 Alexandre J. Kennang Ouamba1,2
Alexandre J. Kennang Ouamba1,2 Mérilie Gagnon1,2
Mérilie Gagnon1,2 Thibault Varin1P. Yvan Chouinard2,3
Thibault Varin1P. Yvan Chouinard2,3 Gisèle LaPointe2,4
Gisèle LaPointe2,4 Denis Roy1,2*
Denis Roy1,2*- 1Département des Sciences des Aliments, Laboratoire de Génomique Microbienne, Université Laval, Québec, QC, Canada
- 2Regroupement de Recherche pour Un Lait de Qualité Optimale (Op+Lait), Saint-Hyacinthe, QC, Canada
- 3Département des Sciences Animales, Université Laval, Québec, QC, Canada
- 4Department of Food Science, University of Guelph, Guelph, ON, Canada
The microbiota of silage is a key determinant of its quality. Although commercial inoculants are often used to improve silage quality, studies to analyze their impact on the microbiota of preserved forage at farm-scale facilities are scarce. We assessed the diversity of viable bacterial communities of hay (unfermented dry forage) and grass or legume (GL) and corn (C) silage to deepen our knowledge of how inoculant addition drives microbial occurrence patterns on dairy farms. Forage samples were collected from 24 dairy farms over two sampling periods. Samples were analyzed by high-throughput sequencing and quantitative PCR after being treated with propidium monoazide to account for viable cells. We found consistent significant differences between hay and silage community structures across sampling periods. Silage was generally dominated by lactic acid bacteria (LAB), while Pantoea and Sphingomonas were the main co-dominant genera in hay. The GL silage dominated by Pediococcus, Weissella, and Bacillus was phylogenetically different from C silage enriched in Acetobacter. The use of inoculants including Lentilactobacillus buchneri either alone or in combination with Lactiplantibacillus plantarum, Lacticaseibacillus casei, Pediococcus pentosaceus, or Enterococcus faecium did not systematically prevent the occurrence of undesirable bacteria, especially when corn-based, probably because of factors that can mitigate the effect of inoculation on the microbiota. The core Lactobacillales constituted the dominant LAB in silage with up to 96% relative abundance, indicating either the ubiquity of inoculants or the high competitiveness of epiphytes. Silage chemical profiles varied inconsistently with sampling periods and the use of inoculants. Multivariate multi-table analyses allowed the identification of bacterial clusters mainly driven by moisture and magnesium content in hay, while pH, lactic, and fatty acids were the main drivers for silage. Bacterial network analyses showed considerable variations in the topological roles with the use of inoculants. These results may help evaluate the effectiveness of forage management practices implemented on dairy farms and, therefore, are useful for fine-tuning the search for new additives. Such knowledge can be used by forage makers to adjust processing routines to improve the hygienic quality, nutritional potential, and aerobic stability of preserved forage.
Introduction
Forage preservation is critical to ensure proper yearlong availability of feed for dairy cattle. This is particularly important in cold-weather areas characterized by rough winter conditions and limited growing seasons such as those prevailing in many parts of North America (Bernardes et al., 2018). Although the origin and patterns of the phyllosphere community assembly are unclear (Rastogi et al., 2013), the fate of microorganisms dwelling on fresh plants throughout the preservation processes contributes to the quality of preserved forage and determines associated potential risks to animal and human health (Driehuis et al., 2018). Due to the development and the application of molecular techniques to gain insight into the microbiome associated with preserved forage (McAllister et al., 2018), management systems have gained substantial upgrades intended to improve safety and production yields, as well as beneficial effects for animals (Dunière et al., 2013; Ogunade et al., 2018a; Borreani et al., 2018; Muck et al., 2018; Ávila and Carvalho, 2020). Management practices and environmental factors (Bernardes et al., 2018) unavoidably alter the microbial content of preserved forage. These feeds therefore constitute important vehicles for various microorganisms, including bacteria and fungi from the growing fields to dairy barns and, ultimately, to milk and dairy products (Julien et al., 2008; Driehuis, 2013; Dunière et al., 2013).
Hay and silage are the main forms of forage preservation in dairy production systems (Muck and Shinners, 2001). Although haymaking consists of drying forage crops to suppress enzymatic and microbial activities, hay still harbors a viable microbiota, for which the composition and structure are not well known (Daniels et al., 2020; Moore-Colyer et al., 2020). On the other hand, ensiling is based on the fermentative properties of epiphytic microorganisms, particularly lactic acid bacteria (LAB), which metabolize water soluble carbohydrates (WSC) into organic acids under anaerobic conditions where the rapid decline in pH is a key determinant of silage quality. The genera Lactiplantibacillus, Lacticaseibacillus, Lentilactobacillus (formerly Lactobacillus), other lactobacilli, Pediococcus, Weissella, Leuconostoc, Enterococcus, Streptococcus, and Lactococcus are generally associated with silage (Dunière et al., 2013; Ávila and Carvalho, 2020).
However, not all LAB strains can induce a fast pH decrease during the early stages of fermentation, and in naturally fermenting forage crops, they may be outcompeted by undesirable acid-tolerant bacteria including Enterobacteriaceae, acetic acid bacteria (AAB), and spore-forming bacteria associated with poor quality silage (Borreani et al., 2018; Ávila and Carvalho, 2020). Microbial additives encompassing homofermentative or facultative heterofermentative LAB (Lactiplantibacillus plantarum, Lacticaseibacillus casei, and Pediococcus spp.), obligate heterofermentative LAB (Lentilactobacillus buchneri and Lentilactobacillus hilgardii), combination inoculants, and non-LAB inoculants (Bacillus subtilis) have been proposed to enhance forage crop fermentation and improve the aerobic stability of silage, as well as its safety and nutritional value (Muck et al., 2018; Ávila and Carvalho, 2020). From a microbiological viewpoint, the efficacy of these commercial inoculants, either as single or as multi-species formulae, has been generally assessed in controlled laboratory conditions that do not mimic the various management practices and changing environmental factors observed in large-scale ensiling (Muck and Shinners, 2001; Kraut-Cohen et al., 2016; Ni et al., 2017). Consequently, few studies have evaluated the microbial communities populating silage stored in farm-scale silo types (Kraut-Cohen et al., 2016; Dos Santos et al., 2020), and none of this kind has focused on associations between physicochemical characteristics and the viable microbiota at feed-out, while contrasting uninoculated versus inoculated silage.
Gagnon et al. (2020) recently used a culture-based approach to analyze LAB communities occurring in hay and grass/legume and corn silage produced with and without inoculation at selected dairy farms. They revealed that while L. casei (or Lacticaseibacillus paracasei) and L. plantarum were common among all forage types, Enterococcus mundtii, Lactiplantibacillus pentosus, Companilactobacillus tucceti, Lactococcus lactis, and Leuconostoc mesenteroides (or Leuconostoc pseudomesenteroides) were only identified in hay, whereas the L. buchneri group was specific to silage regardless of the type and inoculation status. The current study complements that from Gagnon et al. (2020) by implementing a viability high-throughput sequencing approach combined with viability-PCR (Nocker et al., 2010; Kennang Ouamba et al., 2020) on the same samples to provide a comparative analysis of the microbial ecology of hay and grass/legume or corn silage produced with or without inoculants. Therefore, this study aimed to assess the diversity of microbial communities occurring in three types of preserved forage and gain insight into how inoculants shape the microbiota of mature silage in commercial settings.
Materials and methods
Dairy farm selection and sample collection
Farm recruitment and sampling were carried out as previously described (Gagnon et al., 2020). Briefly, the 24 tie-stall herds from the province of Quebec (Canada) were selected based on the forage harvested as hay or silage. Accordingly, farms were grouped into five feeding typologies comprising herds fed with either hay as the unique type of forage (H) or silage as the main forage source, the latter including grass/legume silage (GL), grass/legume silage with corn silage (C) as supplement, grass/legume silage with corn silage inoculated at harvest (CI) as supplement, and grass/legume silage inoculated at harvest (GLI) with CI as supplement.
Sampling of the five feed types (H, GL, GLI, C, and CI) was carried out twice: in fall 2015 and spring 2016. Silage samples were collected after at least 45 days of fermentation. On each farm, forage samples were collected during the same visit as previously described (Gagnon et al., 2020). Within the farms enrolled in the study, eight storage forms or silo types included loose and baled hay as well as wrapped square/round bales, concrete-stave silo, oxygen-limiting silo, pressed bag silo, bunker silo, and stack silo for silage (Table 1). For inoculated silage, the commercial inoculants included 11C33, 11CFT, and 11G22 (Pioneer, Johnston, and IA), as well as Biotal Buchneri 500 and Biotal Supersile (Lallemand Animal Nutrition, Milwaukee, WI). From the 500 g of each forage sample collected, a subsample was sent to Lactanet Laboratories (Sainte-Anne-de-Bellevue, Qc, Canada) for infrared quantification of organic compounds, moisture, pH, and minerals, as well as the estimation of fermentation acids when applicable. All the above-mentioned sample and farm information composed the sample metadata that were further integrated in the dataset and used where applicable as categorical or quantitative variables for data analysis.

TABLE 1. Distribution of forage storage forms and silo types.
DNA extraction and quantitative PCR
For forage samples, 30 g of each were homogenized in 270 ml of peptone buffer solution, as previously described (Gagnon et al., 2020). Aliquots of 2 ml immediately taken from the suspension were centrifuged at 12,000 × g for 15 min at 4°C. The pellets were washed twice with 1 ml sucrose buffer (sucrose 12% [w/v], 25 mM Tris-HCl pH 8.0). Half of the cell suspension was treated with propidium monoazide (PMA) to account for viable cells, as previously described (Kennang Ouamba et al., 2020). All PMA-free and PMA-treated cells were stored at –80°C. Genomic DNA was then extracted using the DNeasy PowerFood Microbial Kit (QIAGEN, Hilden, Germany) combined with enzymatic lysis with mutanolysin from Streptomyces (MilliporeSigma), lysozyme (MilliporeSigma), and proteinase K (MilliporeSigma) following the steps previously described (Kennang Ouamba et al., 2020).
Quantitative polymerase chain reaction (qPCR) was performed to determine copy numbers of specifically targeted genes of L. buchneri, L. plantarum, total LAB, AAB, Enterobacteriaceae, Pseudomonas, total bacteria, and total fungi in all PMA-free and PMA-treated forage, using specific primer pairs as previously described (Kennang Ouamba et al., 2020). Amplification reactions were carried out in duplicate using a ViiA7 system (Thermo Fisher Scientific, Burlington, ON, Canada). Each reaction mixture of a total volume of 10 µL was composed of 3.6 μL UltraPure DNAse and RNAse-free distilled water (Thermo Fisher Scientific), 5 μL PowerUp SYBR Green master mix (Thermo Fisher Scientific), 0.2 μL of each primer at 10 nM, and 1 μL DNA template. Bacterial loads were reported as gene copy number per Gram of forage.
High-throughput sequencing of the 16S rRNA gene pool and bioinformatic and statistical analyses
Sequencing was performed in a single run on Illumina MiSeq at the Plateforme d’Analyses Génomiques, Université Laval (Québec, Canada). Primer pairs 347 F (5′-GGAGGCAGCAGTRRGGAAT)/803R (5′-CTACCRGGGTATCTAATCC) were used to amplify the V3-V4 region of the 16S rRNA gene.
Cutadapt (version 2.3) software (Martin, 2011) was used to remove adapters and primers from demultiplexed sequences. Sequencing reads were modeled and denoised using the DADA2 (version 1.14) pipeline (Callahan et al., 2016a) developed for R. After constructing the merged sequence table and removing chimeras, the Silva version 132 DADA2-formatted reference databases (down to genus and species levels) were used for taxonomy assignment. Sequence alignment and phylogenetic tree construction were performed using the DECIPHER (version 2.14.0) package (Wright, 2015) and phangorn (version 2.5.5) package (Schliep, 2011), respectively, as previously described (Callahan et al., 2016b). The resulting amplicon sequence variants (ASVs) and phylogenetic tree were further processed using the phyloseq (version 1.30.0) package (McMurdie and Holmes, 2013) for alpha- and beta-diversity analyses of the forage microbiota.
As a preprocessing step in the data analysis, community differences between PMA-free and PMA-treated samples were compared, as well as between sampling periods, by computing alpha- and beta-diversity measures using the phyloseq package. The centered log-ratio (CLR) and phylogenetic isomeric log-ratio transform (PhILR) were applied to the ASV table prior to beta-diversity analyses for which Aitchison and Euclidean distances were used, respectively, to account for the compositional nature of microbial data (Gloor et al., 2017; Silverman et al., 2017).
