 Debolina Banerjee
Debolina Banerjee Jun Feng
Jun Feng Frank W. Sellke
Frank W. Sellke
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Surg. , 03 July 2024
Sec. Cardiovascular Surgery
Volume 11 - 2024 | https://doi.org/10.3389/fsurg.2024.1224068
This article is part of the Research Topic Inflammation and Heart Surgery View all 12 articles
Cardiopulmonary bypass (CPB) initiates an intense inflammatory response due to various factors: conversion from pulsatile to laminar flow, cold cardioplegia, surgical trauma, endotoxemia, ischemia-reperfusion injury, oxidative stress, hypothermia, and contact activation of cells by the extracorporeal circuit. Redundant and overlapping inflammatory cascades amplify the initial response to produce a systemic inflammatory response, heightened by coincident activation of coagulation and fibrinolytic pathways. When unchecked, this inflammatory response can become maladaptive and lead to serious postoperative complications. Concerted research efforts have been made to identify technical refinements and pharmacologic interventions that appropriately attenuate the inflammatory response and ultimately translate to improved clinical outcomes. Surface modification of the extracorporeal circuit to increase biocompatibility, miniaturized circuits with sheer resistance, filtration techniques, and minimally invasive approaches have improved clinical outcomes in specific populations. Pharmacologic adjuncts, including aprotinin, steroids, monoclonal antibodies, and free radical scavengers, show real promise. A multimodal approach incorporating technical, circuit-specific, and pharmacologic strategies will likely yield maximal clinical benefit.
The advent of cardiopulmonary bypass (CPB) revolutionized cardiac surgery and dramatically improved patient outcomes. CPB activates various inflammation, coagulation, fibrinolysis, apoptosis, and oxidation pathways. Exposure to nonphysiologic conditions during CPB leads to an intense immunologic response that can affect the function and recovery of multiple organ systems (1).
The body's immunologic response is designed to sequester and destroy what it recognizes as foreign. The initial stimulus or signal undergoes amplification due to redundant and synergistic inflammatory cascades. Frequently, this results in activation of both humoral and cellular components of the immune system, and the initial inflammatory response to CPB is no different. The activation of these pathways leads to release of multiple humoral mediators. Leukocytes, chiefly neutrophils, are then drawn to the site of production of these mediators, become activated, and subsequently adhere to endothelial cells by way of receptor interactions with adhesion molecules. Endothelial cells in turn become activated, rendering regional and remote capillary endothelium permeable to further migration of neutrophils and other intravascular molecules. Extravasation of neutrophils en masse results in release of large amounts of cytokines, chemokines, vasoactive substances, proteases of the coagulation and fibrinolysis systems, cytotoxins, and reactive oxygen species (ROS). Elimination of foreign antigen is the final step of this cascade of events.
Immunologic activation leading to inflammation is usually physiologic and protective, often leading to self-limited or subclinical organ dysfunction. However, on occasion, nonimmunologic activation that persists following CPB can be maladaptive and may progress beyond restoring homeostasis to marked fluid shifts and formation of microemboli, particularly in high-risk patients with limited functional reserve. This exuberant systemic inflammatory response to CPB, often characterized as systemic inflammatory response syndrome (SIRS), may manifest as clinically significant increase in capillary permeability, interstitial edema, and organ dysfunction. The link between CPB-induced inflammatory response and adverse clinical outcomes is still not well delineated. Several hypotheses have been proposed. One hypothesis suggests the balance between pro-inflammatory and anti-inflammatory cytokine release correlates with the magnitude of multiorgan injury (2). Temporal and magnitude changes in cytokine production patterns may further influence the clinical presentation and course of SIRS postoperatively (3, 4). A second hypothesis suggests SIRS is the result of a multifaceted response with overall cytokine upregulation leading to both a proinflammatory state (SIRS) as well as homeostatic, compensatory anti-inflammatory response syndrome (CARS) leading to systemic deactivation of the immune response, predisposing the patient to immunosuppression and infectious complications (5). The multi-hit hypothesis suggests CPB primes polymorphonuclear leukocytes such that subsequent exposure to stimuli that otherwise may be self-limiting (i.e., postoperative infection or ongoing ischemia) results in enhanced cytotoxin release and downstream organ dysfunction (5). This priming is thought to occur through various processes, including the secretion of cytokines, leading to pulmonary leucosequestration (5).
These harmful inflammatory effects are due to the interactions of a wide spectrum of compounds, including inflammatory triggers (complement-derived factors), mediators (cytokines and adhesion molecules), or effectors (proteolytic enzymes, oxygen free radicals, arachidonic acid metabolites, and immune cells). Hallmarks of ensuing multiorgan dysfunction include coagulopathy, myocardial dysfunction, pulmonary and renal insufficiency, neurocognitive deficits, hepatic injury, splanchnic bed hypoperfusion and bacterial translocation. Collectively, these multiorgan effects have been linked to post-perfusion/post-pump syndrome and ischemia-reperfusion injury.
Despite drawbacks attributed to the attendant inflammatory response, CPB remains a mainstay technique in cardiothoracic surgery, as it allows for adequate exposure of the lateral and posterior coronary arteries and facilitates a bloodless field in which to operate. While more drastic strategies have been employed to blunt excessive inflammatory response and its sequelae (including off-pump coronary artery bypass and minimally invasive techniques), an integrated approach to modulate the stress response incorporating pharmacologic measures and technical refinements has shown promise. Herein, we characterize the pathophysiology of the inflammatory response, and discuss potential strategies to intercept and attenuate this response.
Compared with off-pump approaches, CPB may trigger an intense physiologic response due to several stimuli (Figure 1) (6, 7):
• Blood contact with synthetic surfaces within the perfusion circuit and multiple tissues within the wound
• Abnormal blood-gas interface
• Pulsatile flow converted to laminar flow
• Hypothermia
• Surgical trauma
• Global myocardial ischemia during cardioplegic protection
• Ischemia-reperfusion injury to end-organs
• Endotoxemia proceeding from splanchnic hypoperfusion and bacterial translocation
The inflammatory response following CPB is multifactorial, and may become generalized and uncontrolled, leading to SIRS. The “early” phase is initiated by blood contact with nonendothelial surfaces of the extracorporeal circuit and ultimately involves both humoral and cellular constituents of the immune system. The “late” phase that perpetuates inflammatory cascades is characterized by ischemia-reperfusion injury, endotoxemia, coagulopathy, and heparin-protamine complex reactions (Table 1). The link between inflammatory, coagulation, and fibrinolytic cascades is complex and may partially be explained by acute phase reactions during CPB similar to those seen in sepsis (8). Another link may be nuclear factor kappa B (NFκB), a ubiquitous and inducible transcription factor that is implicated during all phases of the response but plays a central role in regulating pro-inflammatory genes during the acute phase reaction (9).
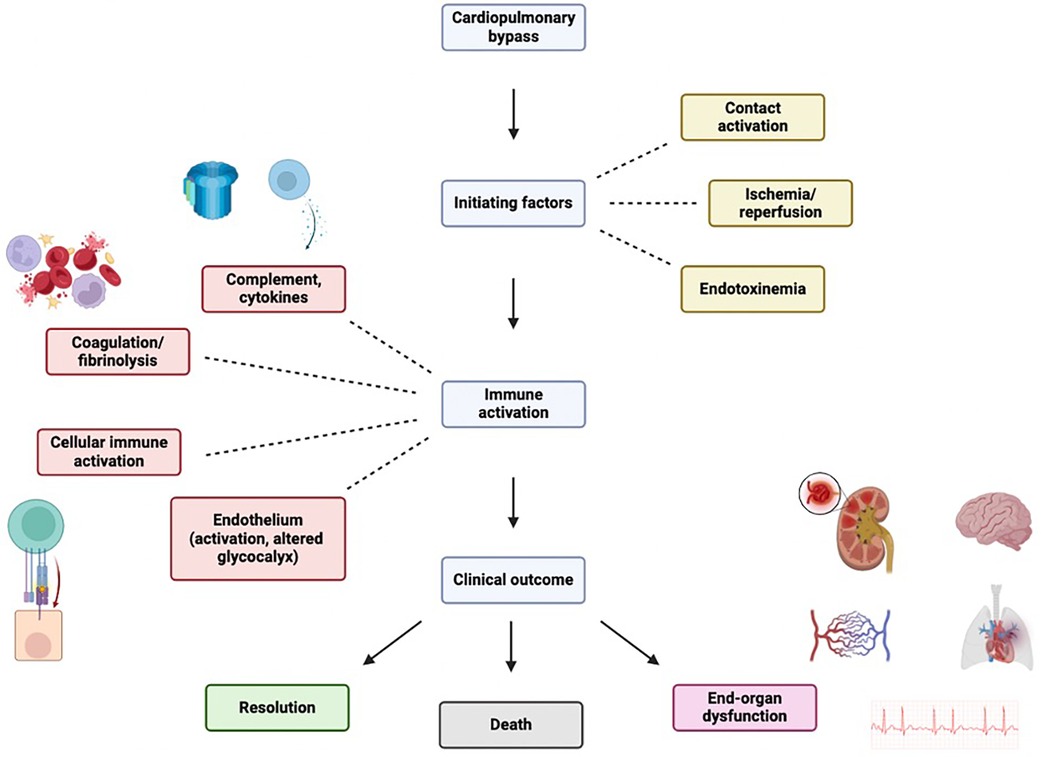
Figure 1 Inflammantory cascade following cardiopulmary bypass initiation. Figure created using Biorender.com.
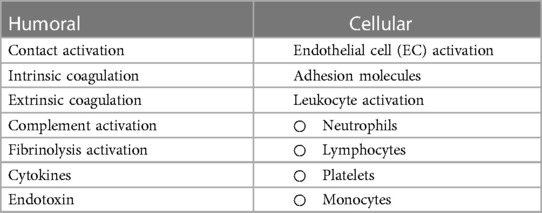
Table 1 Humoral and cellular factors influencing systemic inflammatory response associated with CPB.
The exposure of blood to air and nonphysiologic surfaces of the extracorporeal circuit leads to simultaneous activation of coagulation and fibrinolysis cascades as well as the complement pathways of innate immunity (Figure 2).
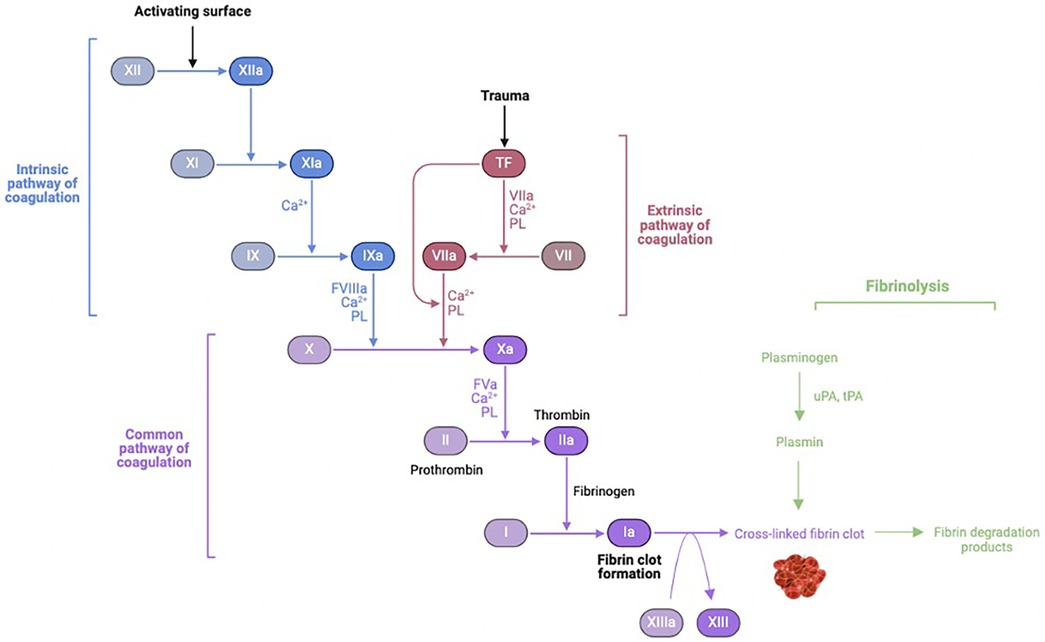
Figure 2 Coagulation-fibrinolysis pathway activation. uPA, urokinase; tPa, tissue plasminogen activator. Figure created using Biorender.com.
Four proteins are involved in the contact activation pathway: factor XII (Hageman factor), factor XI, prekallikrein, and high-molecular-weight kininogen (HMWK). Upon exposure of blood to foreign material of the CPB circuit, factor XII is converted to its active form in the presence of prekallikrein and HMWK. Activated factor XII activates factor XI of the intrinsic pathway, ultimately leading to thrombin formation. Activated factor XII also converts prekallikrein to kallikrein. Kallikrein then: (1) feeds back to facilitate continued activation of factor XII, (2) induces cleavage of HMWK to form bradykinin, (3) potentiates mediators of the alternative complement pathway, and (4) promotes conversion of plasminogen to plasmin. Plasmin is an important link to the fibrinolytic cascade. Plasminogen is activated to plasmin by activated factor XII in the intrinsic pathway during contact activation, and by tPA as part of the extrinsic pathway later during CPB. In addition to thrombin, inflammatory mediators (cytokines and endotoxin) can activate plasminogen. Activated factor XII and plasmin both play a role in stimulating the classical complement pathway (10).
Activation of prothrombin to thrombin by way of the extrinsic pathway contributes significantly to systemic thrombin generation and thrombus formation. Circulating levels of tissue factor and activated factor VII increase following CPB and surgical trauma, correlating with stimulation of pro-inflammatory mediators interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and endotoxin (11–13). CPB necessitates anticoagulation in the form of high-dose heparin to prevent immediate clotting within the circuit. Heparin potentiates antithrombin III to inhibit thrombin and other coagulation factors (activated X, IX, XI, XII, and kallikrein). During CPB, fibrinogen and fibrin are readily deposited onto circuitry, thus creating a surface to which thrombin avidly adheres. Upon binding to deposited fibrinogen/fibrin, thrombin undergoes a conformational change that renders it resistant to inhibition by heparin-activated antithrombin III. Thus, while heparin can inhibit systemic thrombin, it is unable to inhibit surface-bound thrombin. The surface-bound thrombin continues to generate more circulating thrombin, that can in turn activate a number of constituent blood elements, including platelets. Activated platelets can bind fibrin- and fibrinogen-coated surface of CPB circuitry, and also can individually provide a scaffold for prothrombinase complexes to form and convert prothrombin to thrombin. While high-dose heparin can limit fibrin-rich thrombus during CPB, it cannot prevent thrombin generation per se. Thrombin levels, measured as thrombin-antithrombin complex and prothrombin fragment, increase within minutes of initiating CPB, further increase following discontinuation of CPB, and persist for up to 60 days after surgery (14–17). Heparin concentrations, antithrombin III levels, and activated coagulation time are not associated with thrombin generation (10, 18).
Activated factor XII, thrombin, kallikrein, and products of fibrinolysis potentiate the inflammatory response. Activated factor XII and kallikrein stimulate neutrophil aggregation and degranulation. Thrombin induces endothelial cells to express receptors to facilitate neutrophil binding. Fibrinogen fragment D (D-dimer) disrupts the integrity of endothelial cells and stimulates continued complement activation.
Contact activation by biomaterials is a critical inciting event to downstream generation of humoral mediators through its activation of the alternative complement pathway. The CPB circuit lacks inhibitors normally found on endothelial cells that limit cofactor C3 binding and activation. This contact activation, potentiated by kallikrein, leads to activation of C3 and C5 (19). Their active split products, C3a and C5a, are anaphylatoxins that are potent chemoattractants (20). Their activity is mediated by complement receptor type 1 (CR1), a transmembrane glycoprotein expressed on leukocytes that regulates complement pro-inflammatory activity while inhibiting other complement pathways. C3a is a potent stimulator of platelet aggregation. There is a significant rise in plasma C3a levels with CPB associated with its duration (2), which is not observed when CPB is avoided (19, 21–26). C5a avidly binds to neutrophil receptors (19, 24, 27, 28) thereby stimulating them to be chemotactically drawn to sites of C5a production (29, 30), aggregate and adhere to endothelial cells (31–33), degranulate to release proteases (29, 34), and produce ROS (35). C5a levels are more difficult to directly measure as C5a is internalized rapidly upon binding to neutrophil receptors, but several studies have shown decreased available C5a receptors and increased levels of terminal complement complex C5b-C9, which provide evidence for C5a generation during CPB (28, 36).
Endotoxin is a powerful activator of the alternative complement pathway, with a smaller role in activating the classical complement pathway (25). Endotoxin, or lipopolysaccharide Lipid A, arises from cell walls of gram-negative bacteria and is released upon disruption of their cell walls. They may appear in circulation during CPB due to contamination of CPB circuitry, pulmonary arterial catheters, intravenous fluids, or banked blood production. The primary mechanism for endotoxemia, however, is due to splanchnic hypoperfusion and vasoconstriction during aortic cross-clamping and resultant transient gastrointestinal bacterial translocation (37–40). Transient ischemia and laminar flow in the gut increases intestinal permeability (37, 41), facilitating endotoxin release into circulation (42). Here, endotoxin can activate complement and stimulate production of inflammatory cytokines (43, 44). Endotoxin circulates in plasma by binding to LPS-binding protein. This complex binds to CD14 receptors on macrophages, enhancing TNF-α production (25, 45, 46). Endotoxin also stimulates endothelial cells to produce IL-6 (43). Levels of circulating endotoxin rise during and after CPB (25, 40, 42, 47–50), and are correlated with duration of aortic cross-clamping during CPB (48). Elevated LPS levels lead to myocardial dysfunction (46). Increased endotoxin levels in children with congenital heart defects undergoing CPB are associated with increased mortality after CPB (51).
CPB induces the classical complement pathway through contact activation (activated factor XII and plasmin) and heparin reversal with protamine, as evidenced by increased levels of the terminal complex C5b-C9, C3a, and C4a (27, 28, 52, 53). This further augments levels of C3a and C5a. The extent of complement activation has been correlated with duration of CPB (19). It remains unclear whether complement activation portends worse outcomes. While higher levels of C3a have been reported in those requiring prolonged mechanical ventilation, other groups have failed to identify a correlation between complement activation and acute lung injury or adverse hemodynamic responses (54–56). Equivocal findings may be due to the difficulty of parsing out the role of the complement system in the context of the complex inflammatory response associated with CPB (57).
Following initiation of CPB, physiologic pulsatile aortic flow is converted to continuous laminar flow. Endothelial cells sense mechanical stresses through their attachments to the basement membrane and through membrane proteins on their luminal surface. Changes in mechanical stresses result in changes in downstream transcription of genes regulated by promoter regions containing shear stress-responsive elements. Conversion from pulsatile to laminar flow may induce expression of genes related to a pro-inflammatory phenotype. One group showed differential gene expression was responsible for a quiescent endothelial phenotype (lung Kruppel-like factor) after endothelial cells were exposed to pulsatile or laminar flow (58). Antioxidant proteins, including thioredoxin reductase and ferritin, were among shear-regulated gene products. Other studies have shown Mn2+- and Cu2+/Zn2+-superoxide dismutase to be shear-regulated as well (59). Reduced activation of transcription factor NFκB and expression of pro-inflammatory cytokines, including IL-6, TNF-α, and IL-1 (60, 61) as well as decreased endothelial activation (62, 63) in the group undergoing pulsatile perfusion compared to the group undergoing non-pulsatile perfusion has been described.
Free radicals are molecules with unpaired electrons that render them highly reactive. ROS are free radicals derived from oxygen, and can be formed by activated neutrophils during their cytotoxic oxidative burst as a response to C5a stimulation (64–66). Exposure of blood to material of the CPB circuit as well as ischemia-reperfusion both generate various ROS implicated in oxidative damage, including superoxide anion, hydrogen peroxide, hydroxyl radical, peroxynitrite, hypochlorous acid, and singlet oxygen. Natural defense mechanisms to neutralize ROS and restore balance (redox state) include enzymes and free radical scavengers. Antioxidant enzymes include superoxide dismutase, glutathione peroxidase, and catalases. Mitochondrial scavenger complexes integrate thioredoxin and peroxiredoxin proteins (67–70). Should the body's detoxifying capacity be outstripped, ROS can cause direct damage to endothelial cell and fibroblast membrane lipids compromising membrane integrity, render proteins dysfunctional, induce nucleic acid damage that results in downstream changes in transcriptional programs via NFκB modulation, and provide positive feedback to inflammatory cascades.
Direct measurement of ROS is difficult given its short half-life and highly reactive properties, but indirect methods via measurement of more stable intermediates have shown increased ROS activity during and after CPB (24, 71–74). The onset of CPB and aortic cross-clamping creates transient ischemia, subjecting the myocardium and other organs to direct hypoxic cellular damage. CPB itself may induce generation of ROS in the area drained by the inferior vena cava (73, 74) as well as systemic oxidative stress (75). Reperfusion occurs upon removing the aortic cross-clamp, which generates further oxidative stress through recruitment of activated neutrophils to post-ischemic tissue. Following reperfusion, ROS may impair nitric oxide (NO) availability and predispose myocardial vessels to spasm and thrombose (76–78). A correlation has also been observed between timing of lipid peroxidation and degree of complement activation (24). Hypothermia may also influence ROS production by altering neutrophil-endothelium interactions during CPB (79–82).
Cytokines are another major group of humoral mediators that play a central role in inflammation and cell signaling. The body produces cytokines constitutively whereby subsets of immune cells maintain baseline levels of cytokines under normal conditions. Cytokines must bind cell membrane receptors to exert their effects, and the action of one or more cytokines is necessary to mount an immune response. These molecules form an intricate network in the development of inflammation, as the production of one cytokine influences the synthesis or response of others. The overlapping actions of different cytokines can be explained by both pleiotropy (single cytokine: multiple effects) and redundancy (multiple cytokines: same effect). The ability of certain cytokines to signal via more than one type of receptor complex also contributes to their pleiotropic actions wherein separately activated pathways contribute to distinct downstream effects. Simultaneously, redundant actions of cytokines allow for signal amplification; different cytokine receptors with similar motifs mediate coupling to other processes, ultimately leading to activation of converging inflammatory pathways in potentiating the immune response.
