 Anamarija Butković
Anamarija Butković Santiago F. Elena
Santiago F. Elena
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Syst. Biol. , 12 December 2022
Sec. Integrative Genetics and Genomics
Volume 2 - 2022 | https://doi.org/10.3389/fsysb.2022.1005758
Genome-wide association studies (GWAS) have been gaining popularity over the last decade as they provide new insights into the genetic architecture of many disease-related traits. GWAS is based on the common disease common variant hypothesis, allowing identification of alleles associated with susceptibility and symptomatology of most common infectious diseases, such as AIDS, common cold, flu, and many others. It depends on the natural variation in a host population which can help identify genetic variants responsible for virus disease-related traits. Considering the prevalence of viruses in the ecosystem and their societal burden, identification of potential resistance loci or therapeutic targets is of great interest. Here, we highlight the most important points necessary for a successful GWAS of viral infectious diseases, focusing on the study design and various statistical methods used. Finally, we exemplify this application with studies done with human immunodeficiency virus type 1 and turnip mosaic virus.
Viral infections have always had a profound impact on our society throughout history. Viruses causing smallpox, polio, Ebola, flu, AIDS, and the most recent COVID-19 pandemic have ravaged the human society throughout history (Behbehani, 1983; Cohen et al., 2008; WHO Ebola Response Team Aylward et al., 2014; Spreeuwenberg et al., 2018; Sankaran and Weiss, 2021). Epidemics and pandemics not only burden our society directly but also indirectly, by impacting the production of farm animals and crop plants as well. Examples of viruses of farm and wild animals are, e.g., African swine fever, bluetongue, distemper, foot and mouth disease, strains of avian influenza, or rinderpest (Barrett and Rossiter, 1999; Thompson et al., 2002; Rushton Macchioni et al., 2015; Sankaran and Weiss, 2021). While examples for crop plants are, e.g., cucumber mosaic virus, tomato yellow leaf curl viruses, citrus tristeza virus, barley yellow dwarf viruses, or potato virus Y (Jones, 2021). The challenges caused by viruses demand further studies of host defense and resistance or susceptibility mechanisms. A better understanding of the genetic basis of host defenses and viral infections will result in novel antiviral strategies and discovery of naturally resistant species. Due to the fast-increasing availability of data on within-species genetic variability, genome-wide association studies (GWAS) can now be used to identify potential loci involved in susceptibility or resistance to viral infections.
GWAS determine the strength of the association between a phenotype and single-nucleotide polymorphisms (SNPs) or single base changes that vary between individuals. SNPs can be very common or rare, thus leading to different frequencies in the population (Dehghan and Evangelou, 2018). Common diseases will be influenced by common genetic variants as stated in the common disease common variant hypothesis (Bush and Moore, 2012; Uitterlinden, 2016; Chang et al., 2018; Dehghan and Evangelou, 2018). This principle applies to infectious diseases too since they are prevalent and affect many individuals. Henceforth, GWAS would also be a method of choice for detection of common genetic variants responsible for resistance or susceptibility to viral infections.
The rapid development of statistical, computational and genotyping methods has made GWAS easily accessible for genetic analysis. It has proven to be very successful at identifying disease related genes with little previous information on causal variants (non-candidate-gene approach) and allowed identification of genetic determinants in host resistance or susceptibility (Fellay et al., 2007; Butković et al., 2021; Choudhury et al., 2019; COVID-19 Host Genetics Initiative, 2021; Crosslin et al., 2015; Adebamowo et al., 2020; Foresman et al., 2016; Feng et al., 2019; Liu et al., 2021; Garcia-Etxebarria et al., 2015; Montes et al., 2021; Pimenta et al., 2020; Rubio et al., 2019; Tian et al., 2017; Xiao et al., 2019; Yang et al., 2019; Zhang et al., 2020; Zignego et al., 2014). These general GWAS steps, from identification of potential variants to follow-up studies, are highlighted in Figure 1.
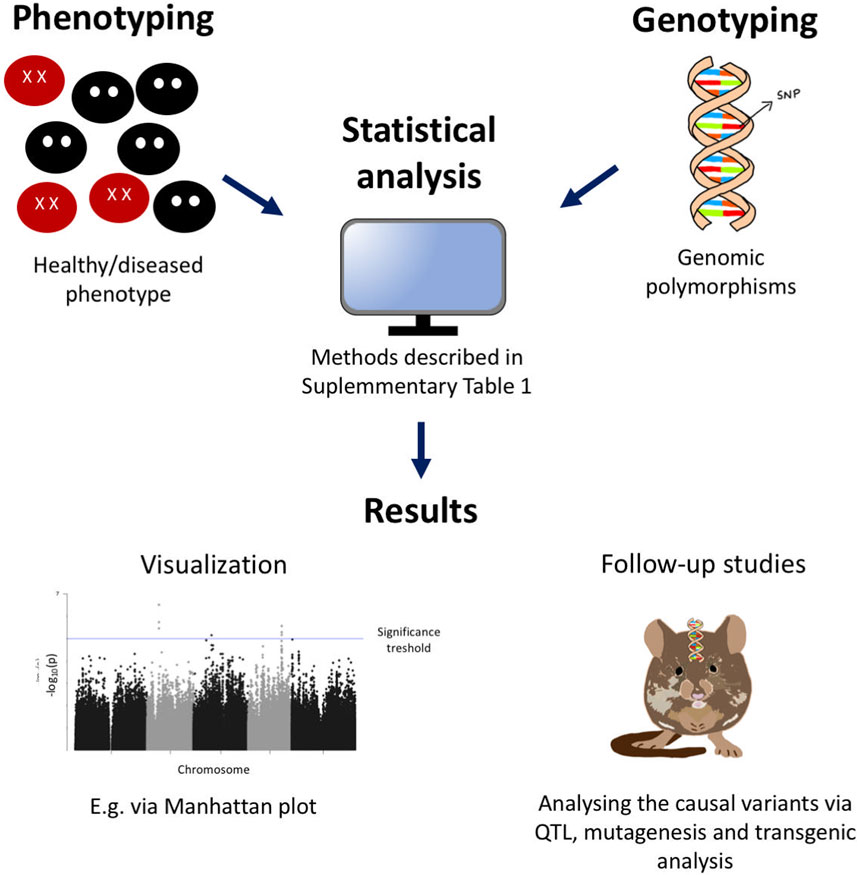
FIGURE 1. Flowchart of the steps taken in a GWAS. After phenotyping and genotyping (SNP arrays) a population of individuals with or without the trait of interest, a statistical analysis is performed and the significant SNPs correlated with the phenotype are visualized in a Manhattan plot. A significance threshold is calculated and all the SNPs with −logp values above this threshold are considered significant. The causal variants can be further analyzed via follow-up studies such as, QTL, mutagenesis and transgenic analysis.