Microbial communities in forage types were characterized by assessing and comparing alpha-diversity measures, including Chao1, Shannon, and inverse Simpson indices, and beta-diversity measures, including the sample local contribution to beta-diversity (LCBD), principal coordinate analysis (PCoA), and principal component analysis (PCA). LCBD is another way of assessing the beta-diversity that provides comparative indicators of the uniqueness of a community profile in a single sample among groups (Legendre and De Cáceres, 2013). LCBD indices were computed on Hellinger-transformed data using the microbiomeSeq R package (Ssekagiri et al., 2020). PCA was performed on CLR and PCoA on PhILR normalized data as described earlier to assess the dissimilarities of community structures among forage types. Significant differences were determined at a threshold of 0.05 from false discovery rate (FDR) corrected p-values after a Kruskal–Wallis test was computed using the R package ggpubr (Kassambara, 2018). Microbial differential abundance testing was performed for paired combinations between uninoculated and inoculated forage types using the R package ALDEx2 4.0 (Fernandes et al., 2014). The functional metagenomic content of forage samples was predicted using Piphillin software (Narayan et al., 2020) through the Piphillin online server (http://piphillin.secondgenome.com/). The differential abundance of microbial functional features was computed using ALDEx2, as described earlier. The abundance and effect size of differentially abundant taxa or functional features between uninoculated and inoculated forage types were visualized using a heatmap constructed using the package ComplexHeatmap 2.4.2 (Gu et al., 2016).
Categorical and quantitative metadata related to farms and forage samples were selectively integrated in multivariate multi-table analyses based on sparse partial least squares regression (sPLS) and canonical correspondence analysis (CCpnA) to determine their association with microbial features. The analysis scheme was partially inspired from a previously described study (Ingham et al., 2019). Relevant metadata variables were selected for multivariate multi-table analyses by permutational multivariate analysis of variance (adonis) on CLR normalized data. The function adonis2() from the vegan (version 2.5–6) R package (Jari et al., 2019) was used on the Aitchison distance matrix computed in the phyloseq package. Significant variables (p < 0.05) identified using adonis test were retained for sPLS-based analyses performed using the MixOmics (version 6.1.1) R package (Rohart et al., 2017). To complement the sPLS approach, general linear models implemented in the MaAsLin2 R package (https://github.com/biobakery/MaAsLin2) were also used to determine multivariate associations between forage metadata and ASVs based on CLR normalized data. For MaAslin2 analyses, ASVs significantly associated with adonis selected variables were limited to those for which BH corrected p-values were lower than 0.25. CCpnA analysis of each forage type was performed, as previously described (Ingham et al., 2019) on a corresponding dataset reduced to all ASVs resulting from both sPLS and MaAsLin2, as well as all variables selected based on adonis test. In addition to the CCnpA triplot result that illustrates how microbiome patterns are related to farm and forage metadata, prevalence, abundance, and distribution of all selected ASVs among each forage type were visualized in a heatmap constructed using the ComplexHeatmap package.
Microbial communities in uninoculated and inoculated forage (grass/legume and corn silage) were further characterized by investigating co-occurrence patterns using network analyses. The network inference was performed by constructing the phylogenetic molecular ecological networks (pMENs) using the online molecular ecological network analysis pipeline (MENAP, http://ieg4.rccc.ou.edu/mena) as previously described (Zhou et al., 2011; Deng et al., 2012). For pMEN construction, only ASVs with at least 40% prevalence within a silage group were considered. The similarity matrix that measures the degree of concordance between the abundance profiles of ASVs across samples was obtained based on Pearson correlation coefficients calculated from CLR-transformed data. Depending on the structure of the dataset corresponding to each silage group, the random matrix theory–based approach (Luo et al., 2006, Luo et al., 2007) was used to automatically determine the appropriate threshold value for the network structure. The fast greedy modularity optimization procedure (Clauset et al., 2004) was used to detect network modules defined as a group of highly interconnected nodes that share few or no connections with other nodes outside the group. In the built network, each node corresponds to a single ASV. The Maslov–Sneppen approach (Maslov and Sneppen, 2002) was used to generate 100 randomly rewired networks for each pMEN obtained. Global network and individual node properties were calculated based on similarity matrices. Node topological roles for a given network were determined based on within-module connectivity (Zi) and among-module connectivity (Pi) indices and visualized with a scatterplot constructed using the ggplot2 (version 3.3.0) R package (Wickham, 2009). Accordingly, nodes were assigned four different topological roles including module hubs, network hubs, peripherals, and connectors, as previously defined (Olesen et al., 2007). Peripheral ASVs, also considered as specialists, have both low Zi and low Pi indices (Zi ≤ 2.5 and Pi ≤ 0.62) and are characterized by few links, almost always with other ASVs within their module. ASVs assigned the connector role have low Zi and high Pi indices (Zi ≤ 2.5 and Pi > 0.62) and are highly connected to several modules. Module hubs have high Zi and low Pi indices (Zi > 2.5 and Pi ≤ 0.62) and are highly connected to several ASVs within their module. Connectors and module hubs are both considered generalist species. Network hubs, also defined as super-generalists, have high Zi and high Pi indices (Zi > 2.5 and Pi > 0.62), thus playing both connector and module hub roles. Habitat generalists refer to species, in this study, ASVs, which are largely distributed across samples within a group, thus having high prevalence, while habitat specialists are restricted to few samples in the group they belong to, thus having low prevalence, but occur at high relative abundance (Barberán et al., 2012). Module hubs, network hubs, and connectors that have high values of either Zi or Pi, or both high connectivity indices, are generally considered keystone species.
In addition to functional analyses, phenotypic traits of forage microbiome were predicted and compared using the BugBase tool (Ward et al., 2017). BugBase’s algorithm relies on software such as PICRUSt (Langille et al., 2013), IMG (Chen et al., 2019), KEGG (Kanehisa et al., 2012), and PATRIC (Snyder et al., 2007) to predict phenotypes and corresponding microbial contributors at the phylum level. Phenotypic traits including biofilm formation, Gram staining, oxygen tolerance, pathogenic potential, mobile element content, and oxidative stress tolerance were then predicted from CLR-transformed data. Prior to BugBase analyses, chimera-free sequences derived from the DADA2 pipeline were mapped to the Greengenes 97% reference database for format compatibility requirements.
Results
Bacterial diversity in forage types
A total of 81 forage samples were collected from 24 dairy farms over two sampling periods (fall and spring). During both periods, H samples exhibited significantly higher Chao1, Shannon, and inverse Simpson indices (p < 0.01) than other forage types which did not significantly differ from each other (Figures 1A,B). However, inconsistent diversity trends were observed between uninoculated and inoculated silage across sampling periods.
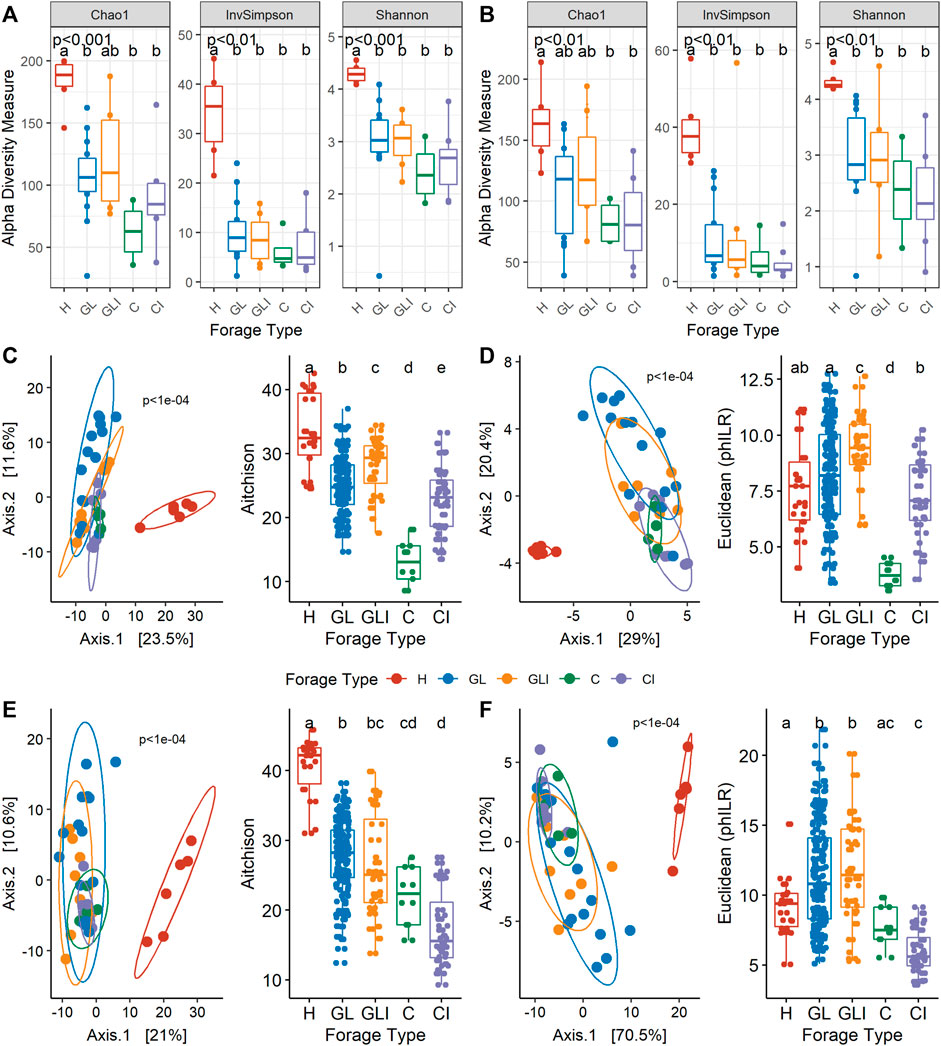
FIGURE 1. Microbial diversity according to the forage type. Alpha-diversity measures of forage types in fall (A) and spring (B). Principal component analysis (left) on CLR (C) or PhILR (D) transformed data with corresponding post hoc tests (right) for fall. Principal component analysis (left) on CLR (E) or PhILR (F) transformed data with corresponding post hoc tests (right) for spring. p-values indicate the significance of differences between groups from the Kruskal–Wallis test. Different lower case letters represent statistically significant differences between forage types.
Forage collected in the fall season (Figures 1C,D) exhibited highly dissimilar community structures between types (p < 1e-04). While there was a clear separation between H and silage, the taxonomic compositions of GL and C were significantly different from those of their inoculated counterparts. Principal coordinate analysis (PCoA) based on PhILR-transformed data revealed significant differences between uninoculated silage and inoculated counterparts (Figure 1D), indicating that taxa occurring in the compared habitats were not phylogenetically related. In contrast, silage microbial communities from the spring season were compositionally similar (Figure 1E) and phylogenetically related (Figure 1F), while being almost all significantly different from H.
Taxonomic profiles were assessed at the genus level, concomitantly with sample LCBD indices. The H samples collected across both periods (Figures 2A,B) showed high LCBD indices. The genera Pantoea, Sphingomonas, Curtobacterium, Methylobacterium, and Pseudomonas were variably dominant across both sampling periods. Among GL samples collected in fall, four showed highly distinctive microbial profiles, with communities variably co-dominated by Bacillus and Saccharopolyspora, Weissella and Pediococcus, or Pediococcus, Methylobacterium, and Enterococcus. Most of the other GL samples were dominated by Lactobacillus, Weissella, Pediococcus, or Lactococcus. In spring, GL samples with high LCBD indices showed dominance of Serratia, Pseudomonas, and an unclassified Enterobacteriaceae or Pediococcus and Weissella (Figure 2B). Other samples were generally dominated by Lactobacillus, Weissella, and Enterococcus. For GLI samples collected in fall, the most distinctive ones showed dominance of either Bacillus or Pediococcus or co-dominance of Lactobacillus, Corynebacterium, and Staphylococcus, compared with a few other samples showing Lactobacillus as the sole dominant genus (Figure 2A).