Broadly, cytokines include tumor necrosis factor, interleukins, interferons, and several growth factors. The production and release of cytokines is induced by complement factors and their degradation products during CPB-related acute phase reaction. A number of other factors may also contribute, including endotoxin, oxidative stress, ischemia-reperfusion, and effects of cytokines themselves. While cytokines are generally considered to be products of mature leukocytes, their secretion may be modulated by other cell types, including platelets and endothelial cells (83–90). The degree of the inflammatory response is strongly influenced by the balance between pro-inflammatory (TNF-α, IL-1, IL-6, and IL-8) and anti-inflammatory (IL-10) cytokines (3).
Tumor necrosis factor α (TNF-α, or cachectin) functions within a complex and tightly regulated cytokine network. It has a role in orchestrating the inflammatory response by inducing expression of other pro-inflammatory cytokines (IL-1 and IL-6) as well as increasing its own production. It functions in cell signaling through interactions with p55 and p75 receptors localized to the myocardium (91). It induces nitric oxide synthase and therefore increases concentrations of nitric oxide following CPB. It usually peaks shortly after surgery and then undergoes rapid degradation, but excessive production may lead to organ dysfunction. In the lung, it induces apoptosis and has been implicated in pulmonary complications. Infusion of antibodies to TNF-α prevented pulmonary edema, improved oxygenation, and significantly reduced markers of inflammation (neutrophil count, plasma TNF-α levels, malondialdehyde concentrations) in a rabbit model (92). TNF-α has been implicated in renal dysfunction as it induces fibrin deposition in the kidney glomerulus, promoting cell infiltration and vasoconstriction, thereby reducing glomerular filtration rate (93). TNF-α and IL-1 synergistically suppress myocardial contractility through a mechanism mediated by sphingosine impeding calcium-induced calcium release from the sarcoplasmic reticulum. This may result in low cardiac output, decreased vascular smooth muscle tone, and development of thrombosis, thereby contributing to dysfunction and hemodynamic instability after CPB (46, 94–96). Trends in TNF-α levels following CPB are variable, as some studies have shown increased levels while others have failed to demonstrate this. Additionally, inter-individual differences in TNF-α production may be attributed to genetic variability (97). In contrast to other cytokines (i.e., IL-6), there is no evidence that indicates TNF-α is released in large amounts following CPB (98).
Interleukins (IL) comprise a broad group of cytokines that function as intermediaries between different leukocytes and regulate various stages of the inflammatory response. IL-1 is an endogenous pyrogen. Levels of IL-1β usually increase after CPB, but this cytokine is difficult to detect due to hemodilutional effects of CPB. The IL-1 response pattern is consistent with its role as a key mediator of inflammation, both through its synergistic actions on TNF-α as well as its induction of other pro-inflammatory cytokines, including IL-6. IL-6 is a pleiotropic cytokine role that chiefly coordinates the acute phase reaction. It also induces the expression of adhesion molecules on cardiac myocytes to facilitate neutrophil adhesion, inhibits apoptosis in various cell types, enhances antibody production by activated B lymphocytes, and has negative inotropic effects on cardiac myocytes through induction of local nitric oxide release (99–104). In this way, it may be more a precise marker for progression of inflammation after CPB. A marked increased in IL-6 occurs during CPB, peaking within a few hours following CPB (105, 106), with a gradual decrease toward preoperative levels within 24 h (28). Peak IL-6 concentrations were a function of aortic cross-clamping duration. This characteristic trend in IL-6 levels has been observed in the setting of bubble and membrane oxygenators (105), after hypothermic and normothermic CPB (107), and with and without heparin-coating CPB circuitry (28). The magnitude of increase in IL-6 levels was positively correlated with duration of CPB but not duration of aortic cross-clamp time (108). In the pediatric population, duration of CPB and aortic cross-clamp time were attributed to pronounced postoperative inflammation, with only a modest influence of the degree of hypothermia (109). Rise in IL-6 after CPB has been correlated with increasing age, as those older than seventy years had a greater increase in plasma IL-6 levels during ischemia and reperfusion than their younger counterparts (110). Postoperative IL-6 levels are significantly higher in patients with complications compared to those without (111). Increased levels of IL-6 have been associated with myocardial dysfunction and wall motion abnormalities (112, 113), while effects on hemodynamics are less clear (114). Rise in IL-6 has not been correlated with complement activation.
IL-8 is a potent chemotactic agent involved in the homing of neutrophils and macrophages sites of inflammation (115). It may also play a role in ischemia-reperfusion injury, as postoperative cardiac troponin-I levels correlate strongly with IL-8 levels in patients following coronary artery bypass grafting (CABG) (116, 117). It has been implicated in precipitating vascular damage, particularly in the lungs and kidneys. Increased levels of IL-6 and IL-8 following CPB have been reported (81, 106, 107, 113, 118–123), and correlate with duration of cardiac ischemia during CPB and regional wall motion abnormalities (113, 118).
IL-10 has anti-inflammatory properties via its downregulation of pro-inflammatory cytokine synthesis by type 1 T helper cells, neutrophils, and monocytes (124–126). It has been associated with decreased production of TNF-α, IL-1, IL-2, and interferon-γ, ROS and nitric oxide derivatives (124, 127, 128). While several pro-inflammatory cytokines (TNF-α, IL-6, and IL-8) have been shown to originate from myocardium (120, 121, 129), the liver has been shown to be the primary source of IL-10 in patients undergoing CPB (120, 121, 130, 131). Rapid and transient secretion of IL-10 has been noted following CPB (132, 133). In vivo kinetics of IL-10 release are similar to those observed in murine endotoxemia experiments following lipopolysaccharide challenge but contrast with in vitro data from human monocytes following lipopolysaccharide stimulation (134, 135). IL-10 produced during CPB may represent an in vivo regulatory mechanism for controlling activation of cells that synthesize pro-inflammatory cytokines.
There are several caveats in relating serum cytokine levels associated with CPB to organ dysfunction. Plasma concentrations often do not reflect local effects. Several cytokines may also avidly bind to other plasma proteins, leading to inaccurate detection. Inter-individual genomic differences also contribute to heterogeneity in cytokine levels, and have implications for identifying therapeutic targets and developing preventative strategies that can be broadly applied. Genetic variants of promoter regions encoding IL-6 (−174 G/C polymorphism, −572 G/C polymorphism), TNF-α (308 G/A polymorphism), and antioxidant response elements (NQO1) have been linked to increased cytokine production and postoperative complications (136–142). A prevalent single-nucleotide polymorphism (SNP) of the gene encoding IL-18 was shown to be associated with increased TNF-α levels and decreased IL-10 levels. Apolipoprotein E4 allele has been correlated with increased IL-8 and TNF-α generation and decreased IL-10 levels, with a speculated link to postoperative cerebral injury (143–145). While the A allele of this SNP was associated with 30-day and 50-year mortality in the INFLACOR study, post-hoc analysis revealed the C allele of −572 G/C polymorphism to be significantly associated with reduced benefit of prophylactic administration of dexamethasone on postoperative IL-6 levels compared to the G allele. Its effects on C-reactive protein (CRP), however, did not appear to be genotype-dependent. These genomic differences pose a challenge to randomized controlled trials (RCTs) evaluating clinically relevant responses to prophylactic measures. Second-generation studies, including genome-wide association studies (GWAS), may be able to more completely evaluate genotype-phenotype relationships.
CPB-induced cellular immune activation plays a key role in the ensuing inflammatory response (146). Recruitment of immune cells is mediated by upregulation of cytokines, chemokines, complement system proteins, and adhesion molecules, including selectins and integrins (147). Primary adhesion occurs when freely moving neutrophils are converted to the “rolling” state following upregulation of P- and E-selectin on endothelium and upregulation of L-selectin on neutrophils. This leads to neutrophils initially traveling in the center of postcapillary venules to tumble along endothelial walls endothelium-neutrophil selectin interactions. C5a, which is released in response to contact activation during CPB, is a potent stimulator of endothelial P-selectin expression (148). E-selectin subsequently replaces P-selectin on endothelium to maintain primary adhesion. Secondary adhesion of neutrophils is mediated by integrins. Integrins CD11a/CD18 and CD11b/CD18 are expressed by neutrophils; IL-8 and C5a are potent stimulators of CD11b/CD18 expression on neutrophils (147, 149, 150). Activated integrins bind adhesion molecules on endothelium (intercellular adhesion molecule-1 and intercellular adhesion molecule-2) and extracellular matrix elements including fibrinogen (147). Both selectins and integrins have been shown to increase following CPB (150). Transmigration of neutrophils follows secondary adhesion (151). CD11b/CD18 binding to endothelium during secondary adhesion and transmigration primes neutrophils to degranulate and undergo respiratory burst for up to 24 h following CPB (49, 147, 151). Elastase, myeloperoxidase, and ROS released from neutrophils result in cytotoxic damage to endothelium and tissues (49, 66, 151). Modulation of adhesion, transmigration, and neutrophil priming is heavily influenced by platelet-activating factor, IL-8, and C5a (147, 152). Normalization of C5a levels limits the extent of CPB-induced inflammatory response (149). Increased secretion of monocyte chemoattractants leads to upregulation of selectins and integrins and further activation of circulating monocytes and macrophages. Naïve monocytes and granulocytes are hyperstimulated following exposure to post-CPB plasma. Impaired oxidative burst and phagocytic activity have been observed 48 h following CPB, suggesting a biphasic course characterized by early tissue cytotoxicity and late neutrophil dysfunction (146, 153, 154).
Clinically, increased cellular immune activation leads to pulmonary leukocyte sequestration that has been associated with severe histologic lung injury (155, 156). Inhibition of CD11/CD18 expression or function improves myocardial function following CPB, and neutrophil adhesion blockade reduces pulmonary injury during CPB (157–159). Reduction of circulating activated leukocytes has been associated with reduced organ injury (160).
The endothelium is central to inflammatory pathophysiology following CPB (161). The endothelial glycocalyx protects the endothelial cell monolayer and is composed primarily of transmembrane heparan sulfate and syndecan proteoglycans. The glycocalyx plays important roles in leukocyte and platelet adhesion following immune activation, vascular permeability facilitating leukocyte transmigration, and regulation of the coagulation cascade on the luminal endothelial surface (161–163). Following CPB, activated metalloproteinases and TNF-α induce shedding of syndecan-1 and heparin sulfate from the glycocalyx; increased plasma levels of these glycocalyx breakdown products following CPB initiation have been extensively documented (164–170). Membrane-bound syndecan-1 reduces cytokine production while soluble syndecan-1 results in neutrophil activation and monocyte chemotaxis (171–173). Pro-inflammatory IL-6 and IL-8 levels at two timepoints (preoperatively and 6 horus postoperatively) and anti-inflammatory IL-10 have been correlated with CPB-induced inflammation (174). Syndecan-1 levels have been shown to prognosticate acute kidney injury following CPB in children though levels vary widely between patients and do not correlate intraoperatively (174, 175).
Innovations and potential therapeutic targets have been investigated to better modulate the inflammatory response to CPB. The main challenge remains to balance mitigating the effects of excessive inflammation and immune activation while still preserving host defenses and wound healing (176). The most clinically effective way to curb CPB-associated inflammation involves targeting multiple inflammatory mediators simultaneously using a combination of surgical, pharmacologic, and mechanical pump approaches as no single intervention is supported by strong level A evidence (Figure 3). Interventions with level A evidence include off-pump surgery, miniaturized CPB circuits, coated circuits to improve biocompatibility, leukocyte filtration, complement (C5) inhibition, preoperative aspirin, and corticosteroid prophylaxis. Interventions with level B evidence include, but not limited to, hemofiltration, aprotinin, nitric oxide donors, C1 esterase inhibition, neutrophil elastase inhibition, N-acetylcysteine, and intensive insulin therapy (177). The precise combinations of studied interventions tailored to specific patient populations have yet to be determined.
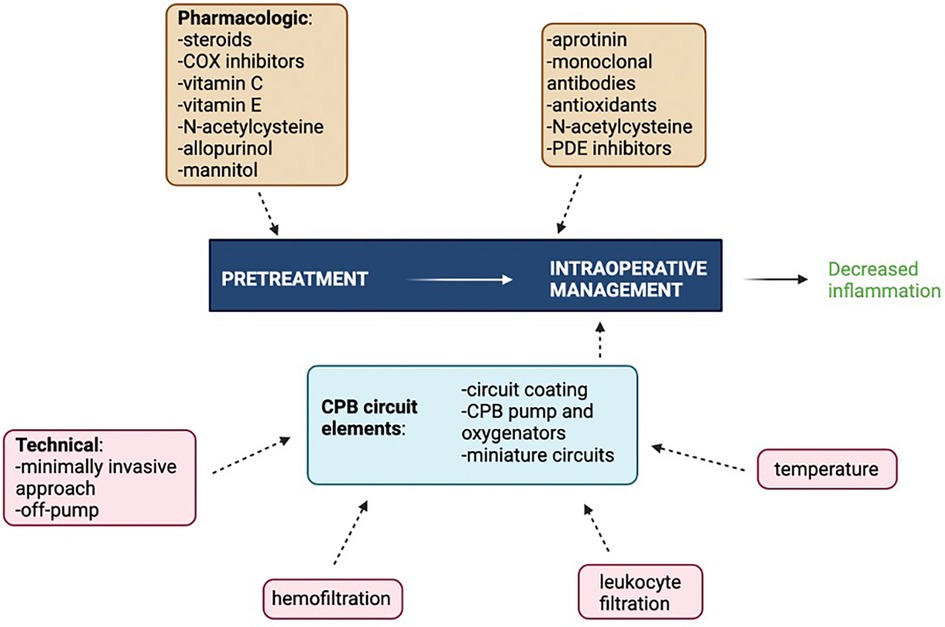
Figure 3 Multimodal approach to attenuating CPB-associated inflammation. COX, cyclooxygenase; PDE, phosphodiesterase. Figure created using Biorender.com.
Heparin-coated circuits improve biocompatibility of the extracorporeal circuit, and translation into clinical benefit has been demonstrated in certain populations (178, 179). Heparin molecules bound to the surface of the CPB circuit resemble heparin sulphate glycosaminoglycans on endothelial cells and reduce direct contact of blood with the otherwise artificial, nonendothelial material lining the CPB circuit. These coated circuits improve biocompatibility through various mechanisms. Heparin-coated circuits reduce cytokine release, complement activation in vivo and in vitro, kallikrein, and leukocyte activation (180–184). They have been shown to reduce platelet adhesion and improve platelet function, as well as inhibit the release of pro-inflammatory cytokines TNF-α, IL-6, IL-8 as well as soluble TNF receptors (180, 185–187). Significant reduction in mean concentrations of polymorphonuclear (PMN) elastase and soluble C5b-9 (p < 0.001 and p = 0.006, respectively) at one hour following initiation of CPB have been reported. Heparin coating also reduced postoperative blood loss (188–190). An RCT conducted in the Netherlands showed reduced complement activation that correlated with improved clinical performance scores in patients who underwent CPB with heparin-coated circuits in combination with full systemic heparinization (191). A large, multicenter RCT demonstrated improved clinical outcomes, shorter length of stay (hospital and ICU), and decreased respiratory and renal dysfunction in high-risk patients (192). A meta-analysis of 41 RCTs showed heparin coating was associated with 40% reduction in re-sternotomy rates (p = 0.002) and 20% reduction in patients requiring blood transfusion but no significant difference in 24-h blood loss or adverse events. Heparin coating reduced average ventilation time by 80 min, ICU length of stay by 9 h (p < 0.001), and average hospital length of stay by 0.5 days (p = 0.02) (193). In cardiac reoperations, use of heparin-coated circuits with full systemic heparinization was associated with decreased rates of reoperation for bleeding (p = 0.058) and lower blood transfusion requirement (p = 0.035) without significant difference in adverse events (189).
A study investigating heparin-coated circuits with administration of reduced systemic heparin dose showed decreased intraoperative platelet counts, maximal intraoperation concentrations of platelet factor 4 and PMN elastase, as well as 12-h blood loss with heparin-coated circuits but no difference adverse events or 30-day mortality (190). A departmental analysis revealed patients undergoing CPB with heparin-coated circuits received lower dose of systemic heparin, showed improvements in clinical parameters, and had a decreased rate of adverse events (10%, p = 0.035) compared to their counterparts (194).
Heparin-albumin and polymer-coated circuits as well as phosphorylcholine-coated circuits have been shown to induce fewer inflammatory responses and oxidative stress compared to other circuits (195, 196). However, heparin-coated circuits are associated with better preservation of endothelial glycocalyx compared with phosphorylcholine-coated circuits (197). An RCT investigating albumin-coated circuits in patients undergoing aortic arch replacement with deep hypothermic circulatory arrest showed mitigation in platelet reduction as evidenced by decreased transfusion requirement but no effect on platelet dysfunction (198). A study investigating hyaluronan-based heparin-coated circuits in various risk cohorts showed improved platelet preservation and better perioperative outcomes; ventilation time, hemorrhage, and degree of inflammation were reduced in high-risk groups, which translated to shorter ICU and hospital length of stay (p = 0.001 and p = 0.006, respectively) (199). New data is still emerging for pediatric populations. A randomized pilot study recently demonstrated application of new ternary polymer, SEC-1 coat™ in pediatric cardiac operations improved biocompatibility with regard to platelet preservation and attenuated coagulation activation and overall inflammatory reaction (200). Another study showed no purported benefit in improving coagulation derangements during pediatric CPB as assessed by primary endpoint of concentration of β-thromboglobulin across all time points, as well as secondary endpoints of other markers of coagulation and platelet function (201).
Multiple studies found reductions in inflammatory markers, marginal improvement in biocompatibility, but minimal or no correlation with improved outcomes (202, 203). One such study showed reduced complement activation and synthesis of pro-inflammatory cytokines but no significant differences in fibrinolysis, platelet activation, time to hemostasis, postoperative blood loss at 12 h, total blood transfusion requirement, or intubation time (202). Another multicenter trial similarly found decreased complement activation but no association with release of specific neutrophil granule enzymes, myeloperoxidase, lactoferrin, or clinical outcome (203). A multimodal approach incorporating heparin-coated circuits, high-dose aprotinin, and pre-CPB hemofiltration reduced inflammatory response and improved clinical outcomes in high-risk patients (179).
Overall, heparin-coated circuits and newer third-generation heparin-polymer-coated circuits induce fewer inflammatory responses and are associated with improved outcomes and, therefore, justify their additional cost. Newer third-generation circuits preserve platelet function and improve perioperative outcomes, including reduced blood loss, reoperation rates, ventilation, time, and length of stay.
Hemofiltration, or ultrafiltration, removes excess fluid and low-molecular-weight substances from plasma through a hydrostatic pressure gradient. It has been shown to improve hemodynamic parameters, cardiac and pulmonary function, and reduce inflammation with greatest benefit in pediatric patients. It has been associated with reduced levels of TNF-α, IL-1, IL-6, IL-8, C3a, and myeloperoxidase levels postoperatively in this population (204–208). Clinical benefits include improved hemodynamic stability and early postoperative oxygenation and decreased postoperative blood loss and duration of mechanical ventilation. It has been associated with decreased endothelin-1, potentially explaining improvement in pulmonary hypertension following congenital heart surgery (204, 206, 207, 209–211). Modified ultrafiltration has been associated with improved left ventricular systolic function and diastolic compliance, increased blood pressure, and reduced inotropic drug administration in the early postoperative period in infants (212–214). Hemofiltration has shown less clinical advantage in adults as it is less effective in removing pro-inflammatory cytokines (215). However, modified hemofiltration has been associated with decreased early morbidity, postoperative bleeding, and blood transfusion requirements in adults and its use was not associated with hemodynamic instability in adults (215–217).
Activated leukocytes form the first line of defense against foreign substances and are critical players in potentiating the inflammatory response. Leukocyte-specific filters that trap activated neutrophils and monocytes implicated in SIRS attenuate inflammation and oxidative stress and have been associated with improved outcomes in the immediate postoperative period (218–222). Filters decrease concentrations of circulating platelets and leukocytes by interfering with endothelial-mediated leukocyte activation and subsequent neutrophil transmigration. This effectively decreases the endothelial-mediated component of the CPB-associated inflammatory response.
Leukocyte filtration may limit postoperative myocardial and pulmonary dysfunction following CPB. Results of a prospective randomized study showed patients undergoing coronary revascularization had decreased total leukocyte counts during and after CPB, and significantly decreased activated leukocyte counts at all timepoints. The rate of alveolar exhaled NO production and alveolar-arterial oxygenation index were significantly increased in the control group, where NO was a marker for lung inflammation. Leukocyte depletion was also associated with lower pro-inflammatory cytokine (IL-6 and IL-8) burst postoperatively in those with normal preoperative oxygenation capacity. There were no differences in intubation time, ICU, or hospital length of stay (223–226). Though, as expected, the rate of alveolar NO production increased in both groups following CPB, absolute NO production was shown to be lower with leukocyte depletion, suggesting filtration may be lung-protective. Leukocyte depletion at early reperfusion in those with limited preoperative oxygenation capacity (mild lung dysfunction and chronic obstructive pulmonary disease) and with increased duration of CPB time was associated with improved oxygenation, shorter intubation time, and shorter ICU and hospital length of stay (227, 228). Filtration may improve postoperative lung function by mitigating pulmonary reperfusion injury. Leukocyte depletion of residual blood prior to re-transfusion also improved lung function. In patients undergoing urgent CABG for unstable angina, leukocyte depletion of re-transfused blood and during CPB reduced markers of myocardial injury, whereas leukocyte depletion did not confer clinical benefit in low-risk patients (229). In patients with decreased left ventricular function, leukocyte depletion of blood cardioplegia alone improved early myocardial function and attenuated myocardial injury (160, 230). In those with left ventricular hypertrophy undergoing valve surgery, terminal blood cardioplegia reduced myocardial injury and improved heart function (231, 232). A large-scale clinical trial showed reduced overall 60-day mortality with leukocyte depletion of transfused blood, mainly attributed to reduction in noncardiac causes of death including multisystem organ failure. In those who received greater than 3 units of blood, postoperative infection rate was lower with leukocyte depletion (233). More recently, a study showed preoperative neutrophil response to in vitro stimuli may predict clinical outcome following CPB, but leukocyte filtration did not offer significant benefit (234).
