Dependent and Greater Than that of Asbestos. Toxicol. Vitro 29 (7), 1513–1528. doi:10.1016/j.tiv.2015.06.012
Brahms, H., Raymackers, J., Union, A., de Keyser, F., Meheus, L., and Lührmann, R. (2000). The C-Terminal RG Dipeptide Repeats of the Spliceosomal Sm Proteins D1 and D3 Contain Symmetrical Dimethylarginines, Which Form a Major B-Cell Epitope for Anti-sm Autoantibodies. J. Biol. Chem. 275 (22), 17122–17129. doi:10.1074/jbc.M000300200
Brieger, K., Schiavone, S., Miller, J., and Krause, K. (2012). Reactive Oxygen Species: from Health to Disease. Swiss Med. Wkly 142, w13659. doi:10.4414/smw.2012.13659
Brody, A. R. (1993). Asbestos-induced Lung Disease. Environ. Health Perspect. 100, 21–30. doi:10.1289/ehp.9310021
Brown, D. M., Kinloch, I. A., Bangert, U., Windle, A. H., Walter, D. M., Walker, G. S., et al. (2007). An In Vitro Study of the Potential of Carbon Nanotubes and Nanofibres to Induce Inflammatory Mediators and Frustrated Phagocytosis. Carbon 45 (9), 1743–1756. doi:10.1016/j.carbon.2007.05.011
Brown, J. M., Archer, A. J., Pfau, J. C., and Holian, A. (2003). Silica Accelerated Systemic Autoimmune Disease in Lupus-Prone New Zealand Mixed Mice. Clin. Exp. Immunol. 131 (3), 415–421. doi:10.1046/j.1365-2249.2003.02094.x
Brown, J. M., Pfau, J. C., and Holian, A. (2004). Immunoglobulin and Lymphocyte Responses Following Silica Exposure in New Zealand Mixed Mice. Inhalation Toxicol. 16 (3), 133–139. doi:10.1080/08958370490270936
Brown, J. M., Pfau, J. C., Pershouse, M. A., and Holian, A. (2005). Silica, Apoptosis, and Autoimmunity. J. Immunotoxicology 1 (3), 177–187. doi:10.1080/15476910490911922
Burbelo, P. D., Iadarola, M. J., Keller, J. M., and Warner, B. M. (2021). Autoantibodies Targeting Intracellular and Extracellular Proteins in Autoimmunity. Front. Immunol. 12 (5), 548469. doi:10.3389/fimmu.2021.548469
Butler, F., Alghubayshi, A., and Roman, Y. (2021). The Epidemiology and Genetics of Hyperuricemia and Gout across Major Racial Groups: A Literature Review and Population Genetics Secondary Database Analysis. Jpm 11 (3), 231. doi:10.3390/jpm11030231
Calder, P. C. (2017). Omega-3 Fatty Acids and Inflammatory Processes: from Molecules to Man. Biochem. Soc. Trans. 45 (5), 1105–1115. doi:10.1042/BST20160474
Campden, R. I., and Zhang, Y. (2019). The Role of Lysosomal Cysteine Cathepsins in NLRP3 Inflammasome Activation. Arch. Biochem. Biophys. 670, 32–42. doi:10.1016/j.abb.2019.02.015
Caronni, N., Piperno, G. M., Simoncello, F., Romano, O., Vodret, S., Yanagihashi, Y., et al. (2021). TIM4 Expression by Dendritic Cells Mediates Uptake of Tumor-Associated Antigens and Anti-tumor Responses. Nat. Commun. 12 (1), 2237. doi:10.1038/s41467-021-22535-z
Caso, F., Costa, L., Nucera, V., Barilaro, G., Masala, I. F., Talotta, R., et al. (2018). From Autoinflammation to Autoimmunity: Old and Recent Findings. Clin. Rheumatol. 37 (9), 2305–2321. doi:10.1007/s10067-018-4209-9
Chang, M.-K., Binder, C. J., Miller, Y. I., Subbanagounder, G., Silverman, G. J., Berliner, J. A., et al. (2004). Apoptotic Cells with Oxidation-specific Epitopes Are Immunogenic and Proinflammatory. J. Exp. Med. 200 (11), 1359–1370. doi:10.1084/jem.20031763
Charoenwoodhipong, P., Harlow, S. D., Marder, W., Hassett, A. L., McCune, W. J., Gordon, C., et al. (2020). Dietary Omega Polyunsaturated Fatty Acid Intake and Patient‐Reported Outcomes in Systemic Lupus Erythematosus: The Michigan Lupus Epidemiology and Surveillance Program. Arthritis Care Res. 72 (7), 874–881. doi:10.1002/acr.23925
Chen, F., Jin, J., Hu, J., Wang, Y., Ma, Z., and Zhang, J. (2019). Endoplasmic Reticulum Stress Cooperates in Silica Nanoparticles-Induced Macrophage Apoptosis via Activation of CHOP-Mediated Apoptotic Signaling Pathway. Ijms 20 (23), 5846. doi:10.3390/ijms20235846
Chen, Q., Wang, N., Zhu, M., Lu, J., Zhong, H., Xue, X., et al. (2018). TiO2 Nanoparticles Cause Mitochondrial Dysfunction, Activate Inflammatory Responses, and Attenuate Phagocytosis in Macrophages: A Proteomic and Metabolomic Insight. Redox Biol. 15, 266–276. doi:10.1016/j.redox.2017.12.011
Chevriaux, A., Pilot, T., Derangère, V., Simonin, H., Martine, P., Chalmin, F., et al. (2020). Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, through NLRP3 Interaction. Front. Cel Dev. Biol. 8, 167. doi:10.3389/fcell.2020.00167
Cho, J. H., and Gregersen, P. K. (2011). Genomics and the Multifactorial Nature of Human Autoimmune Disease. N. Engl. J. Med. 365 (17), 1612–1623. doi:10.1056/NEJMra1100030
Clark, A., Zhao, E. J., Birukova, A., Buckley, E. S., Ord, J. R., Asfaw, Y. G., et al. (2017). Inhaled Silica Induces Autoimmunity in a Strain-dependent Manner. J. Immunol. 198 (1 Suppl. ment), 58–13.
Clément, M., Basatemur, G., Masters, L., Baker, L., Bruneval, P., Iwawaki, T., et al. (2016). Necrotic cell sensor Clec4e promotes a proatherogenic macrophage phenotype through activation of the unfolded protein response. Circulation 134 (14), 1039–1051. doi:10.1161/CIRCULATIONAHA.116.022668
Cohignac, V., Landry, M. J., Ridoux, A., Pinault, M., Annangi, B., Gerdil, A., et al. (2018). Carbon Nanotubes, but Not Spherical Nanoparticles, Block Autophagy by a Shape-Related Targeting of Lysosomes in Murine Macrophages. Autophagy 14 (8), 1323–1334. doi:10.1080/15548627.2018.1474993
Conforti-Andreoni, C., Spreafico, R., Qian, H. L., Riteau, N., Ryffel, B., Ricciardi-Castagnoli, P., et al. (2011). Uric Acid-Driven Th17 Differentiation Requires Inflammasome-Derived IL-1 and IL-18. J.I. 187 (11), 5842–5850. doi:10.4049/jimmunol.1101408
Curtis, L. (2021). PM2.5, NO2, Wildfires, and Other Environmental Exposures Are Linked to Higher Covid 19 Incidence, Severity, and Death Rates. Environ. Sci. Pollut. Res. 28 (39), 54429–54447. doi:10.1007/s11356-021-15556-0
Czajka, M., Sawicki, K., Sikorska, K., Popek, S., Kruszewski, M., and Kapka-Skrzypczak, L. (2015). Toxicity of Titanium Dioxide Nanoparticles in central Nervous System. Toxicol. Vitro 29 (5), 1042–1052. doi:10.1016/j.tiv.2015.04.004
Dai, W., Liu, F., Li, C., Lu, Y., Lu, X., Du, S., et al. (2016). Blockade of Wnt/β-Catenin Pathway Aggravated Silica-Induced Lung Inflammation through Tregs Regulation on Th Immune Responses. Mediators Inflamm. 2016, 1–14. doi:10.1155/2016/6235614
Dai, X.-J., Tao, J.-H., Fang, X., Xia, Y., Li, X.-M., Wang, Y.-P., et al. (2018). Changes of Treg/Th17 Ratio in Spleen of Acute Gouty Arthritis Rat Induced by MSU Crystals. Inflammation 41 (5), 1955–1964. doi:10.1007/s10753-018-0839-y
D’Arcy, M. S. (2019). Cell Death: a Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol Int 43 (6), 582–592. doi:10.1002/cbin.11137
Davidovich, P., Kearney, C. J., and Martin, S. J. (2014). Inflammatory Outcomes of Apoptosis, Necrosis and Necroptosis. Biol. Chem. 395 (10), 1163–1171. doi:10.1515/hsz-2014-0164
De Klerk, N. H., Ambrosini, G. L., and Musk, A. W. (2002). Crystalline Silica Exposure and Major Health Effects in Western Australian Gold Miners. Ann. Occup. Hyg. 46 (Suppl. l_1), 1–3. doi:10.1093/annhyg/46.suppl_1.1
Defesche, J. C., Gidding, S. S., Harada-Shiba, M., Hegele, R. A., Santos, R. D., and Wierzbicki, A. S. (2017). Familial Hypercholesterolaemia. Nat. Rev. Dis. Primers 3, 17093. doi:10.1038/nrdp.2017.93
Delgado-Rizo, V., Martínez-Guzmán, M. A., Iñiguez-Gutierrez, L., García-Orozco, A., Alvarado-Navarro, A., and Fafutis-Morris, M. (2017). Neutrophil Extracellular Traps and its Implications in Inflammation: An Overview. Front. Immunol. 8 (81), 81. doi:10.3389/fimmu.2017.00081
Desai, J., Foresto-Neto, O., Honarpisheh, M., Steiger, S., Nakazawa, D., Popper, B., et al. (2017). Particles of Different Sizes and Shapes Induce Neutrophil Necroptosis Followed by the Release of Neutrophil Extracellular Trap-like Chromatin. Sci. Rep. 7 (1), 15003. doi:10.1038/s41598-017-15106-0
Desaulniers, P., Fernandes, M., Gilbert, C., Bourgoin, S. G., and Naccache, P. H. (2001). Crystal-induced Neutrophil Activation. VII. Involvement of Syk in the Responses to Monosodium Urate Crystals. J. Leukoc. Biol. 70 (4), 659–668. doi:10.1189/jlb.70.4.659
Di Ciaula, A., Wang, D. Q.-H., and Portincasa, P. (2018). An Update on the Pathogenesis of Cholesterol Gallstone Disease. Curr. Opin. Gastroenterol. 34 (2), 71–80. doi:10.1097/MOG.0000000000000423