In this review, we will give general guidelines necessary for a successful GWAS of viral infectious diseases by focusing on careful study design and statistical analysis. We will not go into detail about SNP genotyping step of GWAS since this information is already available for a large number of model organisms. Instead we will provide short general guidelines to keep in mind when selecting the appropriate genotyping methods for GWAS.
Since GWAS require effort and time due to the large number of sampled and phenotyped individuals, it is best to prevent potential pitfalls before starting. Here we present general guidelines that should be taken into account for a good GWAS of viral infectious diseases.
The number of SNPs that are sufficient for GWAS, the cost and the genome coverage are the main determinants when selecting an appropriate genotyping method. Three most commonly used methods are Whole-Genome Sequencing (WGS), Genotyping by sequencing (GBS) and SNP Arrays. WGS aims to capture whole genome variation and is more expensive than other two methods. This method can capture common and rare variants which allows fine-resolution of variant mapping (Höglund et al., 2003). GBS uses restriction enzymes before sequencing in order to reduce genome complexity and price which leads to low coverage (Pavan et al., 2020). It is particularly useful for populations with slowly decaying LD where lower number of genotyped SNPs and their uneven genomic distribution do not affect the questions of the GWAS. Imputation can compensate for low coverage of GBS though it works best in homozygous populations. SNP arrays are the most popular of the genotyping method used in GWAS due to their generally low cost, high SNP density and ability to sequence large number of individuals (Pavan et al., 2020). One major drawback is dependence on the markers present in a population, that is, used as a reference for the development of the array, meaning that SNPs not present in that population will be missed when sequencing new populations. In general, the sequencing costs are decreasing and methods that generate longer reads are becoming more popular (Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT)) because they can discover hidden variation in the genome (De Coster et al., 2021). The decision on which method to use will largely depend on the cost but in general WGS, SNP arrays and long read methods are proving a good choice for viral infectious disease GWAS.
GWAS can be used to study virus-host interactions in any host species. Although model organisms in general have more advantages than non-model organisms (for example humans) when performing GWAS because different individuals: i) can be maintained through inbreeding, as in Arabidopsis thaliana or Caenorhabditis elegans, ii) can be grown in controlled environments under careful observation and iii) can be inoculated with a known viral dose that could be genetically and phenotypically well defined. All these points also highlight the main disadvantages of using non-model host and pathogen organisms with uncontrolled genetic and environmental background. An important advantage of A. thaliana, and many other plants, is self-fertilization where the same genotyped individuals can be maintained through time, thus allowing repeated phenotyping (Korte et al., 2012). Main disadvantages are caused by different breeding strategies that lead to extensive population structure and spurious hits later on in the association analysis (Mozzi et al., 2018). Fortunately, this can be corrected by proper use of statistical models, such as linear mixed models that take into account the population structure thus minimizing spurious hits and leading to recovery of more meaningful associations (Lippert et al., 2011; Zhou and Stephens, 2012; Lippert et al., 2014). In conclusion, the choice of an organism for GWAS should depend on the species of interest and the available information.
When talking about the number of individuals to use in a GWAS of viral infections, the general guideline should be the more the better. However, this is not always feasible due to cost or inability to recruit a large number of individuals. This can be represented as an inverse relationship between sample size and effect size. Where large sample sized studies have more power to detect smaller associations than small sample sized studies (Witte, 2010). Of course, the numbers vary from one organism to another and the studied trait, due to genetic diversity, recombination and ploidy (Genissel et al., 2017). For example, for some traits pretty good sensitivity and meaningful results can be achieved with ∼ 100 lines of A. thaliana; however, thousands of individuals might be needed in the case of human studies (The Wellcome Trust Case Control Consortium, 2007; Manolio et al., 2009; Korte and Farlow, 2013; Butković et al., 2021). This difference can be explained by the number of loci that explain the majority of the phenotypic diversity. In some A. thaliana traits, few loci explain the trait of interest and the diversity can be captured by genotyping less individuals. While in human studies, many small effect loci explain a trait and require genotyping of thousands of individuals in order to detect them (The Wellcome Trust Case Control Consortium, 2007; Korte and Farlow, 2013; Genissel et al., 2017). So far, in infectious disease GWAS an increase in sample sizes identified more meaningful associations (McLaren et al., 2015; Butković et al., 2021; Butković et al., 2022). Since viral infectious diseases are highly prevalent and are governed by common variants, a moderate increase in sample size should lead to enough statistical power needed to identify meaningful associations. However, if there are rare alleles responsible for a studied trait they will not be identified in moderate sample sized studies and would require larger number of samples (The Wellcome Trust Case Control Consortium, 2007; Hong and Park, 2012). Adequate sample sizes can be estimated with the help of Genetic Power or GAS Power calculators (readily available online), mostly intended for case-control studies (Purcell et al., 2003; Johnson and Abecasis, 2017). In summary, sample size should be directly correlated with the target species, the particular trait being studied, and any previous studies on similar topics that might provide even a raw estimate of appropriate sample sizes. Usually, significance thresholds need to be adjusted via correction methods, such as Bonferroni correction. With large sample sizes small effect loci become easier to detect and stricter significance thresholds are recommended to reduce the number of false positives (Fadista et al., 2016; Pulit et al., 2017; Wu et al., 2017).