Leukocyte depletion may provide renal protection. A prospective randomized study of in 40 patients undergoing CABG showed leukocyte filtration decreased indices of glomerular and tubular injury, namely microalbumin/creatinine ratio, urinary excretion of microalbumin, and retinol-binding protein (235). Filtration of neutrophils containing myeloperoxidase decreased apoptosis, caspase-3 activity, and IL-1β activation and effectively improved post-ischemic renal function and structure in a porcine model of isolated kidney perfusion (236).
Extracorporeal blood purification through hemoadsorption utilizes biocompatible highly porous nonpolar polymer sorbent beads to sequester hydrophobic cytokines based on size exclusion and concentration-dependent surface adsorption throughout the beads. Nonspecific adsorptive characteristics allow for reduction in circulating pro-inflammatory cytokines, sequestration of free hemoglobin and bilirubin, and fortifying the endothelial glycocalyx. Clinical benefits include improved hemodynamic and metabolic stabilization postoperatively. No adverse effects of hemolysis or leukocyte removal have been reported. The first RCT investigating hemoadsorption with Cytosorb in cardiac surgical patients found prolonged anti-inflammatory IL-10 effect in the treatment group (237). A trial at a single center in Switzerland showed neither increased nor decreased cytokine levels (pro- or anti-inflammatory) with use of Cytosorb (238). Results showed no change in relevant clinical outcomes though the procedure was both feasible and safe. The REFRESH 1 pilot RCT showed Cytosorb signgicantly reduced C3a and C5a, as well as plasma-free hemoglobin during valve replacement operations (239). When comparing hemoadsorption to glucocorticoids, methylprednisolone was shown to more effectively reduce inflammatory markers (IL-6, TNF-alpha, and IL-8) though no differences in cardiac index or parameters of clinical outcomes were reported (240, 241). An RCT investigating intraoperative hemoadsorption showed clinical benefit as evidenced by reduced incidence of severe acute respiratory distress syndrome and a trend toward shorter ventilation times (242).
Studies comparing the acute phase reaction associated with normothermic vs. hypothermic CPB have conflicting results. This is due in part to inconsistent definitions of hypothermia. Hypothermia has been shown to delay but not completely prevent the expression of inflammatory mediators as increased levels of adhesion molecules and leukocyte proteolytic enzymes were seen at 34°C compared to moderate hypothermia (26°C–28°C) (80–82). Still other studies have shown no difference between patients randomly assigned to undergo CPB at various temperatures: > vs. 27-° in one study; 28°C, 32°C, vs. 37°C in another study (243, 244). Increased levels of NO were seen with CPB at 34°C when compared to CPB at 28°C, resulting in reduced systemic vascular resistance (245). A prospective randomized study in patients undergoing valve operations showed those undergoing normothermic CPB had similar levels of myocardial protection as measured by dynamics of troponin I while those undergoing hypothermic CPB benefitted from significantly lower ventilation times (p = 0.01) (246). A study investigating 262 different proteins using high-throughput technology showed deep hypothermic circulatory arrest (DHCA) and rewarming potentially exert a significant effect on the plasma proteome in patients undergoing aortic operations as evidenced by suppression of complement activation during hypothermia. These findings were confirmed by changes in terminal complement complex (C5b-9) levels. Following rewarming, however, these levels of terminal complement complex were more increased with DHCA than with normothermic CPB while 48 other proteins were significantly downregulated (247). In patients with left ventricular dysfunction, normothermia was found to enable decreased requirement for defibrillation after aortic unclamping and postoperative cardiac pacing, translating to improved myocardial protection. Normothermia had no effect on development of postoperative stroke, atrial fibrillation, renal failure, or mortality (248). A large-animal study recently showed hypothermic CPB attenuated platelet degranulation and coagulopathy and better maintained oxygenator performance in swine (249). Several RCTs studying pediatric populations showed normothermic CPB as noninferior to hypothermic CPB with endpoints including inotrope duration, intubation time, hospital stay, and early neurodevelopmental outcomes in low-risk populations (250, 251).
Studies evaluating the potential benefit of pulsatile pumps have not yielded conclusive results regarding clinical outcomes. Few studies have reported reduced levels of endotoxin and other pro-inflammatory mediators while other studies have not. Routine use of centrifugal pumps for CPB has not shown clear clinical advantages compared to roller pumps, and in fact several studies have showed increased levels of anaphylatoxins, pro-inflammatory cytokines, adhesion molecules, and leukocyte elastase with the use of centrifugal pumps. Radial-flow-patterned oxygenators may limit the extent of inflammation triggered by oxygenators in CPB when compared axial-flow-patterned oxygenators as evidenced by the results of a recent prospective RCT that noted significantly lower levels of humoral inflammatory markers (IL-1, IL-6, and TNF-α) 24 h postoperatively (252). In pediatric patients, controlled reoxygenation during CPB has shown benefit in single-ventricle patients as evidenced by reduced markers of organ damage, inflammation, and oxidative stress when compared to their double-ventricle counterparts (253).
Miniature extracorporeal circulation (MECC) systems have been developed to eliminate blood-foreign surface interface, shorten tubing length, reduce priming volume, eliminate venous reservoirs and cardiotomy suction, minimize hemolytic and consumptive effects on blood cells, and maximize blood re-transfusion (254). These systems consist of a centrifugal pump, membrane oxygenator, and arterial filter with all components of the system coated with heparin to optimize biocompatibility. An RCT confirmed a milder induced inflammatory response compared to conventional CPB with reduced levels of IL-6, TNF-α, and elastase release. A more recent RCT also showed reduced migration inhibitory factor levels associated with MECC systems in addition to decreased release of pro-inflammatory cytokines in the immediate postoperative period. This overall reduction correlated with decreased blood transfusion requirement and shorter mechanical ventilation time on bypass (255). The initially reduced levels of inflammatory markers seen with MECC may not be sustained throughout the postoperative period. An RCT investigating type 2 MECC compared to conventional CPB circuit in 50 patients undergoing aortic valve replacement found significantly lower levels of pro-inflammatory markers at 2 h postoperatively (p = 0.013) but no difference at 24 h (p = 0.990) when adjusting for type of oxygenator and hemoglobin. MECC was still associated with shorter perfusion times and less transfusion requirements (256). A small prospective RCT showed decreased IL-6, decreased hemolysis peaks as evidence by plasma-free hemoglobin levels, higher cardiac index and reduced pulmonary vascular resistance within 30 min postoperatively associated with MECC (257). However, these differences were not significant, and larger prospective RCTs are lacking. Normothermic CPB using MECC systems may be beneficial for perioperative preservation of pulmonary function and hemostasis in low-risk patients (258). These systems offer a promising minimally invasive approach to CPB.
Advances in minimally invasive cardiothoracic surgery, including laparoscopic and thoracoscopic operations, allow for comparable outcomes while avoiding a full median sternotomy. The reduced size of surgical incisions significantly decreases the inflammatory response, but these approaches have not infrequently been associated with increased duration of CPB and aortic cross-clamping time. These results have been seen with minimally invasive valve operations as well as minimally invasive pulmonary embolectomy. While ventilator time and ICU length of stay were similar, minimally invasive operations resulted in decreased overall hospital length of stay by almost 5 days (259). In a retrospective analysis comparing video-assisted thoracoscopic surgery (VATS) vs. open operation for mitral valve disease, VATS was associate with longer duration of CPB and aortic cross-clamping but decreased ventilation time and ICU length of stay. Clearance of lactate was increased while levels of pro-inflammatory C-reactive protein, neutrophil-lymphocyte ratio, and cardiac troponin levels were significantly decreased at 24 h postoperatively in those undergoing VATS (260). These results suggest a totally thoracoscopic approach may be superior to conventional median sternotomy with regard to extent of inflammatory reaction, cardiac injury, and postoperative recovery (260).
Miniature aortic valve replacement (mini-AVR) has been evaluated in recent studies. A prospective RCT showed minimal difference in operative time on CPB, cross-clamp time, and overall operating time when evaluating patients undergoing AVR through median sternotomy compared to right anterolateral thoracotomy (261). However, cosmesis and patient satisfaction were significantly higher with reduced length of incision associated with thoracotomy approach. Ministernotomy for AVR has been associated with significant reduction in intraoperative blood loss compared to counterparts undergoing median sternotomy, though transfusion requirements were unchanged (262). This same prospective randomized study also reported no difference in respiratory function between the two groups, which was supported by results of another prospective RCT comparing outcomes in patients undergoing AVR with partial upper sternotomy vs. median sternotomy (263). This study also found the minimally invasive approach did not affect neurological outcomes or myocardial protection. In contrast, a separate randomized trial showed reduced transfusion requirement, shorter ventilation times, greater sternal stability, improved respiratory function, and earlier extubation and hospital discharge with ministernotomy AVR compared to median sternotomy approach (264).
Off-pump cardiac surgery has been attributed to reduced postoperative SIRS, but operative trauma, regional ischemia/reperfusion injury, and endotoxin release induced even in the absence of CPB and aortic cross-clamping may contribute to postoperative biological derangements and clinical morbidity. The endothelium has been increasingly implicated in multiorgan dysfunction following cardiac surgery, particularly in relation to hemostasis and oxidative stress. In a randomized clinical trial, patients undergoing off-pump cardiac surgery experienced reduction in systemic inflammatory response as measured by decreased plasma TNF-α, IL-10, myeloperoxidase, but avoiding CPB and aortic cross-clamping did not alter circulating levels of endothelial adhesion molecules (265). Differences between on-pump and off-pump cardiac surgery in this context have been limited to the final steps of the operations and early hours thereafter, suggesting that global surgical trauma may play a more important role in activation of systemic inflammatory and coagulation-fibrinolytic pathways (266). Complement activation and release of IL-8 is dependent on extracorporeal CPB circuit, while release of products of endothelial and leukocyte activation are temporally similar but decreased in magnitude in off-pump cardiac surgery. A procoagulant state and no rise in anti-inflammatory IL-10 following off-pump cardiac surgery may offset other benefits. Neurocognitive decline and pulmonary function outcomes following off-pump cardiac surgery are variable (267).
Serine proteases comprise a large portion of effector proteins downstream of pro-inflammatory cytokines, complement activation, and hemolytic cascades. As these proteins amplify the inflammatory reaction, serine protease inhibitors have been investigated as potential therapies to mitigate excessive inflammation. Aprotinin is one such inhibitor and is perhaps one of the most well-studied. Aprotinin dually functions as an inhibitor of thrombin generation via the intrinsic pathway and has also been shown to preserve cellular junctions and reduce myocardial edema following cardioplegia and regional ischemia (268, 269). Its use has been previously associated with reduced intraoperative blood loss, while higher dosages may suppress the inflammatory response (270–272). These effects include attenuation of platelet activation, maintenance of platelet function, decreased complement and leukocyte activation, inhibition of kallikrein production, inhibition of endogenous cytokine-mediated NOS induction, inhibition of upregulation of adhesion molecules, and reduced release of several pro-inflammatory mediators (TNF-α, IL-6, and IL-8) (9, 150, 273–278). High-dose aprotinin has been associated with reduced post-CPB inflammation, myocyte damage, myocardial ischemia, and hospital length of stay in high-risk patients (7, 279, 280). High-dose aprotinin has also been associated with increased pro-inflammatory 8-isoprostane levels in the lungs relative to plasma levels. This effect disappeared with low-dose aprotinin, suggesting its action varies in a dose-dependent manner. The effect of high-dose aprotinin in decreasing circulating 8-isoprostane as estimated by lung passage (based on blood sampled from pulmonary and radial arteries) may signify a shift toward an anti-inflammatory milieu (281). Aprotinin may also decrease postoperative pulmonary and cerebral injury. Initial concerns of lower graft patency and renal dysfunction with aprotinin appear unfounded (7, 282). A meta-analysis revealed aprotinin reduced surgical blood loss, blood transfusion requirement, and need for redo thoracotomy. It also decreased perioperative mortality almost twofold without an associated increase in risk of myocardial infarction (270). A large, international, multi-institutional prospective study comparatively assessing the safety profile of aprotinin and lysine analogs (aminocaproic acid and tranexamic acid) use in patients presenting for coronary artery bypass surgery found that patients administered aprotinin had doubled risk of renal failure requiring dialysis, 55% increase in risk of myocardial infarction or heart failure, 181% increase in risk of stroke or encephalopathy, and increased risk of mortality. Neither of lysine analogs studied was associated with increased risk of cardiac, renal, or cerebral adverse events (283) and reduction in blood loss during surgery was similar for all three drugs. The BART study, a blinded RCT comparing aprotinin and lysine analogs in patients undergoing high-risk cardiac operations had to be prematurely terminated due to increased mortality associated with aprotinin, leading to suspended use of aprotinin in these patients (284). Subsequent initiatives that revisited limitations of these studies resulted in resumed use of aprotinin in select patients in both Canada and the European Union, while use of aprotinin in USA is still restricted.
Pentoxifylline is a nonspecific phosphodiesterase inhibitor with various anti-inflammatory effects, including attenuation of TNF-α and endotoxin release, cytokine synthesis, pulmonary leukocyte sequestration and vascular resistance, and reduction in indices of endothelial injury and permeability (285–289). Pentoxifylline was associated with decreased levels of pro-inflammatory cytokines (TNF-α and IL-6). Its use was also associated with improved left ventricular ejection fraction, decreased ICU length of stay, ventilation time, requirement of inotropic agents, and transfusion requirement (290).
Other phosphodiesterase inhibitors have been evaluated in the context of maximizing splanchnic perfusion as a strategy to attenuate excessive inflammation associated with CPB. Milrinone has been associated with reduction in venous and hepatic endotoxin levels, decrease in gastric intramucosal pH, and decreased IL-6 levels postoperatively in otherwise healthy patients undergoing cardiac operations (291, 292).
Steroids have potent anti-inflammatory effects, and the mechanisms by which they exert their effects are multifactorial. Preoperative administration of glucocorticoids has been shown to attenuate endotoxin release and complement activation in response to CPB (293–295). Methylprednisolone was associated with decreased levels of postoperative pro-inflammatory mediators IL-6, IL-8, and TNF-α along with increased levels of anti-inflammatory IL-10 and IL-1ra (120, 121, 275, 296–300). Corticosteroids blunted the activation of leukocytes, upregulation of neutrophil adhesion molecules, and sequestration of neutrophils in the pulmonary parenchyma and vasculature (275, 294, 301, 302). Combination treatment of patients with methylprednisolone and aprotinin resulted in improved postoperative indices of cardiovascular, pulmonary, hemostatic, and renal function (303). Low-dose aprotinin had similar effect as methylprednisolone in blunting TNF-α release and neutrophil integrin CD11b upregulation (275). Another study showed methylprednisolone pretreatment was associated with improved cardiac performance and decreased bronchial inflammation post-CPB (275, 300). Short course of methylprednisolone has been shown to reduce incidence of postoperative atrial fibrillation (304). Low-dose methylprednisolone in pump priming solutions attenuated degree of myocardial damage (305). Preoperative plus pre-CPB administration may be superior to pre-CPB administration alone. Methylprednisolone prophylaxis was associated with lower levels of neuron-specific enolase, a biomarker for neuronal damage, suggesting it may be useful in reducing post-CPB cerebral injury (296). Benefits of corticosteroid use for reducing pulmonary inflammation, endotoxemia, and complemented activation are still disputed given the results of more recent RCTs, and differences in dosing regimen, formulation, and timing of administration may partially account for conflicting results. The SIRS trial showed no difference in risk of death or major morbidity between those randomized to receive methylprednisolone or placebo at 30 days postoperatively, while the most common adverse effects in both experimental arms were infectious or delirium-related (306). Overall, a clear benefit attributed to corticosteroid treatment in the setting of CPB remains to be demonstrated (293, 302, 306–310). The need for further studies investigating optimal dosage regimens, characterizing adverse events, and optimizing clinical outcomes cannot be overstated.
Myocardial antioxidant enzymes (including glutathione reductase, superoxide dismutase, and catalase) become activated in proportion to the degree of myocardial ischemia and reperfusion injury, but host antioxidants may become depleted after CPB (311–313). When ROS production exceeds host defense scavenging capacity, cellular injury results (314, 315). Increased preoperative total plasma antioxidant status has been associated with decreased levels of lipid peroxidation, which is directly correlated with indices of myocardial injury (313).
Vitamin C and vitamin E levels decline intraoperatively and remain low over two days postoperatively (316). High-dose vitamin C is an effective scavenger of free radicals and has been associated with decreased membrane lipid peroxidation, indices of myocardial injury, improved hemodynamics, and shorter ICU and hospital length of stay (314, 317). High-dose vitamin E has been associated with decreased plasma hydrogen peroxide concentrations and decreased membrane lipid peroxidation after CPB (26, 314). Prophylactic coadministration of vitamin C, vitamin E, and n-PUFAs (eicosapentaenoic acid:docosahexaenoic acid ratio 1:2) has been associated with reduced incidence of postoperative atrial fibrillation (318).
Allopurinol inhibits xanthine oxidase, a pivotal generator of free radicals during reperfusion injury. Some studies have demonstrated allopurinol reduced myocardial formation of cytotoxic free radicals, decreased myocardial injury, and improved myocardial recovery following CPB (315, 319–322). Other studies showed conflicting results, showing no benefit in myocardial injury or function with allopurinol alone (319, 323, 324). Preoperative supplementation of allopurinol in combination with vitamin C and vitamin E reduced cardiovascular dysfunction in both stable and unstable patients undergoing CABG, with unstable patients sustained lesser degree of myocardial injury and lower incidence of perioperative myocardial infarction (325). Results of other studies, however, have refuted effects of vitamin C and vitamin E supplementation on myocardial injury (326). Use of allopurinol in neonates to improve neurodevelopment following cardiac operations for congenital heart disease is an active area of research as it has already demonstrated benefit in infants with hypoplastic left heart syndrome (327, 328).
Preoperative or intraoperative administration of high-dose N-acetylcysteine, another scavenger of free radicals has been shown to reduced neutrophil oxidative burst response and elastase activity (329–331). N-acetylcysteine improved oxygenation and lung mechanics in patients with known acute lung injury, though no change in rates of progression to acute respiratory distress syndrome was noted (332). Clinical outcomes were not significantly affected with regard to mortality, myocardial infarction, bleeding, transfusion requirements, intubation time, and hospital length of stay (333). Low-dose N-acetylcysteine as an adjunct to cardioplegia reduced myocardial oxidative stress in patients undergoing CABG (334). Modified N-acetylcysteine via preparation with activated carbons to create sustained-release microcapsules demonstrated greater cardioprotection than N-acetylcysteine alone in a rat model of myocardial ischemia-reperfusion (335). In another rat model of CPB, N-acetylcysteine was shown to ameliorate CPB-associated intestinal injury via reduction in inflammation and oxidative stress as measured by decreased levels of intestinal malondialdehyde, TNF-α, IL-6, and serum diamine oxidase (336). A number of studies have recently shown N-acetylcysteine to improve pulmonary, hepatic, and renal outcomes in patients undergoing CPB with and without preexisting pulmonary and renal insufficiency (337–342).
Mannitol pretreatment has been associated with decreased myocardial formation of cytotoxic free radicals following CPB (315).
Post-CPB endothelial dysfunction is in large part mediated by ROS. In this way, free-radical scavengers, antioxidants, and iron chelators represent a potential therapeutic adjunct to mitigate deleterious effects of CPB-associated inflammation.
Another approach for decreasing contact activation and downstream inflammation may be utilizing endogenous soluble complement inhibitors. An RCT investigating a monoclonal antibody to human C5 demonstrated its efficacy and safety in the setting of CPB. Inhibition of synthesis of mediators in complement activation and formation of adhesion molecules in a dose-dependent fashion clinically translated to a reduction in coagulopathy, myocardial injury, and postoperative neurocognitive deficits (343). Compstatin, a peptide inhibitor of complement, completely inhibited heparin-protamine-induced complement activation in vivo in non-human primates without associated adverse events (344).
Other promising strategies for complement inhibition therapy include C1 inhibitor, recombinant soluble inhibitor-1 or soluble complement receptor 1, monoclonal antibodies to C3 and C5a, neutrophil elastase inhibitor, membrane-bound regulators of complement, and attenuation of complement receptor-3-mediated adhesion of inflammatory cells to vascular endothelium (7). In unstable patients with acute myocardial infarction undergoing emergency CABG, administration of C1 esterase inhibitor effectively limited complement activation and reduced myocardial ischemia-reperfusion injury as measured by significant reduction in cardiac troponin I. Its use was associated with improved cardiac function and hemodynamic performance without an impact on early mortality (345, 346). Soluble human complement receptor 1 effectively inhibited complement activation during CPB and significantly decreased mortality and myocardial infarction in male patients (347). Neutrophil elastase inhibitor, sivelestat, reduced levels of neutrophil elastase, IL-6, and IL-8 while also attenuating the pattern of physiological deterioration of gas exchange as measured by relative effect on alveolar-arterial oxygen index (348).