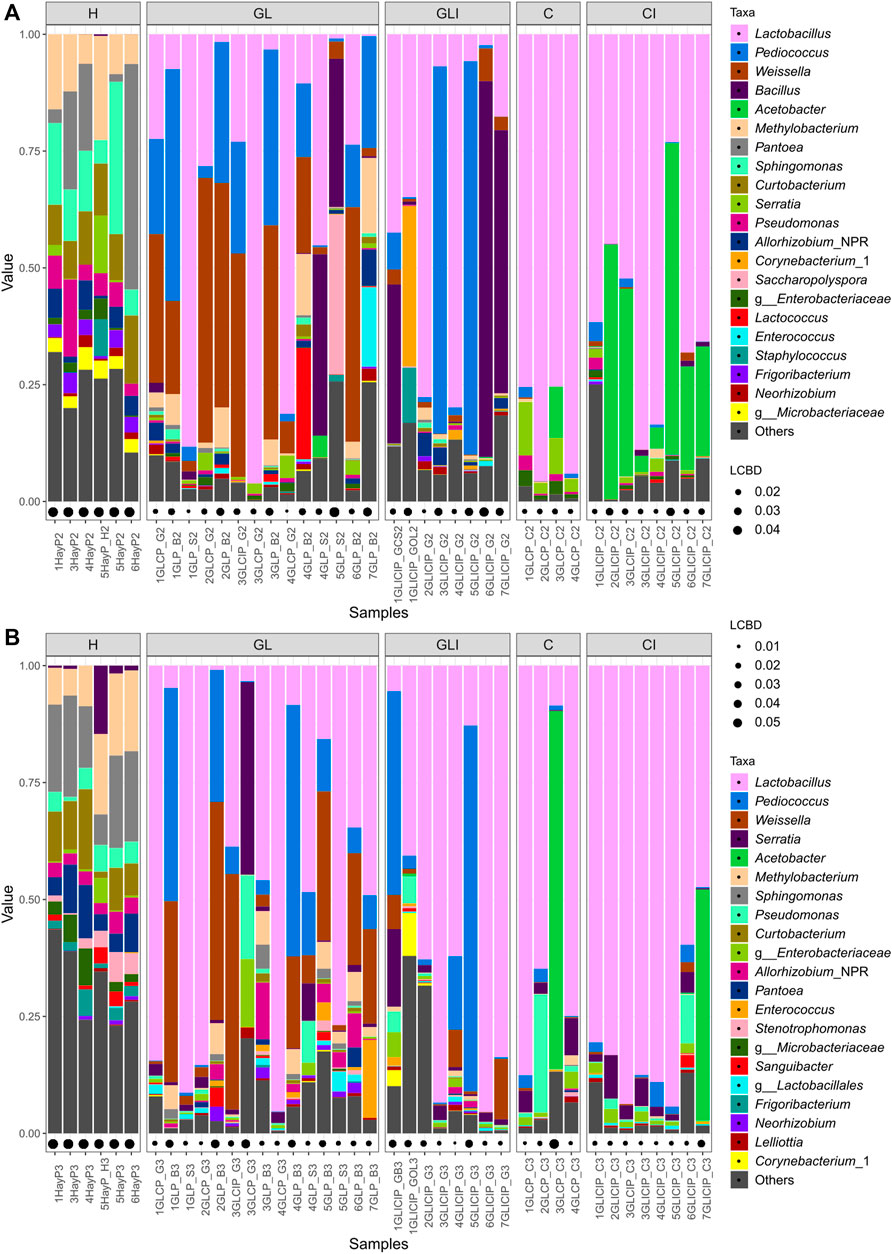
FIGURE 2. Forage microbial profiles across sampling periods. Relative abundance (value) of the top 21 most abundant genera within forage samples collected in fall (A) and spring (B). LCBD indices represent the sample local contribution to the beta-diversity measures between groups. The higher the index, the more unique is the sample microbial profile and the higher its contribution to the beta-diversity measures between groups.
In spring samples, Lactobacillus was the most abundant genus, followed by Pediococcus. Samples with high LCBD indices were dominated by Pediococcus, either alone or in co-dominance with Serratia and Weissella (Figure 2B). C samples collected in fall were the most homogenous with Lactobacillus as the main dominant genus, although Serratia often occurred with considerable relative abundance (Figure 2A). Among C samples collected in spring, one exhibited high abundance of Acetobacter, other samples showing large proportions of Lactobacillus or Pseudomonas as dominant or subdominant genera, respectively (Figure 2B). Finally, CI samples collected during fall were broadly dominated by Lactobacillus, although some with higher LCBD indices exhibited large proportions of Acetobacter as either dominant or co-dominant (Figure 2A). For CI samples collected in spring, Lactobacillus was almost the sole dominant genus, except in one sample showing Acetobacter as co-dominant (Figure 2B).
A differential abundance analysis performed to identify significantly enriched taxa between uninoculated and inoculated silage revealed that in fall, phylotypes (ASV) of P. pentosaceus and Weissella were significantly more abundant in GL than in GLI samples, except for two GLI samples inoculated with Biotal Buchneri 500 (Figure 3A). In spring, Lactobacillus and some Proteobacteria were significantly more abundant in GLI samples (Figure 3B). For corn silage, Loigolactobacillus coryniformis and Lactobacillus phylotypes exhibiting high prevalence (83%–90%) were significantly more abundant in C than in CI samples in fall (Figure 3C). Surprisingly, in spring, while some highly prevalent (83%–100%) phylotypes of Proteobacteria were significantly more abundant in CI samples, phylotypes of Lactobacillus and Proteobacteria were significantly more abundant in C samples (Figure 3D).
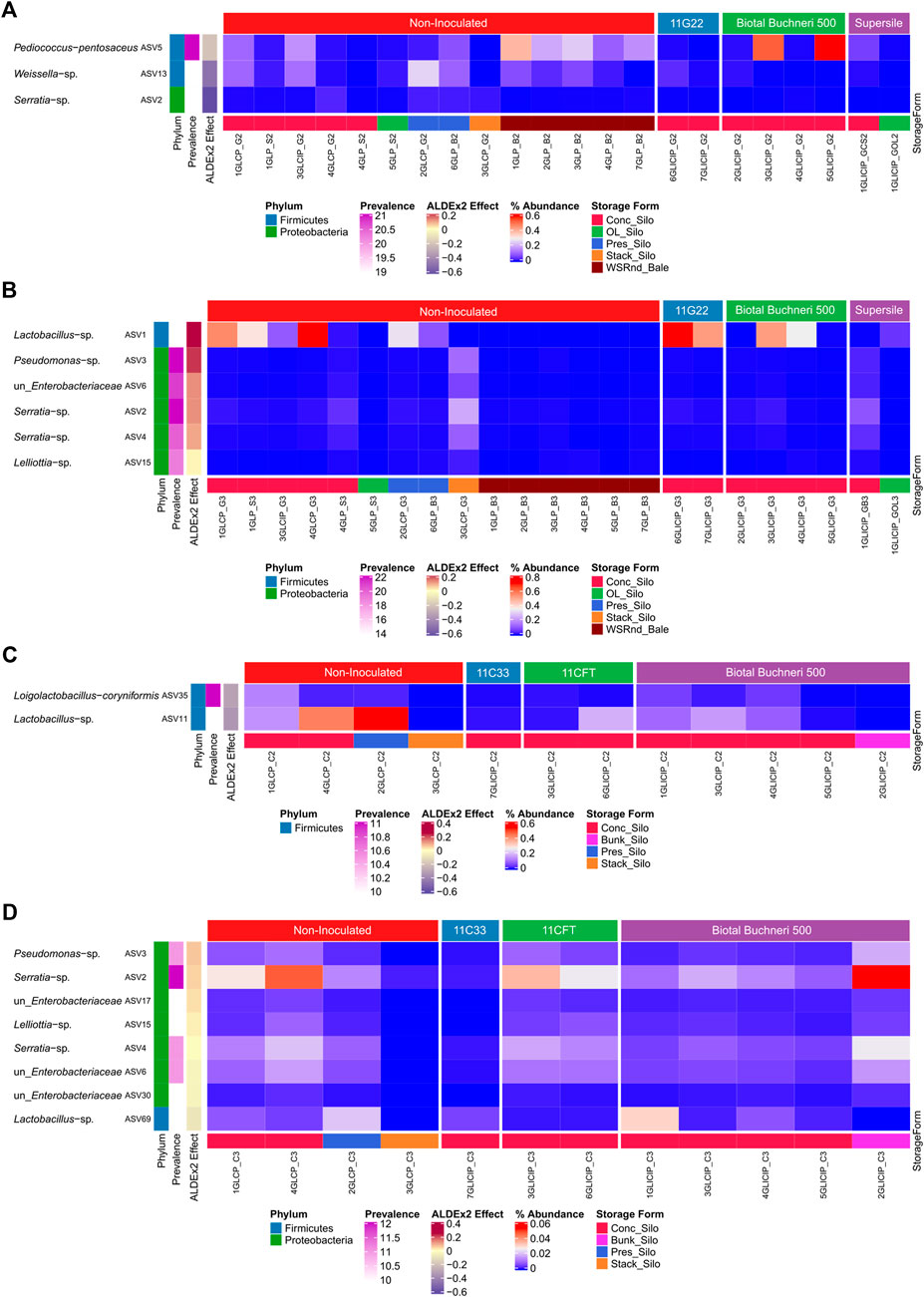
FIGURE 3. Distribution of differentially abundant ASV between uninoculated and inoculated silage. Relative abundance of ASV significantly enriched between uninoculated and inoculated grass/legume silage in fall (A) and spring (B). Relative abundance of ASV significantly enriched between uninoculated and inoculated corn silage in fall (C) and spring (D).
LAB including L. buchneri and L. plantarum were significantly enriched (p < 0.001) in ensiled forage compared with H samples (Figures 4A,B). However, inconsistent enrichment of LAB was noted among silage across sampling periods. Ensiling significantly reduced AAB loads compared with H (p < 0.05), but differences between inoculated silage and uninoculated counterparts were inconsistent across both periods. Ensiling significantly reduced Pseudomonas levels compared with H (p < 0.05) and loads of this group tended to increase with inoculation, as did Enterobacteriaceae levels. While total bacteria load tended to increase with inoculation between silage across both sampling periods, contrasting patterns of significant variations (p < 0.05) of fungi loads were noted (Figures 4A,B). We found similar patterns of bacterial load variations between forage types within PMA-treated (Figures 4A,B) and PMA-free (data not shown) samples, although the latter group broadly exhibited higher load levels.
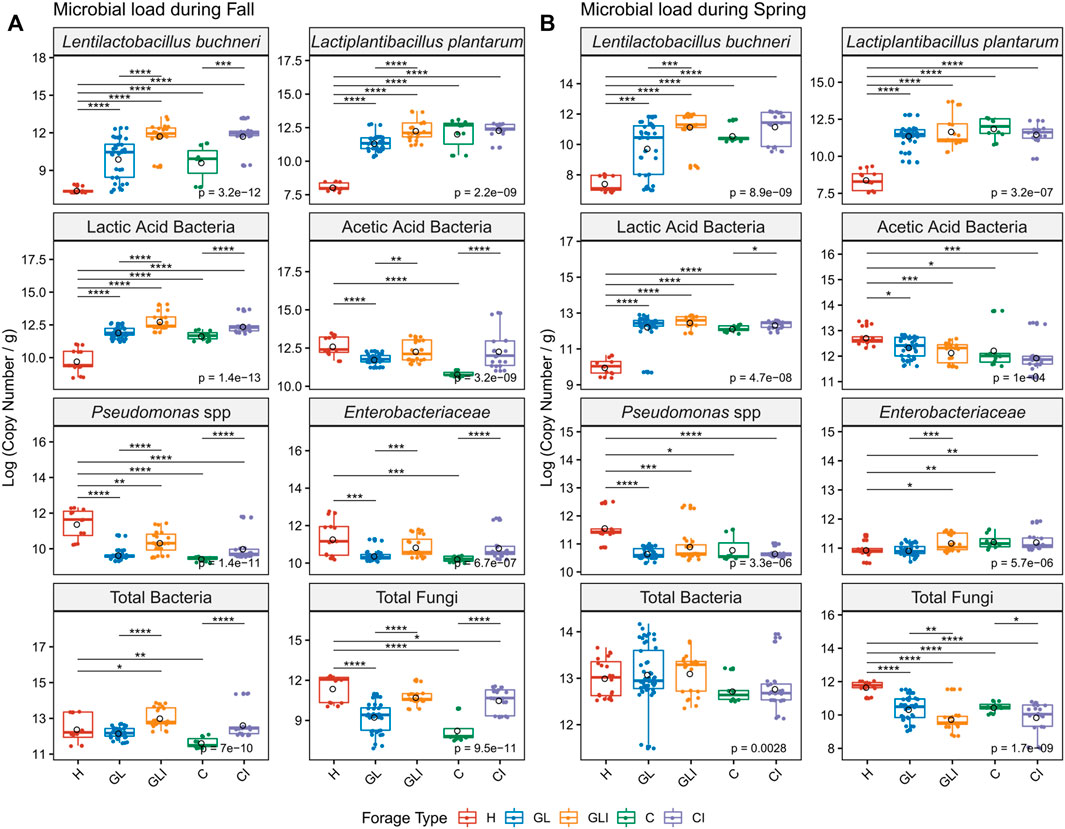
FIGURE 4. Quantification of viable microbial groups across sampling periods. Copy numbers are compared between H and each silage type and between uninoculated and inoculated silage in fall (A) and spring (B) using the Kruskal–Wallis test. Asterisks above boxes indicate significant differences and flag p-values from a Wilcoxon rank test as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
Abundance profile of the core phylotypes from the order Lactobacillales in uninoculated and inoculated silage
In addition to differential abundance testing, specific and shared LAB phylotypes were identified within ensiled forage to understand how they differentially occur between uninoculated and inoculated silage. The analysis was limited to the order Lactobacillales as it is the lowest taxonomic classification level gathering lactobacilli, pediococci, and enterococci. Broadly, of the 2,980 unique ASVs composing the whole PMA-treated dataset, about 30.67% (914) were assigned to the order Lactobacillales. From the 913 Lactobacillales occurring in silage, 836 were found in grass/legume silage, of which 356 were specific to GL, 223 to GLI, and 257 were shared between both, while from the 241 unique ASVs that occurred in corn silage, 33 were specific to C, 123 to CI, and 85 were shared between both (data not shown).
