 Laura-Nanna Lohkamp1*
Laura-Nanna Lohkamp1* Patricia Parkin2†Allan Puran2
Patricia Parkin2†Allan Puran2 Ute Katharina Bartels3Eric Bouffet3
Ute Katharina Bartels3Eric Bouffet3 Uri Tabori3
Uri Tabori3 James Thomas Rutka1†
James Thomas Rutka1†
- 1Division of Neurosurgery, Department of Surgery, The Hospital for Sick Children, Toronto, Ontario, Canada
- 2Division of Paediatric Medicine, Department of Paediatrics, The Hospital for Sick Children, Toronto, Ontario, Canada
- 3Division of Haematology/Oncology, Department of Paediatrics, The Hospital for Sick Children, Toronto, Ontario, Canada
Introduction: Neurofibromatosis type 1 (NF1) has an incidence of 1 in 2,000 to 3,000 individuals and in 15% is associated with optic pathway glioma (OPG). Given the variability in clinical presentation and related morbidity, a multidisciplinary approach for diagnosis and management of children with NF1 and OPG is required, but often lacks coordination and regular information exchange. Herein we summarize our experience and describe the care pathways/network provided by a multidisciplinary team. The role of the distinct team members is elucidated as well as the care amendments made over time.
Methods: We performed a retrospective single-center observational study, including children treated at our institution between 1990 and 2021. Inclusion criteria were clinical diagnosis of NF1, radiographic and/or histopathological diagnosis of OPG and age below 18 years. Patients being treated elsewhere were excluded from the study. Data was abstracted from each child’s health record using a standardized data collection form. Characteristics of children with NF1 and OPG were described using means (SD) and percentages. Outcomes were determined using Kaplan-Meier estimates.
Results: From 1990 to 2021, 1,337 children were followed in our institution. Of those, 195 were diagnosed with OPG (14.6%), including 94 (48.21%) females and 101 (51.79%) males. Comprehensive data were available in 150 patients. The mean (SD) age at diagnosis was 5.31(4.08) years (range: 0.8–17.04 years). Sixty-two (41.3%) patients remained stable and did not undergo treatment, whereas 88 (58.7%) patients required at least one treatment. The mean (SD) duration of follow up was 8.14 (5.46) years (range: 0.1–25.9 years; median 6.8 years). Overall survival was of 23.6 years (±1.08), comprising 5 deaths. A dedicated NF clinic, including pediatricians and a nurse, provides regular follow up and plays a central role in the management of children with NF1, identifying those at risk of OPG, coordinating referrals to Neuroradiology and other specialists as indicated. All children are assessed annually by Ophthalmology. Comprehensive care was provided by a multidisciplinary team consisting of Dermatology, Genetics, Neuro-oncology, Neuroradiology, Neurosurgery, Ophthalmology and Pediatrics.
Conclusions: The care of children with NF1 and OPG is optimized with a multidisciplinary team approach, coordinated by a central specialty clinic.
Introduction
Neurofibromatosis type 1 (NF1) is an inherited neurocutaneous disorder occurring in about 1 in 2,000 to 1 in 3,000 individuals (1–4). Clinical features of NF1 are usually apparent in childhood, most often with multiple café au lait macules, and affect multiple organs, childhood development and neurocognitive status in a variable and age-dependant manner (5). As a familial tumor predisposition syndrome, it is characterized by the presence of multisystem tumors, which carry a risk of malignant transformation, and which are the major determinants of mortality in this population (6, 7). Optic Pathway Glioma (OPG) occur in 15%–20% of children with NF1 (8). They are found along the optic pathway and may involve one or both optic nerves, the chiasm, retro-chiasmatic structures including the hypothalamus, and the optic radiations (6). Although these tumors are typically low-grade gliomas, their behavior can be aggressive and therefore the clinical course may be highly variable, making treatment paradigms difficult (9, 10). Many children with NF1 and OPG remain asymptomatic. However, some children experience symptoms including vision loss and precocious puberty, highlighting the importance of early identification (11). Unfortunately, despite frequent imaging and ophthalmologic evaluations, some children experience progressive vision loss already before treatment, which makes their management even more challenging and emphasizes the need for optimized screening and follow-up protocols (12). Current management options include observation, surgery, chemotherapy, and radiation, all based on the current consensus to initiate treatment, when the tumor shows a substantial progression on MRI and/or becomes a threat to vision (13, 14). Given that OPG are not amenable to complete resection, the main treatment relies on chemotherapy (14). Other sequalae of OPG may include endocrinological deficits as well as primary or secondary neuropsychological limitations. Because of the varying features and clinical heterogeneity inherent to this condition, advocacy for a multidisciplinary approach to care and complex decision making has increased recently (8, 15, 16). However, the number of specialists involved may interfere with coordinated information exchange, referrals and follow up, thus posing dilemmas in the diagnosis and management of these patients. In 2007, the Children’s Tumor Foundation established a Neurofibromatosis Clinic Network in the United States, which aimed to standardize the level of care for individuals with NF and currently consists of 63 accredited clinics within the United States (17). Having established an NF specialty clinic in our institution in 1990, we seek to describe our experience, the evolution of care standards for children with NF1 related OPG over the last 30 years, the observed treatment paradigm changes as well as the characteristics of our patient cohort.
Materials and Methods
Study Design and Patient Cohort
We performed a single center, retrospective observational study at the Hospital for Sick Children (HSC) in Toronto, Canada, including children with NF1 and OPG diagnosed and treated between 1990 and 2021. Study subjects were identified from our NF1 registry, and their medical record number cross-referenced against information held in a second database, the Pediatric Brain Tumor Program registry. The study was approved by the local Research Ethics Board and conducted in accordance with the guidelines of the University of Toronto. Patient/parent consent was waived as de-identified data was abstracted from health records.
Eligibility
Patients were included in the study when fulfilling the following criteria: (1) National Institutes of Health clinical diagnostic criteria (18) and/or NF1 genetic testing (19), (2) confirmed OPG by neuro-imaging (MRI and/or CT), (3) age below 18 years and (4) received care at HSC. Patients, who did not meet these inclusion criteria or whose records were incomplete for more than 50% of the data points were excluded from the study.
Data Extraction
Data was extracted from electronic patient records and included demographic, clinical, radiographic and follow up data at different time points over the entire course of each child’s care. Retrieved data aimed to describe the characteristics of children with NF1 and OPG, including presentation, clinical course (general and ophthalmologic examination), molecular genetic results, neuroimaging, management (observation, chemotherapy, surgery, radiotherapy) and outcome. De-identified data were collected using a standardized data collection form.
Diagnosis and Treatment
A dedicated review of diagnostic and therapeutic procedures was performed, including the date and type of initial neuroimaging, the sequence of follow up examinations, duration of observation and treatment periods, treatment modalities and specific type (type of chemotherapy, surgery, or radiation) and treatment related complications. In case of surgical intervention, the patients’ charts were assessed for histopathological results. Post-treatment outcomes, indications for treatment changes or re-initiation were recorded and correlated with neuroimaging, ophthalmological, as well as clinical results. Molecular genetic testing is offered upon initial diagnosis and pursued according to parents’ preference. Neuropsychological assessments are provided either during neuro-oncological treatment or initiated via the NF clinic when learning difficulties or other neurodevelopmental deficits are identified during clinical follow up.
