
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Surg., 30 January 2023
Sec. Thoracic Surgery
Volume 9 - 2022 | https://doi.org/10.3389/fsurg.2022.1007760
 Santiago A. Endara1*Gerardo A. Dávalos1
Santiago A. Endara1*Gerardo A. Dávalos1 Gabriel A. Molina2
Gabriel A. Molina2 Christian A. Armijos3
Christian A. Armijos3 D. Sebastian Narvaez4
D. Sebastian Narvaez4 Nelson Montalvo5
Nelson Montalvo5
Desmoid fibromatosis is a rare, aggressive borderline lesion arising from soft tissues. Treatment will depend on the structures that the tumor has involved. Surgery with negative margins is the recommended strategy as it can usually achieve disease control; however, the tumor's location sometimes does not allow it. Therefore, a combination of medical therapies along with strict surveillance is crucial. We present the case of a 6-month-old boy with a chest mass. After further evaluation, a rapidly growing mediastinal mass involving the sternum and costal cartilage was detected. Desmoid fibromatosis was the final diagnosis.
Desmoid fibromatosis is a rare soft tissue neoplasm that does not metastasize but presents an aggressive growth and local invasion (1). It can affect all age groups and occur throughout the body (1, 2). In children, there is limited information on managing these complex tumors. We present the case of a young boy in whom a rapidly growing mass was detected in his chest. Desmoid fibromatosis was the final diagnosis.
Patient is a 6-month-old who was born with clinodactyly in both hands, and without a history of familial adenomatous polyposis. He presented with a two-month history of a small, hard, nonmobile, and painless mass in his chest. The mass was initially small and did not disturb the child's development. However, a couple of months later, as the child began to crawl, his mother noticed that he had some discomfort and realized that the mass had doubled its size, so she visited his pediatrician.
On clinical examination, an otherwise healthy young boy was found. Over the xiphoid process, a 2 × 2 cm solid mass with rounded edges was discovered. The mass was attached to the xiphoid process and nearby structures. With these findings, an ultrasound was requested, discovering an echogenic mass measuring 2.3 × 1.8 cm without vascularization and cardiac involvement (Figures 1A,B).
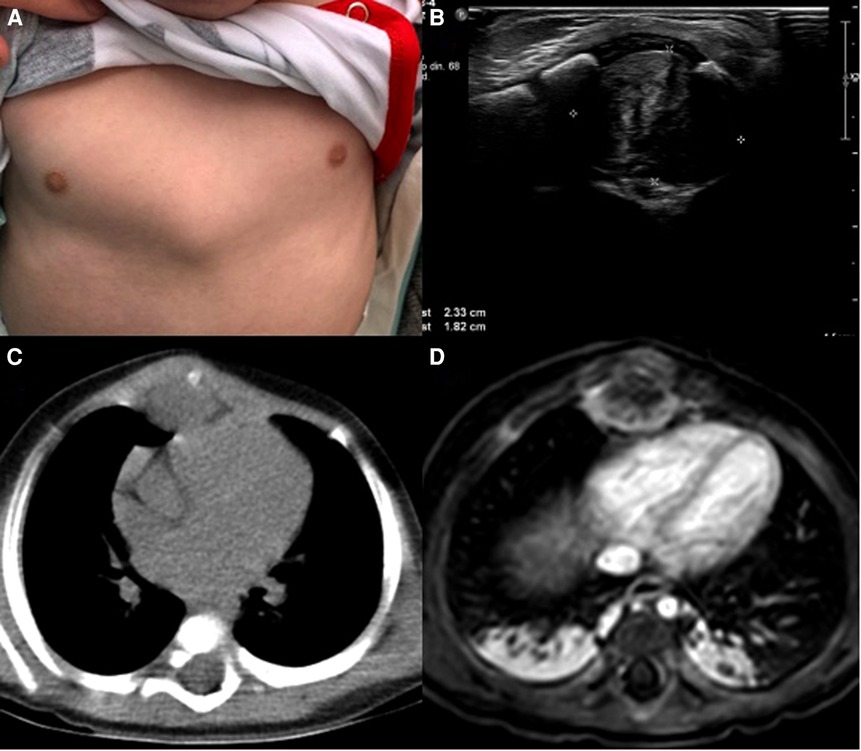
Figure 1. (A) chest mass, (B) ultrasound, echogenic mass in the anterior mediastinum. (C) CT, The mass is in close contact with the xiphoid process. (D) MRI, the mass is near the pericardium.