C5 complement inhibitor, pexelizumab, may offer mortality benefit. Results of an RCT enrolling over 3,000 patients showed a significant risk reduction of death or postoperative myocardial infarction within 30 days postoperatively in patients undergoing CABG with or without valve surgery. However, the study was not powered to detection reduction in mortality alone (349). These results were reproduced in a more recent RCT. Additionally, an exploratory analysis showed a significant mortality benefit in high-risk patients (350).
Selective inhibition of vascular endothelial activation may reduce deleterious effects of uncontrolled inflammation. Adhesion molecular blockade may interfere with adherence within 24 h following CPB, thereby preventing neutrophil-mediated widespread organ damage. Blockade of neutrophil and endothelial selectin molecules resulted in notable attenuation of cerebral injury in an animal model of CPB and DHCA, while inhibition of neutrophil adhesion markedly decreased pulmonary injury in a swine model of CPB (351, 352). A caveat to adhesion molecule blockade is increased susceptibility to infection due to impaired neutrophil demargination and recruitment to sites of infection (353). Strategies to prevent nuclear localization of transcriptional activator NFκB, a key mediator of pro-inflammatory signaling, have also showed promise but studies demonstrating efficacy and safety are pending (151).
Constitutive cyclooxygenase 1 (COX-1) and its inducible isoform, cyclooxygenase 2 (COX-2), are sensible targets for modulating immune activation in response to CPB. Antiplatelet agents, including inhibitors of COX-1 and COX-2, prevent platelet aggregation. Acetylsalicylic acid, or aspirin (ASA), is one of the most commonly prescribed medications for the prevention of cardiovascular diseases. It irreversibly acetylates a serine residue of COX-1, thereby preventing release of thromboxane A2 and its downstream effects on platelet aggregation. The beneficial effects of ASA are not confined to platelet aggregation, as other mechanisms including attenuating inflammation, reducing oxidative stress, inhibiting prostaglandin formation, and inhibition of thromboxane-mediated vasoconstriction may be modulated for clinical benefit. Continued ASA treatment until the time of CABG has been shown to reduce inflammation as demonstrated by lower levels of plasma high-sensitivity CRP at all time points, though no change in cytokines was observed (354). Aspirin and clopidogrel in combination with aprotinin did not significantly affect clinical outcomes (355).
Over the past several decades since modern extracorporeal circulation was first conceived of by Gibbon, strategies for controlling CPB-associated inflammation had some success but have fallen short of controlling SIRS. Surface modification of the extracorporeal circuit, technical advances including control of flow dynamics in the CPB circuit, and mechanical refinements in pumps, oxygenators, tubing, filters, and other material components of the CPB circuit have reduced adverse sequelae and shown clinical benefit. Initial studies completed several decades prior investigating strategies that reduced circulating interleukins and other pro-inflammatory mediators showed limited translational benefit or showed contradictory results; more recent studies investigating these strategies are few as new technologies and therapies have emerged. Appropriate application of adsorptive blood purification techniques or use of immunomodulatory pharmacologics to mitigate hyperinflammatory states following CPB still remains uncertain given inconclusive efficacy and safety results from several studies. Though initially promising, aprotinin has been associated with a significant adverse event profile in target populations reported in several large studies, leading to restricted use in several countries. Postoperative SIRS may delay diagnosis of sepsis and septic shock following cardiac surgery, particularly in high-risk patients. The Sequential Organ Failure Assessment (SOFA) score may be more sensitive for predicting physiologic effects of infection, while Sepsis-3 criteria may be a useful tool for early identification and management of sepsis in patients following cardiac surgery (356). Overall, improving biocompatibility of the CPB circuit and more minimally invasive techniques may lead to improved myocardial preservation. Investigations into pharmacological adjuncts to more specifically and effectively attenuate inflammation continue. A multimodal approach incorporating technical, circuit-specific, and pharmacologic strategies will likely yield maximal clinical benefit.
DB contributed to drafting, design, and revision of the manuscript. JF contributed to revision of the manuscript. FS contributed to revision of the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by NIH T32GM065085-18 (Trauma and Inflammation Research Training Grant, Division of Surgical Research at Brown University) to DB; RO1-HL46716 and RO1HL128831 to FS; 1RO1HL127072-01A1, 1RO1HL136347-01, and RO1HL136347-04S1 to JF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Warren OJ, Smith AJ, Alexiou C, Rogers PL, Jawad N, Vincent C, et al. The inflammatory response to cardiopulmonary bypass: part 1–mechanisms of pathogenesis. J Cardiothorac Vasc Anesth. (2009) 23(2):223–31. doi: 10.1053/j.jvca.2008.08.007
2. Kirklin JK, Westaby S, Blackstone EH, Kirklin JW, Chenoweth DE, Pacifico AD. Complement and the damaging effects of cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1983) 86(6):845–57. doi: 10.1016/S0022-5223(19)39061-0
3. McBride WT, Armstrong MA, Crockard AD, McMurray TJ, Rea JM. Cytokine balance and immunosuppressive changes at cardiac surgery: contrasting response between patients and isolated CPB circuits. Br J Anaesth. (1995) 75(6):724–33. doi: 10.1093/bja/75.6.724
4. Gasz B, Lenard L, Racz B, Benko L, Borsiczky B, Cserepes B, et al. Effect of cardiopulmonary bypass on cytokine network and myocardial cytokine production. Clin Cardiol. (2006) 29(7):311–5. doi: 10.1002/clc.4960290708
5. Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. (1996) 24(7):1125–8. doi: 10.1097/00003246-199607000-00010
6. Larmann J, Theilmeier G. Inflammatory response to cardiac surgery: cardiopulomonary bypass versus non-cardiopulmonary bypass surgery. Best Pract Res Clin Anaesthesiol. (2004) 18(3):425–38. doi: 10.1016/j.bpa.2003.12.004
7. Raja SG, Dreyfus GD. Modulation of systemic inflammatory response after cardiac surgery. Asian Cardiovasc Thorac Ann. (2005) 13(4):382–95. doi: 10.1177/021849230501300422
8. Cate JW, van der Poll T, Levi M, ten Cate H, van Deventer SJ. Cytokines: triggers of clinical thrombotic disease. Thromb Haemost. (1997) 78(1):415–9. doi: 10.1055/s-0038-1657562
9. Hill GE, Robbins RA. Aprotinin but not tranexamic acid inhibits cytokine-induced inducible nitric oxide synthase expression. Anesth Analg. (1997) 84(6):1198–202. doi: 10.1097/00000539-199706000-00005
10. Paparella D, Brister SJ, Buchanan MR. Coagulation disorders of cardiopulmonary bypass: a review. Intensive Care Med. (2004) 30(10):1873–81. doi: 10.1007/s00134-004-2388-0
11. Chung JH, Gikakis N, Rao AK, Drake TA, Colman RW, Edmunds LH Jr. Pericardial blood activates the extrinsic coagulation pathway during clinical cardiopulmonary bypass. Circulation. (1996) 93(11):2014–8. doi: 10.1161/01.cir.93.11.2014
12. Nemerson Y. Tissue factor and hemostasis. Blood. (1988) 71(1):1–8. Erratum in: Blood 1988 71(4):1178. doi: 10.1182/blood.V71.1.1.1
13. De Somer F, Van Belleghem Y, Caes F, François K, Van Overbeke H, Arnout J, et al. Tissue factor as the main activator of the coagulation system during cardiopulmonary bypass. J Thorac Cardiovasc Surg. (2002) 123(5):951–8. doi: 10.1067/mtc.2002.120334
14. Brister SJ, Ofosu FA, Heigenhauser GJ, Gianese F, Buchanan MR. Is heparin the ideal anticoagulant for cardiopulmonary bypass? Dermatan sulphate may be an alternate choice. Thromb Haemost. (1994) 71(4):468–73. doi: 10.1055/s-0038-1642462
15. Brister SJ, Ofosu FA, Buchanan MR. Thrombin generation during cardiac surgery: is heparin the ideal anticoagulant? Thromb Haemost. (1993) 70(2):259–62. doi: 10.1055/s-0038-1649561
16. Boisclair MD, Lane DA, Philippou H, Sheikh S, Hunt B. Thrombin production, inactivation and expression during open heart surgery measured by assays for activation fragments including a new ELISA for prothrombin fragment F1+2. Thromb Haemost. (1993) 70(2):253–8. doi: 10.1055/s-0038-1649480
17. Parolari A, Colli S, Mussoni L, Eligini S, Naliato M, Wang X, et al. Coagulation and fibrinolytic markers in a two-month follow-up of coronary bypass surgery. J Thorac Cardiovasc Surg. (2003) 125(2):336–43. doi: 10.1067/mtc.2003.2
18. Knudsen L, Hasenkam JM, Kure HH, Hughes P, Bellaiche L, Ahlburg P, et al. Monitoring thrombin generation with prothrombin fragment 1.2 assay during cardiopulmonary bypass surgery. Thromb Res. (1996) 84(1):45–54. doi: 10.1016/0049-3848(96)00160-0
19. Chenoweth DE, Cooper SW, Hugli TE, Stewart RW, Blackstone EH, Kirklin JW. Complement activation during cardiopulmonary bypass: evidence for generation of C3a and C5a anaphylatoxins. N Engl J Med. (1981) 304(9):497–503. doi: 10.1056/NEJM198102263040901
20. Ruddy S, Gigle I, Austen KF. The complement system of man (first of four parts). N Engl J Med. (1972) 287:489–94. doi: 10.1056/NEJM197209072871005
21. Matata BM, Sosnowski AW, Galiñanes M. Off-pump bypass graft operation significantly reduces oxidative stress and inflammation. Ann Thorac Surg. (2000) 69(3):785–91. doi: 10.1016/s0003-4975(99)01420-4
22. Gu YJ, Mariani MA, van Oeveren W, Grandjean JG, Boonstra PW. Reduction of the inflammatory response in patients undergoing minimally invasive coronary artery bypass grafting. Ann Thorac Surg. (1998) 65(2):420–4. doi: 10.1016/s0003-4975(97)01127-2
23. Collett B, Alhaq A, Abdullah NB, Korjtsas L, Ware RJ, Dodd NJ, et al. Pathways to complement activation during cardiopulmonary bypass. Br Med J (Clin Res Ed). (1984) 289(6454):1251–4. doi: 10.1136/bmj.289.6454.1251
24. Cavarocchi NC, Pluth JR, Schaff HV, Orszulak TA, Homburger HA, Solis E, et al. Complement activation during cardiopulmonary bypass. Comparison of bubble and membrane oxygenators. J Thorac Cardiovasc Surg. (1986) 91(2):252–8. doi: 10.1016/S0022-5223(19)36089-1
25. Jansen NJ, van Oeveren W, Gu YJ, van Vliet MH, Eijsman L, Wildevuur CR. Endotoxin release and tumor necrosis factor formation during cardiopulmonary bypass. Ann Thorac Surg. (1992) 54(4):744–7; discussion 747–8. doi: 10.1016/0003-4975(92)91021-z
26. Cavarocchi NC, England MD, O'Brien JF, Solis E, Russo P, Schaff HV, et al. Superoxide generation during cardiopulmonary bypass: is there a role for vitamin E? J Surg Res. (1986) 40(6):519–27. doi: 10.1016/0022-4804(86)90093-4
27. Moore FD Jr, Warner KG, Assousa S, Valeri CR, Khuri SF. The effects of complement activation during cardiopulmonary bypass. Attenuation by hypothermia, heparin, and hemodilution. Ann Surg. (1988) 208(1):95–103. doi: 10.1097/00000658-198807000-00014
28. Steinberg JB, Kapelanski DP, Olson JD, Weiler JM. Cytokine and complement levels in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1993) 106(6):1008–16. doi: 10.1016/S0022-5223(19)33971-6
29. Chenoweth DE, Hugli TE. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc Natl Acad Sci U S A. (1978) 75(8):3943–7. doi: 10.1073/pnas.75.8.3943
30. Ward PA, Cochrane CG, Mueller-Eberhard HJ. The role of Serum complement in chemotaxis of leukocytes in vitro. J Exp Med. (1965) 122(2):327–46. doi: 10.1084/jem.122.2.327
31. Craddock PR, Hammerschmidt D, White JG, Dalmosso AP, Jacob HS. Complement (C5-a)-induced granulocyte aggregation in vitro. A possible mechanism of complement-mediated leukostasis and leukopenia. J Clin Invest. (1977) 60(1):260–4. doi: 10.1172/JCI108763
32. Hammerschmidt DE, Harris PD, Wayland JH, Craddock PR, Jacob HS. Complement-induced granulocyte aggregation in vivo. Am J Pathol. (1981) 102(2):146–50.7468764
33. Tonnesen MG, Smedly LA, Henson PM. Neutrophil-endothelial cell interactions. Modulation of neutrophil adhesiveness induced by complement fragments C5a and C5a des arg and formyl-methionyl-leucyl-phenylalanine in vitro. J Clin Invest. (1984) 74(5):1581–92. doi: 10.1172/JCI111574
34. Nilsson L, Brunnkvist S, Nilsson U, Nyström SO, Tydén H, Venge P, et al. Activation of inflammatory systems during cardiopulmonary bypass. Scand J Thorac Cardiovasc Surg. (1988) 22(1):51–3. doi: 10.3109/14017438809106051
35. Jacob HS, Craddock PR, Hammerschmidt DE, Moldow CF. Complement-induced granulocyte aggregation: an unsuspected mechanism of disease. N Engl J Med. (1980) 302(14):789–94. doi: 10.1056/NEJM198004033021407
36. Howard RJ, Crain C, Franzini DA, Hood CI, Hugli TE. Effects of cardiopulmonary bypass on pulmonary leukostasis and complement activation. Arch Surg. (1988) 123(12):1496–501. doi: 10.1001/archsurg.1988.01400360066010
37. Tao W, Zwischenberger JB, Nguyen TT, Vertrees RA, McDaniel LB, Nutt LK, et al. Gut mucosal ischemia during normothermic cardiopulmonary bypass results from blood flow redistribution and increased oxygen demand. J Thorac Cardiovasc Surg. (1995) 110(3):819–28. doi: 10.1016/S0022-5223(95)70116-8
38. Baue AE. The role of the gut in the development of multiple organ dysfunction in cardiothoracic patients. Ann Thorac Surg. (1993) 55(4):822–9. doi: 10.1016/0003-4975(93)90098-3
39. Sinclair DG, Haslam PL, Quinlan GJ, Pepper JR, Evans TW. The effect of cardiopulmonary bypass on intestinal and pulmonary endothelial permeability. Chest. (1995) 108(3):718–24. doi: 10.1378/chest.108.3.718
40. Riddington DW, Venkatesh B, Boivin CM, Bonser RS, Elliott TS, Marshall T, et al. Intestinal permeability, gastric intramucosal pH, and systemic endotoxemia in patients undergoing cardiopulmonary bypass. JAMA. (1996) 275(13):1007–12. doi: 10.1001/jama.1996.03530370045029
41. Ohri SK, Bjarnason I, Pathi V, Somasundaram S, Bowles CT, Keogh BE, et al. Cardiopulmonary bypass impairs small intestinal transport and increases gut permeability. Ann Thorac Surg. (1993) 55(5):1080–6. doi: 10.1016/0003-4975(93)90011-6
42. Andersen LW, Landow L, Baek L, Jansen E, Baker S. Association between gastric intramucosal pH and splanchnic endotoxin, antibody to endotoxin, and tumor necrosis factor-alpha concentrations in patients undergoing cardiopulmonary bypass. Crit Care Med. (1993) 21(2):210–7. doi: 10.1097/00003246-199302000-00011
43. Jirik FR, Podor TJ, Hirano T, Kishimoto T, Loskutoff DJ, Carson DA, et al. Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J Immunol. (1989) 142(1):144–7. doi: 10.4049/jimmunol.142.1.144
44. Taggart DP, Sundaram S, McCartney C, Bowman A, McIntyre H, Courtney JM, et al. Endotoxemia, complement, and white blood cell activation in cardiac surgery: a randomized trial of laxatives and pulsatile perfusion. Ann Thorac Surg. (1994) 57(2):376–82. doi: 10.1016/0003-4975(94)91000-6
45. Giroir BP. Mediators of septic shock: new approaches for interrupting the endogenous inflammatory cascade. Crit Care Med. (1993) 21(5):780–9. doi: 10.1097/00003246-199305000-00024
46. te Velthuis H, Jansen PG, Oudemans-van Straaten HM, Sturk A, Eijsman L, Wildevuur CR. Myocardial performance in elderly patients after cardiopulmonary bypass is suppressed by tumor necrosis factor. J Thorac Cardiovasc Surg. (1995) 110(6):1663–9. doi: 10.1016/S0022-5223(95)70028-5
47. Andersen LW, Baek L, Degn H, Lehd J, Krasnik M, Rasmussen JP. Presence of circulating endotoxins during cardiac operations. J Thorac Cardiovasc Surg. (1987) 93(1):115–9. doi: 10.1016/S0022-5223(19)36483-9
48. Rocke DA, Gaffin SL, Wells MT, Koen Y, Brock-Utine JG. Endotoxemia associated with cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1987) 93(6):832–7. doi: 10.1016/S0022-5223(19)37043-6
49. Kharazmi A, Andersen LW, Baek L, Valerius NH, Laub M, Rasmussen JP. Endotoxemia and enhanced generation of oxygen radicals by neutrophils from patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1989) 98(3):381–5. doi: 10.1016/S0022-5223(19)34384-3
50. Nilsson L, Kulander L, Nyström SO, Eriksson O. Endotoxins in cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1990) 100(5):777–80. doi: 10.1016/S0022-5223(19)35477-7
51. Lequier LL, Nikaidoh H, Leonard SR, Bokovoy JL, White ML, Scannon PJ, et al. Preoperative and postoperative endotoxemia in children with congenital heart disease. Chest. (2000) 117(6):1706–12. doi: 10.1378/chest.117.6.1706
52. Cavarocchi NC, Schaff HV, Orszulak TA, Homburger HA, Schnell WA Jr, Pluth JR. Evidence for complement activation by protamine-heparin interaction after cardiopulmonary bypass. Surgery. (1985) 98(3):525–31.3875906
53. Kirklin JK, Chenoweth DE, Naftel DC, Blackstone EH, Kirklin JW, Bitran DD, et al. Effects of protamine administration after cardiopulmonary bypass on complement, blood elements, and the hemodynamic state. Ann Thorac Surg. (1986) 41(2):193–9. doi: 10.1016/s0003-4975(10)62668-9
54. Zupancich E, Paparella D, Turani F, Munch C, Rossi A, Massaccesi S, et al. Mechanical ventilation affects inflammatory mediators in patients undergoing cardiopulmonary bypass for cardiac surgery: a randomized clinical trial. J Thorac Cardiovasc Surg. (2005) 130(2):378–83. doi: 10.1016/j.jtcvs.2004.11.061
55. Durukan AB, Gurbuz HA, Salman N, Unal EU, Ucar HI, Yorgancioglu CE. Ventilation during cardiopulmonary bypass did not attenuate inflammatory response or affect postoperative outcomes. Cardiovasc J Afr. (2013) 24(6):224–30. doi: 10.5830/CVJA-2013-041
56. Kankılıç N, Aydın MS, Göz M. The effect of low tidal volume ventilation on inflammatory cytokines during cardiopulmonary bypass. Braz J Cardiovasc Surg. (2022) 37(5):694–701. doi: 10.21470/1678-9741-2020-0466
57. Paparella D, Yau TM, Young E. Cardiopulmonary bypass induced inflammation: pathophysiology and treatment. An update. Eur J Cardiothorac Surg. (2002) 21:232–44. doi: 10.1016/S1010-7940(01)01099-5
58. Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, et al. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood. (2002) 100(5):1689–98. doi: 10.1182/blood-2002-01-0046
59. Inoue N, Ramasamy S, Fukai T, Nerem RM, Harrison DG. Shear stress modulates expression of Cu/Zn superoxide dismutase in human aortic endothelial cells. Circ Res. (1996) 79(1):32–7. doi: 10.1161/01.res.79.1.32
60. Siepe M, Goebel U, Mecklenburg A, Doenst T, Benk C, Stein P, et al. Pulsatile pulmonary perfusion during cardiopulmonary bypass reduces the pulmonary inflammatory response. Ann Thorac Surg. (2008) 86(1):115–22. doi: 10.1016/j.athoracsur.2008.03.062
61. Orime Y, Shiono M, Hata H, Yagi S, Tsukamoto S, Okumura H, et al. Cytokine and endothelial damage in pulsatile and nonpulsatile cardiopulmonary bypass. Artif Organs. (1999) 23(6):508–12. doi: 10.1046/j.1525-1594.1999.06392.x
62. Sezai A, Shiono M, Nakata K, Hata M, Iida M, Saito A, et al. Effects of pulsatile CPB on interleukin-8 and endothelin-1 levels. Artif Organs. (2005) 29(9):708–13. doi: 10.1111/j.1525-1594.2005.29112.x
63. Onorati F, Santarpino G, Tangredi G, Palmieri G, Rubino AS, Foti D, et al. Intra-aortic balloon pump induced pulsatile perfusion reduces endothelial activation and inflammatory response following cardiopulmonary bypass. Eur J Cardiothorac Surg. (2009) 35(6):1012–9; discussion 1019. doi: 10.1016/j.ejcts.2008.12.037
64. Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. (1999) 43(4):860–78. doi: 10.1016/s0008-6363(99)00187-x
65. Rossi F. The O2- -forming NADPH oxidase of the phagocytes: nature, mechanisms of activation and function. Biochim Biophys Acta. (1986) 853(1):65–89. doi: 10.1016/0304-4173(86)90005-4
66. Kawahito K, Kobayashi E, Ohmori M, Harada K, Kitoh Y, Fujimura A, et al. Enhanced responsiveness of circulatory neutrophils after cardiopulmonary bypass: increased aggregability and superoxide producing capacity. Artif Organs. (2000) 24(1):37–42. doi: 10.1046/j.1525-1594.2000.06381.x
67. Saugstad OD. Bronchopulmonary dysplasia-oxidative stress and antioxidants. Semin Neonatol. (2003) 8(1):39–49. doi: 10.1016/s1084-2756(02)00194-x
68. Nakamura H, Vaage J, Valen G, Padilla CA, Björnstedt M, Holmgren A. Measurements of plasma glutaredoxin and thioredoxin in healthy volunteers and during open-heart surgery. Free Radic Biol Med. (1998) 24(7-8):1176–86. doi: 10.1016/s0891-5849(97)00429-2
69. Zitta K, Meybohm P, Gruenewald M, Cremer J, Zacharowski KD, Scholz J, et al. Profiling of cell stress protein expression in cardiac tissue of cardiosurgical patients undergoing remote ischemic preconditioning: implications for thioredoxin in cardioprotection. J Transl Med. (2015) 13:34. doi: 10.1186/s12967-015-0403-6
70. Cherry AD. Mitochondrial dysfunction in cardiac surgery. Anesthesiol Clin. (2019) 37(4):769–85. doi: 10.1016/j.anclin.2019.08.003
71. Prasad K, Kalra J, Bharadwaj B, Chaudhary AK. Increased oxygen free radical activity in patients on cardiopulmonary bypass undergoing aortocoronary bypass surgery. Am Heart J. (1992) 123(1):37–45. doi: 10.1016/0002-8703(92)90744-g
72. Davies SW, Duffy JP, Wickens DG, Underwood SM, Hill A, Alladine MF, et al. Time-course of free radical activity during coronary artery operations with cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1993) 105(6):979–87. doi: 10.1016/S0022-5223(19)33769-9
73. Toivonen HJ, Ahotupa M. Free radical reaction products and antioxidant capacity in arterial plasma during coronary artery bypass grafting. J Thorac Cardiovasc Surg. (1994) 108(1):140–7. doi: 10.1016/S0022-5223(94)70230-6
74. Pesonen EJ, Korpela R, Peltola K, Leijala M, Sairanen H, Raivio KO, et al. Regional generation of free oxygen radicals during cardiopulmonary bypass in children. J Thorac Cardiovasc Surg. (1995) 110(3):768–73. doi: 10.1016/S0022-5223(95)70110-9
75. Clermont G, Vergely C, Jazayeri S, Lahet JJ, Goudeau JJ, Lecour S, et al. Systemic free radical activation is a major event involved in myocardial oxidative stress related to cardiopulmonary bypass. Anesthesiology. (2002) 96(1):80–7. doi: 10.1097/00000542-200201000-00019
76. Seccombe JF, Pearson PJ, Schaff HV. Oxygen radical-mediated vascular injury selectively inhibits receptor-dependent release of nitric oxide from canine coronary arteries. J Thorac Cardiovasc Surg. (1994) 107(2):505–9. doi: 10.1016/S0022-5223(94)70096-6
77. Pearson PJ, Lin PJ, Schaff HV. Global myocardial ischemia and reperfusion impair endothelium-dependent relaxations to aggregating platelets in the canine coronary artery. A possible cause of vasospasm after cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1992) 103(6):1147–54. doi: 10.1016/S0022-5223(19)34880-9
78. Matheis G, Sherman MP, Buckberg GD, Haybron DM, Young HH, Ignarro LJ. Role of L-arginine-nitric oxide pathway in myocardial reoxygenation injury. Am J Physiol. (1992) 262(2 Pt 2):H616–20. doi: 10.1152/ajpheart.1992.262.2.H616
79. Haniuda M, Dresler CM, Mizuta T, Cooper JD, Patterson GA. Free radical-mediated vascular injury in lungs preserved at moderate hypothermia. Ann Thorac Surg. (1995) 60(5):1376–81. doi: 10.1016/0003-4975(95)00620-Z
80. Menasché P, Peynet J, Larivière J, Tronc F, Piwnica A, Bloch G, et al. Does normothermia during cardiopulmonary bypass increase neutrophil-endothelium interactions? Circulation. (1994) 90(5 Pt 2):II275–9.