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L., and Falzoni, S. (2017). The P2X7 Receptor in Infection and Inflammation. Immunity 47 (1), 15–31. doi:10.1016/j.immuni.2017.06.020
Dominguez-Gutierrez, P. R., Kusmartsev, S., Canales, B. K., and Khan, S. R. (2018). Calcium Oxalate Differentiates Human Monocytes into Inflammatory M1 Macrophages. Front. Immunol. 9, 1863. doi:10.3389/fimmu.2018.01863
Dominici, F., Wang, Y., Correia, A. W., Ezzati, M., Pope, C. A., and Dockery, D. W. (2015). Chemical Composition of Fine Particulate Matter and Life Expectancy. Epidemiology 26 (4), 556–564. doi:10.1097/EDE.0000000000000297
Donaldson, K., Murphy, F. A., Duffin, R., and Poland, C. A. (2010). Asbestos, Carbon Nanotubes and the Pleural Mesothelium: a Review and the Hypothesis Regarding the Role of Long Fibre Retention in the Parietal Pleura, Inflammation and Mesothelioma. Part. Fibre Toxicol. 7 (1), 5. doi:10.1186/1743-8977-7-5
Donaldson, K., and Seaton, A. (2012). A Short History of the Toxicology of Inhaled Particles. Part. Fibre Toxicol. 9 (1), 13. doi:10.1186/1743-8977-9-13
Dong, J., and Ma, Q. (2018). Macrophage Polarization and Activation at the Interface of Multi-Walled Carbon Nanotube-Induced Pulmonary Inflammation and Fibrosis. Nanotoxicology 12 (2), 153–168. doi:10.1080/17435390.2018.1425501
Doria, A., Zen, M., Bettio, S., Gatto, M., Bassi, N., Nalotto, L., et al. (2012). Autoinflammation and Autoimmunity: Bridging the divide. Autoimmun. Rev. 12 (1), 22–30. doi:10.1016/j.autrev.2012.07.018
Dorrington, M. G., and Fraser, I. D. C. (2019). NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 10 (705), 705. doi:10.3389/fimmu.2019.00705
Dostert, C., Pétrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate Immune Activation through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 320 (5876), 674–677. doi:10.1126/science.1156995
Doyle, H. A., Aswad, D. W., and Mamula, M. J. (2013). Autoimmunity to Isomerized Histone H2B in Systemic Lupus Erythematosus. Autoimmunity 46 (1), 6–13. doi:10.3109/08916934.2012.710859
Doyle, H. A., Yang, M.-L., Raycroft, M. T., Gee, R. J., and Mamula, M. J. (2014). Autoantigens: Novel Forms and Presentation to the Immune System. Autoimmunity 47 (4), 220–233. doi:10.3109/08916934.2013.850495
Dron, J. S., and Hegele, R. A. (2017). Genetics of Triglycerides and the Risk of Atherosclerosis. Curr. Atheroscler. Rep. 19 (7), 31. doi:10.1007/s11883-017-0667-9
Duarte-García, A., Myasoedova, E., Karmacharya, P., Hocaoğlu, M., Murad, M. H., Warrington, K. J., et al. (2020). Effect of omega-3 Fatty Acids on Systemic Lupus Erythematosus Disease Activity: A Systematic Review and Meta-Analysis. Autoimmun. Rev. 19 (12), 102688. doi:10.1016/j.autrev.2020.102688
Dubois, C. M., Bissonnette, E., and Rola-Pleszczynski, M. (1989). Asbestos Fibers and Silica Particles Stimulate Rat Alveolar Macrophages to Release Tumor Necrosis Factor: Autoregulatory Role of Leukotriene B4. Am. Rev. Respir. Dis. 139 (5), 1257–1264. doi:10.1164/ajrccm/139.5.1257
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 464 (7293), 1357–1361. doi:10.1038/nature08938
Duncan, J. A., and Canna, S. W. (2018). The NLRC4 Inflammasome. Immunol. Rev. 281 (1), 115–123. doi:10.1111/imr.12607
Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia, J., Thaiss, C. A., Booth, C. J., et al. (2011). NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell 145 (5), 745–757. doi:10.1016/j.cell.2011.04.022
Elliott, J. H. (1923). Silicosis in Ontario Gold Miners. Trans. Am. Climatol Clin. Assoc. 39, 24–43.
Elmore, S. (2007). Apoptosis: a Review of Programmed Cell Death. Toxicol. Pathol. 35 (4), 495–516. doi:10.1080/01926230701320337
Fazen, L. E., Linde, B., and Redlich, C. A. (2020). Occupational Lung Diseases in the 21st century. Curr. Opin. Pulm. Med. 26 (2), 142–148. doi:10.1097/MCP.0000000000000658
Fenoglio, I., Aldieri, E., Gazzano, E., Cesano, F., Colonna, M., Scarano, D., et al. (2012). Thickness of Multiwalled Carbon Nanotubes Affects Their Lung Toxicity. Chem. Res. Toxicol. 25 (1), 74–82. doi:10.1021/tx200255h
Ferreira, H. B., Pereira, A. M., Melo, T., Paiva, A., and Domingues, M. R. (2019). Lipidomics in Autoimmune Diseases with Main Focus on Systemic Lupus Erythematosus. J. Pharm. Biomed. Anal. 174, 386–395. doi:10.1016/j.jpba.2019.06.005
Finckh, A., Cooper, G. S., Chibnik, L. B., Costenbader, K. H., Watts, J., Pankey, H., et al. (2006). Occupational Silica and Solvent Exposures and Risk of Systemic Lupus Erythematosus in Urban Women. Arthritis Rheum. 54 (11), 3648–3654. doi:10.1002/art.22210
Flach, T. L., Ng, G., Hari, A., Desrosiers, M. D., Zhang, P., Ward, S. M., et al. (2011). Alum Interaction with Dendritic Cell Membrane Lipids Is Essential for its Adjuvanticity. Nat. Med. 17 (4), 479–487. doi:10.1038/nm.2306
Foster, M. H., Ord, J. R., Zhao, E. J., Birukova, A., Fee, L., Korte, F. M., et al. (2019). Silica Exposure Differentially Modulates Autoimmunity in Lupus Strains and Autoantibody Transgenic Mice. Front. Immunol. 10, 2336. doi:10.3389/fimmu.2019.02336
Frank, D., and Vince, J. E. (2019). Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ 26 (1), 99–114. doi:10.1038/s41418-018-0212-6
Freeman, T. L., and Swartz, T. H. (2020). Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 11, 1518. doi:10.3389/fimmu.2020.01518
Fukakusa, J., Rosenblat, J., Jang, B., Ribeiro, M., Kudla, I., and Tarlo, S. M. (2011). Factors Influencing Respirator Use at Work in Respiratory Patients. Occup. Med. 61 (8), 576–582. doi:10.1093/occmed/kqr132
Gaidt, M. M., and Hornung, V. (2017). Alternative Inflammasome Activation Enables IL-1β Release from Living Cells. Curr. Opin. Immunol. 44, 7–13. doi:10.1016/j.coi.2016.10.007
Gallo, P. M., and Gallucci, S. (2013). The Dendritic Cell Response to Classic, Emerging, and Homeostatic Danger Signals. Implications for Autoimmunity. Front. Immunol. 4, 138. doi:10.3389/fimmu.2013.00138
Gallucci, S., and Matzinger, P. (2001). Danger Signals: SOS to the Immune System. Curr. Opin. Immunol. 13 (1), 114–119. doi:10.1016/s0952-7915(00)00191-6
Galluzzi, L., Kepp, O., Chan, F. K.-M., and Kroemer, G. (2017). Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 12, 103–130. doi:10.1146/annurev-pathol-052016-100247
Gasteiger, G., D'Osualdo, A., Schubert, D. A., Weber, A., Bruscia, E. M., and Hartl, D. (2017). Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 9 (2), 111–125. doi:10.1159/000453397
Geng, Y.-J., Phillips, J. E., Mason, R. P., and Casscells, S. W. (2003). Cholesterol Crystallization and Macrophage Apoptosis: Implication for Atherosclerotic Plaque Instability and Rupture. Biochem. Pharmacol. 66 (8), 1485–1492. doi:10.1016/s0006-2952(03)00502-1
Gherardi, R., and Authier, F. (2012). Macrophagic Myofasciitis: Characterization and Pathophysiology. Lupus 21 (2), 184–189. doi:10.1177/0961203311429557
Glenny, A. T., Pope, C. G., Waddington, H., and Wallace, U. (1926). Immunological Notes. XVII-XXIV. J. Pathol. 29 (1), 31–40. doi:10.1002/path.1700290106
Goldstein, J. L., and Brown, M. S. (2015). A century of Cholesterol and Coronaries: from Plaques to Genes to Statins. Cell 161 (1), 161–172. doi:10.1016/j.cell.2015.01.036
Goswami, E., Craven, V., Dahlstrom, D., Alexander, D., and Mowat, F. (2013). Domestic Asbestos Exposure: a Review of Epidemiologic and Exposure Data. Ijerph 10 (11), 5629–5670. doi:10.3390/ijerph10115629
Gough, S. C., and Simmonds, M. J. (2007). The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genomics 8 (7), 453–465. doi:10.2174/138920207783591690
Gozal, E., Ortiz, L. A., Zou, X., Burow, M. E., Lasky, J. A., and Friedman, M. (2002). Silica-Induced Apoptosis in Murine Macrophage. Am. J. Respir. Cel Mol Biol 27 (1), 91–98. doi:10.1165/ajrcmb.27.1.4790
Grazioli, S., and Pugin, J. (2018). Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 9, 832. doi:10.3389/fimmu.2018.00832
Green, D. R., and Llambi, F. (2015). Cell Death Signaling. Cold Spring Harb Perspect. Biol. 7 (12), a006080. doi:10.1101/cshperspect.a006080
Gren, S. T., Rasmussen, T. B., Janciauskiene, S., Håkansson, K., Gerwien, J. G., and Grip, O. (2015). A Single-Cell Gene-Expression Profile Reveals Inter-cellular Heterogeneity within Human Monocyte Subsets. PLoS One 10 (12), e0144351. doi:10.1371/journal.pone.0144351