Another important selection criteria are the geographical distribution and kinship of the samples. More heterogeneous samples can increase the genetic variability, allow replication in independent populations and may detect SNPs associated with the trait of interest in mixed populations (Korte and Farlow, 2013; Li and Keating, 2014). There are downsides to making samples more heterogeneous since this can decrease the power to recover significant variants; causing a non-causative marker to become more significant than the causal one and lead to the problem of handling linkage disequilibrium across heterogeneous populations (Korte and Farlow, 2013; Li and Keating, 2014). For example, a naturally occurring plant population can be previously exposed to a virus and gain resistance alleles while other populations lack them. This difference can lead to overestimation of the impact these resistance alleles have in the global diversity panel since they will become significant when comparing different populations. To avoid all the problems that population heterogeneity can cause, one solution is to densely genotype a local population. Although this approach leads to underrepresentation or absence of some variants of interest that may be relevant to global phenotypic diversity (Korte and Farlow, 2013). Also, focusing on a local population can lead to insufficient statistical power if the size of the population is small. Again, there is no golden rule when selecting how geographically or kinship diverse the samples shall be, the answer mostly depends on the goal of the GWAS. For example, more geographically heterogeneous samples would allow mapping of variants relevant to a global response to infection while less heterogeneous samples can map variants specific to a certain population. But appropriate measures should be taken into account where false positives would be avoided. For example, performing principal component analysis will account for genetic differences between populations which can later on be included as a covariate in the association analysis (Price et al., 2006; The Wellcome Trust Case Control Consortium, 2007; Cook and Morris, 2016). Another good measure that can be used is admixture mapping, a method that tries to associate an allele with a disease based on ancestry information between different populations (Shriner, 2013).
Before starting a GWAS, it is essential to define the infection-related trait to be studied and the method of virus delivery. When measuring viral infection, it is typical to measure the development of symptoms during a specified time period, frequency of infected individuals, viral accumulation, or presence/absence of symptoms related to infection (Fellay et al., 2007; Butković et al., 2021; Choudhury et al., 2019; COVID-19 Host Genetics Initiative, 2021; Crosslin et al., 2015; Adebamowo et al., 2020; Foresman et al., 2016; Feng et al., 2019; Liu et al., 2021; Garcia-Etxebarria et al., 2015; Montes et al., 2021; Pimenta et al., 2020; Rubio et al., 2019; Tian et al., 2017; Xiao et al., 2019; Yang et al., 2019; Zhang et al., 2020; Zignego et al., 2014). All these traits can be divided into categorical (binary case-control studies where diseased individuals get compared to disease-free controls or categories of disease symptoms on a scale ranging from asymptomatic to death) or quantitative measures (frequency or percentage of infected individual, viral accumulation). Generally speaking, quantitative traits are preferred because they improve the power of GWAS and can be better interpreted while case-control studies can lead to large measurement errors (Bush and Moore, 2012).
Expertise is necessary to be able to discern symptoms of infection as opposed to other conditions similar in appearance. Viral accumulation is not a visual trait and therefore, is not dependent on the trained eye of the observer, making it easier to measure. Although viral accumulation is not always directly correlated with the strength of the symptoms and might not involve the same variants for the two traits. This is of special interest if the goal of the GWAS is to discover genetic regions related to development of detrimental symptoms in the host. A common problem when visually detecting infected individuals are asymptomatic infections, mostly caused by errors in the inoculation procedure as opposed to resistant individuals that have no phenotypic symptoms. Another problem in visual evaluation of symptoms is resistance (no symptoms and no virus multiplication), tolerance (prevention of symptom onset despite virus accumulation) or recovery of individuals from infection during the observation period, that may confuse the observer and lead to incorrect measurements. A possible way to deal with these problems is to detect the presence of a virus (RT-PCR, RT-qPCR, PCR … ) in order to confirm or reject the infection status of an individual.
An important factor contributing to development of symptoms is the inoculation procedure. There are many different ways to inoculate individuals, for example, in plants using mechanical inoculation (rubbing using abrasive material) and Agrobacterium-delivery, adding virus infected medium to a cell culture, and vector dispersal. All of the methods except vector dispersal, lead to a controlled administration of the virus amount and remove bias due to large differences in initial virus concentrations between individuals (Gokhale and Bald, 1987). Still, these inoculation methods might lead to different responses of the plant to the virus compared to natural modes of infection and should be taken into account when interpreting results.
A good predictor of how much a trait is explained by genetic factors is heritability (Zaitlen and Kraft, 2012). Due to the presence of many small effect or rare variants, epistasis and phenotypic plasticity, it is impossible to identify all of the components influencing a complex trait in any chosen method (Frazer et al., 2009; Aschard et al., 2012). One solution to this problem might be to study the trait of interest in the context of the environment which might explain some of the missing heritability.
There are a variety of methods available for statistical analysis (Supplementary Table S1) of viral infection GWAS, and the most common ones will be discussed here. The univariate GWAS methods analyze the associations between a single phenotypic trait and the genetic polymorphisms while the multivariate GWAS evaluate the associations between multiple correlated phenotypic traits and the genetic polymorphisms (Höglund et al., 2003). Both approaches account for the population structure by including it in the model and thereby reducing the false positive rate. Before getting into trait analysis using a univariate or multivariate model, the distribution of phenotype data should be checked and transformed (log, square root or Box-Cox) to achieve normality since this helps to avoid spurious hits driven by outliers (Dehghan and Evangelou, 2018).
The most commonly used approach in GWAS is the univariate statistical model. There are different classes of univariate GWAS and we will present the ones most commonly used in viral infection GWAS. The methods using them are presented in Supplementary Table S1:
Logistic regression is a type of linear models used to model binary data scoring infected/non-infected individuals such as case/control study because this data is not normally distributed and cannot be modelled by linear regression (Balding, 2006; Gumpinger et al., 2018).
Linear mixed models (LMM) are the most commonly used models in GWAS for quantitative traits (virus accumulation, symptoms intensity, number of infected individuals per number of inoculated individuals, disease progression over time) and have gained popularity over the years. They require Gaussian distributed residuals and combine fixed and random effects to account for the phenotypic variation, the genetic variation, covariates (fixed effects) and genetic similarity (kinship) or noise (random effect that follows a Gaussian distribution) (Balding, 2006; Gumpinger et al., 2018).
While multivariate mixed models evaluate the association between two or more phenotypes with the genetic variants, and can be a better choice than univariate models when there is a strong linkage disequilibrium between causal loci, or the measured phenotypes are correlated due to pleiotropy and shared environment (phenotyping different virus strains in the same genotype panel or combining different phenotypes from the same virus) (Höglund et al., 2003; Yang et al., 2014). By taking these correlations into account in a statistical GWAS model, the power of the analysis can be increased. There are quite a few software programs implementing this approach (Supplementary Table S1): GCTA (Yang et al., 2011), GEMMA (Zhou and Stephens, 2012), EMMAX (Zhou and Stephens, 2014), LIMIX (Lippert et al., 2014), or MTMM (Höglund et al., 2003). By no means should the two approaches compete against each other; rather they should be used in complement in the search for causal associations.