The dominating core LAB among grass/legume silage (i.e., LAB occurring both in GL and GLI) totaled up to 96% of the LAB communities and included lactobacilli represented by 138 phylotypes, P. pentosaceus represented by two phylotypes, Weissella parasenteroides counting two phylotypes, and other Weissella and Pediococcus spp. represented by 16 and 46 phylotypes, respectively (Figure 5A). For corn silage, the core LAB phylotypes in both silage types dominated the LAB communities with up to 95% relative abundance (Figure 5B), except for samples where LAB were not the sole dominant taxa (Figures 2A,B). Within corn silage, the dominant LAB populations were composed of lactobacilli represented by 67 phylotypes, L. coryniformis represented by three phylotypes, and, to a certain extent, Latilactobacillus sakei and Paucilactobacillus hokkaidonensis each counting one phylotype (Figure 5B).
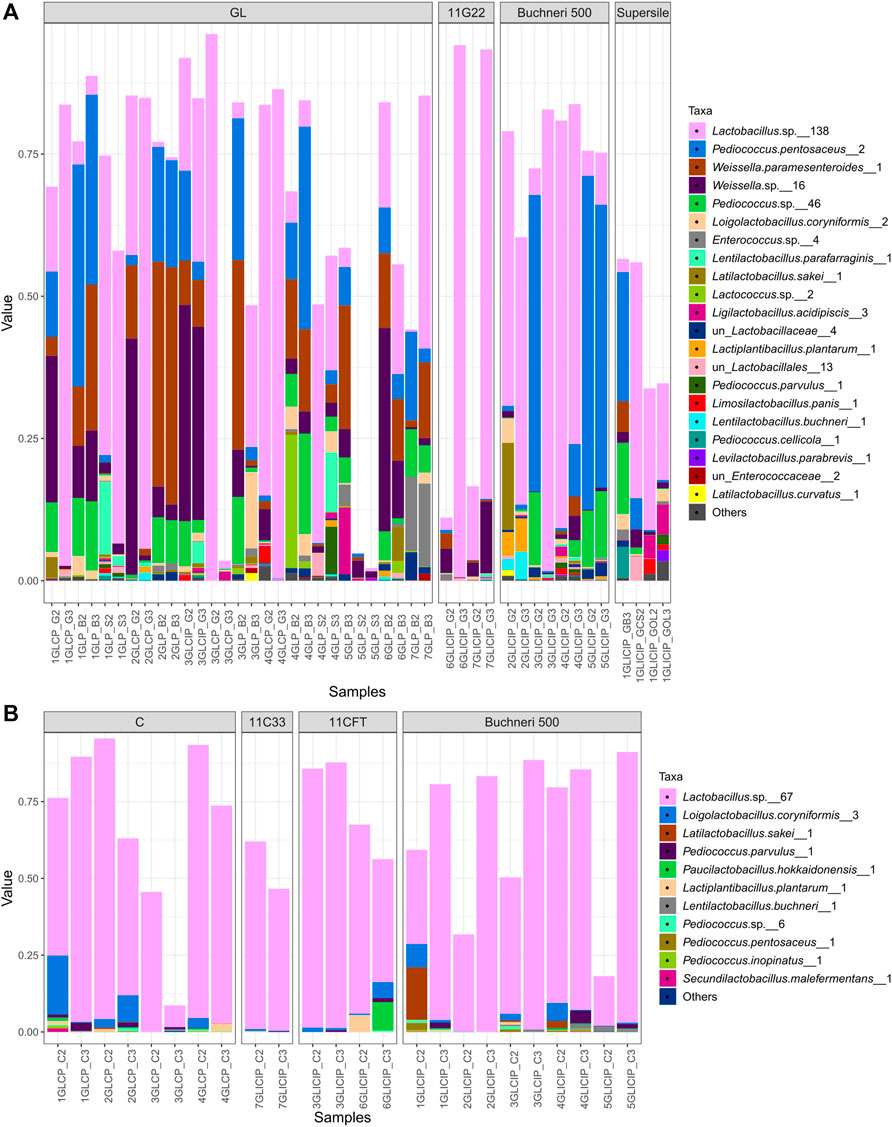
FIGURE 5. Core LAB community profiling. Relative abundance (value) of the dominating core ASV between uninoculated and inoculated grass/legume (A) or corn (B) silage. Numbers following taxonomic names represent corresponding ASV numbers.
Prediction of phenotypic traits and function pathways
The analysis of microbial phenotypic traits using BugBase showed that anaerobic metabolism, presence of mobile elements, biofilm-forming capacity, Gram-negative, Gram-positive, potentially pathogenic, and stress-tolerant phenotypes were differentially distributed among forage types (Figures 6A,B). Across both sampling periods, biofilm-forming capacity and Gram-negative phenotypes were significantly enriched in H compared with silage (GL, GLI, C, and CI). This could be attributable to higher relative abundances of Actinobacteria and Proteobacteria in H samples for biofilm-forming capacity and Gram-negative phenotypes, respectively (Supplementary Figures S1A,B). A significant enrichment of Gram-positive phenotypes was observed in silage compared with H across both sampling periods, a difference attributable to the higher abundance of Firmicutes in silage (Supplementary Figures S1A,B). Analyzing the predicted function pathways, we found that biofilm-forming associated features were significantly enriched in uninoculated grass/legume silage (Supplementary Figures S2,3), while other features associated with the metabolism of macromolecules (Supplementary Material S1) were significantly enriched in the inoculated counterparts (Supplementary Figures S4A,B).
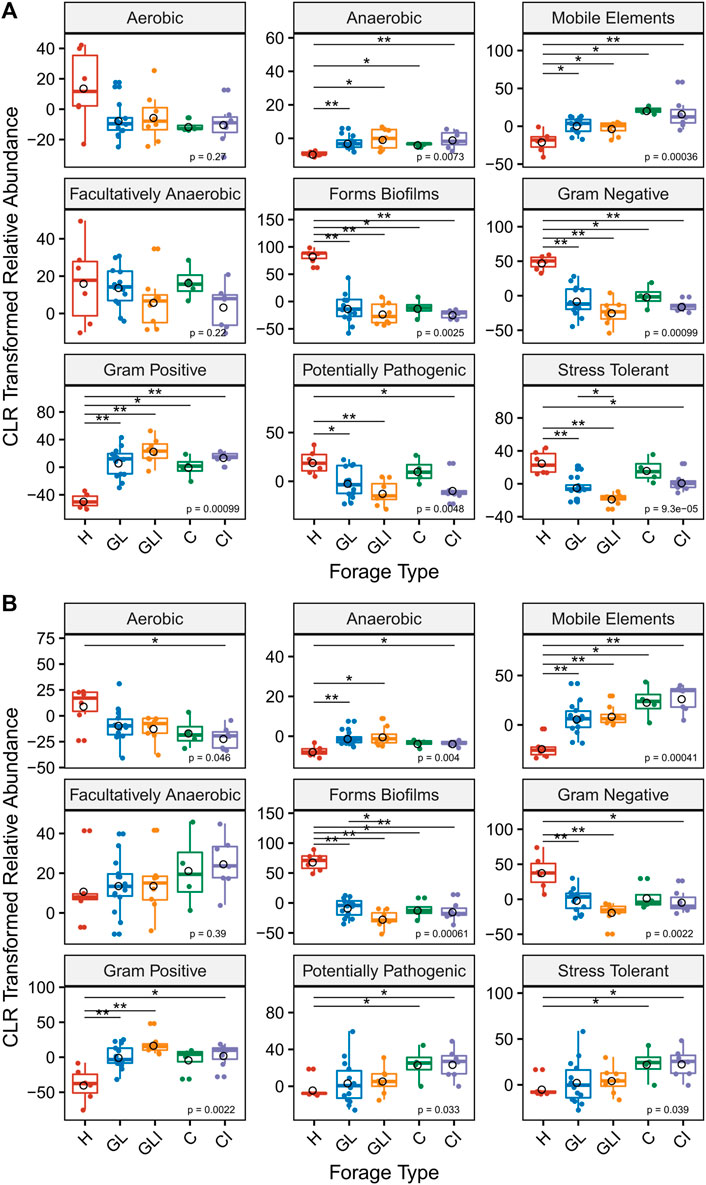
FIGURE 6. Microbial phenotypic composition predicted by BugBase. Proportions of microbial phenotypic traits compared between H and each silage type and between uninoculated and inoculated silage in fall (A) and spring (B) using the Kruskal–Wallis test. Asterisks above boxes indicate significant differences and flag p-values from a Wilcoxon rank test as follows: *, p < 0.05; and **, p < 0.01.
Forage physicochemical characteristics associated with microbial communities
Near infrared spectroscopy analysis of forage showed that the physicochemical traits of H samples were broadly consistent across the sampling periods (Supplementary Table S1). Comparing uninoculated silage with inoculated counterparts, ammonia content was significantly (p < 0.05) higher in GLI and CI than in GL and C silage in spring, respectively, as were acetic acid in GLI in spring and butyric acid in CI in fall compared with GL and C silage, respectively (Supplementary Tables S2, S3). The dry matter content of grass/legume silage ranged between 29% and 65% for GL samples and between 32% and 57% for GLI. pH values ranged between 3.7 and 5.3 for GL samples and between 3.8 and 4.7 for GLI. Butyric acid was detected in less than 38% of GL samples with amounts ranging between 0.42% and 1.1% and in more than 62% of GLI samples with amounts ranging from 0.42% to 0.99% dry matter. In contrast, butyric acid was detected in only 25% of C samples with amounts of butyric acid ranging between 1.09% and 1.96% dry matter and none among CI samples. However, corn silage exhibited lower dry matter content, ranging between 20% and 41%, as well as lower pH values ranging from 3.5 to 3.9.
To gain insight into how forage end products and characteristics associate with microbial communities, categorical and quantitative metadata related to farms and forage samples were selectively integrated in multivariate multi-table analyses based on sPLS and CCpnA. General linear models implemented in MaAsLin2 were also used to determine multivariate associations between forage metadata and ASVs using CLR normalized data. This analysis scheme was implemented for all forage types separately. H microbial communities were grouped into two clusters (Figure 7A; Supplementary Figure S5A). Moisture content and positively correlated phylotypes of Serratia, Enterobacteriaceae, Nocardioides, and Yersinia, as well as those negatively correlated, including Spirosoma, Intrasporangiaceae, and Methylobacterium, were the main contributors to the formation of cluster 1 (Supplementary Figures S5B,C, S6). Magnesium content and positively (Frigoribacterium, Curtobacterium, Pantoea, Rhizobiaceae, Microbacteriaceae, Allorhizobium group, and Pantoea agglomerans) or negatively (Pseudomonas) correlated phylotypes contributed the most to the formation of cluster 2 (Supplementary Figures S5B,C, S6). Among all the taxa found to associate with the selected H variables, phylotypes of Methylobacterium (cluster 1) or Curtobacterium, Pantoea, and Serratia (cluster 2) were the most prevalent and abundant (Supplementary Material S1).