Gross, C. J., Mishra, R., Schneider, K. S., Médard, G., Wettmarshausen, J., Dittlein, D. C., et al. (2016). K + Efflux-independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 45 (4), 761–773. doi:10.1016/j.immuni.2016.08.010
Halade, G. V., Rahman, M. M., Bhattacharya, A., Barnes, J. L., Chandrasekar, B., and Fernandes, G. (2010). Docosahexaenoic Acid-Enriched Fish Oil Attenuates Kidney Disease and Prolongs Median and Maximal Life Span of Autoimmune Lupus-Prone Mice. J.I. 184 (9), 5280–5286. doi:10.4049/jimmunol.0903282
Halade, G. V., Williams, P. J., Veigas, J. M., Barnes, J. L., and Fernandes, G. (2013). Concentrated Fish Oil (Lovaza) Extends Lifespan and Attenuates Kidney Disease in Lupus-Prone Short-Lived (NZBxNZW)F1 Mice. Exp. Biol. Med. (Maywood) 238 (6), 610–622. doi:10.1177/1535370213489485
Hall, N. B., Halldin, C. N., Blackley, D. J., and Laney, A. S. (2020). Assessment of Pneumoconiosis in Surface Coal Miners after Implementation of a National Radiographic Surveillance Program, United States, 2014-2019. Am. J. Ind. Med. 63 (12), 1104–1108. doi:10.1002/ajim.23184
Hamilton, R. F., Iyer, L. L., and Holian, A. (1996). Asbestos Induces Apoptosis in Human Alveolar Macrophages. Am. J. Physiology-Lung Cell Mol. Physiol. 271 (5 Pt 1), L813–L819. doi:10.1152/ajplung.1996.271.5.L813
Hamilton, R. F., Buford, M., Xiang, C., Wu, N., and Holian, A. (2012). NLRP3 Inflammasome Activation in Murine Alveolar Macrophages and Related Lung Pathology Is Associated with MWCNT Nickel Contamination. Inhalation Toxicol. 24 (14), 995–1008. doi:10.3109/08958378.2012.745633
Hamilton, R. F., Wu, N., Porter, D., Buford, M., Wolfarth, M., and Holian, A. (2009). Particle Length-dependent Titanium Dioxide Nanomaterials Toxicity and Bioactivity. Part. Fibre Toxicol. 6 (1), 35. doi:10.1186/1743-8977-6-35
Hamilton, R., Wu, Z., Mitra, S., and Holian, A. (2018). The Effects of Varying Degree of MWCNT Carboxylation on Bioactivity in Various In Vivo and In Vitro Exposure Models. Ijms 19 (2), 354. doi:10.3390/ijms19020354
Haneklaus, M., and O'Neill, L. A. J. (2015). NLRP3 at the Interface of Metabolism and Inflammation. Immunol. Rev. 265 (1), 53–62. doi:10.1111/imr.12285
Harijith, A., Ebenezer, D. L., and Natarajan, V. (2014). Reactive Oxygen Species at the Crossroads of Inflammasome and Inflammation. Front. Physiol. 5, 352. doi:10.3389/fphys.2014.00352
He, Y., Hara, H., and Núñez, G. (2016). Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 41 (12), 1012–1021. doi:10.1016/j.tibs.2016.09.002
Heffernan, P. (1929). Some Notes on the Biophysics of Silica and the Etiology of Silicosis. Bmj 2 (3584), 489–492. doi:10.1136/bmj.2.3584.489
Heid, M. E., Keyel, P. A., Kamga, C., Shiva, S., Watkins, S. C., and Salter, R. D. (2013). Mitochondrial Reactive Oxygen Species Induces NLRP3-dependent Lysosomal Damage and Inflammasome Activation. J.I. 191 (10), 5230–5238. doi:10.4049/jimmunol.1301490
Heilig, R., and Broz, P. (2018). Function and Mechanism of the Pyrin Inflammasome. Eur. J. Immunol. 48 (2), 230–238. doi:10.1002/eji.201746947
Heneka, M. T., McManus, R. M., and Latz, E. (2018). Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat. Rev. Neurosci. 19 (10), 610–621. doi:10.1038/s41583-018-0055-7
Hennig, P., Garstkiewicz, M., Grossi, S., Di Filippo, M., French, L., and Beer, H.-D. (2018). The Crosstalk between Nrf2 and Inflammasomes. Ijms 19 (2), 562. doi:10.3390/ijms19020562
Hilpert, C., Sitte, S., Arnold, H., Lehmann, C. H. K., Dudziak, D., Mattner, J., et al. (2019). Dendritic Cells Control Regulatory T Cell Function Required for Maintenance of Intestinal Tissue Homeostasis. J.I. 203 (11), 3068–3077. doi:10.4049/jimmunol.1900320
Hirano, S., Zhou, Q., Furuyama, A., and Kanno, S. (2017). Differential Regulation of IL-1β and IL-6 Release in Murine Macrophages. Inflammation 40 (6), 1933–1943. doi:10.1007/s10753-017-0634-1
Holdt, L. M., and Teupser, D. (2013). From Genotype to Phenotype in Human Atherosclerosis - Recent Findings. Curr. Opin. Lipidol. 24 (5), 410–418. doi:10.1097/MOL.0b013e3283654e7c
Honarpisheh, M., Foresto-Neto, O., Desai, J., Steiger, S., Gómez, L. A., Popper, B., et al. (2017). Phagocytosis of Environmental or Metabolic Crystalline Particles Induces Cytotoxicity by Triggering Necroptosis across a Broad Range of Particle Size and Shape. Sci. Rep. 7 (1), 15523. doi:10.1038/s41598-017-15804-9
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 9 (8), 847–856. doi:10.1038/ni.1631
Hu, S., Zhao, H., Al-Humadi, N. H., Yin, X. J., and Ma, J. K. H. (2006). Silica-induced Apoptosis in Alveolar Macrophages: Evidence of In Vivo Thiol Depletion and the Activation of Mitochondrial Pathway. J. Toxicol. Environ. Health A 69 (13), 1261–1284. doi:10.1080/15287390500361875
Huh, H., Pearce, S., Yesner, L., Schindler, J., and Silverstein, R. (1996). Regulated Expression of CD36 during Monocyte-To-Macrophage Differentiation: Potential Role of CD36 in Foam Cell Formation. Blood 87 (5), 2020–2028.
Ingersoll, M. A., Spanbroek, R., Lottaz, C., Gautier, E. L., Frankenberger, M., Hoffmann, R., et al. (2010). Comparison of Gene Expression Profiles between Human and Mouse Monocyte Subsets. Blood 115 (3), e10–e19. doi:10.1182/blood-2009-07-235028
Ioannou, G. N. (2016). The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 27 (2), 84–95. doi:10.1016/j.tem.2015.11.008
Ito, F., Yanatori, I., Maeda, Y., Nimura, K., Ito, S., Hirayama, T., et al. (2020). Asbestos Conceives Fe(II)-dependent Mutagenic Stromal Milieu through Ceaseless Macrophage Ferroptosis and β-catenin Induction in Mesothelium. Redox Biol. 36, 101616. doi:10.1016/j.redox.2020.101616
Iyer, R., Hamilton, R. F., Li, L., and Holian, A. (1996). Silica-induced Apoptosis Mediated via Scavenger Receptor in Human Alveolar Macrophages. Toxicol. Appl. Pharmacol. 141 (1), 84–92. doi:10.1006/taap.1996.0263
Janowski, A. M., and Sutterwala, F. S. (2016). Atypical Inflammasomes. Methods Mol. Biol. 1417, 45–62. doi:10.1007/978-1-4939-3566-6_2
Jego, G., Pascual, V., Palucka, A. K., and Banchereau, J. (2004). Dendritic Cells Control B Cell Growth and Differentiation. Curr. Dir. Autoimmun. 8, 124–139. doi:10.1159/000082101
Jin, M., Yang, F., Yang, I., Yin, Y., Luo, J. J., Wang, H., et al. (2012). Uric Acid, Hyperuricemia and Vascular Diseases. Front. Biosci. 17, 656–669. doi:10.2741/3950
Jorch, S. K., and Kubes, P. (2017). An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat. Med. 23 (3), 279–287. doi:10.1038/nm.4294
Joshi, G. N., and Knecht, D. A. (2013). Silica Phagocytosis Causes Apoptosis and Necrosis by Different Temporal and Molecular Pathways in Alveolar Macrophages. Apoptosis 18 (3), 271–285. doi:10.1007/s10495-012-0798-y
Ka, S.-M., Lin, J.-C., Lin, T.-J., Liu, F.-C., Chao, L. K., Ho, C.-L., et al. (2015). Citral Alleviates an Accelerated and Severe Lupus Nephritis Model by Inhibiting the Activation Signal of NLRP3 Inflammasome and Enhancing Nrf2 Activation. Arthritis Res. Ther. 17, 331. doi:10.1186/s13075-015-0844-6
Kalampogias, A., Siasos, G., Oikonomou, E., Tsalamandris, S., Mourouzis, K., Tsigkou, V., et al. (2016). Basic Mechanisms in Atherosclerosis: The Role of Calcium. Mc 12 (2), 103–113. doi:10.2174/1573406411666150928111446
Karasawa, T., and Takahashi, M. (2017). Role of NLRP3 Inflammasomes in Atherosclerosis. Jat 24 (5), 443–451. doi:10.5551/jat.RV17001
Karasawa, T., and Takahashi, M. (2019). Saturated Fatty Acid-Crystals Activate NLRP3 Inflammasome. Aging 11 (6), 1613–1614. doi:10.18632/aging.101892
Kawamura, Y., Nakaoka, H., Nakayama, A., Okada, Y., Yamamoto, K., Higashino, T., et al. (2019). Genome-wide Association Study Revealed Novel Loci Which Aggravate Asymptomatic Hyperuricaemia into Gout. Ann. Rheum. Dis. 78 (10), 1430–1437. doi:10.1136/annrheumdis-2019-215521
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Ijms 20 (13), 3328. doi:10.3390/ijms20133328
Kelly, F. J., and Fussell, J. C. (2020). Toxicity of Airborne Particles-Established Evidence, Knowledge Gaps and Emerging Areas of Importance. Phil. Trans. R. Soc. A. 378 (2183), 20190322. doi:10.1098/rsta.2019.0322
Kerntke, C., Nimmerjahn, F., and Biburger, M. (2020). There Is (Scientific) Strength in Numbers: A Comprehensive Quantitation of Fc Gamma Receptor Numbers on Human and Murine Peripheral Blood Leukocytes. Front. Immunol. 11 (118), 118. doi:10.3389/fimmu.2020.00118