After the statistical analysis of data, significant associations must be selected. To achieve this, p-values (how strongly is a variant associated with the trait) that fall below a significance threshold are chosen. Usually, the significance level is set to 0.05 which results in a false association nearly 5% of the time (Pearson and Manolio, 2008; Bush and Moore, 2012). The problem is that this false positive rate is true when running a single statistical test; however, GWAS analysis usually involves from thousands to millions of tests. This large number of tests increases the false positive rate and calls for adjustment of the significance threshold to account for multiple testing (Manolio et al., 2009; Bush and Moore, 2012; Tam et al., 2019), where Bonferroni correction (Bush and Moore, 2012) is the most commonly used correction method in GWAS.
In conclusion, there are various methods and online resources developed for each stage of a GWAS, thus making it an easy and available feat. The most commonly used methods for association analysis are LMMs and they have proven to be an indispensable tool in GWAS. There are many software options available that implement these methods (Supplementary Table S1) and the choice of the right software depends on the questions being answered, ease of use and speed of computation. Since running a GWAS analysis can be computationally demanding, the recent surge of online tools overcoming this burden has made GWAS analysis even easier (e.g., GWA-Portal, easyGWAS, GWASPro, or GWAPP).
GWAS has been a useful tool in the investigation of resistance or susceptibility loci for numerous viral infectious diseases (Fellay et al., 2007; Butković et al., 2021; Choudhury et al., 2019; COVID-19 Host Genetics Initiative, 2021; Crosslin et al., 2015; Adebamowo et al., 2020; Foresman et al., 2016; Feng et al., 2019; Liu et al., 2021; Garcia-Etxebarria et al., 2015; Montes et al., 2021; Pimenta et al., 2020; Rubio et al., 2019; Tian et al., 2017; Xiao et al., 2019; Yang et al., 2019; Zhang et al., 2020; Zignego et al., 2014). Identification of important factors in viral infection is crucial for understanding of disease dynamics and prevention since there is evidence that infection outcomes depend on host-pathogen interaction (for a thorough review on host-pathogen interactions and virulence see (Casadevall and Pirofski, 2000; Casadevall and Pirofski, 2001)).
We would like to more closely illustrate the accomplished GWAS in virology by focusing on virulence-related traits of a human viral pathogen, human immunodeficiency virus type 1 (HIV-1; species Human immunodeficiency virus 1, genus Lentivirus, family Retroviridae), and a plant pathogen, turnip mosaic virus (TuMV; species Turnip mosaic virus, genus Potyvirus, family Potyviridae). These studies highlight the importance of choosing well-defined phenotypes, sample sizes, host genetics, virus strains, environment and the power to detect variants with small effect sizes.
The first ever GWAS analyzing viral infection traits was reported for this virus (Fellay et al., 2007), wherein host genetic variants associated with HIV-1 RNA viral load, as a determinant of disease progression, were found. This study was performed on 486 European individuals and identified two significant polymorphisms for viral load and seven for disease progression. In the case of the viral load, the first one was SNP rs2395029 near the human leukocyte antigen (HLA) complex P5 (HCP5) gene that is localized within the major histocompatibility complex (MHC) class I region. This SNP is known to be in strong linkage disequilibrium with the HLA-B gene allele 57:01, previously described as associated with restriction of HIV-1 viral replication and low viral load (Migueles et al., 2000; Altfeld et al., 2003). The other significant SNP, rs9264942, is located 35 kb from the transcription initiation of the HLA-C gene and 156 kbp from the HCP5 and might be involved in the control of HIV-1 disease progression (Fellay et al., 2007; Thomas et al., 2009; Kulkarni et al., 2011). For disease progression, Fellay et al. (Fellay et al., 2007) identified seven significant SNPs near the ring finger protein 39 and zinc ribbon domain–containing one genes that can have an effect on HIV-1 disease progression. Following-up, Fellay et al. (Fellay et al., 2009) repeated a GWAS on more than 2,500 individuals. They confirmed the association with the HCP5 gene and the 35 kbp HLA-C region along with some other independent loci in the MHC region. Both studies by Fellay et al., however, failed to identify previously known HIV-1 resistance genes such as CC chemokine receptor type 5 (CCR5) (Lodowski and Palczewski, 2009) and transportin-3 (Rodríguez-Mora et al., 2019) which could result from the stringent statistical criteria in the GWAS, dozens of SNPs explaining only a small proportion of the variance, presence of rare variants or misclassification of cases and controls (Fellay et al., 2009; Sebastiani et al., 2009). Nonetheless, the CCR5 and HLA associations were found in a meta-analysis of 6,315 individuals (McLaren et al., 2015). Also, subsequent studies confirmed the previously identified associations (Fellay et al., 2007; Fellay et al., 2009) and discovered new ones associated with HIV-1 plasma levels in seroconverters (Dalmasso et al., 2008), rapid progression (Limou et al., 2009) and non-progression of AIDS (Le Clerc et al., 2009). The studies done on HIV-1 demonstrate the importance of sample size and correct labelling of cases and controls, where larger studies are able to find more meaningful associations.
TuMV causes significant loses of Brassicaceae crops around the world and ranks as one of the most damaging vegetable viruses worldwide (Tomlinson, 1987; Walsh and Jenner, 2002). It causes a variety of symptoms from chlorosis, stunting and necrosis (Butković et al., 2021).