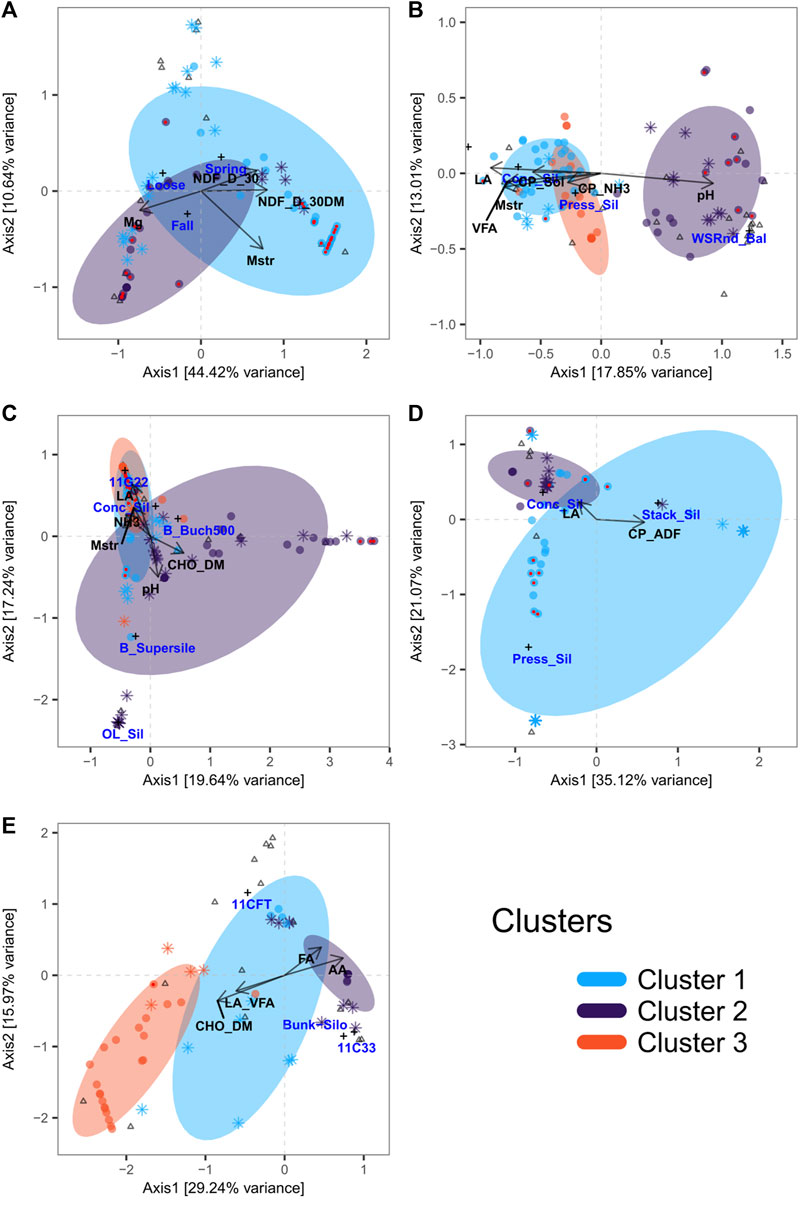
FIGURE 7. Canonical correspondence analysis. Triplots illustrating canonical relationships between bacterial ASV (round and star-shaped points) and physicochemical parameters (arrows), inoculants ( + ), forage storage form ( + ), or sampling periods ( + ) for H (A), GL (B), GLI (C), C (D), and CI (E). Sampling periods include fall 2015 (Fall) and spring 2016 (Spring). Storage forms include loose hay (Loose), wrapped/square round bales (WSRnd_Bal), concrete silo (Conc_Sil), pressed silo (Press_Sil), stack silo (Stack_Sil), and oxygen-limiting silo (OL_Sil). Inoculants include Biotal Buchneri 500 (B_Buch500), Biotal Supersile (B_supersile), 11G22, 11C33, and 11CFT. Forage physicochemical parameters include amino acids (AA), ethanol soluble carbohydrate as % dry matter (CHO_DM), acid detergent fiber as % of crude protein (CP_ADF), ammonia as % crude protein (CP_NH3), soluble crude protein (CP_Sol), fatty acids (FA), lactic acid (LA), LA as % volatile fatty acids (LA_VFA), magnesium (Mg), moisture (Mstr), neutral detergent fiber digestibility at 30 h (NDF_D_30), NDF_D_30 as % dry matter (NDF_D_30DM), ammonia (NH3), pH (pH), and volatile fatty acids (VFA). Round points depict taxa selected by sPLS and star-shaped points those selected by MaAsLin approaches. Round points with red centers are taxa selected using both methods. Triangles represent samples.
Selected taxa of the GL microbiota formed three clusters (Figure 7A; Supplementary Figure S7A). Cluster one was mainly driven by the variables lactic acid (LA), moisture, fatty acid (FA), crude fat (CF), and volatile fatty acid (VFA) and the positively associated taxa including phylotypes of Lactobacillus, Weissella, and Carnobacterium (Figure 7B; Supplementary Figures S7B,C, S8). For cluster 2, the pH and positively correlated taxa including phylotypes of Methylobacterium, Sphingomonas, Curtobacterium, Allorhizobium group, Methylobacterium adhaesivum, Lactobacillus, and Pediococcus were the main contributors. The two variables ammonia expressed as percentage of crude proteins (CP_NH3) and soluble crude proteins (CP_Sol) and positively associated taxa including phylotypes of Weissella, Aeriscardovia, Lactobacillus, Corynebacterium, Pediococcus, Lactobacillus kefiranofaciens, Serratia, Brevibacterium, and Brachybacterium were the main contributors to cluster 3 (Figure 7B; Supplementary Figures S7B,C, S8). Phylotypes of Lactobacillus, Pediococcus, and Weissella paramesenteroides were the most abundant and prevalent of all the GL selected taxa (Supplementary Material S1).
For GLI, ASVs differentially associated with silage parameters also formed three clusters as for GL (Figure 7C; Supplementary Figure S9A). The phylotypes of Lactobacillus, Lentilactobacillus parafarraginis, Weissella, Ligilactobacillus acidipiscis, and Pediococcus that are positively associated with the variables CP_NH3, ammonia (NH3), ammonia expressed as percentage of soluble protein (SP_NH3), and acetic acid (AA) all constituted the main contributors to cluster 1 (Figure 7C; Supplementary Figures S9B,C, S10). The variables ethanol soluble carbohydrates as percentage of dry matter (ESC_DM) and ESC as percentage of non-fiber carbohydrate (ESC_NFC) that are positively correlated with phylotypes of Methylobacterium, Pediococcus, Lactobacillus, Luteimonas aestuarii, Sphingomonas phyllosphaerae, Neorhizobium, and Rhodococcus all contributed the most to cluster 2. Cluster 3 was mainly driven by the variables LA, LA expressed as percentage of VFA (LA_VFA), and CP_Sol, together with the positively associated phylotypes of Lactobacillus, Clostridiaceae, Enterobacteriaceae, Lactobacillales, Frigoribacterium faeni, Microbacteriaceae, and L. plantarum (Figure 7C; Supplementary Figures S9B,C, S10). The most prevalent of the selected ASVs included phylotypes of Lelliotia, Serratia, L. acidipiscis, and Weissella (Supplementary Material S1).
In the case of C, we obtained two clusters (Figure 7D; Supplementary Figure S11A) driven by both LA and acid detergent fiber-crude protein (CP_ADF). Phylotypes of Lactobacillus, Acetobacter, and Serratia positively correlated with LA, and those negatively correlated, including Pediococcus and P. hokkaidonensis, mostly contributed to the formation of cluster 1. Taxa mostly contributing to the formation of cluster two included phylotypes of Lactobacillus, among which are L. sakei, all positively associated with CP_ADF (Figure 7D; Supplementary Figures S11B,C, S12). Phylotypes of Lactobacillus and Serratia were among the more prevalent and abundant (Supplementary Material S1).
Finally, for CI, the selected ASVs formed three clusters (Figure 7E; Supplementary Figure S13A). The parameters AA and FA mainly contributed to clusters 1 and 2. While a few phylotypes of Lactobacillus positively correlated with AA and FA mainly drove cluster 1, those of Comamonas jiangduensis, Ameyamaea, and Acinetobacter gerneri were the main contributors to cluster 2 (Figure 7E; Supplementary Figures S13B,C, S14). For cluster 3, the parameters carbohydrates (CHO_DM and CHO_NFC), CP_DM, and LA_VFA and positively associated phylotypes of Lelliottia, Enterobacter, Raoultella terrigena, Enterobacteriaceae, and Vagococcus fluvialis were the main drivers (Figure 7E; Supplementary Figures S13B,C, S14). Among selected taxa, phylotypes of Lactobacillus, Acetobacter, and Serratia were the most abundant and prevalent.
Molecular ecological network analyses
To investigate how silage bacteria co-occur in the presence or absence of inoculants, a network was constructed for each silage type. The analysis of topological properties revealed that the GLI network had a higher average degree (avgK) and a lower average geodesic path (distance between nodes) than those of GL, thus appearing more complex and denser (Table 2). The 80 nodes composing the GL network totaled eight modules (group of ASVs sharing more links among themselves than with others outside the group) and 405 links, of which only 18.8% were positive (Supplementary Figure S15). Some species of Lactobacillales including phylotypes of Pediococcus, Enterococcus, Weissella, L. sakei, L. coryniformis, and unidentified Lactobacillaceae co-occurred with those of Proteobacteria including Methylobacterium, Pantoea, Sphingomonas, Stenetrophomonas, and Allorhizobium or Actinobacteria comprising Rhodococcus and Curtobacterium (modules 1, 3, and 6). Module 5 exclusively composed of Lactobacillaceae exhibited co-occurrence between phylotypes of L. buchneri and those of lactobacilli (Supplementary Figure S15). On the other hand, the GLI network was composed of 47 nodes grouped into two modules and 848 links from which only 7.2% were positive (Table 2). In contrast to that of GL, the GLI network involved higher amounts of positive relationships among Firmicutes (Supplementary Figure S16). Curiously, phylotypes corresponding to L. buchneri and L. plantarum were found in distinct modules, sharing no relationships, although both co-occurred with the same phylotype of Lactobacillus. In addition, fewer phylotypes of Proteobacteria shared positive interactions with Firmicutes. Another particularity of the GLI network is the co-occurrence between some phylotypes of Weissella and those of Bacillus (Supplementary Material S1). Analyzing taxa topological roles, we identified 47 keystone phylotypes distributed as network hubs and connectors in the GL pMEN, of which 64% were Firmicutes, 34% Proteobacteria, and the remaining Actinobacteria (Supplementary Table S4). Among network hubs were phylotypes of Weissella and Sphingomonas, while connectors mostly included Firmicutes such as L. plantarum, L. buchneri, Bacillus, Weissella, Lactococcus, and other lactobacilli phylotypes (Supplementary Material S1). Conversely, no keystone species was observed in the GLI network. However, compared with the GL network, there was a shift of topological roles in GLI so that nodes formed a cluster around the value of 0.5 along the Pi axis (Figure 8). Consequently, the topological roles of all keystone phylotypes of the GL network also found in GLI were changed to peripherals (Figure 8).
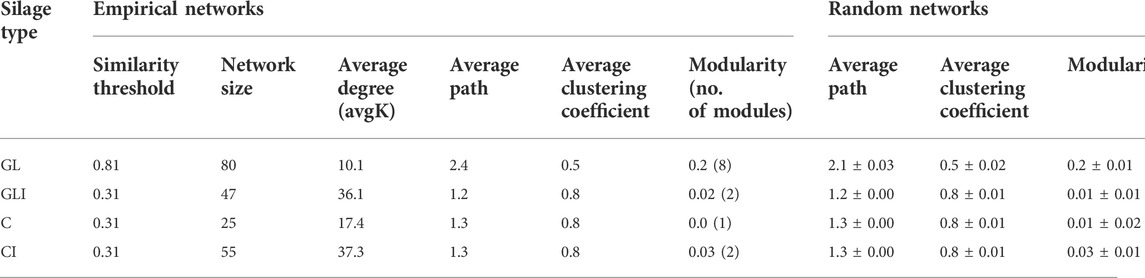
TABLE 2. Topological properties of the empirical pMENs in grass/legume and corn silage microbial communities and their associated random pMENs.
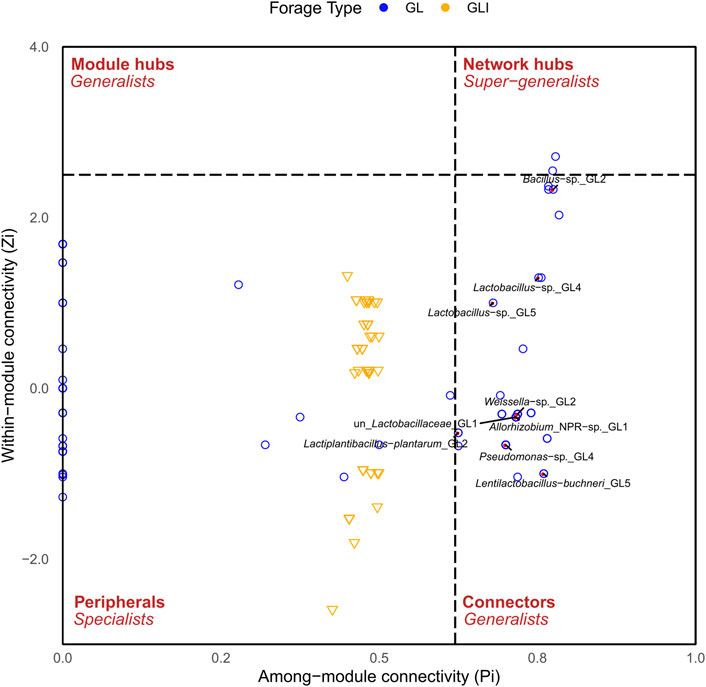
FIGURE 8. Distribution of network topological roles of grass/legume silage. Labeled ASVs (red points) depict those that exhibited different topological roles in GL compared to GLI.
For corn silage, microbial communities formed a larger and more complex network in CI than in C (Table 2). The 25 nodes composing the C network formed a single module, involving 217 links of which 22% were positive (Supplementary Figure S17A). All Proteobacteria phylotypes (Serratia, Pseudomonas, and unclassified Enterobacteriaceae) involved in this network positively interacted with each other and co-occurred with Firmicutes including L. coryniformis, and two Lactobacillus phylotypes. Neither L. buchneri nor L. plantarum were involved in this network. The CI network totaled 55 nodes comprising two modules and 1,026 links of which 9.8% were positive (Supplementary Figure S17B). Unlike in the GLI network, L. buchneri and L. plantarum found in different modules (modules 1 and 2, respectively) co-occurred and were both involved in positive relationships with Pediococcus parvulus and some species of lactobacilli. However, most phylotypes of Proteobacteria including Serratia, Pseudomonas, Acetobacter, and Yersinia variably co-occurred among themselves and with some species of lactobacilli (Supplementary Material S1). No keystone phylotypes were found in the C and CI networks.