Khameneh, H. J., Ho, A. W. S., Spreafico, R., Derks, H., Quek, H. Q. Y., and Mortellaro, A. (2017). The Syk-NFAT-IL-2 Pathway in Dendritic Cells Is Required for Optimal Sterile Immunity Elicited by Alum Adjuvants. J.I. 198 (1), 196–204. doi:10.4049/jimmunol.1600420
Khan, S. R., Pearle, M. S., Robertson, W. G., Gambaro, G., Canales, B. K., Doizi, S., et al. (2016). Kidney Stones. Nat. Rev. Dis. Primers 2, 16008. doi:10.1038/nrdp.2016.8
Khare, S., Dorfleutner, A., Bryan, N. B., Yun, C., Radian, A. D., de Almeida, L., et al. (2012). An NLRP7-Containing Inflammasome Mediates Recognition of Microbial Lipopeptides in Human Macrophages. Immunity 36 (3), 464–476. doi:10.1016/j.immuni.2012.02.001
Kim, S.-K., Choe, J.-Y., and Park, K.-Y. (2016). Enhanced P62 Is Responsible for Mitochondrial Pathway-dependent Apoptosis and Interleukin-1β Production at the Early Phase by Monosodium Urate Crystals in Murine Macrophage. Inflammation 39 (5), 1603–1616. doi:10.1007/s10753-016-0387-2
Kim, S., Joe, Y., Jeong, S. O., Zheng, M., Back, S. H., Park, S. W., et al. (2014). Endoplasmic Reticulum Stress Is Sufficient for the Induction of IL-1β Production via Activation of the NF-Κb and Inflammasome Pathways. Innate Immun. 20 (8), 799–815. doi:10.1177/1753425913508593
Kiyotake, R., Oh-Hora, M., Ishikawa, E., Miyamoto, T., Ishibashi, T., and Yamasaki, S. (2015). Human Mincle Binds to Cholesterol Crystals and Triggers Innate Immune Responses. J. Biol. Chem. 290 (42), 25322–25332. doi:10.1074/jbc.M115.645234
Kobayashi, N., Izumi, H., and Morimoto, Y. (2017). Review of Toxicity Studies of Carbon Nanotubes. Jrnl Occup. Health 59 (5), 394–407. doi:10.1539/joh.17-0089-RA
Komada, T., and Muruve, D. A. (2019). The Role of Inflammasomes in Kidney Disease. Nat. Rev. Nephrol. 15 (8), 501–520. doi:10.1038/s41581-019-0158-z
Kool, M., Pétrilli, V., De Smedt, T., Rolaz, A., Hammad, H., van Nimwegen, M., et al. (2008). Cutting Edge: Alum Adjuvant Stimulates Inflammatory Dendritic Cells through Activation of the NALP3 Inflammasome. J. Immunol. 181 (6), 3755–3759. doi:10.4049/jimmunol.181.6.3755
Krainer, J., Siebenhandl, S., and Weinhäusel, A. (2020). Systemic Autoinflammatory Diseases. J. Autoimmun. 109, 102421. doi:10.1016/j.jaut.2020.102421
Kuroda, E., Ishii, K. J., Uematsu, S., Ohata, K., Coban, C., Akira, S., et al. (2011). Silica Crystals and Aluminum Salts Regulate the Production of Prostaglandin in Macrophages via NALP3 Inflammasome-independent Mechanisms. Immunity 34 (4), 514–526. doi:10.1016/j.immuni.2011.03.019
Kusaka, T., Nakayama, M., Nakamura, K., Ishimiya, M., Furusawa, E., and Ogasawara, K. (2014). Effect of Silica Particle Size on Macrophage Inflammatory Responses. PLoS One 9 (3), e92634. doi:10.1371/journal.pone.0092634
Kusiorowski, R., Zaremba, T., Piotrowski, J., and Adamek, J. (2012). Thermal Decomposition of Different Types of Asbestos. J. Therm. Anal. Calorim. 109 (2), 693–704. doi:10.1007/s10973-012-2222-9
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and Functions of Inflammasomes. Cell 157 (5), 1013–1022. doi:10.1016/j.cell.2014.04.007
Lande, R., Pietraforte, I., Mennella, A., Palazzo, R., Spinelli, F. R., Giannakakis, K., et al. (2021). Complementary Effects of Carbamylated and Citrullinated LL37 in Autoimmunity and Inflammation in Systemic Lupus Erythematosus. Ijms 22 (4), 1650. doi:10.3390/ijms22041650
Lawlor, K. E., and Vince, J. E. (2014). Ambiguities in NLRP3 Inflammasome Regulation: Is There a Role for Mitochondria? Biochim. Biophys. Acta (Bba) - Gen. Subjects 1840 (4), 1433–1440. doi:10.1016/j.bbagen.2013.08.014
Lemke, G. (2013). Biology of the TAM Receptors. Cold Spring Harbor Perspect. Biol. 5 (11), a009076. doi:10.1101/cshperspect.a009076
Ley, K., Pramod, A. B., Croft, M., Ravichandran, K. S., and Ting, J. P. (2016). How Mouse Macrophages Sense what Is Going on. Front. Immunol. 7 (204), 204. doi:10.3389/fimmu.2016.00204
Li, K., Neumann, K., Duhan, V., Namineni, S., Hansen, A. L., Wartewig, T., et al. (2019a). The Uric Acid crystal Receptor Clec12A Potentiates Type I Interferon Responses. Proc. Natl. Acad. Sci. USA 116 (37), 18544–18549. doi:10.1073/pnas.1821351116
Li, X., Bi, X., Wang, S., Zhang, Z., Li, F., and Zhao, A. Z. (2019b). Therapeutic Potential of ω-3 Polyunsaturated Fatty Acids in Human Autoimmune Diseases. Front. Immunol. 10, 2241. doi:10.3389/fimmu.2019.02241
Li, Y., Cao, X., Liu, Y., Zhao, Y., and Herrmann, M. (2018). Neutrophil Extracellular Traps Formation and Aggregation Orchestrate Induction and Resolution of Sterile Crystal-Mediated Inflammation. Front. Immunol. 9, 1559. doi:10.3389/fimmu.2018.01559
Li, Y., Li, Y., Yang, T., and Wang, M. (2020). Dioscin Attenuates oxLDL Uptake and the Inflammatory Reaction of Dendritic Cells under High Glucose Conditions by Blocking P38 MAPK. Mol. Med. Rep. 21 (1), 304–310. doi:10.3892/mmr.2019.10806
Lim, Y., Kim, J. H., Kim, K. A., Chang, H. S., Park, Y. M., Ahn, B. Y., et al. (1999). Silica-induced Apoptosis In Vitro and In Vivo. Toxicol. Lett. 108 (2-3), 335–339. doi:10.1016/s0378-4274(99)00107-1
Lison, D., Vietti, G., and van den Brule, S. (2014). Paracelsus in Nanotoxicology. Part. Fibre Toxicol. 11 (1), 35. doi:10.1186/s12989-014-0035-7
Liu, F., Liu, J., Weng, D., Chen, Y., Song, L., He, Q., et al. (2010). CD4+CD25+Foxp3+ Regulatory T Cells Depletion May Attenuate the Development of Silica-Induced Lung Fibrosis in Mice. PLoS One 5 (11), e15404. doi:10.1371/journal.pone.0015404
Lo Re, S., Lecocq, M., Uwambayinema, F., Yakoub, Y., Delos, M., Demoulin, J.-B., et al. (2011). Platelet-Derived Growth Factor-Producing CD4+Foxp3+Regulatory T Lymphocytes Promote Lung Fibrosis. Am. J. Respir. Crit. Care Med. 184 (11), 1270–1281. doi:10.1164/rccm.201103-0516OC
Lobato-Pascual, A., Saether, P. C., Dahle, M. K., Gaustad, P., Dissen, E., Fossum, S., et al. (2013). Rat Macrophage C-type Lectin Is an Activating Receptor Expressed by Phagocytic Cells. PLOS ONE 8 (2), e57406. doi:10.1371/journal.pone.0057406
Long, J., Ma, W., Yu, Z., Liu, H., and Cao, Y. (2019). Multi-walled Carbon Nanotubes (MWCNTs) Promoted Lipid Accumulation in THP-1 Macrophages through Modulation of Endoplasmic Reticulum (ER) Stress. Nanotoxicology 13 (7), 938–951. doi:10.1080/17435390.2019.1597204
Lorenz, E. C., Michet, C. J., Milliner, D. S., and Lieske, J. C. (2013). Update on Oxalate crystal Disease. Curr. Rheumatol. Rep. 15 (7), 340. doi:10.1007/s11926-013-0340-4
Lugrin, J., and Martinon, F. (2018). The AIM2 Inflammasome: Sensor of Pathogens and Cellular Perturbations. Immunol. Rev. 281 (1), 99–114. doi:10.1111/imr.12618
Luna-Gomes, T., Santana, P. T., and Coutinho-Silva, R. (2015). Silica-induced Inflammasome Activation in Macrophages: Role of ATP and P2X7 Receptor. Immunobiology 220 (9), 1101–1106. doi:10.1016/j.imbio.2015.05.004
Luz, H. L., Reichel, M., Unwin, R. J., Mutig, K., Najenson, A. C., Tonner, L. M., et al. (2019). P2X7 Receptor Stimulation Is Not Required for Oxalate Crystal-Induced Kidney Injury. Sci. Rep. 9 (1), 20086. doi:10.1038/s41598-019-56560-2
Lv, L. L., Tang, P. M.-K., Li, C. J., You, Y. K., Li, J., Huang, X.-R., et al. (2017). The Pattern Recognition Receptor, Mincle, Is Essential for Maintaining the M1 Macrophage Phenotype in Acute Renal Inflammation. Kidney Int. 91 (3), 587–602. doi:10.1016/j.kint.2016.10.020
Ma, Q. (2020). Polarization of Immune Cells in the Pathologic Response to Inhaled Particulates. Front. Immunol. 11, 1060. doi:10.3389/fimmu.2020.01060
Maeda, M., Chen, Y., Lee, S., Kumagai-Takei, N., Yoshitome, K., Matsuzaki, H., et al. (2017). Induction of IL-17 Production from Human Peripheral Blood CD4+ Cells by Asbestos Exposure. Int. J. Oncol. 50 (6), 2024–2032. doi:10.3892/ijo.2017.3991
Malireddi, R. K. S., Gurung, P., Kesavardhana, S., Samir, P., Burton, A., Mummareddy, H., et al. (2020). Innate Immune Priming in the Absence of TAK1 Drives RIPK1 Kinase Activity-independent Pyroptosis, Apoptosis, Necroptosis, and Inflammatory Disease. J. Exp. Med. 217 (3). doi:10.1084/jem.20191644
Malireddi, R. K. S., Kesavardhana, S., and Kanneganti, T.-D. (2019). ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/pyroptosis, Apoptosis, and Necroptosis (PAN-Optosis). Front. Cel. Infect. Microbiol. 9, 406. doi:10.3389/fcimb.2019.00406