Due to this, a chest computed tomography (CT) revealed the presence of a 3.5 × 2 cm soft tissue density mass in the anterior mediastinum. It infiltrated the subcutaneous tissue and involved the ribs and sternum. Magnetic resonance imaging also proved that the pericardium was not compromised (Figures 1C,D).
With these findings, surgery was decided, and after partial distal sternotomy, the 3.5 × 2 cm solid whitish mass was discovered; it was in contact with the diaphragm, xiphoid process, and costal cartilages. Biopsy was taken for intraoperative frozen section; however, the results were inconclusive, malignancy was not confirmed, but atypia could be observed; because of this, and considering the risk of malignancy, complete resection of the mass, including the distal third of the body of the sternum, xiphoid process, diaphragm, and four bilateral costal cartilages was completed. A bioabsorbable mesh was used to repair the diaphragmatic defect and rigid fixation was placed on the ribs with stainless steel wires to reconstruct the thoracic wall, since bioabsorbable prosthetic material was unavailable; drains were placed, and the procedure was completed without complications (Figures 2A,B).
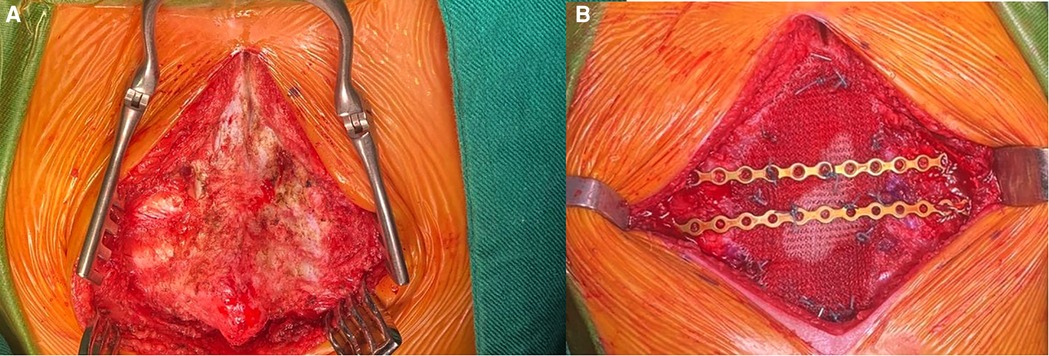
Figure 2. (A) Mass invading the costal cartilages and sternum. (B) Reconstruction with prosthetic material.
Pathology reported a proliferation of fusiform cells without nuclear hyperchromatism or mitosis; some areas showed multiple nodular, hypocellular, and hyalinized areas surrounding and invading muscle fibers. In addition, surgical margins were compromised on the diaphragmatic side. Immunohistochemistry was positive for Beta-catenin in the nuclear cells and negative for CD34, Desmin, and S100. Desmoid fibromatosis (DF) was the final diagnosis (Figure 3).
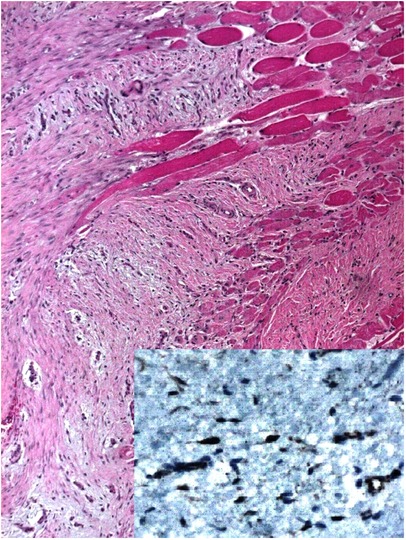
Figure 3. Pathology HE 4X, fusiform cells without nuclear hyperchromatism, hypocellular, and hyalinized areas are seen. Immunohistochemistry positive for Beta-catenin (Inset 40x).
His postoperative course was uneventful. He was discharged on postoperative day five, and he is on close follow-ups a year after surgery, there has been no sign of recurrence or complications, and this disease has not impaired his development. He is entirely normal at this time.