Health Supervision
Children with confirmed or probable NF1 from infancy to 18 years are seen annually in the Pediatric NF Clinic following the health supervision guidelines from the American Academy of Pediatrics (20, 21). Children are also assessed annually in the Pediatric Ophthalmology Clinic. Children are followed by other members of the multidisciplinary team as required. In the early years of the clinic, universal screening and surveillance neuroimaging was recommended; however, currently we recommend neuroimaging as indicated according to medical or ophthalmologic concerns (22).
Figure 1 provides a schematic illustration of all specialties involved in the diagnosis and management of patients with NF1 and associated OPG.
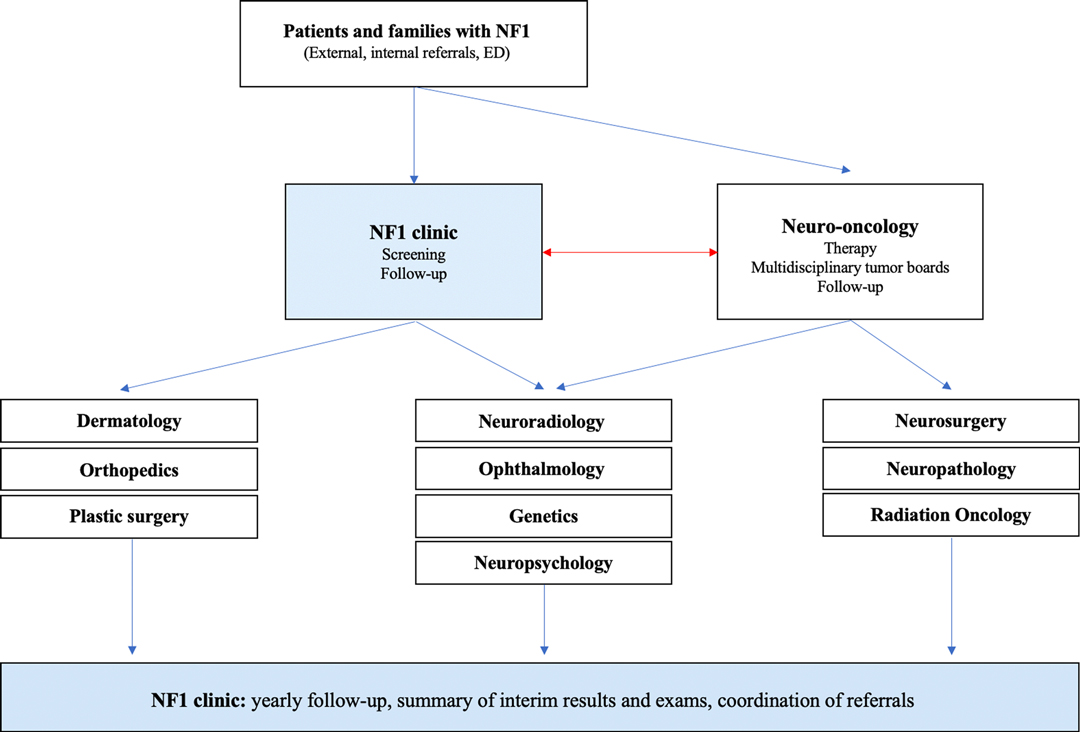
Figure 1. Schematic drawing illustrating the functional network of care for children with NF and OPG at the Hospital for Sick Children. The NF clinic plays a central role in connection, coordination, and integration of directed care for these complex patients.
Outcome
The primary outcome was defined as overall survival (OS), describing the time period between diagnosis and last follow up. Follow up data of our patients, which were obtained from external institutions and available after the age of 18, were also included in the outcome analysis. Secondary outcomes were oncological status at the last visit, as well as presence of persisting visual and/or other sequalae.
Statistical Analysis
Outcome was defined as OS and determined by Kaplan–Meier estimation (23). A confidence interval of 95% was applied and statistical significance was reached at a p-value equal or less than 0.05. All tests were performed using IBM SPSS Statistics Version 26.0 (IBM Corp. Released 2019. IBM SPSS Statistics for Windows, Version 26.0. Armonk, NY: IBM Corp, USA).
Results
General Demographics
Between 1990 and 2021, 1,337 children with NF1 have been followed in the NF clinic and registered in the related database. Of those, 195 (14.6%) patients were radiographically diagnosed with OPG and met the inclusion criteria, including 94 (48.21%) females and 101 (51.79%) males. Forty-five patients were excluded from the study for the following reasons: 33 patients diagnosed between 1990 and 1998 had incomplete records, 9 patients were treated elsewhere, and 3 patients were lost to follow up (Figure 2). The mean (SD) age at diagnosis of the remaining 150 patients was 5.31(4.08) years (range: 0.8–17.04 years).
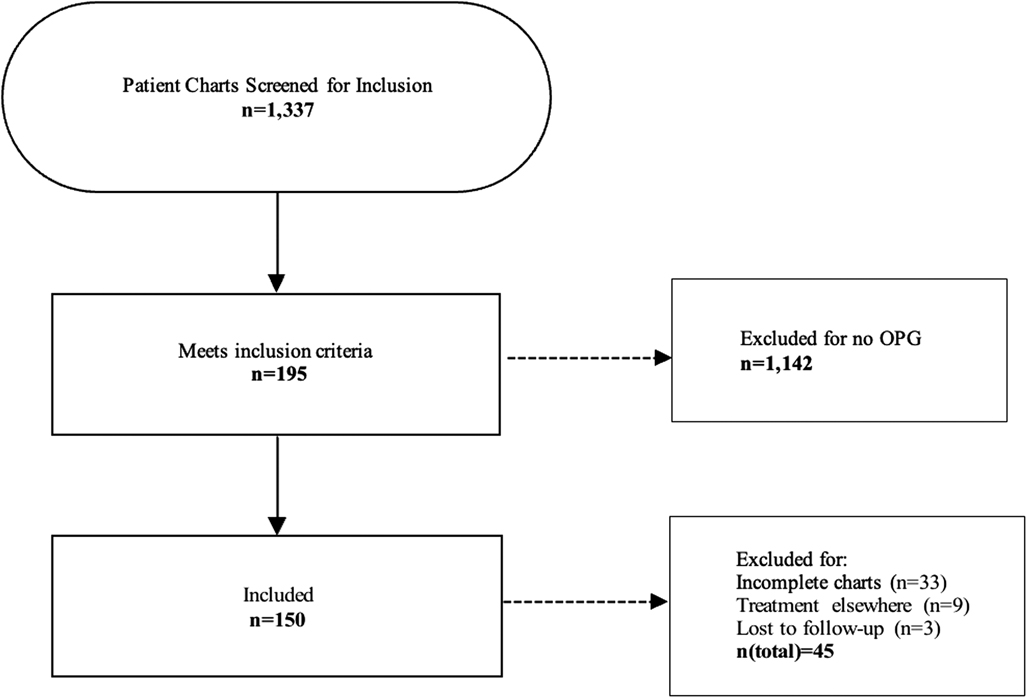
Figure 2. Flowchart illustrating the process of patient screening and the exclusion criteria applied. A final number of 150 patients diagnosed with NF and OPG was included in the study.