Discussion
In this study, we implemented a viability high-throughput sequencing approach combined with viability-PCR (Nocker et al., 2010; Kennang Ouamba et al., 2020) to provide a comprehensive and comparative analysis of the viable microbial ecology of hay and grass/legume or corn silage produced with or without inoculants at commercial farm-scale facilities.
Recently, Daniels et al. (2020) have analyzed the viable microbiota of commercial Meadow and Italian ryegrass hay and revealed Proteobacteria, Cyanobacteria, Bacteroidetes, and Actinobacteria as the predominant phyla. In this study, Cyanobacteria were detected as the rarest taxa and were discarded from the dataset upon abundance filtering, while the phyla Proteobacteria, Actinobacteria, and Bacteroidetes were the most prevalent and abundant in hay. However, the differences found among hay community profiles could be attributable to fluctuations in abundance of predominant genera, including Sphingomonas, Methylobacterium, Pantoea, Curtobacterium, and Pseudomonas. Behrendt et al. (1997) analyzed the microbial community of the grass phyllosphere using a culture-dependent method and identified Pseudomonas, Stenotrophomonas, Pantoea, Clavibacter, and Curtobacterium as the predominant genera. Although not among the most abundant, Stenotrophomonas and Clavibacter were also detected in our study, thus indicating that the microbiota of hay might consistently reflect the epiphytic communities of plants at harvest. Moreover, except Stenotrophomonas, we found that specific phylotypes of the above-mentioned genera correlated with hay moisture content, suggesting bacterial growth in less dried hay. The observed differences between H samples could therefore be also explained by variation in environmental and farm management factors including, but not limited to, plant species, management practices, geographical location, climatic conditions, moisture concentration at harvest, and storage form (Bernardes et al., 2018), which drive the incidence and abundance of epiphytic microorganisms on plants before harvest or during processing. Although LAB were not among the dominant taxa, their prevalence and abundance were found to vary between H samples and across sampling periods as revealed by a concomitant study on the same forage samples, where Gagnon et al. (2020) identified LAB communities through culture-based techniques. W. paramesenteroides (or Weissella thailandensis), P. pentosaceus, L. casei (or L. paracasei), L. mesenteroides (or L. pseudomesenteroides), L. pentosus, and Enterococcus casseliflavus (or Enterococcus gallinarum and E. faecium) were found as the predominant cultivable LAB in hay. In this study, the detection of Weissella, Lactococcus, Enterococcus, and unidentified Lactobacillales as the sole representatives of LAB in hay might be attributed to the other LAB being under the threshold of detection. For survey studies, this emphasizes the relevance of combining both culture-dependent and high-throughput sequencing approaches to deepen our understanding of microbial community composition and function. However, none of the LAB was associated with the hay moisture content. Despite the observed differences among hay samples, their microbial composition and structure clearly discriminated them from those of silage across both sampling periods, revealing haymaking and ensiling as strikingly distinct processes that differentially alter the epiphytic microbiota of fresh forage plants.
Grass/legume forage has higher buffering capacity and lower WSC content than corn forage, offering different habitats for microorganisms (Dunière et al., 2013). These factors differentially modulate the growth of microorganisms depending on their initial abundance on the pre-ensiled forage, thus impacting the rate of pH decrease during the first stages of fermentation (Weinberg and Muck, 1996; Dunière et al., 2013). This could explain the differences in phylogenetic composition and community structure between grass/legumes and corn silage observed in our study. We also found that most GL silage was generally dominated by Lactobacillus alone or in co-dominance with either Weissella or Pediococcus or both, while few samples inconsistently exhibited co-dominance of Bacillus, Saccharopolyspora, Lactococcus, Serratia, Methylobacterium, or Enterococcus across both sampling periods. Previous studies have shown that LAB, preferably lactobacilli, are the main microorganisms expected to dominate the microbiota of good-quality silage (Ávila and Carvalho, 2020). High prevalence and relative abundance of Weissella, Pediococcus, Enterococcus, and Lactococcus were also found by Gagnon et al. (2020) using a culture-dependent approach. In addition to the high buffering capacity, this observation possibly illustrates grass/legume silage for which the fresh forage phyllosphere contained insufficient amounts of Lactobacillus to lead the first stages of the fermentation process (Stevenson et al., 2006; Yang et al., 2019). Such conditions might also favor the growth of undesirable bacteria such as Bacillus and Serratia (Ávila and Carvalho, 2020), as revealed in our study. Although several studies have reported the occurrence of Methylobacterium in silage (Ogunade et al., 2018b; Yang et al., 2020), none have described its role in the fermentation process. The genus Saccharopolyspora represented by three phylotypes of Saccharopolyspora rectivirgula (formerly Micropolyspora faeni), which has been associated with moist hay, compost, or straw (Duchaine et al., 1999; Ranalli et al., 1999), is a thermophilic Actinobacteria identified as a major cause of extrinsic allergic alveolitis (farmer’s lung disease) in dairy barns (Lecours et al., 2012; Schultz, 2016). The occurrence of this pathogen as co-dominant bacteria in GL silage and in lower abundance in GLI samples suggests its fermentative capability. Although this pathogen has already been identified in corn silage (Unaogu et al., 1994), no studies had reported its occurrence in grass/legume silage. In the case of C silage, the microbiota was dominated by either Lactobacillus or Acetobacter or both, while Pseudomonas and Serratia sporadically occurred with considerably high relative abundance. Our results corroborate those of Guan et al. (2018) who found Lactobacillus and Acetobacter as predominant bacteria in naturally fermented corn silage. These authors also highlighted the inconsistent incidence of Acetobacter between laboratory- and large-scale bunker silos. In the current study, two phylotypes of this genus were detected in C silage processed in stack silos. Interestingly, 15 phylotypes of Acetobacter, among which some had higher abundance levels, were identified in CI silage that were inoculated with Biotal Buchneri 500, 11CFT, or 11C33 and were stored in stack, bunker, or conventional silos (concrete-stave silos). These results indicate that species of Acetobacter might outcompete LAB even with the addition of inoculants, perhaps when oxygen infiltration into the forage might have favored their proliferation. Moreover, viable-PCR analyses confirmed inconsistent enrichment of microbial load including that of LAB, AAB, Pseudomonas, Enterobacteriaceae, total bacteria, and total fungi across sampling periods in inoculated silage. These observations suggest that prevailing weather conditions (colder temperatures) might impair the efficiency of the inoculant during ensiling and consequently favor undesirable AAB and acid-tolerant Proteobacteria in mature silage. Except for LAB, the effects of these microorganisms during ensiling are not clear (Dolci et al., 2011; Hu et al., 2018; Guan et al., 2020), and further research is needed to better understand their function and interplays with LAB during silage fermentation. Although other studies had reported a high prevalence of Pseudomonas and Serratia throughout the ensiling process of corn under controlled laboratory conditions (Hu et al., 2018; Keshri et al., 2018), like other Proteobacteria, their effects on the silage fermentation process are not well understood.
A successful ensiling using transplanted epiphytic microbiota as the sole source of microorganisms was recently reported (Ali et al., 2020; Yuan et al., 2020). In this study, uninoculated GL and C silage with good fermentation profiles were observed. However, conditions for successful natural fermentation are not always met and ensiling without additives may therefore most often be associated with an increased risk of economic losses (Ogunade et al., 2018a; Borreani et al., 2018). Consequently, microbial additives that are employed to dominate the communities, minimize the occurrence of undesirable microorganisms, and drive the fermentation process (Wang et al., 2006) are generally recommended (Borreani et al., 2018). However, while this is particularly true for grass or legume silage, microbial additives might not necessarily improve the quality of corn silage, as recently revealed by a meta-analysis (Oliveira et al., 2017; Bernardi et al., 2019). The current study showing dominant or co-dominant Acetobacter in CI silage particularly in fall corroborates results from the above-mentioned meta-analysis, though some C silage exhibited undesirable microbial profiles, mostly in spring.
Our study showed that in GL and GLI silage, specific phylotypes of Lactobacillus, Weissella, Lelliottia, Serratia, and an unidentified Enterobacteriaceae were positively associated with LA and moisture contents and negatively associated with pH. Although some Proteobacteria can produce lactic acid (Ávila and Carvalho, 2020), acetic acid is their primary product during ensiling (Pahlow et al., 2003). Therefore, the correlation of corresponding phylotypes with LA could result from their growth and subsequent acid production in the early stages of fermentation. The relatively low abundance of these phylotypes in mature silage could be due to the inhibitory effect of accumulating lactic acid and lower pH level during ensiling (Da Silva et al., 2017). We also found that phylotypes of P. pentosaceus, W. paramesenteroides, Lactobacillus, Pediococcus, Methylobacterium, and Lactococcus positively correlated with pH and were negatively associated with LA, indicating their sensitivity to lower pH levels or higher amounts of LA. Similar results were obtained by Ni et al. (2017) and Ogunade et al. (2018b). On the other hand, while phylotypes of Pediococcus were co-dominant in some GL samples, this genus largely dominated three GLI samples inoculated with Biotal Buchneri 500 or Biotal Supersile that include P. pentosaceus and P. acidilactici in their formulation, respectively. While the genus Pediococcus is known to dominate the microbiota in the early stages of fermentation, although inconsistently (Stevenson et al., 2006; Ogunade et al., 2018b; Drouin et al., 2019), the factors that favor its dominance in mature silage are not well understood (Nascimento Agarussi et al., 2019). In addition to Methylobacterium, other Proteobacteria, including Pantoea, Sphingomonas, and Stenotrophomonas, and Actinobacteria, including Rhodococcus and Curtobacterium, co-occurred with most of the LAB phylotypes within the GL, as have phylotypes of Pseudomonas and Acetobacter within the C communities. Higher abundance of these Proteobacteria was observed in samples wherein lactobacilli were not predominant. This suggests that the co-occurring LAB probably do not induce a rapid decrease in pH during ensiling as some lactobacilli strains more adapted for ensiling would have done. The decreased rates of co-occurrence between Firmicutes and Proteobacteria observed in inoculated GL and C communities supports this hypothesis. The observed phenomenon could be explained by dominating lactobacilli from inoculation; as more lactic acid is subsequently produced during ensiling, most Proteobacteria are inhibited, and consequently, increased patterns of co-exclusion appear to the detriment of co-occurrence.
However, the enhancement of positive interactions between LAB due to inoculants certainly prompt, in addition to related species, other acid-tolerant bacteria such as Bacillus in GLI, Acetobacter in CI, or Serratia in both. This might explain the observed co-occurrence between phylotypes of Weissella and Bacillus in GLI or Lactobacillus and Acetobacter in CI communities. Although Acetobacter has been frequently identified in corn silage (Dolci et al., 2011; Li and Nishino, 2011; Guan et al., 2018), there is still no consensus on the role played by this genus in silage. While Acetobacter was reported aerobic, Du Toit et al. (2005) demonstrated the effective survival of Acetobacter pasteurianus under anaerobic conditions. Queiroz et al. (2013) found no effect on the aerobic stability, following silage inoculation with AAB. A previous contradictory finding indicated that these bacteria could initiate aerobic spoilage (Spoelstra et al., 1988). In our study, the genus Acetobacter occurred in all 16 CI samples, from which more than 37% showed its dominance or co-dominance without considerable effects on fermentation characteristics. Hence, the conditions under which AAB, specifically Acetobacter species, could drive the silage fermentation process are not clear. Likewise, bacilli are known to produce butyric, acetic, or lactic acids, as well as antibacterial substances. However, beneficial effects of some strains of B. subtilis, Bacillus licheniformis, and Bacillus pumilus on the inhibition of molds and silage aerobic stability have been reported (Ávila and Carvalho, 2020).