So far, three GWAS have focused on TuMV, two studies used A. thaliana (Rubio et al., 2019; Butković et al., 2021) and one used Chinese cabbage (Brassica campestris ssp. Pekinensis) (Zhang et al., 2020) as hosts. The number of individuals greatly varied among these studies, Zhang et al. (Zhang et al., 2020) used 83 cabbage varieties in a controlled experimental setting, measuring virus load by RT-qPCR. Rubio et al. (Rubio et al., 2019) used 317 accessions and measured viral accumulation, frequency of infected plants and symptomatology across 2 years in a natural setting using the UK1 isolate. Butković et al. (Butković et al., 2021) used 450 accessions in a controlled experimental setting with generalist and specialist strains derived from the YC5 isolate, measuring disease progression, symptom severity and percentage of infected plants. There are overlaps between the genetic regions where the significant SNPs were mapped in all three studies but also many differences: e.g., significant SNPs mapped in DNA-J heat shock proteins for all three studies while SNPs mapping in nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins were found by Butković et al. and Zhang et al. and SNPs in F-box family proteins were mapped by Zhang et al. and Rubio et al. The rest of the significant variants were mapped in genes with a wide variety of functions, most of which were previously described as related to disease resistance or susceptibility. Interestingly, Butković et al. described for the first time a factor related to symptom severity within gene AT2G14080, which is an NBS-LRR class protein. These proteins recognize specific pathogen domains and lead to resistance or a hypersensitive immune response that leads to cell death or necrosis (Marone et al., 2013). These three GWAS are a good example of how viral evolutionary history, selected traits, environment and host selection influence the genetic variants associated with the pathogen. Compared to HIV-1, smaller sample sizes in A. thaliana allow a pretty good recovery of meaningful associations. The influence of factors besides host and pathogen genetics should always be considered when planning or analyzing results on GWAS of infectious diseases.
GWAS has proven to be an increasingly useful tool in genetic studies of viral infectious diseases because of its large applicability, analysis of associations between thousands to millions of genetic variants with a phenotype of interest, availability of methods used and simplicity of the underlying assumptions. Based on the hypothesis common gene common variant, it allows identification of common genetic variants associated with disease that can be corroborated in subsequent studies. Genotyping array methods used in GWAS are cheap, reliable and give good genome coverage. There is a wide variety of open-access computational GWAS methods to choose from (Supplementary Table S1), in many cases without the need for large computational power since they are available online. GWAS can serve as a preliminary study that gives information on the genetic architecture of a trait and allows for an informed choice on possible candidates for QTL, mutagenesis and transgenic studies (Figure 1). Virologists should profit from this by being able to shed light on the genetics underlying many viral diseases. Another advantage of GWAS is the possibility of comparing and integrating analyses across experiments into a large meta-analysis that increases statistical power (Seren et al., 2013; Seren et al., 2017; Lee and Lee, 2018; Buniello et al., 2019; Kim et al., 2019; Togninalli et al., 2019).
AB, conceptualization, writing—original draft and editing. SE, writing and editing. All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
AB was supported by Generalitat Valenciana grant GRISOLIAP/2018/005 and Foundation pour la Recherche Mèdicale SPF202110014092 post-doc grant. SE was supported by grants PID 2019-10399 GB-I00 (Spain Agencia Estatal de Investigación-FEDER) and PROMETEU 2019/012 (Generalitat Valenciana).
The authors thank Rubén González for critically reading the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsysb.2022.1005758/full#supplementary-material
Adebamowo, S. N., Adeyemo, A. A., Rotimi, C. N., Olaniyan, O., Offiong, R., Adebamowo, C. A., et al. H3Africa ACCME Research Group (2020). Genome-wide association study of prevalent and persistent cervical high-risk human papillomavirus (HPV) infection. BMC Med. Genet. 21, 231. doi:10.1186/s12881-020-01156-1
Altfeld, M., Addo, M. M., Rosenberg, E. S., Hecht, F. M., Lee, P. K., Vogel, M., et al. (2003). Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17, 2581–2591. doi:10.1097/00002030-200312050-00005
Aschard, H., Chen, J., Cornelis, M. C., Chibnik, L. B., Karlson, E. W., and Kraft, P. (2012). Inclusion of gene-gene and gene-environment interactions unlikely to dramatically improve risk prediction for complex diseases. Am. J. Hum. Genet. 90, 962–972. doi:10.1016/j.ajhg.2012.04.017
WHO Ebola Response Team Aylward, B., Barboza, P., Bawo, L., Bertherat, E., Bilivogui, P., et al. (2014). Ebola virus disease in west Africa — The first 9 months of the epidemic and forward projections. N. Engl. J. Med. 371, 1481–1495. doi:10.1056/NEJMoa1411100
Balding, D. J. (2006). A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 7, 781–791. doi:10.1038/nrg1916
Barrett, T., and Rossiter, P. B. (1999). Rinderpest: The disease and its impact on humans and animals. Adv. Virus Res. 53, 89–110. doi:10.1016/s0065-3527(08)60344-9
Behbehani, A. M. (1983). The smallpox story: Life and death of an old disease. Microbiol. Rev. 47, 455–509. doi:10.1128/mr.47.4.455-509.1983
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). Tassel: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi:10.1093/bioinformatics/btm308
Buniello, A., MacArthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005-D1012. –D1012. doi:10.1093/nar/gky1120
Bush, W. S., and Moore, J. H. (2012). Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 8, e1002822. doi:10.1371/journal.pcbi.1002822
Butković, A., González, R., Rivarez, M. P. S., and Elena, S. F. (2021). A genome-wide association study identifies Arabidopsis thaliana genes that contribute to differences in the outcome of infection with two Turnip mosaic potyvirus strains that differ in their evolutionary history and degree of host specialization. Virus Evol. 30, veab063. veab063. doi:10.1093/ve/veab063
Butković, A., Ellis, T. J., González, R., Jaegle, B., Nordborg, M., and Elena, S. F. (2022). A globally distributed major virus-resistance association in Arabidopsis thaliana (preprint)biorxiv.www.biorxiv.org/content/10.1101/2022.08.02.502433v1.doi:10.1101/2022.08.02.502433
Casadevall, A., and Pirofski, L. (2000). Host-pathogen interactions: Basic concepts of microbial commensalism, colonization, infection, and disease. Infect. Immun. 68, 6511–6518. doi:10.1128/iai.68.12.6511-6518.2000
Casadevall, A., and Pirofski, L. (2001). Host-pathogen interactions: The attributes of virulence. J. Infect. Dis. 184, 337–344. doi:10.1086/322044
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
Chang, M., He, L., and Cai, L. (2018). “An overview of genome-wide association studies” p 97–108.in Computational Systems Biology, methods in molecular Biology. Editor T. Huang (New York, NY: Springer New York).