Diagnosis
Diagnosis of OPG was obtained by neuroimaging, by CT in 19, by MRI in 131 patients. Thirty (20%) patients underwent neuroimaging as part of an NF screening process, whereas in 14 (9.3%) patients the finding of an OPG was incidental (obtained neuroimaging for other reasons) and resulted in further investigation of NF-related manifestations. Clinical presentation and symptoms leading to initial diagnostic imaging of the remaining 106 patients are listed in Table 1. Histopathological diagnosis was obtained in 53 patients: low grade astrocytoma not otherwise specified (NOS) (n = 20); pilocytic astrocytoma, (WHO grade I) (n = 27); pilomyxoid astrocytoma (WHO grade II) (n = 2); fibrillary astrocytoma (WHO grade II) (n = 1); and ganglioglioma (n = 1). Sixty-one patients were confirmed carrying mutations in the NF1 gene by genetic testing.
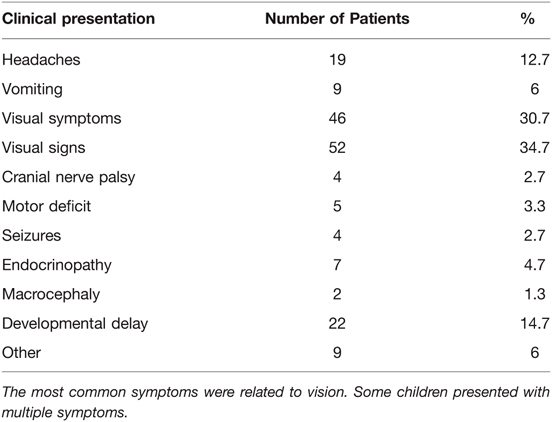
Table 1. Clinical presentation of children leading to initial neuroimaging and diagnosis of OPG.
Management
Being diagnosed with OPG, patients and their management were discussed in our multidisciplinary neuro-oncology rounds. Out of 150 patients, 62 (41.3%) remained stable and did not require any treatment, whereas 88 (58.7%) patients underwent at least one treatment throughout their course of disease. Of the thirty patients who were diagnosed with OPG via MRI screening, 10 (33%) patients required treatment during their clinical course. Four out of 14 (29%) patients whose OPG was an incidental finding were eventually submitted to treatment. Detailed treatment data were available in 150 children with OPG.
Initial management was observation in 84 (56%) patients, followed by chemotherapy (CTX) in 29 (19.3%) patients, and combined therapies in 21 (14%) patients. Upfront surgery alone was performed in 16 (10.7%) patients, including biopsies (n = 3), tumor debulking (n = 7), and gross total resection (GTR) (n = 6). A detailed list of initial management decisions is illustrated in Table 2.
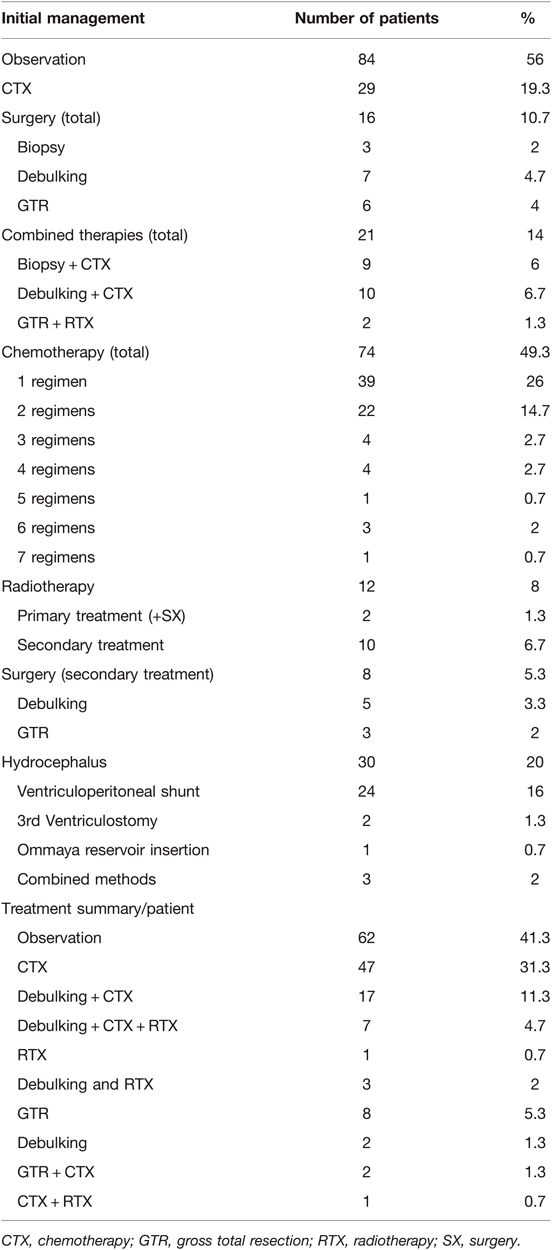
Table 2. Treatment overview.
The main primary treatment was CTX, applied in 74 (49.3%) patients (see Table 3 for chemotherapy protocols used as first-line treatment). Multiple regimens were required in 35 (23.3%) patients, due to tumor progression (n = 27) or CTX-related side effects (n = 8). Radiotherapy (RTX) was performed in a total of 12 (8%) patients, including 10 patients receiving radiation as a second-line treatment. Delayed tumor resection was required as secondary treatment in 8 (5.3%) patients. Clinical and radiographical signs of hydrocephalus were observed in 30 (20%) patients throughout their course of disease, being treated with ventriculoperitoneal (VP) shunts (n = 24), 3rd ventriculostomy (n = 2), Ommaya reservoir insertion (n = 1) and combined methods (n = 3).
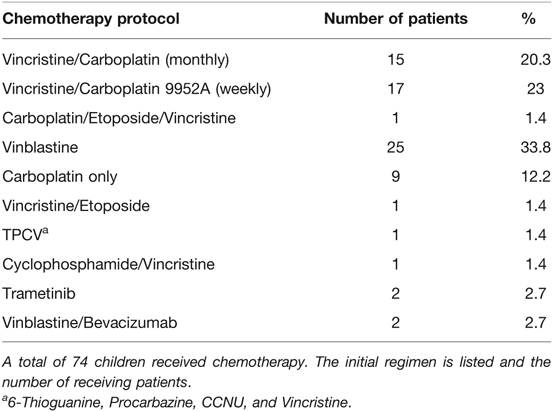
Table 3. First chemotherapy regimen in children with OPG.
Over the decades a shift in treatment modalities was noted from upfront surgery in the 1990s to chemotherapy as first-line treatment in 2000 onwards. Also, the type of chemotherapy underwent changes over the years and shifted from Vincristine/Carboplatin to Vinblastine in the recent years. Radiation therapy has been applied in only 12 out of 150 patients, in 11 before and one after 2000. It has not been administered in any of the patients over the last 15 years. A detailed overview of therapy distributions is given in Table 2 and these observations are more precisely described in the discussion section.
Neuropsychological testing was conducted in 100 (66.7%) patients, identifying 65 (65%) patients with learning disabilities and 29 (29%) with other mental health concerns (eg autism, attention deficit disorder).
Clinical follow up was provided on a 3-monthly basis during tumor treatment by the involved specialties (Neuro-oncology, Neuroradiology, Radiation Oncology, Ophthalmology, Neurosurgery) and for asymptomatic patients on a 6-monthly basis by Ophthalmology as well as on a yearly basis in the NF clinic.