Martínez-García, J. J., Martínez-Banaclocha, H., Angosto-Bazarra, D., de Torre-Minguela, C., Baroja-Mazo, A., Alarcón-Vila, C., et al. (2019). P2X7 Receptor Induces Mitochondrial Failure in Monocytes and Compromises NLRP3 Inflammasome Activation during Sepsis. Nat. Commun. 10 (1), 2711. doi:10.1038/s41467-019-10626-x
Martinon, F., Pétrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 440 (7081), 237–241. doi:10.1038/nature04516
Matsuoka, Y., Yamashita, A., Matsuda, M., Kawai, K., Sawa, T., and Amaya, F. (2019). NLRP2 Inflammasome in Dorsal Root Ganglion as a Novel Molecular Platform that Produces Inflammatory Pain Hypersensitivity. Pain 160 (9), 2149–2160. doi:10.1097/j.pain.0000000000001611
Matzinger, P. (2002). The Danger Model: a Renewed Sense of Self. Science 296 (5566), 301–305. doi:10.1126/science.1071059
Matzinger, P. (1994). Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 12, 991–1045. doi:10.1146/annurev.iy.12.040194.005015
Mayeux, J. M., Escalante, G. M., Christy, J. M., Pawar, R. D., Kono, D. H., and Pollard, K. M. (2018). Silicosis and Silica-Induced Autoimmunity in the Diversity Outbred Mouse. Front. Immunol. 9, 874. doi:10.3389/fimmu.2018.00874
McKee, C. M., and Coll, R. C. (2020). NLRP3 Inflammasome Priming: A riddle Wrapped in a Mystery inside an enigma. J. Leukoc. Biol. 108 (3), 937–952. doi:10.1002/JLB.3MR0720-513R
Mills, E. L., Kelly, B., and O’Neill, L. A. J. (2017). Mitochondria Are the Powerhouses of Immunity. Nat. Immunol. 18 (5), 488–498. doi:10.1038/ni.3704
Mischler, S. E., Cauda, E. G., Di Giuseppe, M., McWilliams, L. J., St. Croix, C., Sun, M., et al. (2016). Differential Activation of RAW 264.7 Macrophages by Size-Segregated Crystalline Silica. J. Occup. Med. Toxicol. 11, 57. doi:10.1186/s12995-016-0145-2
Mitchell, P. S., Sandstrom, A., and Vance, R. E. (2019). The NLRP1 Inflammasome: New Mechanistic Insights and Unresolved Mysteries. Curr. Opin. Immunol. 60, 37–45. doi:10.1016/j.coi.2019.04.015
Mohajerani, A., Burnett, L., Smith, J. V., Kurmus, H., Milas, J., Arulrajah, A., et al. (2019). Nanoparticles in Construction Materials and Other Applications, and Implications of Nanoparticle Use. Materials 12 (19), 3052. doi:10.3390/ma12193052
Moossavi, M., Parsamanesh, N., Bahrami, A., Atkin, S. L., and Sahebkar, A. (2018). Role of the NLRP3 Inflammasome in Cancer. Mol. Cancer 17 (1), 158. doi:10.1186/s12943-018-0900-3
Morishige, T., Yoshioka, Y., Inakura, H., Tanabe, A., Yao, X., Narimatsu, S., et al. (2010). The Effect of Surface Modification of Amorphous Silica Particles on NLRP3 Inflammasome Mediated IL-1β Production, ROS Production and Endosomal Rupture. Biomaterials 31 (26), 6833–6842. doi:10.1016/j.biomaterials.2010.05.036
Morotti, A., Sollaku, I., Catalani, S., Franceschini, F., Cavazzana, I., Fredi, M., et al. (2021). Systematic Review and Meta-Analysis of Epidemiological Studies on the Association of Occupational Exposure to Free Crystalline Silica and Systemic Lupus Erythematosus. Rheumatology (Oxford) 60 (1), 81–91. doi:10.1093/rheumatology/keaa444
Mossman, B. T., and Glenn, R. E. (2013). Bioreactivity of the Crystalline Silica Polymorphs, Quartz and Cristobalite, and Implications for Occupational Exposure Limits (OELs). Crit. Rev. Toxicol. 43 (8), 632–660. doi:10.3109/10408444.2013.818617
Mossman, B. T., Lippmann, M., Hesterberg, T. W., Kelsey, K. T., Barchowsky, A., and Bonner, J. C. (2011). Pulmonary Endpoints (Lung Carcinomas and Asbestosis) Following Inhalation Exposure to Asbestos. J. Toxicol. Environ. Health Part B 14 (1-4), 76–121. doi:10.1080/10937404.2011.556047
Moulton, V. R., Suarez-Fueyo, A., Meidan, E., Li, H., Mizui, M., and Tsokos, G. C. (2017). Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 23 (7), 615–635. doi:10.1016/j.molmed.2017.05.006
Mulay, S. R., and Anders, H.-J. (2016). Crystallopathies. N. Engl. J. Med. 374 (25), 2465–2476. doi:10.1056/NEJMra1601611
Mulay, S. R., Herrmann, M., Bilyy, R., Gabibov, A., and Anders, H.-J. (2019a). Editorial: Nano- and Microparticle-Induced Cell Death, Inflammation and Immune Responses. Front. Immunol. 10 (844), 844. doi:10.3389/fimmu.2019.00844
Mulay, S. R., Honarpisheh, M. M., Foresto-Neto, O., Shi, C., Desai, J., Zhao, Z. B., et al. (2019b). Mitochondria Permeability Transition versus Necroptosis in Oxalate-Induced AKI. Jasn 30 (10), 1857–1869. doi:10.1681/ASN.2018121218
Mulay, S. R., Kulkarni, O. P., Rupanagudi, K. V., Migliorini, A., Darisipudi, M. N., Vilaysane, A., et al. (2013). Calcium Oxalate Crystals Induce Renal Inflammation by NLRP3-Mediated IL-1β Secretion. J. Clin. Invest. 123 (1), 236–246. doi:10.1172/JCI63679
Muñoz-Planillo, R., Kuffa, P., Martínez-Colón, G., Smith, B. L., Rajendiran, T. M., and Núñez, G. (2013). K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 38 (6), 1142–1153. doi:10.1016/j.immuni.2013.05.016
Murakami, T., Ockinger, J., Yu, J., Byles, V., McColl, A., Hofer, A. M., et al. (2012). Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc. Natl. Acad. Sci. 109 (28), 11282–11287. doi:10.1073/pnas.1117765109
Murphy, F. A., Poland, C. A., Duffin, R., Al-Jamal, K. T., Ali-Boucetta, H., Nunes, A., et al. (2011). Length-dependent Retention of Carbon Nanotubes in the Pleural Space of Mice Initiates Sustained Inflammation and Progressive Fibrosis on the Parietal Pleura. Am. J. Pathol. 178 (6), 2587–2600. doi:10.1016/j.ajpath.2011.02.040
Musial, J., Krakowiak, R., Mlynarczyk, D. T., Goslinski, T., and Stanisz, B. J. (2020). Titanium Dioxide Nanoparticles in Food and Personal Care Products-What Do We Know about Their Safety. Nanomaterials 10 (6), 1110. doi:10.3390/nano10061110
Nagai, K., Arito, M., Takakuwa, Y., Ooka, S., Sato, T., Kurokawa, M. S., et al. (2012). Altered Posttranslational Modification on U1 Small Nuclear Ribonucleoprotein 68k in Systemic Autoimmune Diseases Detected by 2D Western Blot. Electrophoresis 33 (13), 2028–2035. doi:10.1002/elps.201200058
Nagata, S. (2018). Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 36 (1), 489–517. doi:10.1146/annurev-immunol-042617-053010
Nahle, S., Safar, R., Grandemange, S., Foliguet, B., Lovera‐Leroux, M., Doumandji, Z., et al. (2019). Single wall and Multiwall Carbon Nanotubes Induce Different Toxicological Responses in Rat Alveolar Macrophages. J. Appl. Toxicol. 39 (5), 764–772. doi:10.1002/jat.3765
Negri, V., Pacheco-Torres, J., Calle, D., and López-Larrubia, P. (2020). Carbon Nanotubes in Biomedicine. Top. Curr. Chem. (Z) 378 (1), 15. doi:10.1007/s41061-019-0278-8
Neumann, K., Castiñeiras-Vilariño, M., Höckendorf, U., Hannesschläger, N., Lemeer, S., Kupka, D., et al. (2014). Clec12a Is an Inhibitory Receptor for Uric Acid Crystals that Regulates Inflammation in Response to Cell Death. Immunity 40 (3), 389–399. doi:10.1016/j.immuni.2013.12.015
Newton, K., and Manning, G. (2016). Necroptosis and Inflammation. Annu. Rev. Biochem. 85 (1), 743–763. doi:10.1146/annurev-biochem-060815-014830
Ng, G., Sharma, K., Ward, S. M., Desrosiers, M. D., Stephens, L. A., Schoel, W. M., et al. (2008). Receptor-independent, Direct Membrane Binding Leads to Cell-Surface Lipid Sorting and Syk Kinase Activation in Dendritic Cells. Immunity 29 (5), 807–818. doi:10.1016/j.immuni.2008.09.013
Ngo, S. T., Steyn, F. J., and McCombe, P. A. (2014). Gender Differences in Autoimmune Disease. Front. Neuroendocrinology 35 (3), 347–369. doi:10.1016/j.yfrne.2014.04.004
Nidorf, S. M., Fiolet, A., and Abela, G. S. (2020). Viewing Atherosclerosis through a crystal Lens: How the Evolving Structure of Cholesterol Crystals in Atherosclerotic Plaque Alters its Stability. J. Clin. Lipidol. 14 (5), 619–630. doi:10.1016/j.jacl.2020.07.003
Niemann, B., and Rohrbach, S. (2016). Metabolically Relevant Cell Biology - Role of Intracellular Organelles for Cardiac Metabolism, in The Scientist's Guide to Cardiac Metabolism. Editors M. Schwarzer, and T. Doenst (Boston: Academic Press), 19–38. doi:10.1016/b978-0-12-802394-5.00003-0
Niemi, K., Teirilä, L., Lappalainen, J., Rajamäki, K., Baumann, M. H., Öörni, K., et al. (2011). Serum Amyloid A Activates the NLRP3 Inflammasome via P2X7 Receptor and a Cathepsin B-Sensitive Pathway. J.I. 186 (11), 6119–6128. doi:10.4049/jimmunol.1002843
Nimmerjahn, F., and Ravetch, J. V. (2008). Fcγ Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 8 (1), 34–47. doi:10.1038/nri2206
NIOSH (2019). Silica, amorphous. Available: https://www.cdc.gov/niosh/npg/npgd0552.html (Accessed 2 8, 2021 2021).