81. Menasché P, Peynet J, Haeffner-Cavaillon N, Carreno MP, de Chaumaray T, Dillisse V, et al. Influence of temperature on neutrophil trafficking during clinical cardiopulmonary bypass. Circulation. (1995) 92(9 Suppl):II334–40. doi: 10.1161/01.cir.92.9.334
82. Le Deist F, Menasché P, Kucharski C, Bel A, Piwnica A, Bloch G. Hypothermia during cardiopulmonary bypass delays but does not prevent neutrophil-endothelial cell adhesion. A clinical study. Circulation. (1995) 92(9 Suppl):II354–8. doi: 10.1161/01.cir.92.9.354
83. Soslau G, Morgan DA, Jaffe JS, Brodsky I, Wang Y. Cytokine mRNA expression in human platelets and a megakaryocytic cell line and cytokine modulation of platelet function. Cytokine. (1997) 9(6):405–11. doi: 10.1006/cyto.1996.0182
84. Krishnaswamy G, Smith JK, Mukkamala R, Hall K, Joyner W, Yerra L, et al. Multifunctional cytokine expression by human coronary endothelium and regulation by monokines and glucocorticoids. Microvasc Res. (1998) 55(3):189–200. doi: 10.1006/mvre.1998.2079
85. Ala Y, Palluy O, Favero J, Bonne C, Modat G, Dornand J. Hypoxia/reoxygenation stimulates endothelial cells to promote interleukin-1 and interleukin-6 production. Effects of free radical scavengers. Agents Actions. (1992) 37(1–2):134–9. doi: 10.1007/BF01987902
86. Sterpetti AV, Cucina A, Morena AR, Di Donna S, D'Angelo LS, Cavalarro A, et al. Shear stress increases the release of interleukin-1 and interleukin-6 by aortic endothelial cells. Surgery. (1993) 114(5):911–4.8236014
87. Colotta F, Sironi M, Borrè A, Luini W, Maddalena F, Mantovani A. Interleukin 4 amplifies monocyte chemotactic protein and interleukin 6 production by endothelial cells. Cytokine. (1992) 4(1):24–8. doi: 10.1016/1043-4666(92)90032-m
88. Delneste Y, Lassalle P, Jeannin P, Joseph M, Tonnel AB, Gosset P. Histamine induces IL-6 production by human endothelial cells. Clin Exp Immunol. (1994) 98(2):344–9. doi: 10.1111/j.1365-2249.1994.tb06148.x
89. von Asmuth EJ, Leeuwenberg JF, Ceska M, Buurman WA. LPS and cytokine-induced endothelial cell IL-6 release and ELAM-1 expression; involvement of serum. Eur Cytokine Netw. (1991) 2(4):291–7.1721850
90. Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav Immun. (2006) 20(5):449–55. doi: 10.1016/j.bbi.2005.10.005
91. Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. (1995) 92(6):1487–93. doi: 10.1161/01.cir.92.6.1487
92. Yu Y, Gao M, Li H, Zhang F, Gu C. Pulmonary artery perfusion with anti-tumor necrosis factor alpha antibody reduces cardiopulmonary bypass-induced inflammatory lung injury in a rabbit model. PLoS One. (2013) 8(12):e83236. doi: 10.1371/journal.pone.0083236
93. Meldrum DR, Donnahoo KK. Role of TNF in mediating renal insufficiency following cardiac surgery: evidence of a postbypass cardiorenal syndrome. J Surg Res. (1999) 85(2):185–99. doi: 10.1006/jsre.1999.5660
94. Ohkawa F, Ikeda U, Kanbe T, Kawasaki K, Shimada K. Effects of inflammatory cytokines on vascular tone. Cardiovasc Res. (1995) 30(5):711–5. doi: 10.1016/S0008-6363(95)00101-8
95. Menasché P, Haydar S, Peynet J, Du Buit C, Merval R, Bloch G, et al. A potential mechanism of vasodilation after warm heart surgery. The temperature-dependent release of cytokines. J Thorac Cardiovasc Surg. (1994) 107(1):293–9. doi: 10.1016/S0022-5223(94)70484-8
96. Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, et al. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med. (1999) 27(7):1309–18. doi: 10.1097/00003246-199907000-00018
97. Boehm J, Hauner K, Grammer J, Dietrich W, Wagenpfeil S, Braun S, et al. Tumor necrosis factor-α −863 C/A promoter polymorphism affects the inflammatory response after cardiac surgery. Eur J Cardiothorac Surg. (2011) 40(1):e50–4. doi: 10.1016/j.ejcts.2011.01.084
98. Hansen PR, Svendsen JH, Høyer S, Kharazmi A, Bendtzen K, Haunsø S. Tumor necrosis factor-alpha increases myocardial microvascular transport in vivo. Am J Physiol. (1994) 266(1 Pt 2):H60–7. doi: 10.1152/ajpheart.1994.266.1.H60
99. Youker K, Smith CW, Anderson DC, Miller D, Michael LH, Rossen RD, et al. Neutrophil adherence to isolated adult cardiac myocytes. Induction by cardiac lymph collected during ischemia and reperfusion. J Clin Invest. (1992) 89(2):602–9. doi: 10.1172/JCI115626
100. Umegaki H, Yamada K, Naito M, Kameyama T, Iguchi A, Nabeshima T. Protective effect of interleukin-6 against the death of PC12 cells caused by serum deprivation or by the addition of a calcium ionophore. Biochem Pharmacol. (1996) 52(6):911–6. doi: 10.1016/0006-2952(96)00422-4
101. Schwarze MM, Hawley RG. Prevention of myeloma cell apoptosis by ectopic bcl-2 expression or interleukin 6-mediated up-regulation of bcl-xL. Cancer Res. (1995) 55(11):2262–5.7757973
102. Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. (1992) 257(5068):387–9. doi: 10.1126/science.1631560
103. Ungureanu-Longrois D, Balligand JL, Kelly RA, Smith TW. Myocardial contractile dysfunction in the systemic inflammatory response syndrome: role of a cytokine-inducible nitric oxide synthase in cardiac myocytes. J Mol Cell Cardiol. (1995) 27(1):155–67. doi: 10.1016/s0022-2828(08)80015-6
104. Dreyer WJ, Phillips SC, Lindsey ML, Jackson P, Bowles NE, Michael LH, et al. Interleukin 6 induction in the canine myocardium after cardiopulmonary bypass. J Thorac Cardiovasc Surg. (2000) 120(2):256–63. doi: 10.1067/mtc.2000.108168
105. Butler J, Chong GL, Baigrie RJ, Pillai R, Westaby S, Rocker GM. Cytokine responses to cardiopulmonary bypass with membrane and bubble oxygenation. Ann Thorac Surg. (1992) 53(5):833–8. doi: 10.1016/0003-4975(92)91446-g
106. Kawamura T, Wakusawa R, Okada K, Inada S. Elevation of cytokines during open heart surgery with cardiopulmonary bypass: participation of interleukin 8 and 6 in reperfusion injury. Can J Anaesth. (1993) 40(11):1016–21. doi: 10.1007/BF03009470
107. Frering B, Philip I, Dehoux M, Rolland C, Langlois JM, Desmonts JM. Circulating cytokines in patients undergoing normothermic cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1994) 108(4):636–41. doi: 10.1016/S0022-5223(94)70287-X
108. Whitten CW, Hill GE, Ivy R, Greilich PE, Lipton JM. Does the duration of cardiopulmonary bypass or aortic cross-clamp, in the absence of blood and/or blood product administration, influence the IL-6 response to cardiac surgery? Anesth Analg. (1998) 86(1):28–33. doi: 10.1097/00000539-199801000-00006
109. Eggum R, Ueland T, Mollnes TE, Videm V, Aukrust P, Fiane AE, et al. Effect of perfusion temperature on the inflammatory response during pediatric cardiac surgery. Ann Thorac Surg. (2008) 85(2):611–7. doi: 10.1016/j.athoracsur.2007.10.062
110. Howell KW, Cleveland JC Jr, Meng X, Ao L, Su X, Schwartz RS, et al. Interleukin 6 production during cardiac surgery correlates with increasing age. J Surg Res. (2016) 201(1):76–81. doi: 10.1016/j.jss.2015.10.016
111. Oka Y, Murata A, Nishijima J, Yasuda T, Hiraoka N, Ohmachi Y, et al. Circulating interleukin 6 as a useful marker for predicting postoperative complications. Cytokine. (1992) 4(4):298–304. Erratum in: Cytokine 1993 5(1):89-90. doi: 10.1016/1043-4666(92)90070-8
112. Deng MC, Dasch B, Erren M, Möllhoff T, Scheld HH. Impact of left ventricular dysfunction on cytokines, hemodynamics, and outcome in bypass grafting. Ann Thorac Surg. (1996) 62(1):184–90. doi: 10.1016/0003-4975(96)00230-5
113. Hennein HA, Ebba H, Rodriguez JL, Merrick SH, Keith FM, Bronstein MH, et al. Relationship of the proinflammatory cytokines to myocardial ischemia and dysfunction after uncomplicated coronary revascularization. J Thorac Cardiovasc Surg. (1994) 108(4):626–35. doi: 10.1016/S0022-5223(94)70286-1
114. Preiser JC, Schmartz D, Van der Linden P, Content J, Vanden Bussche P, Buurman W, et al. Interleukin-6 administration has no acute hemodynamic or hematologic effect in the dog. Cytokine. (1991) 3(1):1–4. doi: 10.1016/1043-4666(91)90002-u
115. Smith WB, Gamble JR, Clark-Lewis I, Vadas MA. Interleukin-8 induces neutrophil transendothelial migration. Immunology. (1991) 72:65–72.1997402
116. Metinko AP, Kunkel SL, Standiford TJ, Strieter RM. Anoxia-hyperoxia induces monocyte-derived interleukin-8. J Clin Invest. (1992) 90:791–8. doi: 10.1172/JCI115953
117. Wan S, Izzat MB, Lee TW, Wan IY, Tang NL, Yim AP. Avoiding cardiopulmonary bypass in multivessel CABG reduces cytokine response and myocardial injury. Ann Thorac Surg. (1999) 68(1):52–6; discussion 56–7. doi: 10.1016/s0003-4975(99)00315-x
118. Finn A, Naik S, Klein N, Levinsky RJ, Strobel S, Elliott M. Interleukin-8 release and neutrophil degranulation after pediatric cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1993) 105(2):234–41. doi: 10.1016/S0022-5223(19)33806-1
119. Burns SA, Newburger JW, Xiao M, Mayer JE Jr, Walsh AZ, Neufeld EJ. Induction of interleukin-8 messenger RNA in heart and skeletal muscle during pediatric cardiopulmonary bypass. Circulation. (1995) 92(9 Suppl):II315–21. doi: 10.1161/01.cir.92.9.315
120. Wan S, DeSmet JM, Barvais L, Goldstein M, Vincent JL, LeClerc JL. Myocardium is a major source of proinflammatory cytokines in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1996) 112(3):806–11. doi: 10.1016/S0022-5223(96)70068-5
121. Wan S, DeSmet JM, Antoine M, Goldman M, Vincent JL, LeClerc JL. Steroid administration in heart and heart-lung transplantation: is the timing adequate? Ann Thorac Surg. (1996) 61(2):674–8. doi: 10.1016/0003-4975(95)01059-9
122. Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL. Tumor necrosis factor-alpha gene and protein expression in adult feline myocardium after endotoxin administration. J Clin Invest. (1995) 96(2):1042–52. doi: 10.1172/JCI118090
123. Liangos O, Kolyada A, Tighiouart H, Perianayagam MC, Wald R, Jaber BL. Interleukin-8 and acute kidney injury following cardiopulmonary bypass: a prospective cohort study. Nephron Clin Pract. (2009) 113(3):c148–54. doi: 10.1159/000232595
124. Wang P, Wu P, Anthes JC, Siegel MI, Egan RW, Billah MM. Interleukin-10 inhibits interleukin-8 production in human neutrophils. Blood. (1994) 83:2678–83. doi: 10.1182/blood.V83.9.2678.2678
125. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. (1991) 147:3815–22. doi: 10.4049/jimmunol.147.11.3815
126. Cassatella MA, Meda L, Gasperini S, Calzetti F, Bonora S. Interleukin-10 (IL-10) upregulates IL-1 receptor antagonist production from lipopolysaccharide-stimulated human polymorphonuclear leukocytes by delaying mRNA degradation. J Exp Med. (1994) 179:1695–9. doi: 10.1084/jem.179.5.1695
127. Thompson K, Wright S, Sheron N. Chemotactic and suppressor cytokine networks. Hepatogastroenterology. (1996) 43:15–31.8682452
128. Ayala A, Deol DK, Lehman DL, Herdon CD, Chaudry IH. Polymicrobial sepsis but not low-dose endotoxin infusion causese decreased splenocyte IL-1/IFN-gamma release while increasing IL-4/IL-10 production. J Surg Res. (1994) 56:579–85. doi: 10.1006/jsre.1994.1092
129. Oz MC, Liao H, Naka Y, Seldomridge A, Becker DN, Michler RE, et al. Ischemia-induced interleukin-8 release after human heart transplantation: a potential role for endothelial cells. Circulation. (1995) 92(Suppl II):II-428–32. doi: 10.1161/01.cir.92.9.428
130. Drazan KE, Wu L, Olthoff KM, Jurim O, Busuttil RW, Shaked A. Transduction of hepatic allografts achieves local levels of viral IL-10 which suppress alloreactivity in vitro. J Surg Res. (1995) 59:219–23. doi: 10.1006/jsre.1995.1157
131. Le Moine O, Marchant A, Durand F, Ickx B, Pradier O, Belghiti J, et al. Systemic release of interleukin-10 during orthotopic liver transplantation. Hepatology. (1994) 20:889–92. doi: 10.1002/hep.1840200417
132. Tabardel Y, Duchateau J, Schmartz D, Marécaux G, Shahla M, Barvais L, et al. Corticosteroids increase blood interleukin-10 levels during cardiopulmonary bypass in men. Surgery. (1996) 119(1):76–80. doi: 10.1016/s0039-6060(96)80217-0
133. Dehoux M, Philip I, Chollet-Martin S, Boutten A, Hvass U, Desmots JM, et al. Early production of interleukin-10 during normothermic cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1995) 110:286–7. doi: 10.1016/S0022-5223(05)80052-2
134. Marchant A, Bruyns C, Vandenabeele P, Ducarme M, Gérard C, Delvaux A, et al. Interleukin-10 controls interferon-gamma and tumor necrosis factor production during experimental endotoxemia. Eur J Immunol. (1994) 24:1167–71. doi: 10.1002/eji.1830240524
135. de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin-10 inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. (1991) 174:1209–20. doi: 10.1084/jem.174.5.1209
136. Brull DJ, Montgomery HE, Sanders J, Dhamrait S, Luong L, Rumley A, et al. Interleukin-6 gene −174g>c and −572g>c promoter polymorphisms are strong predictors of plasma interleukin-6 levels after coronary artery bypass surgery. Arterioscler Thromb Vasc Biol. (2001) 21(9):1458–63. doi: 10.1161/hq0901.094280
137. Gaudino M, Di Castelnuovo A, Zamparelli R, Andreotti F, Burzotta F, Iacoviello L, et al. Genetic control of postoperative systemic inflammatory reaction and pulmonary and renal complications after coronary artery surgery. J Thorac Cardiovasc Surg. (2003) 126(4):1107–12. doi: 10.1016/s0022-5223(03)00396-9
138. Grocott HP, White WD, Morris RW, Podgoreanu MV, Mathew JP, Nielsen DM, et al. Genetic polymorphisms and the risk of stroke after cardiac surgery. Stroke. (2005) 36(9):1854–8. doi: 10.1161/01.STR.0000177482.23478.dc
139. Podgoreanu MV, White WD, Morris RW, Mathew JP, Stafford-Smith M, Welsby IJ, et al. Inflammatory gene polymorphisms and risk of postoperative myocardial infarction after cardiac surgery. Circulation. (2006) 114(1 Suppl):I275–81. doi: 10.1161/CIRCULATIONAHA.105.001032
140. Burzotta F, Iacoviello L, Di Castelnuovo A, Glieca F, Luciani N, Zamparelli R, et al. Relation of the −174 G/C polymorphism of interleukin-6 to interleukin-6 plasma level s and to length of hospitalization after surgical coronary revascularization. Am J Cardiol. (2001) 88(10):1125–8. doi: 10.1016/s0002-9149(01)02046-x
141. Yoon SZ, Jang IJ, Choi YJ, Kang MH, Lim HJ, Lim YJ, et al. Association between tumor necrosis factor alpha 308G/A polymorphism and increased proinflammatory cytokine release after cardiac surgery with cardiopulmonary bypass in the Korean population. J Cardiothorac Vasc Anesth. (2009) 23(5):646–50. doi: 10.1053/j.jvca.2009.03.004
142. Isbir CS, Ergen A, Tekeli A, Zeybek U, Gormus U, Arsan S. The effect of NQO1 polymorphism on the inflammatory response in cardiopulmonary bypass. Cell Biochem Funct. (2008) 26(4):534–8. doi: 10.1002/cbf.1456
143. MacKensen GB, Swaminathan M, Ti LK, Grocott HP, Phillips-Bute BG, Mathew JP, et al. Preliminary report on the interaction of apolipoprotein E polymorphism with aortic atherosclerosis and acute nephropathy after CABG. Ann Thorac Surg. (2004) 78(2):520–6. doi: 10.1016/j.athoracsur.2004.02.106
144. Drabe N, Zünd G, Grünenfelder J, Sprenger M, Hoerstrup SP, Bestmann L, et al. Genetic predisposition in patients undergoing cardiopulmonary bypass surgery is associated with an increase of inflammatory cytokines. Eur J Cardiothorac Surg. (2001) 20(3):609–13. doi: 10.1016/s1010-7940(01)00842-9
145. Grünenfelder J, Umbehr M, Plass A, Bestmann L, Maly FE, Zünd G, et al. Genetic polymorphisms of apolipoprotein E4 and tumor necrosis factor beta as predisposing factors for increased inflammatory cytokines after cardiopulmonary bypass. J Thorac Cardiovasc Surg. (2004) 128(1):92–7. doi: 10.1016/j.jtcvs.2004.02.022
146. Patrick DA, Moore EE, Fullerton DA, Barnett CC Jr, Meldrum DR, Silliman CC. Cardiopulmonary bypass renders patients at risk for multiple organ failure via early neutrophil priming and late neutrophil disability. J Surg Res. (1999) 86(1):42–9. doi: 10.1006/jsre.1999.5702
147. Elliott MJ, Finn AH. Interaction between neutrophils and endothelium. Ann Thorac Surg. (1993) 56(6):1503–8. doi: 10.1016/0003-4975(93)90741-y
148. Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. (1994) 94(3):1147–55. doi: 10.1172/JCI117430
149. Boyle EM Jr, Pohlman TH, Johnson MC, Verrier ED. Endothelial cell injury in cardiovascular surgery: the systemic inflammatory response. Ann Thorac Surg. (1997) 63(1):277–84. doi: 10.1016/s0003-4975(96)01061-2
150. Gilliland HE, Armstrong MA, Uprichard S, Clarke G, McMurray TJ. The effect of aprotinin on interleukin-8 concentration and leukocyte adhesion molecule expression in an isolated cardiopulmonary bypass system. Anaesthesia. (1999) 54(5):427–33. doi: 10.1046/j.1365-2044.1999.00656.x
151. Tönz M, Mihaljevic T, von Segesser LK, Fehr J, Schmid ER, Turina MI. Acute lung injury during cardiopulmonary bypass. Are the neutrophils responsible? Chest. (1995) 108(6):1551–6. doi: 10.1378/chest.108.6.1551
152. Boyle EM Jr, Morgan EN, Kovacich JC, Canty TG Jr, Verrier ED. Microvascular responses to cardiopulmonary bypass. J Cardiothorac Vasc Anesth. (1999) 13(4 Suppl 1):30–5; discussion 36–7. doi: 10.1016/S1053-0770(21)00595-4
153. Shimono T, Yada I, Kanamori Y, Sato T, Kondo C, Tanaka K, et al. Reticuloendothelial function following cardiopulmonary bypass: laboratory and electron microscopy findings. J Surg Res. (1994) 56(5):446–51. doi: 10.1006/jsre.1994.1070
154. Shimono T, Yada I, Kusagawa M, Nosé Y. Oversaturation status of reticuloendothelial system following cardiopulmonary bypass. Artif Organs. (1994) 18(8):596–602. doi: 10.1111/j.1525-1594.1994.tb03384.x
155. Ratliff NB, Young WG Jr, Hackel DB, Mikat E, Wilson JW. Pulmonary injury secondary to extracorporeal circulation. An ultrastructural study. J Thorac Cardiovasc Surg. (1973) 65(3):425–32. doi: 10.1016/S0022-5223(19)40771-X