Clinical Outcome
The mean (SD) duration of follow up was 8.14 (5.46) years (range: 0.1–25.9 years; median 6.8 years). Five patients died during the study period; one after widespread dissemination of disease to the meninges and the brainstem after exhaustion of all therapy options; one from a radiotherapy induced secondary PNET which did not respond to treatment; two patients who were severely neurologically disabled from the effects of their tumor, progressed after completing one and three courses of chemotherapy, respectively, and no further treatment was attempted. The fifth patient, being severely compromised by multiple comorbidities died after a fatal tumor hemorrhage. For the remaining patients the status at last follow up was determined radiographically and/or clinically as stable in 132 (88%), in progression in 6 (4%), and in complete remission in 7 (4.7%) patients. Complete remission was defined as complete radiographic resolution of the OPG.
Tumor progression (ophthalmological: n = 9; radiographical: n = 18; both: n = 24; not documented: n = 10) after the first line treatment requiring further management was observed in 61 (40.7%) and included 22 (26.2%) out of 84 patients with initial observation only and 15 (51.7%) out 29 patients with initial chemotherapy. Decisions about second line treatments were made in our multidisciplinary tumor rounds and included start or change of CTX in 43 (28.6%), RTX in 10 (6.7%) and SX in 8 (5.3%) patients, respectively. A summary of all treatments administered per patient are indicated in Table 2.
Overall survival was 25.23 years (±0.47) (mean; 95% CI of 24.3–26.16) in the treatment group and 24.49 years (±1.07) (mean; 95% CI of 18.4–22.57) in the observation group (Figure 3). The overall survival of the entire group was calculated with a mean of 23.6 years (±1.08) (95% CI of 21.48–25.72). Cumulative survival was 96.7 ± 2.3% in the treatment group and 97.1 ± 2.9% in the non-treatment group at 10 years of follow up. A log rank test comparing the OS of the treated and non-treated group did not confirm significance (p = 0.455).
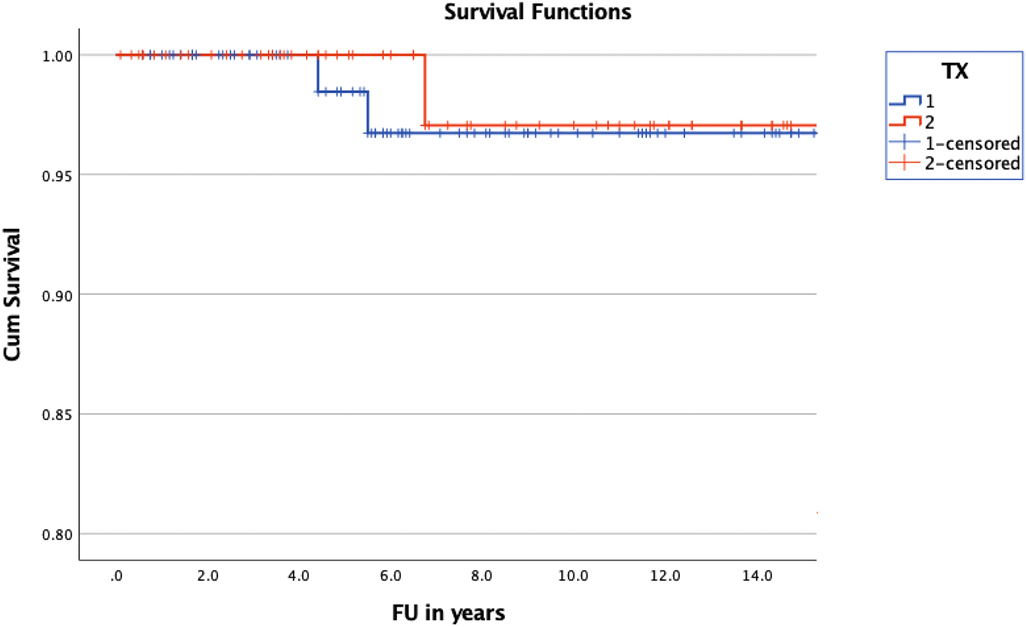
Figure 3. Kaplan-Meier curve illustrating the OS of children with NF1 and OPG. The difference of OS between treated (blue) and non-treated (red) children was statistically not significant (p = 0.455).
Persisting visual deficits were noted in 29 (19.3%) patients at their last follow up, including 11 (7.3%) patients with unilateral and 5 (3.3%) patients with bilateral vision loss. A comprehensive list of all clinical features of NF1 patients with OPG is provided in Table 4.
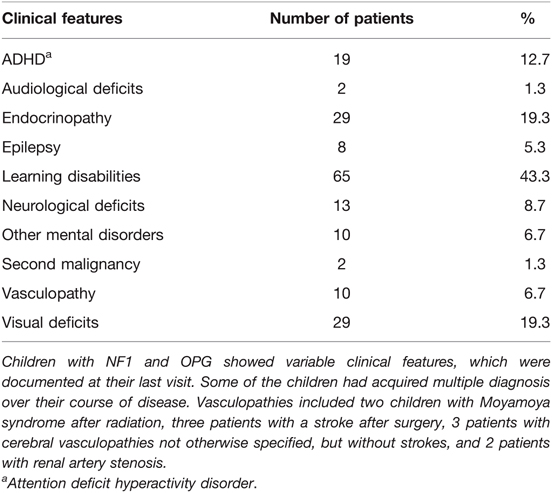
Table 4. Clinical features of children with NF1 and OPG.
Discussion
In this study we reviewed the clinical course of 150 children with NF1 and OPG, treated at HSC between 1990 and 2021. We analyzed the clinical outcome of both children who required treatment for OPG and those who underwent observation only. Despite many clinical features leading to an increased morbidity in this population, the estimated OS was 25.23 years (±0.467) in the treatment group and 24.487 years (±1.065) in the observation group, including 5 deaths. Treatment paradigm changes became visible in this retrospective study and will be discussed in the following. Furthermore, detailed analysis allowed us to shed light on the established treatment pathways at our institution, their evolution over time and integrative role within a multidisciplinary setting.