Nirmala, J. G., and Lopus, M. (2020). Cell Death Mechanisms in Eukaryotes. Cell Biol Toxicol 36 (2), 145–164. doi:10.1007/s10565-019-09496-2
Nishijima, N., Hirai, T., Misato, K., Aoyama, M., Kuroda, E., Ishii, K. J., et al. (2017). Human Scavenger Receptor A1-Mediated Inflammatory Response to Silica Particle Exposure Is Size Specific. Front. Immunol. 8, 379. doi:10.3389/fimmu.2017.00379
Niyonzima, N., Halvorsen, B., Sporsheim, B., Garred, P., Aukrust, P., Mollnes, T. E., et al. (2017). Complement Activation by Cholesterol Crystals Triggers a Subsequent Cytokine Response. Mol. Immunol. 84, 43–50. doi:10.1016/j.molimm.2016.09.019
Omori, S., Tsugita, M., Hoshikawa, Y., Morita, M., Ito, F., Yamaguchi, S.-I., et al. (2021). Tim4 Recognizes Carbon Nanotubes and Mediates Phagocytosis Leading to Granuloma Formation. Cel Rep. 34 (6), 108734. doi:10.1016/j.celrep.2021.108734
Orlowski, G. M., Colbert, J. D., Sharma, S., Bogyo, M., Robertson, S. A., and Rock, K. L. (2015). Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J.I. 195 (4), 1685–1697. doi:10.4049/jimmunol.1500509
Palomäki, J., Välimäki, E., Sund, J., Vippola, M., Clausen, P. A., Jensen, K. A., et al. (2011). Long, Needle-like Carbon Nanotubes and Asbestos Activate the NLRP3 Inflammasome through a Similar Mechanism. ACS Nano 5 (9), 6861–6870. doi:10.1021/nn200595c
Papayannopoulos, V. (2018). Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 18 (2), 134–147. doi:10.1038/nri.2017.105
Parackova, Z., Zentsova, I., Vrabcova, P., Klocperk, A., Sumnik, Z., Pruhova, S., et al. (2020). Neutrophil Extracellular Trap Induced Dendritic Cell Activation Leads to Th1 Polarization in Type 1 Diabetes. Front. Immunol. 11, 661. doi:10.3389/fimmu.2020.00661
Park, M., Joo, H. S., Lee, K., Jang, M., Kim, S. D., Kim, I., et al. (2018). Differential Toxicities of fine Particulate Matters from Various Sources. Sci. Rep. 8 (1), 17007. doi:10.1038/s41598-018-35398-0
Parks, C. G., Cooper, G. S., Nylander-French, L. A., Sanderson, W. T., Dement, J. M., Cohen, P. L., et al. (2002). Occupational Exposure to Crystalline Silica and Risk of Systemic Lupus Erythematosus: a Population-Based, Case-Control Study in the southeastern United States. Arthritis Rheum. 46 (7), 1840–1850. doi:10.1002/art.10368
Pavan, C., Santalucia, R., Leinardi, R., Fabbiani, M., Yakoub, Y., Uwambayinema, F., et al. (2020). Nearly Free Surface Silanols Are the Critical Molecular Moieties that Initiate the Toxicity of Silica Particles. Proc. Natl. Acad. Sci. USA 117, 27836–27846. doi:10.1073/pnas.2008006117
Pazár, B., Ea, H.-K., Narayan, S., Kolly, L., Bagnoud, N., Chobaz, V., et al. (2011). Basic Calcium Phosphate Crystals Induce Monocyte/Macrophage IL-1β Secretion through the NLRP3 Inflammasome In Vitro. J.I. 186 (4), 2495–2502. doi:10.4049/jimmunol.1001284
Peixoto, M. S., de Oliveira Galvão, M. F., and Batistuzzo de Medeiros, S. R. (2017). Cell Death Pathways of Particulate Matter Toxicity. Chemosphere 188, 32–48. doi:10.1016/j.chemosphere.2017.08.076
Perkins, R. C., Scheule, R. K., Hamilton, R., Gomes, G., Freidman, G., and Holian, A. (1993). Human Alveolar Macrophage Cytokine Release in Response to In Vitro and In Vivo Asbestos Exposure. Exp. Lung Res. 19 (1), 55–65. doi:10.3109/01902149309071080
Perlman, D. M., and Maier, L. A. (2019). Occupational Lung Disease. Med. Clin. North America 103 (3), 535–548. doi:10.1016/j.mcna.2018.12.012
Pestka, J. J., Akbari, P., Wierenga, K. A., Bates, M. A., Gilley, K. N., Wagner, J. G., et al. (2021). Omega-3 Polyunsaturated Fatty Acid Intervention against Established Autoimmunity in a Murine Model of Toxicant-Triggered Lupus. Front. Immunol. 12, 653464. doi:10.3389/fimmu.2021.653464
Pestka, J. J., Vines, L. L., Bates, M. A., He, K., and Langohr, I. (2014). Comparative Effects of N-3, N-6 and N-9 Unsaturated Fatty Acid-Rich Diet Consumption on Lupus Nephritis, Autoantibody Production and CD4+ T Cell-Related Gene Responses in the Autoimmune NZBWF1 Mouse. PLoS One 9 (6), e100255. doi:10.1371/journal.pone.0100255
Pira, E., Donato, F., Maida, L., and Discalzi, G. (2018). Exposure to Asbestos: Past, Present and Future. J. Thorac. Dis. 10 (2), S237–S245. doi:10.21037/jtd.2017.10.126
Polizzi, S., Pira, E., Ferrara, M., Bugiani, M., Papaleo, A., Albera, R., et al. (2002). Neurotoxic Effects of Aluminium Among Foundry Workers and Alzheimer's Disease. Neurotoxicology 23 (6), 761–774. doi:10.1016/S0161-813X(02)00097-9
Pollard, K. M. (2012). Gender Differences in Autoimmunity Associated with Exposure to Environmental Factors. J. Autoimmun. 38 (2-3), J177–J186. doi:10.1016/j.jaut.2011.11.007
Pollard, K. M. (2016). Silica, Silicosis, and Autoimmunity. Front. Immunol. 7, 97. doi:10.3389/fimmu.2016.00097
Porter, D. W., Wu, N., Hubbs, A. F., Mercer, R. R., Funk, K., Meng, F., et al. (2013). Differential Mouse Pulmonary Dose and Time Course Responses to Titanium Dioxide Nanospheres and Nanobelts. Toxicol. Sci. 131 (1), 179–193. doi:10.1093/toxsci/kfs261
Pownall, H. J., and Gotto, Jr., A. M. (2019). Cholesterol: Can't Live with it, Can't Live without it. Methodist Debakey Cardiovasc. J. 15 (1), 9–15. doi:10.14797/mdcj-15-1-9
Prabhudas, M., Bowdish, D., Drickamer, K., Febbraio, M., Herz, J., Kobzik, L., et al. (2014). Standardizing Scavenger Receptor Nomenclature. J.I. 192 (5), 1997–2006. doi:10.4049/jimmunol.1490003
Rabolli, V., Lison, D., and Huaux, F. (2016). The Complex cascade of Cellular Events Governing Inflammasome Activation and IL-1β Processing in Response to Inhaled Particles. Part. Fibre Toxicol. 13 (1), 40. doi:10.1186/s12989-016-0150-8
Rabolli, V., Thomassen, L. C. J., Princen, C., Napierska, D., Gonzalez, L., Kirsch-Volders, M., et al. (2010). Influence of Size, Surface Area and Microporosity on Thein Vitrocytotoxic Activity of Amorphous Silica Nanoparticles in Different Cell Types. Nanotoxicology 4 (3), 307–318. doi:10.3109/17435390.2010.482749
Rada, B. (2017). Neutrophil Extracellular Traps and Microcrystals. J. Immunol. Res. 2017, 1–7. doi:10.1155/2017/2896380
Rai, V., and Agrawal, D. K. (2017). The Role of Damage- and Pathogen-Associated Molecular Patterns in Inflammation-Mediated Vulnerability of Atherosclerotic Plaques. Can. J. Physiol. Pharmacol. 95 (10), 1245–1253. doi:10.1139/cjpp-2016-0664
Rajamäki, K., Lappalainen, J., Öörni, K., Välimäki, E., Matikainen, S., Kovanen, P. T., et al. (2010). Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: a Novel Link between Cholesterol Metabolism and Inflammation. PLoS One 5 (7), e11765. doi:10.1371/journal.pone.0011765
Rajasinghe, L. D., Li, Q.-Z., Zhu, C., Yan, M., Chauhan, P. S., Wierenga, K. A., et al. (2020). Omega-3 Fatty Acid Intake Suppresses Induction of Diverse Autoantibody Repertoire by Crystalline Silica in Lupus-Prone Mice. Autoimmunity 53 (7), 415–433. doi:10.1080/08916934.2020.1801651
Raker, V. K., Domogalla, M. P., and Steinbrink, K. (2015). Tolerogenic Dendritic Cells for Regulatory T Cell Induction in Man. Front. Immunol. 6 (569), 569. doi:10.3389/fimmu.2015.00569
Rello, S., Stockert, J. C., Moreno, V., Gamez, A., Pacheco, M., Juarranz, A., et al. (2005). Morphological Criteria to Distinguish Cell Death Induced by Apoptotic and Necrotic Treatments. Apoptosis 10 (1), 201–208. doi:10.1007/s10495-005-6075-6
Reus, T. L., Marcon, B. H., Paschoal, A. C. C., Ribeiro, I. R. S., Cardoso, M. B., Dallagiovanna, B., et al. (2020). Dose-dependent Cell Necrosis Induced by Silica Nanoparticles. Toxicol. Vitro 63, 104723. doi:10.1016/j.tiv.2019.104723
Rheinheimer, J., de Souza, B. M., Cardoso, N. S., Bauer, A. C., and Crispim, D. (2017). Current Role of the NLRP3 Inflammasome on Obesity and Insulin Resistance: A Systematic Review. Metabolism 74, 1–9. doi:10.1016/j.metabol.2017.06.002
Riedhammer, C., and Weissert, R. (2015). Antigen Presentation, Autoantigens, and Immune Regulation in Multiple Sclerosis and Other Autoimmune Diseases. Front. Immunol. 6, 322. doi:10.3389/fimmu.2015.00322