Childs, L. H., Lisec, J., and Walther, D. (2012). Matapax: An online high-throughput genome-wide association study pipeline. Plant Physiol. 158, 1534–1541. doi:10.1104/pp.112.194027
Choudhury, S., Larkin, P., Xu, R., Hayden, M., Forrest, K., Meinke, H., et al. (2019). Genome wide association study reveals novel QTL for barley yellow dwarf virus resistance in wheat. BMC Genomics 20, 891. doi:10.1186/s12864-019-6249-1
Cohen, M. S., Hellmann, N., Levy, J. A., DeCock, K., and Lange, J. (2008). The spread, treatment, and prevention of HIV-1: Evolution of a global pandemic. J. Clin. Invest. 118, 1244–1254. doi:10.1172/JCI34706
Cook, J. P., and Morris, A. P. (2016). Multi-ethnic genome-wide association study identifies novel locus for type 2 diabetes susceptibility. Eur. J. Hum. Genet. 24, 1175–1180. doi:10.1038/ejhg.2016.17
COVID-19 Host Genetics Initiative (2021). The mutation that helps Delta spread like wildfire. Nature 600, 472–473. doi:10.1038/d41586-021-02275-2
Crosslin, D. R., Carrell, D. S., Burt, A., Kim, D. S., Underwood, J. G., Hanna, D. S., et al. (2015). Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 16, 1–7. doi:10.1038/gene.2014.51
Dalmasso, C., Carpentier, W., Meyer, L., Rouzioux, C., Goujard, C., Chaix, M-L., et al. (2008). Distinct genetic loci control plasma HIV-RNA and cellular HIV-DNA levels in HIV-1 infection: The ANRS Genome Wide Association 01 Study. PLoS ONE 3, e3907. doi:10.1371/journal.pone.0003907
De Coster, W., Weissensteiner, M. H., and Sedlazeck, F. J. (2021). Towards population-scale long-read sequencing. Nat. Rev. Genet. 22, 572–587. doi:10.1038/s41576-021-00367-3
Dehghan, A. (2018). “Genome-wide association studies, p 37-49,” in Genetic epidemiology, methods in molecular Biology. Editor E. Evangelou (New York, NY: Springer New York).
Fadista, J., Manning, A. K., Florez, J. C., and Groop, L. (2016). The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 24, 1202–1205. doi:10.1038/ejhg.2015.269
Fellay, J., Ge, D., Shianna, K. V., Colombo, S., Ledergerber, B., Cirulli, E. T., et al. (2009). Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 5, e1000791. doi:10.1371/journal.pgen.1000791
Fellay, J., Shianna, K. V., Ge, D., Colombo, S., Ledergerber, B., Weale, M., et al. (2007). A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947. doi:10.1126/science.1143767
Feng, Z., Kang, H., Li, M., Zou, L., Wang, X., Zhao, J., et al. (2019). Identification of new rice cultivars and resistance loci against rice black-streaked dwarf virus disease through genome-wide association study. Rice 12, 49. doi:10.1186/s12284-019-0310-1
Foresman, B. J., Oliver, R. E., Jackson, E. W., Chao, S., Arruda, M. P., and Kolb, F. L. (2016). Genome-wide association mapping of Barley yellow dwarf virus tolerance in spring oat (Avena sativa L.). PLoS ONE 11, e0155376. doi:10.1371/journal.pone.0155376
Frazer, K. A., Murray, S. S., Schork, N. J., and Topol, E. J. (2009). Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251. doi:10.1038/nrg2554
Garcia-Etxebarria, K., Bracho, M. A., Galán, J. C., Pumarola, T., Castilla, J., Ortiz de Lejarazu, R., et al. (2015). CIBERESP Cases and Controls in Pandemic Influenza Working GroupNo major host genetic risk factor contributed to A(H1N1)2009 influenza severity. PLoS ONE 10, e0135983. doi:10.1371/journal.pone.0135983
Genissel, A., Confais, J., Lebrun, M-H., and Gout, L. (2017). Association genetics in plant pathogens: Minding the gap between the natural variation and the molecular function. Front. Plant Sci. 8, 1301. doi:10.3389/fpls.2017.01301
Gokhale, D. V., and Bald, J. G. (1987). Relationship between plant virus concentration and infectivity: A ‘growth curve’ model. J. Virol. Methods 18, 225–232. doi:10.1016/0166-0934(87)90084-x
Grimm, D. G., Roqueiro, D., Salomé, P. A., Kleeberger, S., Greshake, B., Zhu, W., et al. (2017). easyGWAS: a cloud-based platform for comparing the results of genome-wide association studies. Plant Cell 29, 5–19. doi:10.1105/tpc.16.00551
Gumpinger, A. C., Roqueiro, D., Grimm, D. G., and Borgwardt, K. M. (2018). “Methods and tools in genome-wide association studies” p 93–136.in Computational cell Biology, methods in molecular Biology. Editors L. von Stechow, and A. Santos Delgado (New York, NY: Springer New York).