156. Royston D, Fleming JS, Desai JB, Westaby S, Taylor KM. Increased production of peroxidation products associated with cardiac operations. Evidence for free radical generation. J Thorac Cardiovasc Surg. (1986) 91(5):759–66. doi: 10.1016/S0022-5223(19)35998-7
157. Wilson I, Gillinov AM, Curtis WE, DiNatale J, Burch RM, Gardner TJ, et al. Inhibition of neutrophil adherence improves postischemic ventricular performance of the neonatal heart. Circulation. (1993) 88(5 Pt 2):II372–9.8222181
158. Mayers I, Hurst T, Johnson D, Cujec B, Ang LC, Thomson D, et al. Anti-CD18 antibodies improve cardiac function following cardiopulmonary bypass in dogs. J Crit Care. (1996) 11(4):189–96. doi: 10.1016/s0883-9441(96)90030-1
159. Friedman M, Wang SY, Sellke FW, Cohn WE, Weintraub RM, Johnson RG. Neutrophil adhesion blockade with NPC 15669 decreases pulmonary injury after total cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1996) 111(2):460–8. doi: 10.1016/s0022-5223(96)70457-9
160. Roth M, Kraus B, Scheffold T, Reuthebuch O, Klövekorn WP, Bauer EP. The effect of leukocyte-depleted blood cardioplegia in patients with severe left ventricular dysfunction: a randomized, double-blind study. J Thorac Cardiovasc Surg. (2000) 120(4):642–50. doi: 10.1067/mtc.2000.109707
161. Hill GE, Whitten CW, Landers DF. The influence of cardiopulmonary bypass on cytokines and cell-cell communication. J Cardiothorac Vasc Anesth. (1997) 11(3):367–75. doi: 10.1016/s1053-0770(97)90107-5
162. Cicala C, Cirino G. Linkage between inflammation and coagulation: an update on the molecular basis of the crosstalk. Life Sci. (1998) 62(20):1817–24. doi: 10.1016/s0024-3205(97)01167-3
163. Mulivor AW, Lipowsky HH. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am J Physiol Heart Circ Physiol. (2002) 283(4):H1282–91. doi: 10.1152/ajpheart.00117.2002
164. Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. (2007) 116(17):1896–906. doi: 10.1161/CIRCULATIONAHA.106.684852
165. Myers GJ, Wegner J. Endothelial glycocalyx and cardiopulmonary bypass. J Extra Corpor Technol. (2017) 49(3):174–81. doi: 10.1051/ject/201749174
166. Yao Y, Rabodzey A, Dewey CF Jr. Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol. (2007) 293(2):H1023–30. doi: 10.1152/ajpheart.00162.2007
167. Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol. (2015) 80(3):389–402. doi: 10.1111/bcp.12629
168. Madjid M, Willerson JT. Inflammatory markers in coronary heart disease. Br Med Bull. (2011) 100:23–38. doi: 10.1093/bmb/ldr043
169. Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. (2003) 278:40764–70. doi: 10.1074/jbc.M306736200
170. Robich M, Ryzhov S, Kacer D, Palmeri M, Peterson SM, Quinn RD, et al. Prolonged cardiopulmonary bypass is associated with endothelial glycocalyx degradation. J Surg Res. (2020) 251:287–95. doi: 10.1016/j.jss.2020.02.011
171. Zhang Y, Wang Z, Liu J, Zhang S, Fei J, Li J, et al. Cell surface-anchored syndecan-1 ameliorates intestinal inflammation and neutrophil transmigration in ulcerative colitis. J Cell Mol Med. (2017) 21:13–25. doi: 10.1111/jcmm.12934
172. Gill SE, Nadler ST, Li Q, Frevert CW, Park PW, Chen P, et al. Shedding of syndecan-1/CXCL1 complexes by matrix metalloproteinase 7 functions as an epithelial checkpoint of neutrophil activation. Am J Respir Cell Mol Biol. (2016) 55:243–51. doi: 10.1165/rcmb.2015-0193OC
173. Mosheimer BA, Kaneider NC, Feistritzer C, Djanani AM, Sturn DH, Patsch JR, et al. Syndecan-1 is involved in osteoprotegerin-induced chemotaxis in human peripheral blood monocytes. J Clin Endocrinol Metab. (2005) 90:2964–71. doi: 10.1210/jc.2004-1895
174. Pesonen E, Passov A, Andersson S, Suojaranta R, Niemi T, Raivio P, et al. Glycocalyx degradation and inflammation in cardiac surgery. J Cardiothorac Vasc Anesth. (2019) 33(2):341–5. doi: 10.1053/j.jvca.2018.04.007
175. de Melo Bezerra Cavalcante CT, Castelo Branco KM, Pinto Júnior VC, Meneses GC, de Oliveira Neves FM, de Souza NM, et al. Syndecan-1 improves severe acute kidney injury prediction after pediatric cardiac surgery. J Thorac Cardiovasc Surg. (2016) 152(1):178–186.e2. doi: 10.1016/j.jtcvs.2016.03.079
176. Warren OJ, Watret AL, de Wit KL, Alexiou C, Vincent C, Darzi AW, et al. The inflammatory response to cardiopulmonary bypass: part 2-anti-inflammatory therapeutic strategies. J Cardiothorac Vasc Anesth. (2009) 23(3):384–93. doi: 10.1053/j.jvca.2008.09.007
177. Landis RC, Brown JR, Fitzgerald D, Likosky DS, Shore-Lesserson L, Baker RA, et al. Attenuating the systemic inflammatory response to adult cardiopulmonary bypass: a critical review of the evidence base. J Extra Corpor Technol. (2014) 46(3):197–211. doi: 10.1051/ject/201446197
178. Videm V, Mollnes TE, Bergh K, Fosse E, Mohr B, Hagve TA, et al. Heparin-coated cardiopulmonary bypass equipment. II mechanism for reduced complement activation in vivo. J Thorac Cardiovasc Surg. (1999) 117(4):803–9. doi: 10.1016/S0022-5223(99)70302-8
179. Goudeau JJ, Clermont G, Guillery O, Lemaire-Ewing S, Musat A, Vernet M, et al. In high-risk patients, combination of antiinflammatory procedures during cardiopulmonary bypass can reduce incidences of inflammation and oxidative stress. J Cardiovasc Pharmacol. (2007) 49(1):39–45. doi: 10.1097/FJC.0b013e31802c0cd0
180. Gu YJ, van Oeveren W, Akkerman C, Boonstra PW, Huyzen RJ, Wildevuur CR. Heparin-coated circuits reduce the inflammatory response to cardiopulmonary bypass. Ann Thorac Surg. (1993) 55(4):917–22. doi: 10.1016/0003-4975(93)90117-z
181. Videm V, Mollnes TE, Fosse E, Mohr B, Bergh K, Hagve TA, et al. Heparin-coated cardiopulmonary bypass equipment. I biocompatability markers and development of complications in high-risk population. J Thorac Cardiovasc Surg. (1999) 117(4):794–802. doi: 10.1016/s0022-5223(99)70301-6
182. Wan S, LeClerc JL, Antoine M, DeSmet JM, Yim AP, Vincent JL. Heparin-coated circuits reduce myocardial injury in heart or heart-lung transplantation: a prospective, randomized trial. Ann Thorac Surg. (1999) 68(4):1230–5. doi: 10.1016/s0003-4975(99)00701-8
183. te Velthuis H, Baufreton C, Jansen PG, Thijs CM, Hack CE, Sturk A, et al. Heparin coating of extracorporeal circuits inhibits contact activation during cardiac operations. J Thorac Cardiovasc Surg. (1997) 114(1):117–22. doi: 10.1016/S0022-5223(97)70124-7
184. Moen O, Høgåsen K, Fosse E, Dregelid E, Brockmeier V, Venge P, et al. Attenuation of changes in leukocyte surface markers and complement activation with heparin-coated cardiopulmonary bypass. Ann Thorac Surg. (1997) 63(1):105–11. doi: 10.1016/s0003-4975(96)00743-6
185. Plötz FB, van Oeveren W, Hultquist KA, Miller C, Bartlett RH, Wildevuur CR. A heparin-coated circuit reduces complement activation and the release of leukocyte inflammatory mediators during extracorporeal circulation in a rabbit. Artif Organs. (1992) 16(4):366–70. doi: 10.1111/j.1525-1594.1992.tb00533.x
186. Weerwind PW, Maessen JG, van Tits LJ, Stad RK, Fransen EJ, de Jong DS, et al. Influence of Duraflo II heparin-treated extracorporeal circuits on the systemic inflammatory response in patients having coronary bypass. J Thorac Cardiovasc Surg. (1995) 110(6):1633–41. doi: 10.1016/S0022-5223(95)70024-2
187. Steinberg BM, Grossi EA, Schwartz DS, McLoughlin DE, Aguinaga M, Bizekis C, et al. Heparin bonding of bypass circuits reduces cytokine release during cardiopulmonary bypass. Ann Thorac Surg. (1995) 60(3):525–9. doi: 10.1016/0003-4975(95)00482-z
188. Lindholm L, Westerberg M, Bengtsson A, Ekroth R, Jensen E, Jeppsson A. A closed perfusion system with heparin coating and centrifugal pump improves cardiopulmonary bypass biocompatibility in elderly patients. Ann Thorac Surg. (2004) 78(6):2131–8. doi: 10.1016/j.athoracsur.2004.06.011
189. McCarthy PM, Yared JP, Foster RC, Ogella DA, Borsh JA, Cosgrove DM. A prospective randomized trial of Duraflo II heparin-coated circuits in cardiac reoperations. Ann Thorac Surg. (1999) 67(5):1268–73. doi: 10.1016/s0003-4975(99)00136-8
190. Mirow N, Zittermann A, Koertke H, Maleszka A, Knobl H, Coskun T, et al. Heparin-coated extracorporeal circulation in combination with low dose systemic heparinization reduces early postoperative blood loss in cardiac surgery. J Cardiovasc Surg (Torino). (2008) 49(2):277–84.18431350
191. Jansen PG, te Velthuis H, Huybregts RA, Paulus R, Bulder ER, van der Spoel HI, et al. Reduced complement activation and improved postoperative performance after cardiopulmonary bypass with heparin-coated circuits. J Thorac Cardiovasc Surg. (1995) 110(3):829–34. doi: 10.1016/S0022-5223(95)70117-6
192. Ranucci M, Mazzucco A, Pessotto R, Grillone G, Casati V, Porreca L, et al. Heparin-coated circuits for high risk patients: a multicenter, prospective, randomized trial. Ann Thorac Surg. (1999) 67(4):994–1000. doi: 10.1016/s0003-4975(99)00062-4
193. Mangoush O, Purkayastha S, Haj-Yahia S, Kinross J, Hayward M, Bartolozzi F, et al. Heparin-bonded circuits versus nonheparin-bonded circuits: an evaluation of their effect on clinical outcomes. Eur J Cardiothorac Surg. (2007) 31(6):1058–69. doi: 10.1016/j.ejcts.2007.01.029
194. Svenmarker S, Häggmark S, Jansson E, Lindholm R, Appelblad M, Sandström E, et al. Use of heparin-bonded circuits in cardiopulmonary bypass improves clinical outcome. Scand Cardiovasc J. (2002) 36(4):241–6. doi: 10.1080/14017430260180418
195. Thiara AS, Andersen VY, Videm V, Mollnes TE, Svennevig K, Hoel TN, et al. Comparable biocompatibility of phisio- and bioline-coated cardiopulmonary bypass circuits indicated by the inflammatory response. Perfusion. (2010) 25(1):9–16. doi: 10.1177/0267659110362822
196. Sohn N, Marcoux J, Mycyk T, Krahn J, Meng Q. The impact of different biocompatible coated cardiopulmonary bypass circuits on inflammatory response and oxidative stress. Perfusion. (2009) 24(4):231–7. doi: 10.1177/0267659109351218
197. Dekker NAM, Veerhoek D, van Leeuwen ALI, Vonk ABA, van den Brom CE, Boer C. Microvascular alterations during cardiac surgery using a heparin or phosphorylcholine-coated circuit. J Cardiothorac Vasc Anesth. (2020) 34(4):912–9. doi: 10.1053/j.jvca.2019.10.012
198. Chen L, Lv L, Long C, Lou S. Effects of circuit albumin coating on coagulation and inflammatory response for patients receiving aortic arch replacement: a randomized controlled trial. Perfusion. (2016) 31(7):576–83. doi: 10.1177/0267659116645662
199. Gunaydin S, McCusker K, Sari T, Onur MA, Zorlutuna Y. Clinical performance and biocompatibility of hyaluronan-based heparin-bonded extracorporeal circuits in different risk cohorts. Interact CardioVasc Thorac Surg. (2010) 10(3):371–6. doi: 10.1510/icvts.2009.220756
200. Hasegawa T, Oshima Y, Yokoyama S, Akimoto A, Misaka Y, Akiyama S. Clinical application of a new ternary polymer, SEC-1 coat™, for pediatric cardiopulmonary bypass circuits: a prospective randomized pilot study. Perfusion. (2020) 35(8):826–32. doi: 10.1177/0267659120915387
201. Giorni C, Pezzella C, Bojan M, Ricci Z, Pouard P, Raisky O, et al. Impact of heparin- or nonheparin-coated circuits on platelet function in pediatric cardiac surgery. Ann Thorac Surg. (2019) 107(4):1241–7. doi: 10.1016/j.athoracsur.2018.09.032
202. Tayama E, Hayashida N, Akasu K, Kosuga T, Fukunaga S, Akashi H, et al. Biocompatibility of heparin-coated extracorporeal bypass circuits: new heparin bonded bioline system. Artif Organs. (2000) 24(8):618–23. doi: 10.1046/j.1525-1594.2000.06615.x
203. Fosse E, Thelin S, Svennevig JL, Jansen P, Mollnes TE, Hack E, et al. Duraflo II coating of cardiopulmonary bypass circuits reduces complement activastion but does not affect the release of granulocyte enzymes: a European multicenter study. Eur J Cardiothorac Surg. (1997) 11(2):320–7. doi: 10.1016/s1010-7940(96)01062-7
204. Journois D, Israel-Biet D, Pouard P, Rolland B, Silvester W, Vouhé P, et al. High-volume, zero-balanced hemofiltration to reduce delayed inflammatory response to cardiopulmonary bypass in children. Anesthesiology. (1996) 85(5):965–76. doi: 10.1097/00000542-199611000-00003
205. Huang H, Yao T, Wang W, Zhu D, Zhang W, Chen H, et al. Combination of balanced ultrafiltration with modified ultrafiltration attenuates pulmonary injury in patients undergoing open heart surgery. Chin Med J (Engl). (2003) 116(10):1504–7.14570610
206. Journois D, Pouard P, Greeley WJ, Mauriat P, Vouhé P, Safran D. Hemofiltration during cardiopulmonary bypass in pediatric cardiac surgery. Effects on hemostasis, cytokines, and complement components. Anesthesiology. (1994) 81(5):1181–9; discussion 26A–27A. doi: 10.1097/00000542-199411000-00011
207. Bando K, Vijay P, Turrentine MW, Sharp TG, Means LJ, Ensing GJ, Lalone BJ, Sekine Y, Szekely L, Brown JW. Dilutional and modified ultrafiltration reduces pulmonary hypertension after operations for congenital heart disease: a prospective randomized study. J Thorac Cardiovasc Surg. (1998) 115(3):517–25; discussion 525–7. doi: 10.1016/S0022-5223(98)70313-7
208. Garau I, März A, Sehner S, Reuter DA, Reichenspurner H, Zöllner C, et al. Hemadsorption during cardiopulmonary bypass reduces interleukin 8 and tumor necrosis factor α serum levels in cardiac surgery: a randomized controlled trial. Minerva Anestesiol. (2019) 85(7):715–23. doi: 10.23736/S0375-9393.18.12898-7
209. Boodhwani M, Williams K, Babaev A, Gill G, Saleem N, Rubens FD. Ultrafiltration reduces blood transfusions following cardiac surgery: a meta-analysis. Eur J Cardiothorac Surg. (2006) 30(6):892–7. doi: 10.1016/j.ejcts.2006.09.014
210. Hiramatsu T, Imai Y, Kurosawa H, Takanashi Y, Aoki M, Shinoka T, et al. Effects of dilutional and modified ultrafiltration in plasma endothelin-1 and pulmonary vascular resistance after the Fontan procedure. Ann Thorac Surg. (2002) 73(3):861–5.11899192
211. Kosour C, Dragosavac D, Antunes N, Almeida de Oliveira RA, Martins Oliveira PP, Wilson Vieira R. Effect of ultrafiltration on pulmonary function and interleukins in patients undergoing cardiopulmonary bypass. J Cardiothorac Vasc Anesth. (2016) 30(4):884–90. doi: 10.1053/j.jvca.2015.10.009
212. Naik SK, Knight A, Elliott MJ. A successful modification of ultrafiltration for cardiopulmonary bypass in children. Perfusion. (1991) 6(1):41–50. doi: 10.1177/026765919100600106
213. Davies MJ, Nguyen K, Gaynor JW, Elliott MJ. Modified ultrafiltration improves left ventricular systolic function in infants after cardiopulmonary bypass. J Thorac Cardiovasc Surg. (1998) 115(2):361–9; discussion 369–70. doi: 10.1016/S0022-5223(98)70280-6
214. Bierer J, Stanzel R, Henderson M, Sett S, Horne D. Ultrafiltration in pediatric cardiac surgery review. World J Pediatr Congenit Heart Surg. (2019) 10(6):778–88. doi: 10.1177/2150135119870176
215. Watanabe T, Sakai Y, Mayumi T, Shimomura T, Song MH, Tajima K, et al. Effect of ultrafiltration during cardiopulmonary bypass for pediatric cardiac surgery. Artif Organs. (1998) 22(12):1052–5. doi: 10.1046/j.1525-1594.1998.06192.x
216. Torina AG, Petrucci O, Oliveira PP, Severino ES, Vilarinho KA, Lavagnoli CF, et al. The effects of modified ultrafiltration on pulmonary function and transfusion requirements in patients underwent coronary artery bypass graft surgery. Rev Bras Cir Cardiovasc. (2010) 25(1):59–65. doi: 10.1590/s0102-76382010000100014
217. Luciani GB, Menon T, Vecchi B, Auriemma S, Mazzucco A. Modified ultrafiltration reduces morbidity after adult cardiac operations: a prospective, randomized clinical trial. Circulation. (2001) 104(12 Suppl 1):I253–9. doi: 10.1161/hc37t1.094931
218. Apostolakis E, Filos KS, Koletsis E, Dougenis D. Lung dysfunction following cardiopulmonary bypass. J Card Surg. (2010) 25(1):47–55. doi: 10.1111/j.1540-8191.2009.00823.x
219. Onorati F, Santini F, Mariscalco G, Bertolini P, Sala A, Faggian G, et al. Leukocyte filtration ameliorates the inflammatory response in patients with mild to moderate lung dysfunction. Ann Thorac Surg. (2011) 92(1):111–21; discussion 121. doi: 10.1016/j.athoracsur.2011.03.087
220. Rubino AS, Serraino GF, Mariscalco G, Marsico R, Sala A, Renzulli A. Leukocyte depletion during extracorporeal circulation allows better organ protection but does not change hospital outcomes. Ann Thorac Surg. (2011) 91(2):534–40. doi: 10.1016/j.athoracsur.2010.09.077
221. Zhang X, Zhou C, Zhuang J, Xiao X, Zheng S, Xiong W, et al. Effects of leukocyte depletion on cardiopulmonary protection and inflammation after valve surgery. Int J Artif Organs. (2010) 33(11):812–8. PMID: 21140357.21140357
222. Koskenkari J, Rimpiläinen J, Biancari F, Surcel HM, Kaukoranta P, Kiviluoma K, et al. Leukocyte depleting filter attenuates myocardial injury during elective coronary artery bypass surgery. Scand Cardiovasc J. (2005) 39(6):358–68. doi: 10.1080/14017430510035943
223. Alexiou C, Tang AT, Sheppard SV, Haw MP, Gibbs R, Smith DC. A prospective randomized study to evaluate the effect of leukodepletion on the rate of alveolar production of exhaled nitric oxide during cardiopulmonary bypass. Ann Thorac Surg. (2004) 78(6):2139–45; discussion 2145. doi: 10.1016/j.athoracsur.2004.05.087
224. Alexiou C, Tang AA, Sheppard SV, Smith DC, Gibbs R, Livesey SA, et al. The effect of leucodepletion on leucocyte activation, pulmonary inflammation and respiratory index in surgery for coronary revascularization: a prospective randomized study. Eur J Cardiothorac Surg. (2004) 26(2):294–300. doi: 10.1016/j.ejcts.2004.04.017