Rifat, S. L., Eastwood, M. R., Crapper McLachlan, D. R., and Corey, P. N. (1990). Effect of Exposure of Miners to Aluminium Powder. The Lancet 336 (8724), 1162–1165. doi:10.1016/0140-6736(90)92775-d
Rock, K. L., Kataoka, H., and Lai, J.-J. (2013). Uric Acid as a Danger Signal in Gout and its Comorbidities. Nat. Rev. Rheumatol. 9 (1), 13–23. doi:10.1038/nrrheum.2012.143
Ros, U., Pedrera, L., and Garcia-Saez, A. J. (2020). Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis. Ijms 21 (7), 2412. doi:10.3390/ijms21072412
Rosales, C., and Uribe-Querol, E. (2017). Phagocytosis: A Fundamental Process in Immunity. Biomed. Res. Int. 2017, 1–18. doi:10.1155/2017/9042851
Rosen, A., and Casciola-Rosen, L. (2016). Autoantigens as Partners in Initiation and Propagation of Autoimmune Rheumatic Diseases. Annu. Rev. Immunol. 34, 395–420. doi:10.1146/annurev-immunol-032414-112205
Rosenthal, A. K., and Ryan, L. M. (2016). Calcium Pyrophosphate Deposition Disease. N. Engl. J. Med. 374 (26), 2575–2584. doi:10.1056/NEJMra1511117
Russ, K. A., Thompson, J. A., Reynolds, J. S., Mercer, R. R., Porter, D. W., McKinney, W., et al. (2020). Biological Effects of Inhaled Hydraulic Fracturing Sand Dust. IV. Pulmonary Effects. Toxicol. Appl. Pharmacol. 409, 115284. doi:10.1016/j.taap.2020.115284
Ryan, A. J., Larson-Casey, J. L., He, C., Murthy, S., and Carter, A. B. (2014). Asbestos-induced Disruption of Calcium Homeostasis Induces Endoplasmic Reticulum Stress in Macrophages. J. Biol. Chem. 289 (48), 33391–33403. doi:10.1074/jbc.M114.579870
Sager, T. M., and Castranova, V. (2009). Surface Area of Particle Administered versus Mass in Determining the Pulmonary Toxicity of Ultrafine and fine Carbon Black: Comparison to Ultrafine Titanium Dioxide. Part. Fibre Toxicol. 6 (1), 15. doi:10.1186/1743-8977-6-15
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T Cells and Immune Tolerance. Cell 133 (5), 775–787. doi:10.1016/j.cell.2008.05.009
Samir, P., Malireddi, R. K. S., and Kanneganti, T.-D. (2020). The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cel. Infect. Microbiol. 10, 238. doi:10.3389/fcimb.2020.00238
Santavanond, J. P., Rutter, S. F., Atkin-Smith, G. K., and Poon, I. K. H. (2021). Apoptotic Bodies: Mechanism of Formation, Isolation and Functional Relevance, in New Frontiers: Extracellular Vesicles. Editors S. Mathivanan, P. Fonseka, C. Nedeva, and I. Atukorala (Cham: Springer International Publishing), 61–88. doi:10.1007/978-3-030-67171-6_4
Sayan, M., and Mossman, B. T. (2016). The NLRP3 Inflammasome in Pathogenic Particle and Fibre-Associated Lung Inflammation and Diseases. Part. Fibre Toxicol. 13 (1), 51. doi:10.1186/s12989-016-0162-4
Sayer, J. A. (2017). Progress in Understanding the Genetics of Calcium-Containing Nephrolithiasis. Jasn 28 (3), 748–759. doi:10.1681/ASN.2016050576
Schaftenaar, F., Frodermann, V., Kuiper, J., and Lutgens, E. (2016). Atherosclerosis. Curr. Opin. Lipidol. 27 (3), 209–215. doi:10.1097/MOL.0000000000000302
Scheiblich, H., Schlütter, A., Golenbock, D. T., Latz, E., Martinez-Martinez, P., and Heneka, M. T. (2017). Activation of the NLRP3 Inflammasome in Microglia: the Role of Ceramide. J. Neurochem. 143 (5), 534–550. doi:10.1111/jnc.14225
Schnurr, M., Then, F., Galambos, P., Scholz, C., Siegmund, B., Endres, S., et al. (2000). Extracellular ATP and TNF-α Synergize in the Activation and Maturation of Human Dendritic Cells. J. Immunol. 165 (8), 4704–4709. doi:10.4049/jimmunol.165.8.4704
Schwarzer, R., Laurien, L., and Pasparakis, M. (2020). New Insights into the Regulation of Apoptosis, Necroptosis, and Pyroptosis by Receptor Interacting Protein Kinase 1 and Caspase-8. Curr. Opin. Cel Biol. 63, 186–193. doi:10.1016/j.ceb.2020.02.004
Segawa, K., and Nagata, S. (2015). An Apoptotic 'eat Me' Signal: Phosphatidylserine Exposure. Trends Cel Biol. 25 (11), 639–650. doi:10.1016/j.tcb.2015.08.003
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 36 (3), 401–414. doi:10.1016/j.immuni.2012.01.009
So, A., Ives, A., Joosten, L. A. B., and Busso, N. (2013). Targeting Inflammasomes in Rheumatic Diseases. Nat. Rev. Rheumatol. 9 (7), 391–399. doi:10.1038/nrrheum.2013.61
Sollberger, G., Tilley, D. O., and Zychlinsky, A. (2018). Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Develop. Cel 44 (5), 542–553. doi:10.1016/j.devcel.2018.01.019
Stephen, S. L., Freestone, K., Dunn, S., Twigg, M. W., Homer-Vanniasinkam, S., Walker, J. H., et al. (2010). Scavenger Receptors and Their Potential as Therapeutic Targets in the Treatment of Cardiovascular Disease. Int. J. Hypertens. 2010, 1–21. doi:10.4061/2010/646929
Stranges, P. B., Watson, J., Cooper, C. J., Choisy-Rossi, C.-M., Stonebraker, A. C., Beighton, R. A., et al. (2007). Elimination of Antigen-Presenting Cells and Autoreactive T Cells by Fas Contributes to Prevention of Autoimmunity. Immunity 26 (5), 629–641. doi:10.1016/j.immuni.2007.03.016
Strowig, T., Henao-Mejia, J., Elinav, E., and Flavell, R. (2012). Inflammasomes in Health and Disease. Nature 481 (7381), 278–286. doi:10.1038/nature10759
Su, Z., Yang, Z., Xie, L., DeWitt, J. P., and Chen, Y. (2016). Cancer Therapy in the Necroptosis Era. Cel Death Differ 23 (5), 748–756. doi:10.1038/cdd.2016.8
Sukhanova, A., Bozrova, S., Sokolov, P., Berestovoy, M., Karaulov, A., and Nabiev, I. (2018). Dependence of Nanoparticle Toxicity on Their Physical and Chemical Properties. Nanoscale Res. Lett. 13 (1), 44. doi:10.1186/s11671-018-2457-x
Sutton, C., Brereton, C., Keogh, B., Mills, K. H. G., and Lavelle, E. C. (2006). A Crucial Role for Interleukin (IL)-1 in the Induction of IL-17-producing T Cells that Mediate Autoimmune Encephalomyelitis. J. Exp. Med. 203 (7), 1685–1691. doi:10.1084/jem.20060285
Tai, V., Merriman, T. R., and Dalbeth, N. (2019). Genetic Advances in Gout: Potential Applications in Clinical Practice. Curr. Opin. Rheumatol. 31 (2), 144–151. doi:10.1097/BOR.0000000000000571
Tanaka, M., Saka-Tanaka, M., Ochi, K., Fujieda, K., Sugiura, Y., Miyamoto, T., et al. (2020). C-type Lectin Mincle Mediates Cell Death-Triggered Inflammation in Acute Kidney Injury. J. Exp. Med. 217 (11). doi:10.1084/jem.20192230
Terzoglou, A. G., Routsias, J. G., Moutsopoulos, H. M., and Tzioufas, A. G. (2006). Post-translational Modifications of the Major Linear Epitope 169?190aa of Ro60 kDa Autoantigen Alter the Autoantibody Binding. Clin. Exp. Immunol. 146 (1), 60–65. doi:10.1111/j.1365-2249.2006.03192.x
Thakur, S. A., Beamer, C. A., Migliaccio, C. T., and Holian, A. (2009). Critical Role of MARCO in Crystalline Silica-Induced Pulmonary Inflammation. Toxicol. Sci. 108 (2), 462–471. doi:10.1093/toxsci/kfp011
Thibodeau, M., Giardina, C., and Hubbard, A. K. (2003). Silica-induced Caspase Activation in Mouse Alveolar Macrophages Is Dependent upon Mitochondrial Integrity and Aspartic Proteolysis. Toxicol. Sci. 76 (1), 91–101. doi:10.1093/toxsci/kfg178
Thibodeau, M. S., Giardina, C., Knecht, D. A., Helble, J., and Hubbard, A. K. (2004). Silica-induced Apoptosis in Mouse Alveolar Macrophages Is Initiated by Lysosomal Enzyme Activity. Toxicol. Sci. 80 (1), 34–48. doi:10.1093/toxsci/kfh121
Toldo, S., and Abbate, A. (2018). The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat. Rev. Cardiol. 15 (4), 203–214. doi:10.1038/nrcardio.2017.161
Tomljenovic, L., and A. Shaw, C. (2011). Aluminum Vaccine Adjuvants: Are They Safe? Cmc 18 (17), 2630–2637. doi:10.2174/092986711795933740
Trapani, J. A., and Smyth, M. J. (2002). Functional Significance of the Perforin/granzyme Cell Death Pathway. Nat. Rev. Immunol. 2 (10), 735–747. doi:10.1038/nri911
Tsai, P.-Y., Ka, S.-M., Chang, J.-M., Chen, H.-C., Shui, H.-A., Li, C.-Y., et al. (2011). Epigallocatechin-3-gallate Prevents Lupus Nephritis Development in Mice via Enhancing the Nrf2 Antioxidant Pathway and Inhibiting NLRP3 Inflammasome Activation. Free Radic. Biol. Med. 51 (3), 744–754. doi:10.1016/j.freeradbiomed.2011.05.016