Paradigm Shift in Treatment
Whereas surgery and radiation therapy were the initial mainstay therapies for OPG in 1990s, especially diagnostic biopsies followed by radiation and/or chemotherapy, the number of surgical interventions for OPG decreased significantly after 2000. This change occurred simultaneously with the introduction of our brain tumor program, which aimed to reduce treatment related side-effects in low-grade gliomas (LGG), and to establish standardized treatment approaches for children with NF1 and OPG according to their age and symptoms (14). Multiple studies were published around that time revisiting the optimal treatment strategy for these complex patients by investigating the role and risks of surgery. A report by Nicolin et al., showed that aggressive debulking surgery had a negative impact on the neurocognitive outcomes of children with OPG. Patients treated with upfront debulking and chemotherapy displayed lower full and verbal scale IQ, than those treated with chemotherapy alone (24). Other studies confirmed the higher risk of neurological and/or endocrinological sequalae related to surgical therapy. Valdueza et al. reported their surgical experience in 20 patients with large hypothalamic/chiasmatic OPG (including 6 NF1 patients). Ten patients underwent debulking with resection of more than 50% of the tumor volume, 6 underwent partial resections, and 4 biopsies. The outcome was favorable in five patients with visual improvement following surgery. However, 4 patients had a visual decline, 1 patient developed a large cerebral infarction post-operatively and 4 patients suffered from endocrine complications (25). Similar observations were made by Sawaruma et al. They performed GTR in 7 patients with OPG, of which 5 patients experienced significant complications, which led the authors to the conclusion that the benefit of initial resection is questionable (26). Other studies, such as the report of Ahn et al. confirmed the surgery-related morbidity in a total of 33 patients, including 27 patients with GTR and 6 patients with debulking. Two patients in their study died of post-operative pulmonary embolism and diffuse cerebral infarction, 5 patients had a transient hemiparesis and 7 a visual deterioration (27). Furthermore, the benefit of aggressive surgery in OPG has been difficult to demonstrate with respect to tumor progression when compared to other upfront treatments (28). Different observations were made by Liu et al., who observed in their study that residual tumor size was inversely associated with PFS in 165 patients with OPG and that adjuvant treatment could be avoided or deferred by maximized resection (29). Our cohort included 16 patients, who underwent upfront surgical treatment. Ten of them (GTR in 8 patients with isolated optic nerve glioma, debulking in 2 patients) did not require any further treatments. However, this number is by far too small for drawing comparative conclusions about the impact of tumor size reduction on PFS. With respect to surgical morbidity, we report 8 out of a total of 39 patients, who underwent either GTR or debulking, who suffered from significant neurological sequalae after surgery, including hemiparesis, focal deficits, and epilepsy, underlining the previously reported risk of surgical morbidity. Nevertheless, the role of surgery in NF 1 patients with OPG remains controversial and until recently there was no consensus for surgical management of these tumors except for unilateral optic nerve lesions associated with severe proptosis and/or complete unilateral blindness allowing for radical resection (14, 30). Surgical interventions for the treatment of related hydrocephalus or salvage surgeries follow a different argumentation and should be discussed individually for each patient.
Radiation therapy declined more drastically in its application for children with NF1 and OPG over the last 2 decades. It was considered an effective treatment for OPG in the past with high progression free survival rates (PFS) of up to 90% at 10 years (31–34). However, multiple studies have reported long-term side effects of radiation in patients with OPG, including endocrine abnormalities (33, 35), cerebrovascular disease (36–38), poor visual outcomes (31, 33, 39), secondary malignancies (40, 41), and neurocognitive deficits, particularly in young patients with developing brains (31, 42). Capelli et al. found that cerebrovascular complications after radiation were five times more frequent in children with OPG and NF1 than in those without NF1, and therefore calling for new treatment strategies in these patients (31). In a retrospective study Ulrich et al. analysed risk factors for the development of Moyamoya after radiation for primary brain tumors in 345 children. They observed that patients with NF1 carried a threefold increased risk of developing Moyamoya (HR = 3.07, 95% CI, 0.90–10.46, p = 0.07) besides an earlier onset of Moyamoya compared to non-NF1 patients (median of 38 vs 55 months) (38). Because patients with NF1 have a greater risk of neoplasms and cerebrovascular disease at baseline, these adverse outcomes are of particular concern in the NF1 population. Hence radiation therapy has become a therapy of last resort, reserved for adolescent patients and those with no remaining chemotherapeutic or targeted treatment options (43). In our institution, a total of 12 children with NF1 and OPG underwent radiation therapy over the last 31 years, 11 of them irradiated before 2000, when our brain tumor program was initiated. Two of those patients developed Moyamoya post radiation and required a cerebral revascularization procedure. However, there are newer methods of radiation, such as proton beam radiation therapy and stereotactic radiosurgery. These have shown positive short-term results in NF1 patients with OPG and may be potential (salvage) treatment options in the future, depending on their currently investigated long-term outcomes (43–45).
Beyond the shift of primary treatment towards chemotherapy, we observed additional changes within the first line chemotherapy regimens administered in children with NF1 and OPG. In line with the former recommendations, the regimen we mainly used in the 1990s was a combination of Vincristine and Carboplatin (46). Other regimens followed until a cutoff was noted in 2007 with predominant prescription of either Vinblastine alone or recently also Vinblastine in combination with Bevacizumab. This change was a response to the common adverse effects of other chemotherapies, including neurotoxicity (vincristine), hypersensitivity (carboplatin) (47, 48), ototoxicity (49) (cisplatin), infertility, and other long-term toxicities with alkylating agents (procarbazine and lomustine) or increased risk of secondary malignancies with etoposide (50). Lassaletta et al. reported a multicenter study, which investigated the comparative effectiveness and safety of a single-agent first-line chemotherapy with Vinblastine in pediatric low-grade glioma (LGG), including 13 patients with NF1. They showed that Vinblastine was well tolerated by all children while representing a less toxic treatment with similar efficacy, in terms of OS, PFS, and visual outcomes, as other regimens. Interestingly, patients with NF1 had a significantly better PFS (85.1%; 95% CI, 68.0%–100%) when compared to patients without NF1 (42.0%; 95% CI, 29.1%–60.7%; P = .012), identifying Vinblastine as an optimal agent for children with NF1 and OPG (51). Most recently, combinations of Vinblastine and Bevacizumab have emerged as promising regimens for OPG (52, 53). Bevacizumab is an anti-vascular endothelial growth factor (VEGF) monoclonal antibody, which reduces neovascularization as well as tumor growth by targeting VEGF. It has been previously used in other childhood malignancies and demonstrated good tolerability (54, 55). Current applications in patients with NF1 and OPG are refractory patients until further studies elucidate its comprehensive effect on clinical outcomes (52).
Screening (MRI Algorithm)
Routine MRI screening for OPG in children with NF1 has been a topic of controversy over the last decade. Despite the high incidence of OPG in the NF1 population, brain MRI is not recommended as a screening tool, as treatment is not indicated in the absence of visual symptoms or proptosis (14). Serial visual assessment is currently considered the standard screening tool for OPG, including yearly visual assessment up to the age of 8 years and every other year until the age of 18 years (22). However, ophthalmological exams in the youngest children, which are also at highest risk for developing OPG can be challenging and requires relevant expertise (14, 56). Furthermore, in the context of a tumor that involves the optic pathway, loss of visual acuity may be a late occurrence and efforts to detect a threat to vision should ideally be identified earlier. Prevention of visual deficits is one of the major goals in following NF1 patients with OPG and awaiting the ophthalmologist to detect a visual decline as evidence for tumor progression may be a justified critique. However, early detection of an asymptomatic OPG has not been proven to reduce the incidence of visual loss, nor does an initial normal MRI exclude the development of a subsequent OPG (57, 58). But there are more factors to consider. Similar to the observation in our cohort, a significant number of children with NF1 and OPG remain asymptomatic and do not require any treatment throughout their entire clinical course (16). A study by Listernick et al. in 1989 demonstrated that, if all children with NF1 undergo screening neuroimaging, 15% of them will have radiographic evidence of OPG with only half of these children developing symptoms or signs related to the OPG (59). We followed 1337 children with NF in our specialty clinic, of which 195 (14.6%) were diagnosed with OPG. Unfortunately, complete data were only available for 150 patients (11.2%), including 88 treated and 62 asymptomatic patients, who never required any intervention. This corresponds roughly to the results of Listernick et al. and underlines that only a small group of patients would benefit from serial MRI screening – in our study 88 (6.6%) patients out of 1,337. Given that MR Imaging is an expensive, time-consuming exam, which requires the use of anesthetics in young patients (mostly until the age of 5–6 years), the costs, risk of general anesthesia and recourse control outweigh the benefit of serial screening MR Imaging in NF1 patients. Additionally, few studies reported spontaneous regression of low-grade OPG (60, 61). Taking these aspects into consideration, the recommendation of primary ophthalmological and clinical screening for OPG was implemented as diagnostic standard in our institution and is coordinated by our NF clinic.