Höglund, J., Rafati, N., Rask-Andersen, M., Enroth, S., Karlsson, T., Ek, W. E., et al. (2003). Improved power and precision with whole genome sequencing data in genome-wide association studies of inflammatory biomarkers. Sci. Rep. 9, 16844. doi:10.1038/s41598-019-53111-7
Hong, E. P., and Park, J. W. (2012). Sample size and statistical power calculation in genetic association studies. Genomics Inf. 10, 117–122. doi:10.5808/GI.2012.10.2.117
Johnson, J. L., and Abecasis, G. R. (2017). GAS power calculator: Web-based power calculator for genetic association studies (preprint) biorxiv, https://www.biorxiv.org/content/10.1101/164343v1.Bioinformatics. doi:10.1101/164343
Jones, R. A. C. (2021). Global plant virus disease pandemics and epidemics. Plants 10, 233. doi:10.3390/plants10020233
Kim, B., Dai, X., Zhang, W., Zhuang, Z., Sanchez, D. L., Lübberstedt, T., et al. (2019). GWASpro: A high-performance genome-wide association analysis server. Bioinformatics 35, 2512–2514. doi:10.1093/bioinformatics/bty989
Korte, A., and Farlow, A. (2013). The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 9, 29. doi:10.1186/1746-4811-9-29
Korte, A., Vilhjálmsson, B. J., Segura, V., Platt, A., Long, Q., and Nordborg, M. (2012). A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat. Genet. 44, 1066–1071. doi:10.1038/ng.2376
Kulkarni, S., Savan, R., Qi, Y., Gao, X., Yuki, Y., Bass, S. E., et al. (2011). Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 472, 495–498. doi:10.1038/nature09914
Le Clerc, S., Limou, S., Coulonges, C., Carpentier, W., Dina, C., Taing, L., et al. (2009). Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03). J. Infect. Dis. 200, 1194–1201. doi:10.1086/605892
Lee, T., and Lee, I. (2018). araGWAB: Network-based boosting of genome-wide association studies in Arabidopsis thaliana. Sci. Rep. 8, 2925. doi:10.1038/s41598-018-21301-4
Li, Y. R., and Keating, B. J. (2014). Trans-ethnic genome-wide association studies: Advantages and challenges of mapping in diverse populations. Genome Med. 6, 91. doi:10.1186/s13073-014-0091-5
Limou, S., Le Clerc, S., Coulonges, C., Carpentier, W., Dina, C., Delaneau, O., Labib, T., Taing, L., Sladek, R., Deveau, C., Ratsimandresy, R., Montes, M., Spadoni, J., Lelièvre, J., Lévy, Y., Therwath, A., Schächter, F., Matsuda, F., Gut, I., Froguel, P., Delfraissy, J., Hercberg, S., and Zagury, J.ANRS Genomic Group (2009). Genome wide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02). J. Infect. Dis. 199, 419–426. doi:10.1086/596067
Lippert, C., Casale, F. P., Rakitsch, B., and Stegle, O. (2014). Limix: Genetic analysis of multiple traits (preprint). bioRxiv. doi:10.1101/003905
Lippert, C., Listgarten, J., Liu, Y., Kadie, C. M., Davidson, R. I., and Heckerman, D. (2011). FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835. doi:10.1038/nmeth.1681
Liu, Q., Lan, G., Zhu, Y., Chen, K., Shen, C., Zhao, X., et al. (2021). Genome-wide association study on resistance to rice black-streaked dwarf disease caused by Rice black-streaked dwarf virus. Plant Dis. 105, 607–615. doi:10.1094/PDIS-10-19-2263-RE
Lodowski, D. T., and Palczewski, K. (2009). Chemokine receptors and other G protein-coupled receptors. Curr. Opin. HIV AIDS 4, 88–95. doi:10.1097/COH.0b013e3283223d8d
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi:10.1038/nature08494
Marone, D., Russo, M., Laidò, G., De Leonardis, A., and Mastrangelo, A. (2013). Plant nucleotide binding site–leucine-rich repeat (NBS-lrr) genes: Active guardians in host defense responses. Int. J. Mol. Sci. 14, 7302–7326. doi:10.3390/ijms14047302
McLaren, P. J., Coulonges, C., Bartha, I., Lenz, T. L., Deutsch, A. J., Bashirova, A., et al. (2015). Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. U. S. A. 112, 14658–14663. doi:10.1073/pnas.1514867112
Migueles, S. A., Sabbaghian, M. S., Shupert, W. L., Bettinotti, M. P., Marincola, F. M., Martino, L., et al. (2000). HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. U. S. A. 97, 2709–2714. doi:10.1073/pnas.050567397
Montes, N., Cobos, A., Gil-Valle, M., Caro, E., and Pagán, I. (2021). Arabidopsis thaliana genes associated with Cucumber mosaic virus virulence and their link to virus seed transmission. Microorganisms 9, 692. doi:10.3390/microorganisms9040692
Mozzi, A., Pontremoli, C., and Sironi, M. (2018). Genetic susceptibility to infectious diseases: Current status and future perspectives from genome-wide approaches. Infect. Genet. Evol. 66, 286–307. doi:10.1016/j.meegid.2017.09.028
Pavan, S., Delvento, C., Ricciardi, L., Lotti, C., Ciani, E., and D’Agostino, N. (2020). Recommendations for choosing the genotyping method and best practices for Quality control in crop genome-wide association studies. Front. Genet. 5, 447. doi:10.3389/fgene.2020.00447
Pearson, T. A., and Manolio, T. A. (2008). How to interpret a genome-wide association study. JAMA 299, 1335–1344. doi:10.1001/jama.299.11.1335
Pimenta, R. J. G., Aono, A. H., Villavicencio Burbano, R. C., Coutinho, A. E., da Silva, C. C., dos Anjos, I. A., et al. (2020). Genome-wide approaches for the identification of markers and genes associated with sugarcane yellow leaf virus resistance. Sci. Rep. 11, 15730. doi:10.1038/s41598-021-95116-1
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi:10.1038/ng1847
Pulit, S. L., de With, S. A. J., and de Bakker, P. I. W. (2017). Resetting the bar: Statistical significance in whole-genome sequencing-based association studies of global populations. Genet. Epidemiol. 41, 145–151. doi:10.1002/gepi.22032
Purcell, S., Cherny, S. S., and Sham, P. C. (2003). Genetic power calculator: Design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150. doi:10.1093/bioinformatics/19.1.149
Rodríguez-Mora, S., De Wit, F., García-Perez, J., Bermejo, M., López-Huertas, M. R., Mateos, E., et al. (2019). The mutation of Transportin 3 gene that causes limb girdle muscular dystrophy 1F induces protection against HIV-1 infection. PLoS Pathog. 15, e1007958. doi:10.1371/journal.ppat.1007958
Rubio, B., Cosson, P., Caballero, M., Revers, F., Bergelson, J., Roux, F., et al. (2019). Genome-wide association study reveals new loci involved in Arabidopsis thaliana and Turnip mosaic virus (TuMV) interactions in the field. New Phytol. 221, 2026–2038. doi:10.1111/nph.15507
Rushton Macchioni, F., Chelucci, L., and Torracca, B. (2015). Economic impact of bluetongue: A review of the effects on production. Vet. Ital. 51, 401–406. doi:10.12834/VetIt.646.3183.1
Sankaran, N., and Weiss, R. A. (20212021). Viruses: Impact on science and society. Encycl. Virology, 2021 671–680. doi:10.1016/b978-0-12-814515-9.00075-8
Sebastiani, P., Timofeev, N., Dworkis, D. A., Perls, T. T., and Steinberg, M. H. (2009). Genome-wide association studies and the genetic dissection of complex traits. Am. J. Hematol. 84, 504–515. doi:10.1002/ajh.21440
Segura, V., Vilhjálmsson, B. J., Platt, A., Korte, A., Seren, Ü., Long, Q., et al. (2012). An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830. doi:10.1038/ng.2314
Seren, Ü., Grimm, D., Fitz, J., Weigel, D., Nordborg, M., Borgwardt, K., et al. (2017). AraPheno: A public database for Arabidopsis thaliana phenotypes. Nucleic Acids Res. 45, D1054-D1059–D1059. doi:10.1093/nar/gkw986
Seren, Ü. (2018). “GWA-portal: Genome-wide association studies made easy p 303–319,” in Root development, methods in molecular Biology. Editors D. Ristova, and E. Barbez (New York, NY: Springer New York).