The addition of inoculants to grass/legume and corn silage drastically changed bacterial interconnection patterns compared with uninoculated counterparts, resulting in increased network density and complexity levels, as well as a modified modularity and taxa topological roles. As suggested by Ma et al. (2020), in addition to beta-diversity, microbial co-occurrence networks could be used to characterize community assemblage depending on the environment. The observed network modules that have been interpreted as microbial niches (Faust and Raes, 2012; Röttjers and Faust, 2018) generally contained desirable and undesirable bacteria interconnected with positive or negative links. While positive interactions among bacterial phylotypes might indicate cooperation, nutritional cross-feeding, co-colonization, or co-survival in similar environments, negative associations might result from bacteriocin or other substance production, competition, changing environment, or overpopulation of a niche (Faust et al., 2012). These modules could reflect heterogeneity of the fermentation process occurring during ensiling or reveal the main players of that fermentation as pictured at feed-out of mature silage community composition. If keystone species identified in the GL network can be essential to its stability (Faust and Raes, 2012), the interpretation of their ecological relevance is not evident (Layeghifard et al., 2017; Röttjers and Faust, 2018). In the context of silage, owing to the plethora of studies conducted on the fermentation process and subsequent effects on animal performance (Bernardes et al., 2018; Muck et al., 2018; Wilkinson and Rinne, 2018), there is no doubt of the type of microbial community expected in successful mature silage, although this is not always obtained despite inoculation. For instance, our study revealed phylotypes of L. buchneri and L. plantarum as keystone species of the GL network that do not show any type of cooperation with undesirable bacteria. This finding confirms the ecological importance of the two taxa from which specific strains are currently used as inoculants. Other keystone species from the same network, among which are phylotypes of L. coryniformis, Weissella sp., and Lactobacillus spp., were found to cooperate with at least one undesirable bacterial phylotype. Isolates of such keystone species, although belonging to the LAB community, could not be theoretically selected as candidate inoculants. Obviously, keystone species such as Serratia spp., Pantoea spp., or Pseudomonas spp. are undesirable in silage and therefore illustrate key taxa to inhibit or suppress during fermentation. On the other hand, phylotypes of the genus Methylobacterium, of which some were identified as keystone species, shared positive and negative interactions with other bacteria regardless of the phylum they belong to. Species of the genus Methylobacterium are methanotrophic bacteria commonly associated with pre-ensiled forage plants (Knief et al., 2012; Ni et al., 2017). In this study, the positive correlation of this genus with pH is in accordance with its neutrophilic characteristics. Since Methylobacterium species are facultative aerobes (Knief et al., 2012), their occurrence in GL samples as co-dominant taxa might be explained by the presence of oxygen during ensiling or the low abundance of lactobacilli strains required to drive the early stages of the fermentation process. Rigorous experiments should therefore be carried out to link keystone species derived from co-occurrence network topological roles to a particular role in the ecosystem dynamics and stability of preserved forage. In this study, the observed disparities of taxa occurrence, abundance, or dominance, as well as differential associations with forage metadata within and across the identified forms of forage storage, depict the relevance of microbial additives and management conditions to the end products and microbiome structure of mature silage. Bacterial occurrence was shown to be highly variable, particularly LAB communities in farm-scale mature silage. Integrating silage associated parameters mentioned previously in a time-varying network analysis approach (Faust et al., 2015; Layeghifard et al., 2017) to decipher the temporal variations of microbial interactions would help fill the gaps in the current knowledge of microbial interplays and complex succession throughout ensiling.
Conclusion
In summary, hay microbiota characterized with high abundance of Sphingomonas, Methylobacterium, Curtobacterium, and Pantoea is significantly different from that of ensiled forage. At commercial farm-scale facilities, the use of inoculants may unpredictably counteract the effects of factors such as competing endogenous epiphytic populations, inadequate pressing, or oxygen penetration into the silo on the microbiota composition of mature silage. While LAB were underrepresented in hay, their abundance was inconsistent in inoculated compared with uninoculated silage. Since LAB were ubiquitous, they probably originated mostly from epiphytic LAB instead of from commercial additives. The microbiota of grass/legume silage was variably dominated or co-dominated by Lactobacillus or Pediococcus, specifically exhibiting higher abundance of Weissella or Bacillus in uninoculated and inoculated silage, respectively. On the other hand, Lactobacillus and Acetobacter inconsistently dominated the microbial communities of corn silage, regardless of inoculation. Based on microbiota composition and structure, the analysis of co-occurrence and co-exclusion patterns among community assemblage clearly distinguished uninoculated from inoculated silage. This study provides a better knowledge of how inoculants used for ensiling might modulate bacterial communities populating preserved mature forage at feed-out on commercial dairy farms. Further investigations integrating management practices and silage physicochemical parameters with microbial dynamics and interactions throughout silage fermentation and post-feedout periods are needed to fully understand biological processes involved in maintaining high-quality silage.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/ PRJNA846858.
Author Contributions
Conceptualization, DR, GL, and PYC; methodology, AKO and MG; software, AKO and TV; validation, DR, GL, and PYC; investigation, AKO; resources, DR; data curation, AKO; writing—original draft preparation, AKO; writing—review and editing, AKO, MG, DR, GL, and PYC; visualization, AKO; supervision and project administration, DR; funding acquisition, DR, GL, and PYC. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
The authors thank the Natural Sciences and Engineering Research Council of Canada (Ottawa) [Canada Research Chair on Lactic Cultures Biotechnology for Dairy and Probiotic Industries, 2003–2017], Novalait (Quebec, Canada), Agriculture and Agri-Food Canada (Ottawa), and the Fonds de recherche du Québec—Nature et technologies (Quebec, Canada) for their financial contributions. They also specifically thank the Op + Lait research group (Saint-Hyacinthe, Canada) for scholarships awarded to AK. Ouamba and MéG. The authors finally thank Marie Verheyde, Myriam Laberge, and Halimatou Diallo for their kind assistance during sampling and laboratory experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsysb.2022.955611/full#supplementary-material
References
Ali, N., Wang, S., Zhao, J., Dong, Z., Li, J., Nazar, M., et al. (2020). Microbial diversity and fermentation profile of red clover silage inoculated with reconstituted indigenous and exogenous epiphytic microbiota. Bioresour. Technol. 314, 123606. doi:10.1016/j.biortech.2020.123606
Ávila, C. L. S., and Carvalho, B. F. (2020). Silage fermentation - updates focusing on the performance of micro-organisms. J. Appl. Microbiol. 128, 966–984. doi:10.1111/jam.14450
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi:10.1038/ismej.2011.119
Behrendt, U., Müller, T., and Seyfarth, W. (1997). The influence of extensification in grassland management on the populations of micro-organisms in the phyllosphere of grasses. Microbiol. Res. 152, 75–85. doi:10.1016/S0944-5013(97)80026-2
Bernardes, T. F., Daniel, J. L. P., Adesogan, A. T., McAllister, T. A., Drouin, P., Nussio, L. G., et al. (2018). Silage review: Unique challenges of silages made in hot and cold regions. J. Dairy Sci. 101, 4001–4019. doi:10.3168/jds.2017-13703
Bernardi, A., Härter, C. J., Silva, A. W. L., Reis, R. A., and Rabelo, C. H. S. (2019). A meta-analysis examining lactic acid bacteria inoculants for maize silage: Effects on fermentation, aerobic stability, nutritive value and livestock production. Grass Forage Sci. 74, 596–612. doi:10.1111/gfs.12452
Borreani, G., Tabacco, E., Schmidt, R. J., Holmes, B. J., and Muck, R. E. (2018). Silage review: Factors affecting dry matter and quality losses in silages. J. Dairy Sci. 101, 3952–3979. doi:10.3168/jds.2017-13837
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., Holmes, S. P., et al. (2016a). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi:10.1038/nmeth.3869
Callahan, B. J., Sankaran, K., Fukuyama, J. A., McMurdie, P. J., and Holmes, S. P. (2016b). Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Res. 5, 1492. doi:10.12688/f1000research.8986.2
Chen, I.-M. A., Chu, K., Palaniappan, K., Pillay, M., Ratner, A., Huang, J., et al. (2019). IMG/M v.5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677. doi:10.1093/nar/gky901
Clauset, A., Newman, M. E. J., and Moore, C. (2004). Finding community structure in very large networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 70, 066111. doi:10.1103/PhysRevE.70.066111
Da Silva, T. C., da Silva, L. D., Santos, E. M., and Oliveira, J. S. (2017). “Importance of the fermentation to produce high-quality silage,” in Fermentation processes (London, United Kingdom: InTech). doi:10.5772/64887
Daniels, S., Hepworth, J., and Moore-Colyer, M. (2020). The haybiome: Characterising the viable bacterial community profile of four different hays for horses following different pre-feeding regimens. PLoS One 15, e0242373. doi:10.1371/journal.pone.0242373
Deng, Y., Jiang, Y. H., Yang, Y., He, Z., Luo, F., Zhou, J., et al. (2012). Molecular ecological network analyses. BMC Bioinforma. 13, 113. doi:10.1186/1471-2105-13-113
Dolci, P., Tabacco, E., Cocolin, L., and Borreani, G. (2011). Microbial dynamics during aerobic exposure of corn silage stored under oxygen barrier or polyethylene films. Appl. Environ. Microbiol. 77, 7499–7507. doi:10.1128/AEM.05050-11
Dos Santos, A. D. O., Junior, G. S. D., Pereira, M. N., Schwan, R. F., and Da Silva Ávila, C. L. (2020). A survey of whole-plant corn silages from Minas Gerais dairy farms. Sci. Agric. 77, 2020. doi:10.1590/1678-992x-2018-0080
Driehuis, F. (2013). Silage and the safety and quality of dairy foods: A review. AFSci. 22, 16–34. doi:10.23986/afsci.6699
Driehuis, F., Wilkinson, J. M., Jiang, Y., Ogunade, I., and Adesogan, A. T. (2018). Silage review: Animal and human health risks from silage. J. Dairy Sci. 101, 4093–4110. doi:10.3168/jds.2017-13836
Drouin, P., Tremblay, J., and Chaucheyras-Durand, F. (2019). Dynamic succession of microbiota during ensiling of whole plant corn following inoculation with Lactobacillus buchneri and Lactobacillus hilgardii alone or in combination. Microorganisms 7, 595. doi:10.3390/microorganisms7120595
Du Toit, W. J., Pretorius, I. S., and Lonvaud-Funel, A. (2005). The effect of sulphur dioxide and oxygen on the viability and culturability of a strain of Acetobacter pasteurianus and a strain of Brettanomyces bruxellensis isolated from wine. J. Appl. Microbiol. 98, 862–871. doi:10.1111/j.1365-2672.2004.02549.x
Duchaine, C., Mériaux, A., Brochu, G., Bernard, K., and Cormier, Y. (1999). Saccharopolyspora rectivirgula from Quebec dairy barns: Application of simplified criteria for the identification of an agent responsible for farmer’s lung disease. J. Med. Microbiol. 48, 173–180. doi:10.1099/00222615-48-2-173
Dunière, L., Sindou, J., Chaucheyras-Durand, F., Chevallier, I., and Thévenot-Sergentet, D. (2013). Silage processing and strategies to prevent persistence of undesirable microorganisms. Anim. Feed Sci. Technol. 182, 1–15. doi:10.1016/j.anifeedsci.2013.04.006
Faust, K., Lahti, L., Gonze, D., de Vos, W. M., and Raes, J. (2015). Metagenomics meets time series analysis: Unraveling microbial community dynamics. Curr. Opin. Microbiol. 25, 56–66. doi:10.1016/j.mib.2015.04.004
Faust, K., and Raes, J. (2012). Microbial interactions: From networks to models. Nat. Rev. Microbiol. 10, 538–550. doi:10.1038/nrmicro2832
Faust, K., Sathirapongsasuti, J. F., Izard, J., Segata, N., Gevers, D., Raes, J., et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 8, e1002606. doi:10.1371/journal.pcbi.1002606
Fernandes, A. D., Reid, J. N. S., Macklaim, J. M., McMurrough, T. A., Edgell, D. R., Gloor, G. B., et al. (2014). Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15. doi:10.1186/2049-2618-2-15
Gagnon, M., Ouamba, A. J. K., LaPointe, G., Chouinard, P. Y., and Roy, D. (2020). Prevalence and abundance of lactic acid bacteria in raw milk associated with forage types in dairy cow feeding. J. Dairy Sci. 103, 5931–5946. doi:10.3168/jds.2019-17918
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V., and Egozcue, J. J. (2017). Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 8, 2224. doi:10.3389/fmicb.2017.02224
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi:10.1093/bioinformatics/btw313
Guan, H., Shuai, Y., Yan, Y., Ran, Q., Wang, X., Li, D., et al. (2020). Microbial community and fermentation dynamics of corn silage prepared with heat-resistant lactic acid bacteria in a hot environment. Microorganisms 8, 719. doi:10.3390/microorganisms8050719
Guan, H., Yan, Y., Li, X., Li, X., Shuai, Y., Feng, G., et al. (2018). Microbial communities and natural fermentation of corn silages prepared with farm bunker-silo in Southwest China. Bioresour. Technol. 265, 282–290. doi:10.1016/j.biortech.2018.06.018
Hu, Z., Chang, J., Yu, J., Li, S., and Niu, H. (2018). Diversity of bacterial community during ensiling and subsequent exposure to air in whole-plant maize silage. Asian-Australas. J. Anim. Sci. 31, 1464–1473. doi:10.5713/ajas.17.0860
Ingham, A. C., Kielsen, K., Cilieborg, M. S., Lund, O., Holmes, S., Aarestrup, F. M., et al. (2019). Specific gut microbiome members are associated with distinct immune markers in pediatric allogeneic hematopoietic stem cell transplantation. Microbiome 7, 131. doi:10.1186/s40168-019-0745-z
Jari, O., Guillaume, F., Michael, F., Roeland, K., Pierre, L., Dan, M., et al. (2019). Vegan: Community ecology package. R package version 2.5-6, 1–298. Available at: https://cran.r-project.org/package=vegan.
Julien, M.-C., Dion, P., Lafrenière, C., Antoun, H., and Drouin, P. (2008). Sources of clostridia in raw milk on farms. Appl. Environ. Microbiol. 74, 6348–6357. doi:10.1128/AEM.00913-08
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. doi:10.1093/nar/gkr988
Kassambara, A. (2018). Ggpubr: “ggplot2” based publication ready plots. R package version 0.2. Available at: https://cran.r-project.org/web/packages/ggpubr/index.html (Accessed June 12, 2019).