Tsugita, M., Morimoto, N., and Nakayama, M. (2017a). SiO2 and TiO2 Nanoparticles Synergistically Trigger Macrophage Inflammatory Responses. Part. Fibre Toxicol. 14 (1), 11. doi:10.1186/s12989-017-0192-6
Tsugita, M., Morimoto, N., Tashiro, M., Kinoshita, K., and Nakayama, M. (2017b). SR-B1 Is a Silica Receptor that Mediates Canonical Inflammasome Activation. Cel Rep. 18 (5), 1298–1311. doi:10.1016/j.celrep.2017.01.004
Tummers, B., and Green, D. R. (2017). Caspase-8: Regulating Life and Death. Immunol. Rev. 277 (1), 76–89. doi:10.1111/imr.12541
Turci, F., Pavan, C., Leinardi, R., Tomatis, M., Pastero, L., Garry, D., et al. (2016). Revisiting the Paradigm of Silica Pathogenicity with Synthetic Quartz Crystals: the Role of Crystallinity and Surface Disorder. Part. Fibre Toxicol. 13 (1), 32. doi:10.1186/s12989-016-0136-6
Tveita, A. A. (2010). The Danger Model in Deciphering Autoimmunity. Rheumatology 49 (4), 632–639. doi:10.1093/rheumatology/keq004
Vadasz, Z., Haj, T., Kessel, A., and Toubi, E. (2013). Age-related Autoimmunity. BMC Med. 11, 94. doi:10.1186/1741-7015-11-94
Vaillancourt, M., Desaulniers, P., Paré, G., Pagé, N., Lachhab, A., Kerever, A., et al. (2021). Expression of the Myeloid Inhibitory Receptor CLEC12A Correlates with Disease Activity and Cytokines in Early Rheumatoid Arthritis. Sci. Rep. 11 (1), 11248. doi:10.1038/s41598-021-90631-7
van Bavel, C. C., Dieker, J., Muller, S., Briand, J.-P., Monestier, M., Berden, J. H., et al. (2009). Apoptosis-associated Acetylation on Histone H2B Is an Epitope for Lupus Autoantibodies. Mol. Immunol. 47 (2-3), 511–516. doi:10.1016/j.molimm.2009.08.009
Varsano, N., Beghi, F., Elad, N., Pereiro, E., Dadosh, T., Pinkas, I., et al. (2018). Two Polymorphic Cholesterol Monohydrate crystal Structures Form in Macrophage Culture Models of Atherosclerosis. Proc. Natl. Acad. Sci. USA 115 (30), 7662–7669. doi:10.1073/pnas.1803119115
Venkataraman, A., Amadi, E. V., Chen, Y., and Papadopoulos, C. (2019). Carbon Nanotube Assembly and Integration for Applications. Nanoscale Res. Lett. 14 (1), 220. doi:10.1186/s11671-019-3046-3
Vladimer, G. I., Weng, D., Paquette, S. W. M., Vanaja, S. K., Rathinam, V. A. K., Aune, M. H., et al. (2012). The NLRP12 Inflammasome Recognizes Yersinia pestis. Immunity 37 (1), 96–107. doi:10.1016/j.immuni.2012.07.006
Voskoboinik, I., Dunstone, M. A., Baran, K., Whisstock, J. C., and Trapani, J. A. (2010). Perforin: Structure, Function, and Role in Human Immunopathology. Immunol. Rev. 235 (1), 35–54. doi:10.1111/j.0105-2896.2010.00896.x
Voskoboinik, I., Whisstock, J. C., and Trapani, J. A. (2015). Perforin and Granzymes: Function, Dysfunction and Human Pathology. Nat. Rev. Immunol. 15 (6), 388–400. doi:10.1038/nri3839
Wada, J., and Makino, H. (2016). Innate Immunity in Diabetes and Diabetic Nephropathy. Nat. Rev. Nephrol. 12 (1), 13–26. doi:10.1038/nrneph.2015.175
Walsh, J. G., Cullen, S. P., Sheridan, C., Luthi, A. U., Gerner, C., and Martin, S. J. (2008). Executioner Caspase-3 and Caspase-7 Are Functionally Distinct Proteases. Proc. Natl. Acad. Sci. 105 (35), 12815–12819. doi:10.1073/pnas.0707715105
Wang, S., and Zhang, Y. (2020). HMGB1 in Inflammation and Cancer. J. Hematol. Oncol. 13 (1), 116. doi:10.1186/s13045-020-00950-x
Watad, A., Bragazzi, N. L., Adawi, M., Amital, H., Toubi, E., Porat, B.-S., et al. (2017). Autoimmunity in the Elderly: Insights from Basic Science and Clinics - A Mini-Review. Gerontology 63 (6), 515–523. doi:10.1159/000478012
Westerfield, B. T. (1992). Asbestos-related Lung Disease. South. Med. J. 85 (6), 616–620. doi:10.1097/00007611-199206000-00009
Westerterp, M., Gautier, E. L., Ganda, A., Molusky, M. M., Wang, W., Fotakis, P., et al. (2017). Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cel Metab. 25 (6), 1294–1304. e1296. doi:10.1016/j.cmet.2017.04.005
Wierenga, K. A., Harkema, J. R., and Pestka, J. J. (2019). Lupus, Silica, and Dietary omega-3 Fatty Acid Interventions. Toxicol. Pathol. 47 (8), 1004–1011. doi:10.1177/0192623319878398
Wierenga, K. A., Strakovsky, R. S., Benninghoff, A. D., Rajasinghe, L. D., Lock, A. L., Harkema, J. R., et al. (2020). Requisite omega-3 HUFA Biomarker Thresholds for Preventing Murine Lupus Flaring. Front. Immunol. 11 (1796), 1796. doi:10.3389/fimmu.2020.01796
Winter, M., Beer, H.-D., Hornung, V., Krämer, U., Schins, R. P. F., and Förster, I. (2011). Activation of the Inflammasome by Amorphous Silica and TiO2nanoparticles in Murine Dendritic Cells. Nanotoxicology 5 (3), 326–340. doi:10.3109/17435390.2010.506957
Wong, K., Valdez, P. A., Tan, C., Yeh, S., Hongo, J.-A., and Ouyang, W. (2010). Phosphatidylserine Receptor Tim-4 Is Essential for the Maintenance of the Homeostatic State of Resident Peritoneal Macrophages. Proc. Natl. Acad. Sci. 107 (19), 8712–8717. doi:10.1073/pnas.0910929107
Yang, C.-A., and Chiang, B.-L. (2015). Inflammasomes and Human Autoimmunity: A Comprehensive Review. J. Autoimmun. 61, 1–8. doi:10.1016/j.jaut.2015.05.001
Yang, D., Chen, Q., Yang, H., Tracey, K. J., Bustin, M., and Oppenheim, J. J. (2007). High Mobility Group Box-1 Protein Induces the Migration and Activation of Human Dendritic Cells and Acts as an Alarmin. J. Leukoc. Biol. 81 (1), 59–66. doi:10.1189/jlb.0306180
Yi, Y.-S. (2017). Caspase-11 Non-canonical Inflammasome: a Critical Sensor of Intracellular Lipopolysaccharide in Macrophage-Mediated Inflammatory Responses. Immunology 152 (2), 207–217. doi:10.1111/imm.12787
Yipp, B. G., and Kubes, P. (2013). NETosis: How Vital Is it? Blood 122 (16), 2784–2794. doi:10.1182/blood-2013-04-457671
Zani, I., Stephen, S., Mughal, N., Russell, D., Homer-Vanniasinkam, S., Wheatcroft, S., et al. (2015). Scavenger Receptor Structure and Function in Health and Disease. Cells 4 (2), 178–201. doi:10.3390/cells4020178
Zhang, A., and Lieber, C. M. (2016). Nano-Bioelectronics. Chem. Rev. 116 (1), 215–257. doi:10.1021/acs.chemrev.5b00608
Zhao, C.-N., Xu, Z., Wu, G.-C., Mao, Y.-M., Liu, L.-N., Qian-Wu, W., et al. (2019). Emerging Role of Air Pollution in Autoimmune Diseases. Autoimmun. Rev. 18 (6), 607–614. doi:10.1016/j.autrev.2018.12.010
Zhao, L., Zhu, Y., Chen, Z., Xu, H., Zhou, J., Tang, S., et al. (2018). Cardiopulmonary Effects Induced by Occupational Exposure to Titanium Dioxide Nanoparticles. Nanotoxicology 12 (2), 169–184. doi:10.1080/17435390.2018.1425502
Zheng, D., Liwinski, T., and Elinav, E. (2020). Inflammasome Activation and Regulation: toward a Better Understanding of Complex Mechanisms. Cell Discov 6 (1), 36. doi:10.1038/s41421-020-0167-x
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X.-j., et al. (2018). New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 560 (7717), 198–203. doi:10.1038/s41586-018-0372-z
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 469 (7329), 221–225. doi:10.1038/nature09663
Zhou, Y., Tong, Z., Jiang, S., Zheng, W., Zhao, J., and Zhou, X. (2020). The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation. Cells 9 (5), 1219. doi:10.3390/cells9051219
Keywords: endogenous and exogenous particles, inflammation, autoimmunity, myeloid-lineage phagocytes, inflammasome activity, immunogenic cell death
Citation: Favor OK, Pestka JJ, Bates MA and Lee KSS (2021) Centrality of Myeloid-Lineage Phagocytes in Particle-Triggered Inflammation and Autoimmunity. Front. Toxicology 3:777768. doi: 10.3389/ftox.2021.777768
Received: 15 September 2021; Accepted: 19 October 2021;
Published: 04 November 2021.
Edited by:
Valérie Lecureur, University of Rennes 1, FranceReviewed by:
Yasuo Yoshioka, Osaka University, Japan`Alessandro Venosa, The University of Utah, United States
Copyright © 2021 Favor, Pestka, Bates and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivia K. Favor, ZmF2b3JvbGlAbXN1LmVkdQ==; James J. Pestka, cGVzdGthQG1zdS5lZHU=