225. Chen YF, Tsai WC, Lin CC, Tsai LY, Lee CS, Huang CH, Pan PC, Chen ML. Effect of leukocyte depletion on endothelial cell activation and transendothelial migration of leukocytes during cardiopulmonary bypass. Ann Thorac Surg. (2004) 78(2):634–42; discussion 642–3. doi: 10.1016/j.athoracsur.2004.02.091
226. de Amorim CG, Malbouisson LM, da Silva FC Jr, Fiorelli AI, Murakami CK, Carmona MJ. Leukocyte depletion during CPB: effects on inflammation and lung function. Inflammation. (2014) 37(1):196–204. doi: 10.1007/s10753-013-9730-z
227. Karaiskos TE, Palatianos GM, Triantafillou CD, Kantidakis GH, Astras GM, Papadakis EG, et al. Clinical effectiveness of leukocyte filtration during cardiopulmonary bypass in patients with chronic obstructive pulmonary disease. Ann Thorac Surg. (2004) 78(4):1339–44. doi: 10.1016/j.athoracsur.2004.04.040
228. Sheppard SV, Gibbs RV, Smith DC. Does leucocyte depletion during cardiopulmonary bypass improve oxygenation indices in patients with mild lung dysfunction? Br J Anaesth. (2004) 93(6):789–92. doi: 10.1093/bja/aeh267
229. Hurst T, Johnson D, Cujec B, Thomson D, Mycyk T, Burbridge B, et al. Depletion of activated neutrophils by a filter during cardiac valve surgery. Can J Anaesth. (1997) 44(2):131–9. doi: 10.1007/BF03013000
230. De Vecchi E, Paroni R, Pala MG, Di Credico G, Agape V, Gobbi C, et al. Role of leucocytes in free radical production during myocardial revascularization. Heart. (1997) 77(5):449–55. doi: 10.1136/hrt.77.5.449
231. Sawa Y, Taniguchi K, Kadoba K, Nishimura M, Ichikawa H, Amemiya A, et al. Leukocyte depletion attenuates reperfusion injury in patients with left ventricular hypertrophy. Circulation. (1996) 93(9):1640–6. doi: 10.1161/01.cir.93.9.1640
232. Hayashi Y, Sawa Y, Fukuyama N, Miyamoto Y, Takahashi T, Nakazawa H, et al. Leukocyte-depleted terminal blood cardioplegia provides superior myocardial protective effects in association with myocardium-derived nitric oxide and peroxynitrite production for patients undergoing prolonged aortic crossclamping for more than 120 min. J Thorac Cardiovasc Surg. (2003) 126(6):1813–21. doi: 10.1016/s0022-5223(03)01282-0
233. van de Watering LM, Hermans J, Houbiers JG, van den Broek PJ, Bouter H, Boer F, Harvey MS, Huysmans HA, Brand A. Beneficial effects of leukocyte depletion of transfused blood on postoperative complications in patients undergoing cardiac surgery: a randomized clinical trial. Circulation. (1998) 97(6):562–8. doi: 10.1161/01.cir.97.6.562
234. Soo AW, Maher BM, Daly L, Wood AE, Watson WR. Preoperative neutrophil response as a predictive marker of clinical outcome following open heart surgery and the impact of leukocyte filtration. Interact Cardiovasc Thorac Surg. (2010) 11(5):604–11. doi: 10.1510/icvts.2009.228056
235. Tang AT, Alexiou C, Hsu J, Sheppard SV, Haw MP, Ohri SK. Leukodepletion reduces renal injury in coronary revascularization: a prospective randomized study. Ann Thorac Surg. (2002) 74(2):372–7; discussion 377. doi: 10.1016/s0003-4975(02)03715-3.12173815
236. Yang B, Hosgood SA, Harper SJ, Nicholson ML. Leucocyte depletion improves renal function in porcine kidney hemoreperfusion through reduction of myeloperoxidase+ cells, caspase-3, IL-1β, and tubular apoptosis. J Surg Res. (2010) 164(2):e315–24. doi: 10.1016/j.jss.2010.07.044
237. Bernardi MH, Rinoesl H, Dragosits K, Ristl R, Hoffelner F, Opfermann P, et al. Effect of hemoadsorption during cardiopulmonary bypass surgery—a blinded, randomized, controlled pilot study using a novel adsorbent. Crit Care. (2016) 20:96. doi: 10.1186/s13054-016-1270-0
238. Poli EC, Alberio L, Bauer-Doerries A, Marcucci C, Roumy A, Kirsch M, et al. Cytokine clearance with CytoSorb® during cardiac surgery: a pilot randomized controlled trial. Crit Care. (2019) 23(1):108. doi: 10.1186/s13054-019-2399-4
239. Gleason TG, Argenziano M, Bavaria JE, Kane LC, Coselli JS, Engelman RM, et al. Hemoadsorption to reduce plasma-free hemoglobin during cardiac surgery: results of REFRESH I pilot study. Semin Thorac Cardiovasc Surg. (2019) 31(4):783–93. doi: 10.1053/j.semtcvs.2019.05.006
240. Taleska Stupica G, Sostaric M, Bozhinovska M, Rupert L, Bosnic Z, Jerin A, et al. Extracorporeal hemadsorption versus glucocorticoids during cardiopulmonary bypass: a prospective, randomized, controlled trial. Cardiovasc Ther. (2020) 2020:7834173. doi: 10.1155/2020/7834173
241. Bourbon A, Vionnet M, Leprince P, Vaissier E, Copeland J, McDonagh P, et al. The effect of methylprednisolone treatment on the cardiopulmonary bypass-induced systemic inflammatory response. Eur J Cardiothorac Surg. (2004) 26(5):932–8. doi: 10.1016/j.ejcts.2004.07.044
242. Doukas P, Hellfritsch G, Wendt D, Magliani M, Barbati ME, Jalaie H, et al. Intraoperative hemoadsorption (cytosorb™) during open thoracoabdominal aortic repair: a pilot randomized controlled trial. J Clin Med. (2023) 12(2):546. doi: 10.3390/jcm12020546
243. Boldt J, Osmer C, Linke LC, Görlach G, Hempelmann G. Hypothermic versus normothermic cardiopulmonary bypass: influence on circulating adhesion molecules. J Cardiothorac Vasc Anesth. (1996) 10(3):342–7. doi: 10.1016/s1053-0770(96)80094-2
244. Birdi I, Caputo M, Underwood M, Bryan AJ, Angelini GD. The effects of cardiopulmonary bypass temperature on inflammatory response following cardiopulmonary bypass. Eur J Cardiothorac Surg. (1999) 16(5):540–5. doi: 10.1016/s1010-7940(99)00301-2
245. Ohata T, Sawa Y, Kadoba K, Kagisaki K, Suzuki K, Matsuda H. Role of nitric oxide in a temperature dependent regulation of systemic vascular resistance in cardiopulmonary bypass. Eur J Cardiothorac Surg. (2000) 18(3):342–7. doi: 10.1016/s1010-7940(00)00455-3
246. Lomivorotov VV, Shmirev VA, Efremov SM, Ponomarev DN, Moroz GB, Shahin DG, et al. Hypothermic versus normothermic cardiopulmonary bypass in patients with valvular heart disease. J Cardiothorac Vasc Anesth. (2014) 28(2):295–300. doi: 10.1053/j.jvca.2013.03.009
247. Oda T, Yamaguchi A, Yokoyama M, Shimizu K, Toyota K, Nikai T, et al. Plasma proteomic changes during hypothermic and normothermic cardiopulmonary bypass in aortic surgeries. Int J Mol Med. (2014) 34(4):947–56. doi: 10.3892/ijmm.2014.1855
248. Yuksel V, Canbaz S, Ege T. Comparison between normothermic and mild hypothermic cardiopulmonary bypass in myocardial revascularization of patients with left ventricular dysfunction. Perfusion. (2013) 28(5):419–23. doi: 10.1177/0267659113483798
249. Akeho K, Nakata H, Suehiro S, Shimizu K, Imai K, Yamaguchi A, et al. Hypothermic effects on gas exchange performance of membrane oxygenator and blood coagulation during cardiopulmonary bypass in pigs. Perfusion. (2020) 35(7):687–96. doi: 10.1177/0267659120901413
250. Caputo M, Pike K, Baos S, Sheehan K, Selway K, Ellis L, et al. Normothermic versus hypothermic cardiopulmonary bypass in low-risk paediatric heart surgery: a randomised controlled trial. Heart. (2019) 105(6):455–64. doi: 10.1136/heartjnl-2018-313567
251. Hannon CE, Osman Z, Grant C, Chung EML, Corno AF. Part II. Comparison of neurodevelopmental outcomes between normothermic and hypothermic pediatric cardiopulmonary bypass. Front Pediatr. (2019) 7:447. doi: 10.3389/fped.2019.00447
252. Yildirim F, Amanvermez Senarslan D, Yersel S, Bayram B, Taneli F, Tetik O. Systemic inflammatory response during cardiopulmonary bypass: axial flow versus radial flow oxygenators. Int J Artif Organs. (2022) 45(3):278–83. doi: 10.1177/03913988221075043
253. Caputo M, Mokhtari A, Miceli A, Ghorbel MT, Angelini GD, Parry AJ, Suleiman SM. Controlled reoxygenation during cardiopulmonary bypass decreases markers of organ damage, inflammation, and oxidative stress in single-ventricle patients undergoing pediatric heart surgery. J Thorac Cardiovasc Surg. (2014) 148(3):792–801.e8; discussion 800–1. doi: 10.1016/j.jtcvs.2014.06.001
254. Paparella D, Rotunno C, Guida P, Travascia M, De Palo M, Paradiso A, et al. Minimally invasive heart valve surgery: influence on coagulation and inflammatory response. Interact Cardiovasc Thorac Surg. (2017) 25(2):225–32. doi: 10.1093/icvts/ivx090
255. Farag M, Patil NP, Sabashnikov A, Arif R, Szabó G, Kallenbach K, et al. Comparison of two miniaturized cardiopulmonary bypass systems regarding inflammatory response. Artif Organs. (2017) 41(2):139–45. doi: 10.1111/aor.12750
256. Gygax E, Kaeser HU, Stalder M, Gahl B, Rieben R, Carrel T, et al. Type II minimal-invasive extracorporeal circuit for aortic valve replacement: a randomized controlled trial. Artif Organs. (2018) 42(6):620–9. doi: 10.1111/aor.13093
257. Beghi C, Nicolini F, Agostinelli A, Borrello B, Budillon AM, Bacciottini F, et al. Mini-cardiopulmonary bypass system: results of a prospective randomized study. Ann Thorac Surg. (2006) 81(4):1396–400. doi: 10.1016/j.athoracsur.2005.10.015
258. Starinieri P, Declercq PE, Robic B, Yilmaz A, Van Tornout M, Dubois J, Mees U, Hendrikx M. A comparison between minimized extracorporeal circuits and conventional extracorporeal circuits in patients undergoing aortic valve surgery: is “minimally invasive extracorporeal circulation” just low prime or closed loop perfusion? Perfusion. (2017) 32(5):403–8. doi: 10.1177/0267659117691814
259. Pasrija C, Shah A, Sultanik E, Rouse M, Ghoreishi M, Bittle GJ, et al. Minimally invasive surgical pulmonary embolectomy: a potential alternative to conventional sternotomy. Innovations (Phila). (2017) 12(6):406–10. doi: 10.1097/IMI.0000000000000439
260. Jiang Q, Wang Z, Guo J, Yu T, Zhang X, Hu S. Retrospective comparison of endoscopic versus open procedure for mitral valve disease. J Invest Surg. (2021) 34(9):1000–6. doi: 10.1080/08941939.2020.1726531
261. Ahangar AG, Charag AH, Wani ML, Ganie FA, Singh S, Ahmad Qadri SA, et al. Comparing aortic valve replacement through right anterolateral thoracotomy with median sternotomy. Int Cardiovasc Res J. (2013) 7(3):90–4.24757629
262. Calderon J, Richebe P, Guibaud JP, Coiffic A, Branchard O, Asselineau J, et al. Prospective randomized study of early pulmonary evaluation of patients scheduled for aortic valve surgery performed by ministernotomy or total median sternotomy. J Cardiothorac Vasc Anesth. (2009) 23(6):795–801. doi: 10.1053/j.jvca.2009.03.011
263. Dogan S, Dzemali O, Wimmer-Greinecker G, Derra P, Doss M, Khan MF, et al. Minimally invasive versus conventional aortic valve replacement: a prospective randomized trial. J Heart Valve Dis. (2003) 12(1):76–80.12578340
264. Moustafa MA, Abdelsamad AA, Zakaria G, Omarah MM. Minimal vs median sternotomy for aortic valve replacement. Asian Cardiovasc Thorac Ann. (2007) 15(6):472–5. doi: 10.1177/021849230701500605
265. Jongman RM, Zijlstra JG, Kok WF, van Harten AE, Mariani MA, Moser J, et al. Off-pump CABG surgery reduces systemic inflammation compared with on-pump surgery but does not change systemic endothelial responses: a prospective randomized study. Shock. (2014) 42(2):121–8. doi: 10.1097/SHK.0000000000000190
266. Lehmann S, Dieterlen MT, Flister A, Klaeske K, Jawad K, Garbade J, et al. Differences of early immunological responses in on-pump versus off-pump cardiac surgery. Perfusion. (2019) 34(5):399–407. doi: 10.1177/0267659118823137
267. Vallely MP, Bannon PG, Kritharides L. The systemic inflammatory response syndrome and off-pump cardiac surgery. Heart Surg Forum. (2001) 4(Suppl 1):S7–13.11178301
268. Khan TA, Bianchi C, Araujo E, Voisine P, Xu SH, Feng J, et al. Aprotinin preserves cellular junctions and reduces myocardial edema after regional ischemia and cardioplegic arrest. Circulation. (2005) 112(9 Suppl):I196–201. doi: 10.1161/CIRCULATIONAHA.104.526053
269. Lisman T, Adelmeijer J, Huskens D, Meijers JCM. Aprotinin inhibits thrombin generation by inhibition of the intrinsic pathway, but is not a direct thrombin inhibitor. TH Open. (2021) 5(3):e363–75. doi: 10.1055/s-0041-1735154
270. Levi M, Cromheecke ME, de Jonge E, Prins MH, de Mol BJ, Briët E, et al. Pharmacological strategies to decrease excessive blood loss in cardiac surgery: a meta-analysis of clinically relevant endpoints. Lancet. (1999) 354(9194):1940–7. doi: 10.1016/S0140-6736(99)01264-7
271. Mössinger H, Dietrich W, Braun SL, Jochum M, Meisner H, Richter JA. High-dose aprotinin reduces activation of hemostasis, allogeneic blood requirement, and duration of postoperative ventilation in pediatric cardiac surgery. Ann Thorac Surg. (2003) 75(2):430–7. doi: 10.1016/s0003-4975(02)04412-0
272. Ray MJ, Marsh NA. Aprotinin reduces blood loss after cardiopulmonary bypass by direct inhibition of plasmin. Thromb Haemost. (1997) 78(3):1021–6. doi: 10.1055/s-0038-1657680
273. Wachtfogel YT, Kucich U, Hack CE, Gluszko P, Niewiarowski S, Colman RW, et al. Aprotinin inhibits the contact, neutrophil, and platelet activation systems during simulated extracorporeal perfusion. J Thorac Cardiovasc Surg. (1993) 106(1):1–9; discussion 9–10. doi: 10.1016/S0022-5223(19)33735-3
274. Mojcik CF, Levy JH. Aprotinin and the systemic inflammatory response after cardiopulmonary bypass. Ann Thorac Surg. (2001) 71(2):745–54. doi: 10.1016/s0003-4975(00)02218-9
275. Hill GE, Snider S, Galbraith TA, Forst S, Robbins RA. Glucocorticoid reduction of bronchial epithelial inflammation during cardiopulmonary bypass. Am J Respir Crit Care Med. (1995) 152(6 Pt 1):1791–5. doi: 10.1164/ajrccm.152.6.8520738
276. Harig F, Feyrer R, Mahmoud FO, Blum U, von der Emde J. Reducing the post-pump syndrome by using heparin-coated circuits, steroids, or aprotinin. Thorac Cardiovasc Surg. (1999) 47(2):111–8. doi: 10.1055/s-2007-1013121
277. Soeparwata R, Hartman AR, Frerichmann U, Stefano GB, Scheld HH, Bilfinger TV. Aprotinin diminishes inflammatory processes. Int J Cardiol. (1996) 53 Suppl:S55–63. doi: 10.1016/0167-5273(96)02573-9
278. Alonso A, Whitten CW, Hill GE. Pump prime only aprotinin inhibits cardiopulmonary bypass-induced neutrophil CD11b up-regulation. Ann Thorac Surg. (1999) 67(2):392–5. doi: 10.1016/s0003-4975(98)01132-1
279. Wendel HP, Heller W, Michel J, Mayer G, Ochsenfahrt C, Graeter U, et al. Lower cardiac troponin T levels in patients undergoing cardiopulmonary bypass and receiving high-dose aprotinin therapy indicate reduction of perioperative myocardial damage. J Thorac Cardiovasc Surg. (1995) 109(6):1164–72. doi: 10.1016/S0022-5223(95)70200-8
280. Gott JP, Cooper WA, Schmidt FE Jr, Brown WM 3rd, Wright CE, Merlino JD, Fortenberry JD, Clark WS, Guyton RA. Modifying risk for extracorporeal circulation: trial of four antiinflammatory strategies. Ann Thorac Surg. (1998) 66(3):747–53; discussion 753–4. doi: 10.1016/s0003-4975(98)00695-x
281. Toikkanen V, Rinne T, Nieminen R, Moilanen E, Laurikka J, Porkkala H, et al. Aprotinin impacts 8-isoprostane after coronary artery bypass grafting. Scand J Surg. (2018) 107(4):329–35. doi: 10.1177/1457496918766720
282. Lemmer JH Jr, Dilling EW, Morton JR, Rich JB, Robicsek F, Bricker DL, Hantler CB, Copeland JG 3rd, Ochsner JL, Daily PO, Whitten CW, Noon GP, Maddi R. Aprotinin for primary coronary artery bypass grafting: a multicenter trial of three dose regimens. Ann Thorac Surg. (1996) 62(6):1659–67; discussion 1667–8. doi: 10.1016/s0003-4975(96)00451-1
283. Mangano DT, Tudor IC, Dietzel C, Multicenter Study of Perioperative Ischemia Research Group, Ischemia Research and Education Foundation. The risk associated with aprotinin in cardiac surgery. N Engl J Med. (2006) 354(4):353–65. doi: 10.1056/NEJMoa051379
284. Fergusson DA, Hébert PC, Mazer CD, Fremes S, MacAdams C, Murkin JM, et al. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. N Engl J Med. (2008) 358(22):2319–31. Erratum in: N Engl J Med. 2010 363(13):1290. doi: 10.1056/NEJMoa0802395
285. Doherty GM, Jensen JC, Alexander HR, Buresh CM, Norton JA. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery. (1991) 110(2):192–8.1858029
286. Schade UF. Pentoxifylline increases survival in murine endotoxin shock and decreases formation of tumor necrosis factor. Circ Shock. (1990) 31(2):171–81.2344657
287. Sullivan GW, Carper HT, Novick WJ Jr, Mandell GL. Inhibition of the inflammatory action of interleukin-1 and tumor necrosis factor (alpha) on neutrophil function by pentoxifylline. Infect Immun. (1988) 56(7):1722–9. doi: 10.1128/iai.56.7.1722-1729.1988
288. Tsang GM, Allen S, Pagano D, Wong C, Graham TR, Bonser RS. Pentoxifylline preloading reduces endothelial injury and permeability in cardiopulmonary bypass. ASAIO J. (1996) 42(5):M429–34. doi: 10.1097/00002480-199609000-00025
289. Türköz R, Yörükoğlu K, Akcay A, Yilik L, Baltalarli A, Karahan N, et al. The effect of pentoxifylline on the lung during cardiopulmonary bypass. Eur J Cardiothorac Surg. (1996) 10(5):339–46. doi: 10.1016/s1010-7940(96)80092-3
290. Mansourian S, Bina P, Fehri A, Karimi AA, Boroumand MA, Abbasi K. Preoperative oral pentoxifylline in case of coronary artery bypass grafting with left ventricular dysfunction (ejection fraction equal to/less than 30%). Anatol J Cardiol. (2015) 15(12):1014–9. doi: 10.5152/akd.2014.5883
291. Möllhoff T, Loick HM, Van Aken H, Schmidt C, Rolf N, Tjan TD, et al. Milrinone modulates endotoxemia, systemic inflammation, and subsequent acute phase response after cardiopulmonary bypass (CPB). Anesthesiology. (1999) 90(1):72–80. doi: 10.1097/00000542-199901000-00012
292. McNicol L, Andersen LW, Liu G, Doolan L, Baek L. Markers of splanchnic perfusion and intestinal translocation of endotoxins during cardiopulmonary bypass: effects of dopamine and milrinone. J Cardiothorac Vasc Anesth. (1999) 13(3):292–8. doi: 10.1016/s1053-0770(99)90266-5
293. Andersen LW, Baek L, Thomsen BS, Rasmussen JP. Effect of methylprednisolone on endotoxemia and complement activation during cardiac surgery. J Cardiothorac Anesth. (1989) 3(5):544–9. doi: 10.1016/0888-6296(89)90150-6