Kennang Ouamba, A. J., LaPointe, G., Dufour, S., and Roy, D. (2020). Optimization of preservation methods allows deeper insights into changes of raw milk microbiota. Microorganisms 8, 368. doi:10.3390/microorganisms8030368
Keshri, J., Chen, Y., Pinto, R., Kroupitski, Y., Weinberg, Z. G., Sela, S., et al. (2018). Microbiome dynamics during ensiling of corn with and without Lactobacillus plantarum inoculant. Appl. Microbiol. Biotechnol. 102, 4025–4037. doi:10.1007/s00253-018-8903-y
Knief, C., Delmotte, N., Chaffron, S., Stark, M., Innerebner, G., Wassmann, R., et al. (2012). Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 6, 1378–1390. doi:10.1038/ismej.2011.192
Kraut-Cohen, J., Tripathi, V., Chen, Y., Gatica, J., Volchinski, V., Sela, S., et al. (2016). Temporal and spatial assessment of microbial communities in commercial silages from bunker silos. Appl. Microbiol. Biotechnol. 100, 6827–6835. doi:10.1007/s00253-016-7512-x
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi:10.1038/nbt.2676
Layeghifard, M., Hwang, D. M., and Guttman, D. S. (2017). Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 25, 217–228. doi:10.1016/j.tim.2016.11.008
Lecours, P. B., Veillette, M., Marsolais, D., and Duchaine, C. (2012). Characterization of bioaerosols from dairy barns: Reconstructing the puzzle of occupational respiratory diseases by using molecular approaches. Appl. Environ. Microbiol. 78, 3242–3248. doi:10.1128/AEM.07661-11
Legendre, P., and De Cáceres, M. (2013). Beta diversity as the variance of community data: Dissimilarity coefficients and partitioning. Ecol. Lett. 16, 951–963. doi:10.1111/ele.12141
Li, Y., and Nishino, N. (2011). Monitoring the bacterial community of maize silage stored in a bunker silo inoculated with Enterococcus faecium, Lactobacillus plantarum and Lactobacillus buchneri. J. Appl. Microbiol. 110, 1561–1570. doi:10.1111/j.1365-2672.2011.05010.x
Luo, F., Yang, Y., Zhong, J., Gao, H., Khan, L., Thompson, D. K., et al. (2007). Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinforma. 8, 299. doi:10.1186/1471-2105-8-299
Luo, F., Zhong, J., Yang, Y., Scheuermann, R. H., and Zhou, J. (2006). Application of random matrix theory to biological networks. Phys. Lett. A 357, 420–423. doi:10.1016/j.physleta.2006.04.076
Ma, B., Wang, Y., Ye, S., Liu, S., Stirling, E., Gilbert, J. A., et al. (2020). Earth microbial co-occurrence network reveals interconnection pattern across microbiomes. Microbiome 8, 82. doi:10.1186/s40168-020-00857-2
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10. doi:10.14806/ej.17.1.200
Maslov, S., and Sneppen, K. (2002). Specificity and stability in topology of protein networks. Science 296, 910–913. doi:10.1126/science.1065103
McAllister, T. A., Dunière, L., Drouin, P., Xu, S., Wang, Y., Munns, K., et al. (2018). Silage review: Using molecular approaches to define the microbial ecology of silage. J. Dairy Sci. 101, 4060–4074. doi:10.3168/jds.2017-13704
McMurdie, P. J., and Holmes, S. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. doi:10.1371/journal.pone.0061217
Moore-Colyer, M., Longland, A., Harris, P., Zeef, L., and Crosthwaite, S. (2020). Mapping the bacterial ecology on the phyllosphere of dry and post soaked grass hay for horses. PLoS One 15, e0227151. doi:10.1371/journal.pone.0227151
Muck, R. E., Nadeau, E. M. G., McAllister, T. A., Contreras-Govea, F. E., Santos, M. C., Kung, L., et al. (2018). Silage review: Recent advances and future uses of silage additives. J. Dairy Sci. 101, 3980–4000. doi:10.3168/jds.2017-13839
Muck, R. E., and Shinners, K. J. (2001). Conserved forage (silage and hay): Progress and priorities” in 19th international grassland congress. São Pedro, Brazil, 753–762. Available at: http://library1.nida.ac.th/termpaper6/sd/2554/19755.pdf.
Narayan, N. R., Weinmaier, T., Laserna-Mendieta, E. J., Claesson, M. J., Shanahan, F., Dabbagh, K., et al. (2020). Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genomics 21, 56. doi:10.1186/s12864-019-6427-1
Nascimento Agarussi, M. C., Gomes Pereira, O., Paula, R. A., Santos Roseira, J. P., Fonseca E Silva, F., and Fonseca e Silva, F. (2019). Novel lactic acid bacteria strains as inoculants on alfalfa silage fermentation. Sci. Rep. 9, 8007–8009. doi:10.1038/s41598-019-44520-9
Ni, K., Minh, T. T., Tu, T. T. M., Tsuruta, T., Pang, H., Nishino, N., et al. (2017). Comparative microbiota assessment of wilted Italian ryegrass, whole crop corn, and wilted alfalfa silage using denaturing gradient gel electrophoresis and next-generation sequencing. Appl. Microbiol. Biotechnol. 101, 1385–1394. doi:10.1007/s00253-016-7900-2
Nocker, A., Richter-heitmann, T., Montijn, R., Schuren, F., and Kort, R. (2010). Discrimination between live and dead cells in bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int. Microbiol. 13, 59–65. doi:10.2436/20.1501.01.111
Ogunade, I. M., Martinez-Tuppia, C., Queiroz, O. C. M., Jiang, Y., Drouin, P., Wu, F., et al. (2018a). Silage review: Mycotoxins in silage: Occurrence, effects, prevention, and mitigation. J. Dairy Sci. 101, 4034–4059. doi:10.3168/jds.2017-13788
Ogunade, I. M., Jiang, Y., Pech Cervantes, A. A., Kim, D. H., Oliveira, A. S., Vyas, D., et al. (2018b). Bacterial diversity and composition of alfalfa silage as analyzed by Illumina MiSeq sequencing: Effects of Escherichia coli O157:H7 and silage additives. J. Dairy Sci. 101, 2048–2059. doi:10.3168/jds.2017-12876
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2007). The modularity of pollination networks. Proc. Natl. Acad. Sci. U. S. A. 104, 19891–19896. doi:10.1073/pnas.0706375104
Oliveira, A. S., Weinberg, Z. G., Ogunade, I. M., Cervantes, A. A. P., Arriola, K. G., Jiang, Y., et al. (2017). Meta-analysis of effects of inoculation with homofermentative and facultative heterofermentative lactic acid bacteria on silage fermentation, aerobic stability, and the performance of dairy cows. J. Dairy Sci. 100, 4587–4603. doi:10.3168/jds.2016-11815
Pahlow, G., Muck, R. E., Driehuis, F., and Oude Elferink, S. J. W. H. (2003). “Microbiology of ensiling,” in Silage science and technology. Editors D. R. Buxton, R. E. Muck, J. H. Harrison, and Madison (Wisconsin, USA: American Society of Agronomy), 1–62. doi:10.2134/agronmonogr42.c2
Queiroz, O. C. M., Arriola, K. G., Daniel, J. L. P., and Adesogan, A. T. (2013). Effects of 8 chemical and bacterial additives on the quality of corn silage. J. Dairy Sci. 96, 5836–5843. doi:10.3168/jds.2013-6691
Ranalli, G., Grazia, L., and Roggeri, A. (1999). The influence of hay-packing techniques on the presence of Saccharopolyspora rectivirgula. J. Appl. Microbiol. 87, 359–365. doi:10.1046/j.1365-2672.1999.00826.x
Rastogi, G., Coaker, G. L., and Leveau, J. H. J. (2013). New insights into the structure and function of phyllosphere microbiota through high-throughput molecular approaches. FEMS Microbiol. Lett. 348, 1–10. doi:10.1111/1574-6968.12225
Rohart, F., Gautier, B., Singh, A., and Lê Cao, K. A. (2017). mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 13, e1005752. doi:10.1371/journal.pcbi.1005752
Röttjers, L., and Faust, K. (2018). From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol. Rev. 42, 761–780. doi:10.1093/femsre/fuy030
Schliep, K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi:10.1093/bioinformatics/btq706
Schultz, C. (2016). Comparing silo filler disease with farmer’s lung disease. Gen. Int. Med. Clin. Innov. 1, 76–78. doi:10.15761/gimci.1000123
Silverman, J. D., Washburne, A. D., Mukherjee, S., and David, L. A. (2017). A phylogenetic transform enhances analysis of compositional microbiota data. Elife 6, e21887. doi:10.7554/eLife.21887
Snyder, E. E., Kampanya, N., Lu, J., Nordberg, E. K., Karur, H. R., Shukla, M., et al. (2007). Patric: The VBI PathoSystems resource integration center. Nucleic Acids Res. 35, D401–D406. doi:10.1093/nar/gkl858
Spoelstra, S. F., Courtin, M. G., and Van Beers, J. A. C. (1988). Acetic acid bacteria can initiate aerobic deterioration of whole crop maize silage. J. Agric. Sci. 111, 127–132. doi:10.1017/S0021859600082915
Ssekagiri, A., Sloan, W. T., and Zeeshan Ijaz, U. (2020). Microbiomeseq: An R package for microbial community analysis in an environmental context. Available at: http://userweb.eng.gla.ac.uk/umer.ijaz/projects/microbiomeSeq_Tutorial.html#references (Accessed July 9, 2020).
Stevenson, D. M., Muck, R. E., Shinners, K. J., and Weimer, P. J. (2006). Use of real time PCR to determine population profiles of individual species of lactic acid bacteria in alfalfa silage and stored corn stover. Appl. Microbiol. Biotechnol. 71, 329–338. doi:10.1007/s00253-005-0170-z
Unaogu, I. C., Gugnani, H. C., and Lacey, J. (1994). Occurrence of thermophilic actinomycetes in natural substrates in Nigeria. Ant. Van Leeuwenhoek 65, 1–5. doi:10.1007/BF00878272
Wang, X., Haruta, S., Wang, P., Ishii, M., Igarashi, Y., Cui, Z., et al. (2006). Diversity of a stable enrichment culture which is useful for silage inoculant and its succession in alfalfa silage. FEMS Microbiol. Ecol. 57, 106–115. doi:10.1111/j.1574-6941.2006.00099.x
Ward, T., Larson, J., Meulemans, J., Hillmann, B., Lynch, J., Sidiropoulos, D., et al. (2017). BugBase predicts organism-level microbiome phenotypes. bioRxiv 1, 133462. doi:10.1101/133462
Weinberg, Z. G., and Muck, R. E. (1996). New trends and opportunities in the development and use of inoculants for silage. FEMS Microbiol. Rev. 19, 53–68. doi:10.1111/j.1574-6976.1996.tb00253.x
Wilkinson, J. M., and Rinne, M. (2018). Highlights of progress in silage conservation and future perspectives. Grass Forage Sci. 73, 40–52. doi:10.1111/gfs.12327
Wright, E. S. (2015). Decipher: Harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinforma. 16, 322. doi:10.1186/s12859-015-0749-z
Yang, F., Wang, Y., Zhao, S., and Wang, Y. (2020). Lactobacillus plantarum inoculants delay spoilage of high moisture alfalfa silages by regulating bacterial community composition. Front. Microbiol. 11, 1989. doi:10.3389/fmicb.2020.01989
Yang, L., Yuan, X., Li, J., Dong, Z., and Shao, T. (2019). Dynamics of microbial community and fermentation quality during ensiling of sterile and nonsterile alfalfa with or without Lactobacillus plantarum inoculant. Bioresour. Technol. 275, 280–287. doi:10.1016/j.biortech.2018.12.067
Yuan, X. J., Li, J. F., Dong, Z. H., and Shao, T. (2020). The reconstitution mechanism of napier grass microiota during the ensiling of alfalfa and their contributions to fermentation quality of silage. Bioresour. Technol. 297, 122391. doi:10.1016/j.biortech.2019.122391
Keywords: dairy farm, preserved forage, commercial inoculant, microbiota, viable bacteria, co-occurrence patterns, qPCR, silage quality
Citation: Kennang Ouamba AJ, Gagnon M, Varin T, Chouinard PY, LaPointe G and Roy D (2022) Metataxonomic insights into the microbial ecology of farm-scale hay, grass or legume, and corn silage produced with and without inoculants. Front. Syst. Biol. 2:955611. doi: 10.3389/fsysb.2022.955611
Received: 29 May 2022; Accepted: 07 July 2022;
Published: 12 August 2022.
Edited by:
Liliana Fadul, University of Wisconsin-Madison, United StatesReviewed by:
Soumya Roy Chowdhury, National Institutes of Health (NIH), United StatesJie Zhao, Nanjing Agricultural University, China
Copyright © 2022 Kennang Ouamba, Gagnon, Varin, Chouinard, LaPointe and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denis Roy, RGVuaXMuUm95QGZzYWEudWxhdmFsLmNh