Seren, Ü., Vilhjálmsson, B. J., Horton, M. W., Meng, D., Forai, P., Huang, Y. S., et al. (2013). Gwapp: A web application for genome-wide association mapping in arabidopsis. Plant Cell 24, 4793–4805. doi:10.1105/tpc.112.108068
Shriner, D. (2013). Overview of admixture mapping. Curr. Protoc. Hum. Genet. 76, Unit 1.23. doi:10.1002/0471142905.hg0123s76
Spreeuwenberg, P., Kroneman, M., and Paget, J. (2018). Reassessing the global mortality burden of the 1918 Influenza pandemic. Am. J. Epidemiol. 187, 2561–2567. doi:10.1093/aje/kwy191
Tam, V., Patel, N., Turcotte, M., Bossé, Y., Paré, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484. doi:10.1038/s41576-019-0127-1
The Wellcome Trust Case Control Consortium (2007). Genome-wide association study of 14, 000 cases of seven common diseases and 3, 000 shared controls. Nature 447, 661–678. doi:10.1038/nature05911
Thomas, R., Apps, R., Qi, Y., Gao, X., Male, V., O’hUigin, C., et al. (2009). HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 41, 1290–1294. doi:10.1038/ng.486
Thompson, D. K., Muriel, P., Russell, D., Osborne, P., Bromley, A., Rowland, M., et al. (2002). Economic costs of the foot and mouth disease outbreak in the United Kingdom in 2001. Rev. Sci. Tech. 21, 675–687. doi:10.20506/rst.21.3.1353
Tian, C., Hromatka, B. S., Kiefer, A. K., Eriksson, N., Noble, S. M., Tung, J. Y., et al. (2017). Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599. doi:10.1038/s41467-017-00257-5
Togninalli, M., Seren, Ü., Freudenthal, J. A., Monroe, J. G., Meng, D., Nordborg, M., et al. (2019). AraPheno and the AraGWAS catalog 2020: A major database update including RNA-seq and knockout mutation data for Arabidopsis thaliana. Nucleic Acids Res. 48, D1063-D1068–D1068. doi:10.1093/nar/gkz925
Tomlinson, J. A. (1987). Epidemiology and control of virus diseases of vegetables. Ann. Appl. Biol. 110, 661–681. doi:10.1111/j.1744-7348.1987.tb04187.x
Uitterlinden, A. G. (2016). An introduction to genome-wide association studies: GWAS for dummies. Semin. Reprod. Med. 34, 196–204. doi:10.1055/s-0036-1585406
Walsh, J. A., and Jenner, C. E. (2002). Turnip mosaic virus and the quest for durable resistance. Mol. Plant Pathol. 3, 289–300. doi:10.1046/j.1364-3703.2002.00132.x
Willer, C. J., Li, Y., and Abecasis, G. R. (2010). Metal: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. doi:10.1093/bioinformatics/btq340
Witte, J. S. (2010). Genome-wide association studies and beyond. Annu. Rev. Public Health 31, 9–20 4 p following 20. doi:10.1146/annurev.publhealth.012809.103723
Wu, Y., Zheng, Z., Visscher, P. M., and Yang, J. (2017). Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data. Genome Biol. 18, 86. doi:10.1186/s13059-017-1216-0
Xiao, S., Wang, B., Liu, Y., Miao, T., Zhang, H., Wen, P., et al. (2019). Genome-wide association study and linkage analysis on resistance to rice black-streaked dwarf virus disease. Mol. Breed. 39, 73. doi:10.1007/s11032-019-0980-9
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). Gcta: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi:10.1016/j.ajhg.2010.11.011
Yang, J., Zaitlen, N. A., Goddard, M. E., Visscher, P. M., and Price, A. L. (2014). Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 46, 100–106. doi:10.1038/ng.2876
Yang, X., Sood, S., Luo, Z., Todd, J., and Wang, J. (2019). Genome-wide association studies identified resistance loci to orange rust and yellow leaf virus diseases in sugarcane (Saccharum spp.). Phytopathology 109, 623–631. doi:10.1094/PHYTO-08-18-0282-R
Zaitlen, N., and Kraft, P. (2012). Heritability in the genome-wide association era. Hum. Genet. 131, 1655–1664. doi:10.1007/s00439-012-1199-6
Zhang, R., Liu, C., Song, X., Sun, F., Xiao, D., Wei, Y., et al. (2020). Genome-wide association study of turnip mosaic virus resistance in non-heading Chinese cabbage. 3 Biotech. 10, 363. doi:10.1007/s13205-020-02344-9
Zhou, X., and Stephens, M. (2014). Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 11, 407–409. doi:10.1038/nmeth.2848
Zhou, X., and Stephens, M. (2012). Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824. doi:10.1038/ng.2310
Keywords: GWAS, host genetic variability, proviral genes, antiviral genes, infection
Citation: Butković A and Elena SF (2022) Genome-wide association studies of viral infections—A short guide to a successful experimental and statistical analysis. Front. Syst. Biol. 2:1005758. doi: 10.3389/fsysb.2022.1005758
Received: 29 July 2022; Accepted: 30 November 2022;
Published: 12 December 2022.
Edited by:
Riyan Cheng, University of California, San Diego, United StatesReviewed by:
Bin Guo, Merck, United StatesCopyright © 2022 Butković and Elena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anamarija Butković, YW5hbWFyaWphLmJ1dGtvdmljQHBhc3RldXIuZnI=
†ORCID: Anamarija Butković, orcid.org/0000-0002-1435-0912; Santiago F. Elena, orcid.org/0000-0001-08249-5593
‡Present address: Institute Pasteur, Archaeal Virology Unit, Paris, France
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.