Role of NF Clinic
Multiple factors have led in the past to the establishment and integration of specialized pediatric NF clinics in major therapy centers (15–17, 62, 63). First, the complexity of the disease and its requirements for comprehensive diagnosis and follow up are important and exceed the competences and capacities of single specialists, such as oncologists, neuroradiologists, neurosurgeons, or ophthalmologists (15). Children with NF may present simultaneously with multiple clinical features and are constantly at risk of developing malignancies (64, 65), which may have a significant impact on their life expectancy (17). This results in a high-intensity demand of care, coordination of multiple specialties and integration of continuous exam results. Second, symptoms such as rapid expansion of existing tumors, chronic pain, and neurological deficits require immediate medical attention, dedicated examination, and treatment (66). Age-specific monitoring of symptoms and education of patients and their parents are important in managing NF1 patients and should be provided by a pediatrician specialized in NF (15, 67). Our NF1 clinic was established in 1990, consists of 3 pediatricians and one nurse, having provided care for 1,337 children with NF1 to date. Its role is threefold and includes diagnosis of NF1, regular follow up and screening for new malignancies. Patients undergo a comprehensive diagnostic work-up at presentation, including genetic testing if desired by the parents/patient, followed by yearly follow up visits. Referrals to other specialties are made upon clinical needs and diagnosis, except for ophthalmology, which is part of a regulated follow up schedule. According to the recommendations of the NF1 Optic Pathway Glioma Task Force the children undergo yearly visual assessment until the age of 8 years and assessment every other year until the age of 18 years (22). Having established this central specialty clinic for children with NF1, the coordination of the patient’s care as well as their maintenance of follow-up has improved significantly. Further studies will aim to objectify the impact on care via a dedicated NF1 clinic by reviewing data on diagnosis of malignancies, referral pathways and adherence to follow up. Lastly, we could also observe a significant advantage of the NF1 clinic with respect to reviewing and updating other specialty exam results and with respect to collecting outcome data in a comprehensive manner for all NF associated clinical features. This sort of surveillance may contribute positively to care optimization and institutional quality assessment. Other considerations of NF specialty clinics are their guidance for facilitating transition and continuation of care in adult NF patients. Adult counterpart clinics may provide purposeful follow up while maintaining the same level of expertise and quality of care and therefore represent an equally important institution after transition from pediatrics to adulthood (68).
Limitations
There are several limitations to the study. First, a relevant number of patients had to be excluded from the study due to missing data, which might compromise the significance of our results. Second, we did not report detailed neuro-ophthalmologic results of visual outcome. Although it is one of the most important determinants of OPG management and outcome, we focused on OS and overall clinical status at patients’ discharge to adult care. The reason we did not report visual outcome in detail was to avoid redundance as it was previously described by Nicolin et al (24). However, the overall visual outcome was considered when evaluating the clinical status at last follow up. Patients, who were categorized it as clinically stable, also had a stable exam in their last ophthalmological follow up. The number of patients with persisting visual deficits are summarized in Table 4. Lastly, further limitations of our study are reflected by missing subgroup analysis regarding the different treatment constellations and their impact on OS. Acknowledging the highly variable and to some extent small patient numbers per subgroup (compare treatment summary/patient in Table 2), we did not consider its results as sufficiently robust for generalizability to all pediatric populations with NF1 and OPG.
Conclusion
This report confirms the complexity of the natural history of OPG, its requirement for multidisciplinary management and the beneficial role of a centrally integrated NF1 clinic. Treatment algorithms experienced several changes over the last decades and will be subject to constant optimization. Specialized NF clinics represent a substantial element in the care network for children with NF1 and OPG, supporting coordination of several multidisciplinary clinics, offering specialized and comprehensive care besides serving epidemiological aspects by assembling data of these patients in a centralized manner. Having this integrative role, dedicated NF1 clinics should become a regular component in the multidisciplinary management of patients with NF and OPG.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon request, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Research Ethics Board, Hospital for Sick Children, Toronto, Canada. Written informed consent for participation was not provided by the participants' legal guardians/next of kin due to the retrospective character of the study.
Author Contributions
LNL performed data acquisition, analysis, and drafting of the manuscript. UKB, EB and UT guided the study design and critically reviewed the manuscript. AP provided support with data extraction and analysis. PCP and JTR supervised the study and reviewed the manuscript as senior authors. All authors contributed to the article and approved the submitted version.
Nomenclature
CTComputed TomographyCTXChemotherapyHSCHospital for Sick ChildrenMRIMagnet Resonance ImagingNF1Neurofibromatosis (type 1)OPGOptic Pathway GliomaOSOverall SurvivalPFSProgression Free SurvivalRTXRadiation TherapySDStandard DeviationSXSurgeryVP shuntVentriculoperitoneal ShuntConflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kissil JL, Blakeley JO, Ferner RE, Huson SM, Kalamarides M, Mautner VF, et al. What’s new in neurofibromatosis? Proceedings from the 2009 NF Conference: new frontiers. Am J Med Genet A. 152a(2):269–83. doi: 10.1002/ajmg.a.33189
2. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. (2010) 152a(2):327–32. doi: 10.1002/ajmg.a.33139
3. Uusitalo E, Leppävirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. (2015) 135(3):904–6. doi: 10.1038/jid.2014.465
4. Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. (2005) 141(1):71–4. doi: 10.1001/archderm.141.1.71
5. Evans DGR, Salvador H, Chang VY, Erez A, Voss SD, Schneider KW, et al. Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 1. Clin Cancer Res. (2017) 23(12):e46–e53. doi: 10.1158/1078-0432.Ccr-17-0589
6. Shofty B, Ben Sira L, Constantini S. Neurofibromatosis 1-associated optic pathway gliomas. Childs Nerv Syst. (2020) 36(10):2351–61. doi: 10.1007/s00381-020-04697-1
7. Tadini G, Milani D, Menni F, Pezzani L, Sabatini C, Esposito S. Is it time to change the neurofibromatosis 1 diagnostic criteria? Eur J Intern Med. (2014) 25(6):506–10. doi: 10.1016/j.ejim.2014.04.004
8. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. (2014) 13(8):834–43. doi: 10.1016/s1474-4422(14)70063-8
9. Binning MJ, Liu JK, Kestle JR, Brockmeyer DL, Walker ML. Optic pathway gliomas: a review. Neurosurg Focus. (2007) 23(5):E2. doi: 10.3171/foc-07/11/e2
10. Chong AL, Pole JD, Scheinemann K, Hukin J, Tabori U, Huang A, et al. Optic pathway gliomas in adolescence–time to challenge treatment choices? Neuro-oncology. (2013) 15(3):391–400. doi: 10.1093/neuonc/nos312
11. Karaconji T, Whist E, Jamieson RV, Flaherty MP, Grigg JRB. Neurofibromatosis type 1: review and update on emerging therapies. Asia Pac J Ophthalmol (Phila). (2019) 8(1):62–72. doi: 10.22608/apo.2018182
12. de Blank PMK, Fisher MJ, Liu GT, Gutmann DH, Listernick R, Ferner RE, et al. Optic pathway gliomas in neurofibromatosis type 1: an update: surveillance, treatment indications, and biomarkers of vision. J Neuroophthalmol. (2017) 37 Suppl 1(Suppl 1):S23–s32. doi: 10.1097/wno.0000000000000550
13. Fisher MJ, Loguidice M, Gutmann DH, Listernick R, Ferner RE, Ullrich NJ, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro-oncology. (2012) 14(6):790–7. doi: 10.1093/neuonc/nos076
14. Fried I, Tabori U, Tihan T, Reginald A, Bouffet E. Optic pathway gliomas: a review. CNS Oncol. (2013) 2(2):143–59. doi: 10.2217/cns.12.47
15. Nishida Y, Ikuta K, Natsume A, Ishihara N, Morikawa M, Kidokoro H, et al. Establishment of in-hospital clinical network for patients with neurofibromatosis type 1 in Nagoya University Hospital. Sci Rep. (2021) 11(1):11933. doi: 10.1038/s41598-021-91345-6
16. Kokkinou E, Roka K, Alexopoulos A, Tsina E, Nikas I, Krallis P, et al. Development of a multidisciplinary clinic of neurofibromatosis type 1 and other neurocutaneous disorders in Greece. A 3-year experience. Postgrad Med. (2019) 131(7):445–52. doi: 10.1080/00325481.2019.1659708
17. Merker VL, Dai A, Radtke HB, Knight P, Jordan JT, Plotkin SR. Increasing access to specialty care for rare diseases: a case study using a foundation sponsored clinic network for patients with neurofibromatosis 1, neurofibromatosis 2, and schwannomatosis. BMC Health Serv Res. (2018) 18(1):668. doi: 10.1186/s12913-018-3471-5
18. National Institutes of Health. National institutes of health consensus development conference statement: neurofibromatosis. Bethesda, Md., USA, July 13–15, 1987. Neurofibromatosis. (1988) 1(3):172–8.
19. Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. (2021) 23(8):1506–13. doi: 10.1038/s41436-021-01170-5
20. Hersh JH. Health supervision for children with neurofibromatosis. Pediatrics. (2008) 121(3):633–42. doi: 10.1542/peds.2007-3364
21. Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR. Health supervision for children with neurofibromatosis type 1. Pediatrics. (2019) 143(5): e20190660. doi: 10.1542/peds.2019-0660
22. Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. (1997) 41(2):143–9. doi: 10.1002/ana.410410204
24. Nicolin G, Parkin P, Mabbott D, Hargrave D, Bartels U, Tabori U, et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer. (2009) 53(7):1231–7. doi: 10.1002/pbc.22198
25. Valdueza JM, Lohmann F, Dammann O, Hagel C, Eckert B, Freckmann N. Analysis of 20 primarily surgically treated chiasmatic/hypothalamic pilocytic astrocytomas. Acta Neurochir. (1994) 126(1):44–50. doi: 10.1007/bf01476493
26. Sawamura Y, Kamada K, Kamoshima Y, Yamaguchi S, Tajima T, Tsubaki J, et al. Role of surgery for optic pathway/hypothalamic astrocytomas in children. Neuro-oncology. (2008) 10(5):725–33. doi: 10.1215/15228517-2008-033
27. Ahn Y, Cho BK, Kim SK, Chung YN, Lee CS, Kim IH, et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst. (2006) 22(9):1136–42. doi: 10.1007/s00381-006-0086-7
28. Steinbok P, Hentschel S, Almqvist P, Cochrane DD, Poskitt K. Management of optic chiasmatic/hypothalamic astrocytomas in children. Can J Neurol Sci. (2002) 29(2):132–8.
29. Liu ZM, Liao CH, An X, Zhou WT, Ma ZY, Liu W, et al. The role of imaging features and resection status in the survival outcome of sporadic optic pathway glioma children receiving different adjuvant treatments. Neurosurg Rev. (2022). doi: 10.1007/s10143-022-01743-1
30. Zeid JL, Charrow J, Sandu M, Goldman S, Listernick R. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J AAPOS. (2006) 10(6):534–9. doi: 10.1016/j.jaapos.2006.03.014
31. Cappelli C, Grill J, Raquin M, Pierre-Kahn A, Lellouch-Tubiana A, Terrier-Lacombe MJ, et al. Long-term follow up of 69 patients treated for optic pathway tumours before the chemotherapy era. Arch Dis Child. (1998) 79(4):334–8. doi: 10.1136/adc.79.4.334
32. Grabenbauer GG, Schuchardt U, Buchfelder M, Rodel CM, Gusek G, Marx M, et al. Radiation therapy of optico-hypothalamic gliomas (OHG)–radiographic response, vision and late toxicity. Radiother Oncol. (2000) 54(3):239–45. doi: 10.1016/s0167-8140(00)00149-3
33. Horwich A, Bloom HJ. Optic gliomas: radiation therapy and prognosis. Int J Radiat Oncol Biol Phys. (1985) 11(6):1067–79. doi: 10.1016/0360-3016(85)90052-5
34. Jenkin D, Angyalfi S, Becker L, Berry M, Buncic R, Chan H, et al. Optic glioma in children: surveillance, resection, or irradiation? Int J Radiat Oncol Biol Phys. (1993) 25(2):215–25. doi: 10.1016/0360-3016(93)90342-s
35. Pierce SM, Barnes PD, Loeffler JS, McGinn C, Tarbell NJ. Definitive radiation therapy in the management of symptomatic patients with optic glioma. Survival and long-term effects. Cancer. (1990) 65(1):45–52. doi: 10.1002/1097-0142(19900101)65:1<45::aid-cncr2820650111>3.0.co;2-z
36. Grill J, Couanet D, Cappelli C, Habrand JL, Rodriguez D, Sainte-Rose C, et al. Radiation-induced cerebral vasculopathy in children with neurofibromatosis and optic pathway glioma. Ann Neurol. (1999) 45(3):393–6. doi: 10.1002/1531-8249(199903)45:3<393::aid-ana17>3.0.co;2-b
37. Kestle JR, Hoffman HJ, Mock AR. Moyamoya phenomenon after radiation for optic glioma. J Neurosurg. (1993) 79(1):32–5. doi: 10.3171/jns.1993.79.1.0032
38. Ullrich NJ, Robertson R, Kinnamon DD, Scott RM, Kieran MW, Turner CD, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology. (2007) 68(12):932–8. doi: 10.1212/01.wnl.0000257095.33125.48
39. Bataini JP, Delanian S, Ponvert D. Chiasmal gliomas: results of irradiation management in 57 patients and review of literature. Int J Radiat Oncol Biol Phys. (1991) 21(3):615–23. doi: 10.1016/0360-3016(91)90678-w
40. Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. (2006) 24(16):2570–5. doi: 10.1200/jco.2005.03.8349
41. Tsang DS, Murphy ES, Merchant TE. Radiation therapy for optic pathway and hypothalamic low-grade gliomas in children. Int J Radiat Oncol Biol Phys. (2017) 99(3):642–51. doi: 10.1016/j.ijrobp.2017.07.023
42. Lacaze E, Kieffer V, Streri A, Lorenzi C, Gentaz E, Habrand JL, et al. Neuropsychological outcome in children with optic pathway tumours when first-line treatment is chemotherapy. Br J Cancer. (2003) 89(11):2038–44. doi: 10.1038/sj.bjc.6601410
43. Farazdaghi MK, Katowitz WR, Avery RA. Current treatment of optic nerve gliomas. Curr Opin Ophthalmol. (2019) 30(5):356–63. doi: 10.1097/icu.0000000000000587
44. El-Shehaby AM, Reda WA, Abdel Karim KM, Emad Eldin RM, Nabeel AM. Single-session Gamma Knife radiosurgery for optic pathway/hypothalamic gliomas. J Neurosurg. (2016) 125(Suppl 1):50–57. doi: 10.3171/2016.8.Gks161432
45. Fuss M, Hug EB, Schaefer RA, Nevinny-Stickel M, Miller DW, Slater JM, et al. Proton radiation therapy (PRT) for pediatric optic pathway gliomas: comparison with 3D planned conventional photons and a standard photon technique. Int J Radiat Oncol Biol Phys. (1999) 45(5):1117–26. doi: 10.1016/s0360-3016(99)00337-5
46. Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. (2012) 30(21):2641–7. doi: 10.1200/jco.2011.36.6054
47. Lafay-Cousin L, Sung L, Carret AS, Hukin J, Wilson B, Johnston DL, et al. Carboplatin hypersensitivity reaction in pediatric patients with low-grade glioma: a Canadian Pediatric Brain Tumor Consortium experience. Cancer. (2008) 112(4):892–9. doi: 10.1002/cncr.23249
48. Yu DY, Dahl GV, Shames RS, Fisher PG. Weekly dosing of carboplatin increases risk of allergy in children. J Pediatr Hematol Oncol. (2001) 23(6):349–52. doi: 10.1097/00043426-200108000-00005
49. Orts Alborch M, García Callejo J, Morant Ventura A, Ferrer Baixauli F, Esparcia Navarro M, Marco Algarra J. Clinical study on the ototoxicity of cisplatin with distortion products. Acta Otorrinolaringol Esp. (2000) 51(6):469–77.