DF is an extremely infrequent tumor (2 to 4 cases per million per year) (1, 2). It's categorized as a slowly growing borderline tumor with low malignant potential formed by the monoclonal proliferation of fibroblasts in muscles, tendons, and ligaments (2, 3). This proliferation can lead to local aggressiveness that, depending on its location, can severely affect or impair vital organs, leading to functional impairment and life-threatening conditions (1, 3). Although the pathological pathways are still under investigation, DF is believed to appear from the mesenchymal stromal cells with mutation of the CTNNB1 gene in adults (the β-catenin gene) (1, 4). In children, it's associated with AKT (31%), BRAF (19%), and p53 (9%) mutations, in addition to the CTNNB1 (3–5).
DF can present in two ways; the sporadic form, which more frequently affects women, and the condition associated with familial adenomatous polyposis (FAP) (1, 6). DF clinically appears as a firm, often asymptomatic, slow-growing mass (2, 3). Its course can be indolent (5 to 10% of the cases) or invade nearby structures (3, 4). In children, DF primarily affects the head and neck (1). In adults, the trunk and limbs are more affected (1, 2). In our case, the patient presented with a mass that affected his thoracic wall. Treatment will depend on the course of the tumor; a conservative approach or “watch-and-wait” strategy has been suggested if the tumor doesn't show progression (6, 7). However, surgery is recommended if the tumor is over 5 cm, located in the head or neck, and affects younger patients (<10 years) (3, 5). As we did in our case.
Treatment will be based on the symptoms and the tumor growth, especially in critical locations where an earlier surgical decision may potentially prevent a higher risk of morbidity (9). The primary goal should be a wide (R0) microscopic margin resection; however, in problematic locations like the skull base or when functions or cosmetics is an issue, positive (R1), microscopic margins can also be accepted (8–10). Treatment options other than surgery may be preferred when positive microscopic margins are found (10). If an R1 resection is achieved after the initial surgery, in that case, neither perioperative radiotherapy nor reoperation is necessarily required since the risk of a local recurrence when we compare surgery alone and perioperative radiotherapy is not statistically significantly different (11). In contrast, the morbidity of the two combined modalities is higher (10, 11).
Even with successful surgery the risk of relapse is high especially in children where pediatric DF is associated with a high recurrence rate (24%–76%) (5, 6). Therefore, other treatments have also been proposed (8, 9). Another thing to consider is reconstruction in pediatric patients, as children have unique characteristics related to growth; therefore, the prosthetic material for reconstruction should be chosen carefully. Bioabsorbable materials should be used to avoid complications (8, 9). Since these materials were not available to us, we had to use rigid fixation.
Other therapies include radiation therapy as an adjuvant treatment; however, the risk of adverse effects, such as fibrosis, paresthesia, skin ulcers, and, in children, a significant risk of long-term functional sequelae and radiation-induced cancers, must be taken into account. Several cytotoxic and non-cytotoxic drug treatments have been proposed for desmoid tumors, and several have had partial or complete responses in multiple trials (11, 12). Newer treatments, such as tyrosine kinase inhibitors of vascular endothelial growth factor receptors, have also arisen as potential therapies in the future (6, 7). However, each case must be individualized. In our case, surgery was completed as the mass had aggressive behavior, but we could not achieve negative margins; therefore, even though radiotherapy, chemotherapy, anti-hormonal therapies, and tyrosine kinase inhibitors were considered. After exposing all the treatments with their benefits and risk to the child's parents, and multiple discussions with a pediatric oncologist, surveillance was decided.
When facing a desmoid tumor and its treatment, especially in pediatric patients, there are still many gray areas between surgery, observation, and medical therapies. Deciding between them is not always easy, as we should always avoid or limit any possible harm from the disease and our treatments. Still, quality of life must be the single central dimension to weigh when facing these tumors, a decision that must always be made following the wishes of the parents and the child. This article has been prepared according to CARE Guidelines (13).
His parents found it difficult at first to accept the disease, especially after the pathology and the positive margins of the tumor were explained to them. However, after the patient's successful recovery and normal development, they have come to accept its nature. They have changed their approach and have learned to accept the nature of the disease in a way that has surprised the entire medical team. We are grateful to the parents for allowing us to treat the patient, and we are on close watch.