294. Dernek S, Tünerir B, Sevin B, Aslan R, Uyguç O, Kural T. The effects of methylprednisolone on complement, immunoglobulins and pulmonary neutrophil sequestration during cardiopulmonary bypass. Cardiovasc Surg. (1999) 7(4):414–8. doi: 10.1016/s0967-2109(98)00153-7
295. Wan S, LeClerc JL, Huynh CH, Schmartz D, DeSmet JM, Yim AP, et al. Does steroid pretreatment increase endotoxin release during clinical cardiopulmonary bypass? J Thorac Cardiovasc Surg. (1999) 117(5):1004–8. doi: 10.1016/S0022-5223(99)70382-X
296. Demir T, Demir H, Tansel T, Kalko Y, Tireli E, Dayioglu E, et al. Influence of methylprednisolone on levels of neuron-specific enolase in cardiac surgery: a corticosteroid derivative to decrease possible neuronal damage. J Card Surg. (2009) 24(4):397–403. doi: 10.1111/j.1540-8191.2009.00842.x
297. Liakopoulos OJ, Schmitto JD, Kazmaier S, Bräuer A, Quintel M, Schoendube FA, Dörge H. Cardiopulmonary and systemic effects of methylprednisolone in patients undergoing cardiac surgery. Ann Thorac Surg. 2007 84(1):110–8; discussion 118–9. doi: 10.1016/j.athoracsur.2007.01.003
298. Yared JP, Bakri MH, Erzurum SC, Moravec CS, Laskowski DM, Van Wagoner DR, et al. Effect of dexamethasone on atrial fibrillation after cardiac surgery: prospective, randomized, double-blind, placebo-controlled trial. J Cardiothorac Vasc Anesth. (2007) 21(1):68–75. doi: 10.1053/j.jvca.2005.10.014
299. Sobieski MA 2nd, Graham JD, Pappas PS, Tatooles AJ, Slaughter MS. Reducing the effects of the systemic inflammatory response to cardiopulmonary bypass: can single dose steroids blunt systemic inflammatory response syndrome? ASAIO J. (2008) 54(2):203–6. doi: 10.1097/MAT.0b013e3181640331
300. Kawamura T, Inada K, Okada H, Okada K, Wakusawa R. Methylprednisolone inhibits increase of interleukin 8 and 6 during open heart surgery. Can J Anaesth. (1995) 42(5 Pt 1):399–403. doi: 10.1007/BF03015485
301. Amr YM, Elmistekawy E, El-Serogy H. Effects of dexamethasone on pulmonary and renal functions in patients undergoing CABG with cardiopulmonary bypass. Semin Cardiothorac Vasc Anesth. (2009) 13(4):231–7. doi: 10.1177/1089253209351598
302. Jansen NJ, van Oeveren W, van Vliet M, Stoutenbeek CP, Eysman L, Wildevuur CR. The role of different types of corticosteroids on the inflammatory mediators in cardiopulmonary bypass. Eur J Cardiothorac Surg. (1991) 5(4):211–7. doi: 10.1016/1010-7940(91)90032-f
303. Tassani P, Richter JA, Barankay A, Braun SL, Haehnel C, Spaeth P, et al. Does high-dose methylprednisolone in aprotinin-treated patients attenuate the systemic inflammatory response during coronary artery bypass grafting procedures? J Cardiothorac Vasc Anesth. (1999) 13(2):165–72. doi: 10.1016/s1053-0770(99)90081-2
304. Halonen J, Halonen P, Järvinen O, Taskinen P, Auvinen T, Tarkka M, et al. Corticosteroids for the prevention of atrial fibrillation after cardiac surgery: a randomized controlled trial. JAMA. (2007) 297(14):1562–7. doi: 10.1001/jama.297.14.1562
305. Yilmaz M, Ener S, Akalin H, Sagdic K, Serdar OA, Cengiz M. Effect of low-dose methyl prednisolone on serum cytokine levels following extracorporeal circulation. Perfusion. (1999) 14(3):201–6. doi: 10.1177/026765919901400308
306. Whitlock RP, Devereaux PJ, Teoh KH, Lamy A, Vincent J, Pogue J, et al. Methylprednisolone in patients undergoing cardiopulmonary bypass (SIRS): a randomised, double-blind, placebo-controlled trial. Lancet. (2015) 386(10000):1243–53. doi: 10.1016/S0140-6736(15)00273-1
307. Dieleman JM, van Paassen J, van Dijk D, Arbous MS, Kalkman CJ, Vandenbroucke JP, et al. Prophylactic corticosteroids for cardiopulmonary bypass in adults. Cochrane Database Syst Rev. (2011) 5:CD005566. doi: 10.1002/14651858.CD005566.pub3
308. Jorens PG, De Jongh R, De Backer W, Van Damme J, Van Overveld F, Bossaert L, et al. Interleukin-8 production in patients undergoing cardiopulmonary bypass. The influence of pretreatment with methylprednisolone. Am Rev Respir Dis. (1993) 148(4 Pt 1):890–5. doi: 10.1164/ajrccm/148.4_Pt_1.890
309. van Overveld FJ, De Jongh RF, Jorens PG, Walter P, Bossaert L, De Backer WA. Pretreatment with methylprednisolone in coronary artery bypass grafting influences the levels of histamine and tryptase in serum but not in bronchoalveolar lavage fluid. Clin Sci (Lond). (1994) 86(1):49–53. doi: 10.1042/cs0860049
310. Lodge AJ, Chai PJ, Daggett CW, Ungerleider RM, Jaggers J. Methylprednisolone reduces the inflammatory response to cardiopulmonary bypass in neonatal piglets: timing of dose is important. J Thorac Cardiovasc Surg. (1999) 117(3):515–22. doi: 10.1016/s0022-5223(99)70331-4
311. Mezzetti A, Lapenna D, Pierdomenico SD, Di Giammarco G, Bosco G, Di Ilio C, et al. Myocardial antioxidant defenses during cardiopulmonary bypass. J Card Surg. (1993) 8(2):167–71. doi: 10.1111/j.1540-8191.1993.tb00368.x
312. Ballmer PE, Reinhart WH, Jordan P, Bühler E, Moser UK, Gey KF. Depletion of plasma vitamin C but not of vitamin E in response to cardiac operations. J Thorac Cardiovasc Surg. (1994) 108(2):311–20. Erratum in: J Thorac Cardiovasc Surg 1995 110(4 Pt 1):1072. doi: 10.1016/S0022-5223(94)70013-3
313. McColl AJ, Keeble T, Hadjinikolaou L, Cohen A, Aitkenhead H, Glenville B, Richmond W. Plasma antioxidants: evidence for a protective role against reactive oxygen species following cardiac surgery. Ann Clin Biochem. (1998) 35(Pt 5):616–23. doi: 10.1177/000456329803500504
314. Barta E, Pechán I, Cornák V, Luknárová O, Rendeková V, Verchovodko P. Protective effect of alpha-tocopherol and L-ascorbic acid against the ischemic-reperfusion injury in patients during open-heart surgery. Bratisl Lek Listy. (1991) 92(3-4):174–83.2029659
315. England MD, Cavarocchi NC, O'Brien JF, Solis E, Pluth JR, Orszulak TA, et al. Influence of antioxidants (mannitol and allopurinol) on oxygen free radical generation during and after cardiopulmonary bypass. Circulation. (1986) 74(5 Pt 2):III134–7.3094981
316. Hill A, Borgs C, Fitzner C, Stoppe C. Perioperative vitamin C and E levels in cardiac surgery patients and their clinical significance. Nutrients. (2019) 11(9):2157. doi: 10.3390/nu11092157
317. Dingchao H, Zhiduan Q, Liye H, Xiaodong F. The protective effects of high-dose ascorbic acid on myocardium against reperfusion injury during and after cardiopulmonary bypass. Thorac Cardiovasc Surg. (1994) 42(5):276–8. doi: 10.1055/s-2007-1016504
318. Rodrigo R, Korantzopoulos P, Cereceda M, Asenjo R, Zamorano J, Villalabeitia E, et al. A randomized controlled trial to prevent post-operative atrial fibrillation by antioxidant reinforcement. J Am Coll Cardiol. (2013) 62(16):1457–65. doi: 10.1016/j.jacc.2013.07.014
319. Zoran P, Juraj F, Ivana D, Reik H, Dusan N, Mihailo V. Effects of allopurinol on oxygen stress status during open heart surgery. Int J Cardiol. (1994) 44(2):123–9. doi: 10.1016/0167-5273(94)90015-9
320. Tabayashi K, Suzuki Y, Nagamine S, Ito Y, Sekino Y, Mohri H. A clinical trial of allopurinol (zyloric) for myocardial protection. J Thorac Cardiovasc Surg. (1991) 101(4):713–8. doi: 10.1016/S0022-5223(19)36703-0
321. Castelli P, Condemi AM, Brambillasca C, Fundarò P, Botta M, Lemma M, et al. Improvement of cardiac function by allopurinol in patients undergoing cardiac surgery. J Cardiovasc Pharmacol. (1995) 25(1):119–25. doi: 10.1097/00005344-199501000-00019
322. Bochenek A, Religa Z, Spyt TJ, Mistarz K, Bochenek A, Zembala M, et al. Protective influence of pretreatment with allopurinol on myocardial function in patients undergoing coronary artery surgery. Eur J Cardiothorac Surg. (1990) 4(10):538–42. doi: 10.1016/1010-7940(90)90142-m
323. Coetzee A, Roussouw G, Macgregor L. Failure of allopurinol to improve left ventricular stroke work after cardiopulmonary bypass surgery. J Cardiothorac Vasc Anesth. (1996) 10(5):627–33. doi: 10.1016/s1053-0770(96)80141-8
324. Taggart DP, Young V, Hooper J, Kemp M, Walesby R, Magee P, et al. Lack of cardioprotective efficacy of allopurinol in coronary artery surgery. Br Heart J. (1994) 71(2):177–81. doi: 10.1136/hrt.71.2.177
325. Sisto T, Paajanen H, Metsä-Ketelä T, Harmoinen A, Nordback I, Tarkka M. Pretreatment with antioxidants and allopurinol diminishes cardiac onset events in coronary artery bypass grafting. Ann Thorac Surg. (1995) 59(6):1519–23. doi: 10.1016/0003-4975(95)00197-s
326. Westhuyzen J, Cochrane AD, Tesar PJ, Mau T, Cross DB, Frenneaux MP, et al. Effect of preoperative supplementation with alpha-tocopherol and ascorbic acid on myocardial injury in patients undergoing cardiac operations. J Thorac Cardiovasc Surg. (1997) 113(5):942–8. doi: 10.1016/s0022-5223(97)70268-x
327. Stegeman R, Lamur KD, van den Hoogen A, Breur JMPJ, Groenendaal F, Jansen NJG, et al. Neuroprotective drugs in infants with severe congenital heart disease: a systematic review. Front Neurol. (2018) 9:521. doi: 10.3389/fneur.2018.00521
328. Stegeman R, Nijman M, Breur JMPJ, Groenendaal F, Haas F, Derks JB, et al. Cerebrum and cardiac protection with allopurinol in neonates with critical congenital heart disease requiring cardiac surgery with cardiopulmonary bypass (CRUCIAL): study protocol of a phase III, randomized, quadruple-blinded, placebo-controlled, Dutch multicenter trial. Trials. (2022) 23(1):174. doi: 10.1186/s13063-022-06098-y
329. Andersen LW, Thiis J, Kharazmi A, Rygg I. The role of N-acetylcystein administration on the oxidative response of neutrophils during cardiopulmonary bypass. Perfusion. (1995) 10(1):21–6. doi: 10.1177/026765919501000105
330. De Backer WA, Amsel B, Jorens PG, Bossaert L, Hiemstra PS, van Noort P, et al. N-acetylcysteine pretreatment of cardiac surgery patients influences plasma neutrophil elastase and neutrophil influx in bronchoalveolar lavage fluid. Intensive Care Med. (1996) 22(9):900–8. doi: 10.1007/BF02044114
331. Tossios P, Bloch W, Huebner A, Raji MR, Dodos F, Klass O, et al. N-acetylcysteine prevents reactive oxygen species-mediated myocardial stress in patients undergoing cardiac surgery: results of a randomized, double-blind, placebo-controlled clinical trial. J Thorac Cardiovasc Surg. (2003) 126(5):1513–20. doi: 10.1016/s0022-5223(03)00968-1
332. Suter PM, Domenighetti G, Schaller MD, Laverrière MC, Ritz R, Perret C. N-acetylcysteine enhances recovery from acute lung injury in man. A randomized, double-blind, placebo-controlled clinical study. Chest. (1994) 105(1):190–4. doi: 10.1378/chest.105.1.190
333. El-Hamamsy I, Stevens LM, Carrier M, Pellerin M, Bouchard D, Demers P, et al. Effect of intravenous N-acetylcysteine on outcomes after coronary artery bypass surgery: a randomized, double-blind, placebo-controlled clinical trial. J Thorac Cardiovasc Surg. (2007) 133(1):7–12. doi: 10.1016/j.jtcvs.2006.05.070
334. Köksal H, Rahman A, Burma O, Halifeoğlu I, Bayar MK. The effects of low dose N-acetylcysteine (NAC) as an adjunct to cardioplegia in coronary artery bypass surgery. Anadolu Kardiyol Derg. (2008) 8(6):437–43.
335. Cai Z, Shi T, Zhuang R, Fang H, Jiang X, Shao Y, et al. Protective effect of N-acetylcysteine activated carbon release microcapsule on myocardial ischemia-reperfusion injury in rats. Exp Ther Med. (2018) 15(2):1809–18. doi: 10.3892/etm.2017.5653
336. Xu Z, Jiang G, Lin S, Guan J, Chen G, Chen G. Effect of N-acetylcysteine on intestinal injury induced by cardiopulmonary bypass in rats. Nan Fang Yi Ke Da Xue Xue Bao. (2014) 34(8):1171–5. Chinese.25176089
337. Kumar R, Bansal M, Nath SS, Kumar V, Malviya D, Srivastava D. N-Acetylcysteine supplementation for the prevention of postoperative liver dysfunction after on-pump cardiac surgery. Turk J Anaesthesiol Reanim. (2021) 49(6):460–9. doi: 10.5152/TJAR.2021.21370
338. Onk D, Özçelik F, Onk OA, Günay M, Akarsu Ayazoğlu T, Ünver E. Assessment of renal and hepatic tissue-protective effects of N-acetylcysteine via ammonia metabolism: a prospective randomized study. Med Sci Monit. (2018) 24:1540–6. doi: 10.12659/msm.908172
339. Savluk OF, Guzelmeric F, Yavuz Y, Cevirme D, Gurcu E, Ogus H, et al. N-acetylcysteine versus dopamine to prevent acute kidney injury after cardiac surgery in patients with preexisting moderate renal insufficiency. Braz J Cardiovasc Surg. (2017) 32(1):8–14. doi: 10.21470/1678-9741-2016-0028
340. Tan SI, Brewster DJ, Horrigan D, Sarode V. Pharmacological and non-surgical renal protective strategies for cardiac surgery patients undergoing cardiopulmonary bypass: a systematic review. ANZ J Surg. (2019) 89(4):296–302. doi: 10.1111/ans.14800
341. Erdil N, Eroglu T, Akca B, Disli OM, Yetkin O, Colak MC, et al. The effects of N-acetylcysteine on pulmonary functions in patients Undergoing on-pump coronary artery surgery: a double blind placebo controlled study. Eur Rev Med Pharmacol Sci. (2016) 20(1):180–7.26813472
342. Santana-Santos E, Gowdak LH, Gaiotto FA, Puig LB, Hajjar LA, Zeferino SP, et al. High dose of N-acetylcystein prevents acute kidney injury in chronic kidney disease patients undergoing myocardial revascularization. Ann Thorac Surg. (2014) 97(5):1617–23. doi: 10.1016/j.athoracsur.2014.01.056
343. Fitch JC, Rollins S, Matis L, Alford B, Aranki S, Collard CD, et al. Pharmacology and biological efficacy of a recombinant, humanized, single-chain antibody C5 complement inhibitor in patients undergoing coronary artery bypass graft surgery with cardiopulmonary bypass. Circulation. (1999) 100(25):2499–506. doi: 10.1161/01.cir.100.25.2499
344. Soulika AM, Khan MM, Hattori T, Bowen FW, Richardson BA, Hack CE, et al. Inhibition of heparin/protamine complex-induced complement activation by compstatin in baboons. Clin Immunol. (2000) 96(3):212–21. doi: 10.1006/clim.2000.4903
345. Thielmann M, Marggraf G, Neuhäuser M, Forkel J, Herold U, Kamler M, et al. Administration of C1-esterase inhibitor during emergency coronary artery bypass surgery in acute ST-elevation myocardial infarction. Eur J Cardiothorac Surg. (2006) 30(2):285–93. doi: 10.1016/j.ejcts.2006.04.022
346. Fattouch K, Bianco G, Speziale G, Sampognaro R, Lavalle C, Guccione F, et al. Beneficial effects of C1 esterase inhibitor in ST-elevation myocardial infarction in patients who underwent surgical reperfusion: a randomised double-blind study. Eur J Cardiothorac Surg. (2007) 32(2):326–32. doi: 10.1016/j.ejcts.2007.04.038
347. Lazar HL, Bokesch PM, van Lenta F, Fitzgerald C, Emmett C, Marsh HC Jr, Ryan U. Soluble human complement receptor 1 limits ischemic damage in cardiac surgery patients at high risk requiring cardiopulmonary bypass. Circulation. (2004) 110(11 Suppl 1):II274–9. doi: 10.1161/01.CIR.0000138315.99788.eb
348. Fujii M, Miyagi Y, Bessho R, Nitta T, Ochi M, Shimizu K. Effect of a neutrophil elastase inhibitor on acute lung injury after cardiopulmonary bypass. Interact Cardiovasc Thorac Surg. (2010) 10(6):859–62. doi: 10.1510/icvts.2009.225243
349. Verrier ED, Shernan SK, Taylor KM, Van de Werf F, Newman MF, Chen JC, et al. Terminal complement blockade with pexelizumab during coronary artery bypass graft surgery requiring cardiopulmonary bypass: a randomized trial. JAMA. (2004) 291(19):2319–27. Erratum in: JAMA. 2006 295(2):164. doi: 10.1001/jama.291.19.2319
350. Smith PK, Shernan SK, Chen JC, Carrier M, Verrier ED, Adams PX, et al. Effects of C5 complement inhibitor pexelizumab on outcome in high-risk coronary artery bypass grafting: combined results from the PRIMO-CABG I and II trials. J Thorac Cardiovasc Surg. (2011) 142(1):89–98. doi: 10.1016/j.jtcvs.2010.08.035
351. Shin'oka T, Nagashima M, Nollert G, Shum-Tim D, Laussen PC, Lidov HG, et al. A novel sialyl Lewis X analog attenuates cerebral injury after deep hypothermic circulatory arrest. J Thorac Cardiovasc Surg. (1999) 117(6):1204–11. doi: 10.1016/s0022-5223(99)70260-6
352. Gillinov AM, Redmond JM, Zehr KJ, Wilson IC, Curtis WE, Bator JM, et al. Inhibition of neutrophil adhesion during cardiopulmonary bypass. Ann Thorac Surg. (1994) 57(1):126–33. doi: 10.1016/0003-4975(94)90380-8
353. Sharar SR, Winn RK, Murry CE, Harlan JM, Rice CL. A CD18 monoclonal antibody increases the incidence and severity of subcutaneous abscess formation after high-dose Staphylococcus aureus injection in rabbits. Surgery. (1991) 110(2):213–9; discussion 219–20.1677492
354. Berg K, Langaas M, Ericsson M, Pleym H, Basu S, Nordrum IS, et al. Acetylsalicylic acid treatment until surgery reduces oxidative stress and inflammation in patients undergoing coronary artery bypass grafting. Eur J Cardiothorac Surg. (2013) 43(6):1154–63. doi: 10.1093/ejcts/ezs591
355. Akowuah E, Shrivastava V, Jamnadas B, Hopkinson D, Sarkar P, Storey R, et al. Comparison of two strategies for the management of antiplatelet therapy during urgent surgery. Ann Thorac Surg. (2005) 80(1):149–52. doi: 10.1016/j.athoracsur.2005.01.009
Keywords: cardiac surgery, cardiopulmonary bypass, inflammation, ischemiareperfusion injury, organ damage
Citation: Banerjee D, Feng J and Sellke FW (2024) Strategies to attenuate maladaptive inflammatory response associated with cardiopulmonary bypass. Front. Surg. 11:1224068. doi: 10.3389/fsurg.2024.1224068
Received: 17 May 2023; Accepted: 7 June 2024;
Published: 3 July 2024.
Edited by:
Shahzad Raja, Harefield Hospital, United KingdomReviewed by:
Nobuyuki Ishibashi, Children's National Hospital, United States© 2024 Banerjee, Feng and Sellke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank W. Sellke, ZnJhbmtfc2VsbGtlQGJyb3duLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.