50. Nageswara Rao AA, Packer RJ. Advances in the management of low-grade gliomas. Curr Oncol Rep. (2014) 16(8):398. doi: 10.1007/s11912-014-0398-9
51. Lassaletta A, Scheinemann K, Zelcer SM, Hukin J, Wilson BA, Jabado N, et al. Phase II weekly vinblastine for chemotherapy-naïve children with progressive low-grade glioma: a canadian pediatric brain tumor consortium study. J Clin Oncol. (2016) 34(29):3537–43. doi: 10.1200/jco.2016.68.1585
52. Avery RA, Hwang EI, Jakacki RI, Packer RJ. Marked recovery of vision in children with optic pathway gliomas treated with bevacizumab. JAMA Ophthalmol. (2014) 132(1):111–4. doi: 10.1001/jamaophthalmol.2013.5819
53. Zhukova N, Rajagopal R, Lam A, Coleman L, Shipman P, Walwyn T, et al. Use of bevacizumab as a single agent or in adjunct with traditional chemotherapy regimens in children with unresectable or progressive low-grade glioma. Cancer Med. (2019) 8(1):40–50. doi: 10.1002/cam4.1799
54. Roux C, Revon-Rivière G, Gentet JC, Verschuur A, Scavarda D, Saultier P, et al. Metronomic maintenance with weekly vinblastine after induction with bevacizumab-irinotecan in children with low-grade glioma prevents early relapse. J Pediatr Hematol Oncol. (2021) 43(5):e630–34. doi: 10.1097/mph.0000000000002002
55. Glade Bender JL, Adamson PC, Reid JM, Xu L, Baruchel S, Shaked Y, et al. Phase I trial and pharmacokinetic study of bevacizumab in pediatric patients with refractory solid tumors: a Children’s Oncology Group Study. J Clin Oncol. (2008) 26(3):399–405. doi: 10.1200/jco.2007.11.9230
56. Avery RA, Hwang EI, Ishikawa H, Acosta MT, Hutcheson KA, Santos D, et al. Handheld optical coherence tomography during sedation in young children with optic pathway gliomas. JAMA Ophthalmol. (2014) 132(3):265–71. doi: 10.1001/jamaophthalmol.2013.7649
57. Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. (1997) 278(1):51–7.
58. Cassiman C, Legius E, Spileers W, Casteels I. Ophthalmological assessment of children with neurofibromatosis type 1. Eur J Pediatr. (2013) 172(10): 1327–33. doi: 10.1007/s00431-013-2035-2
59. Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. (1989) 114(5):788–92. doi: 10.1016/s0022-3476(89)80137-4
60. Parsa CF, Hoyt CS, Lesser RL, Weinstein JM, Strother CM, Muci-Mendoza R, et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. (2001) 119(4):516–29. doi: 10.1001/archopht.119.4.516
61. Piccirilli M, Lenzi J, Delfinis C, Trasimeni G, Salvati M, Raco A. Spontaneous regression of optic pathways gliomas in three patients with neurofibromatosis type I and critical review of the literature. Childs Nerv Syst. (2006) 22(10):1332–7. doi: 10.1007/s00381-006-0061-3
62. McKeever K, Shepherd CW, Crawford H, Morrison PJ. An epidemiological, clinical and genetic survey of neurofibromatosis type 1 in children under sixteen years of age. Ulster Med J. (2008) 77(3):160–3.
63. Rosenfeld A, Listernick R, Charrow J, Goldman S. Neurofibromatosis type 1 and high-grade tumors of the central nervous system. Childs Nerv Syst. (2010) 26(5):663–7. doi: 10.1007/s00381-009-1024-2
64. Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. (2009) 9(3):247–53. doi: 10.1007/s11910-009-0036-3
65. Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. (2013) 108(1):193–8. doi: 10.1038/bjc.2012.535
66. Créange A, Zeller J, Rostaing-Rigattieri S, Brugières P, Degos JD, Revuz J, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. (1999) 122(Pt 3):473–81. doi: 10.1093/brain/122.3.473
67. Noble F, Kornberg AJ, Elder JE, Delatycki MB. Retrospective analysis of patients attending a neurofibromatosis type 1 clinic. J Paediatr Child Health. (2007) 43(1–2):55–9. doi: 10.1111/j.1440-1754.2007.01003.x
Keywords: optic pathway glioma, neurofibromatosis type 1, diagnosis, management, multidisciplinary, neurofibromatosis clinic
Citation: Lohkamp L, Parkin P, Puran A, Bartels UK, Bouffet E, Tabori U and Rutka JT (2022) Optic Pathway Glioma in Children with Neurofibromatosis Type 1: A Multidisciplinary Entity, Posing Dilemmas in Diagnosis and Management Multidisciplinary Management of Optic Pathway Glioma in Children with Neurofibromatosis Type 1. Front. Surg. 9:886697. doi: 10.3389/fsurg.2022.886697
Received: 8 February 2022; Accepted: 8 April 2022;
Published: 3 May 2022.
Edited by:
Zohreh Habibi, Tehran University of Medical Sciences, IranReviewed by:
Farideh Nejat, Tehran University of Medical Sciences, Tehran, IranKeyvan Tayebi Meybodi, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran
Copyright © 2022 Lohkamp, Parkin, Puran, Bartels, Buffet, Tabori and Rutka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura-Nanna Lohkamp TGF1cmEtTmFubmEuTG9oa2FtcEBzaWNra2lkcy5jYQ==
†These authors share senior authorship
Speciality section: This article was submitted to Neurosurgery, a section of the journal Frontiers in Surgery