The management of DF is complex and even with treatment, it can still behave like an aggressive disease. Further research is needed to determine which patients will benefit from surgery, medical therapies, or close surveillance. We will always have to weigh the risk of treating the disease with all the complications and recurrences that it entails or waiting and losing the opportunity to offer a chance of a complete cure.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
SA and GD performed the surgery. GM and CA recollected the data and wrote the first manuscript. SN and NM reviewed the manuscript. All authors approved the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Miyashita H, Asoda S, Soma T, Munakata K, Yazawa M, Nakagawa T, et al. Desmoid-type fibromatosis of the head and neck in children: a case report and review of the literature. J Med Case Rep. (2016) 10:173. doi: 10.1186/s13256-016-0949-9
2. Fortunati D, Kaplan J, López Martí J, Ponzone A, Innocenti S, Fiscina S, et al. Desmoid-type fibromatosis in children. Clinical features, treatment response, and long-term follow-up. Fibromatosis tipo desmoide en niños. Características, respuesta al tratamiento y seguimiento a largo plazo. Medicina (B Aires). (2020) 80(5):495–504. PMID: 3304879433048794
3. Paul A, Blouin MJ, Minard-Colin V, Galmiche L, Coulomb A, Corradini N, et al. Desmoid-type fibromatosis of the head and neck in children: a changing situation. Int J Pediatr Otorhinolaryngol. (2019) 123:33–7. doi: 10.1016/j.ijporl.2019.04.037
4. Honeyman JN, Theilen TM, Knowles MA, McGlynn MM, Hameed M, Meyers P, et al. Desmoid fibromatosis in children and adolescents: a conservative approach to management. J Pediatr Surg. (2013) 48(1):62–6. doi: 10.1016/j.jpedsurg.2012.10.017
5. Pounds N, Skapek SX. Desmoid-type fibromatosis in children: a step forward in the cooperative group setting. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting. (2012):593–7. doi: 10.14694/EdBook_AM.2012.32.32
6. Bhat V, Raju P, Rao S, Ramaiah S. Infantile fibromatosis: a rare cause of anterior mediastinal mass in a child. J Clin Imaging Sci. (2015) 5:34. doi: 10.4103/2156-7514.159452
7. Zhang Z, Shi J, Yang T, Liu T, Zhang K. Management of aggressive fibromatosis. Oncol Lett. (2021) 21(1):43. doi: 10.3892/ol.2012.991
8. Makarawo TP, Reynolds RA, Cullen ML. Polylactide bioabsorbable struts for chest wall reconstruction in a pediatric patient. Ann Thorac Surg. (2015) 99(2):689–91. doi: 10.1016/j.athoracsur.2014.03.052
9. Risoud M, Mortuaire G, Leroy X, Leblond P, Fayoux P. Desmoid tumours of the head and neck in children: review of management. Eur Ann Otorhinolaryngol Head Neck Dis. (2017) 134(3):155–60. doi: 10.1016/j.anorl.2016.11.007
10. Greene AC, Van Tine BA. Are the pieces starting to Come together for management of desmoid tumors? Clin Cancer Res. (2022) 28(18):3911–3. doi: 10.1158/1078-0432.CCR-22-0620
11. Desmoid Tumor Working Group. The management of desmoid tumours: a joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer (Oxford, England: 1990). (2020) 127:96–107. doi: 10.1016/j.ejca.2019.11.013
12. Kasper B, Raut CP, Gronchi A. Desmoid tumors: to treat or not to treat, that is the question. Cancer. (2020) 126(24):5213–21. doi: 10.1002/cncr.33233
Keywords: desmoid fibromatosis, mediastinal mass, desmoid tumor, desmoid disease, pediatric
Citation: Endara SA, Dávalos GA, Molina GA, Armijos CA, Narvaez DS and Montalvo N (2023) Case Report: Desmoid fibromatosis in the mediastinum of a 6-month-old toddler, what to do?. Front. Surg. 9:1007760. doi: 10.3389/fsurg.2022.1007760
Received: 30 July 2022; Accepted: 30 December 2022;
Published: 30 January 2023.
Edited by:
Lorenzo Rosso, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, ItalyReviewed by:
Carlos Andrés Carvajal Fierro, National Cancer Institute (Colombia), Colombia© 2023 Endara, Davalos, Molina, Armijos, Narvaez and Montalvo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Santiago A. Endara ZHJzYW50aWFnb2VuZGFyYUBnbWFpbC5jb20=
Specialty Section: This article was submitted to Thoracic Surgery, a section of the journal Frontiers in Surgery
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.