 Wei Wu
Wei Wu Yibo Wu
Yibo Wu- Department of General Surgery, Shanghai Children's Hospital, Shanghai Jiao Tong University, Shanghai, China
Background: One of the most prevalent forms of renal tumors detected among pediatric patients is the Wilms tumor (WT). Teratoid WT is a rare WT subclassification that is characterized by teratoma-like characteristics that include the features of many diverse tissue categories. Less than 70 teratoid Wilms tumor (TWT) cases have been explained up to now.
Methods: Between 2010 and 2020, patients with classical WT and TWT admitted to our hospital were included in this study. Clinicopathological characteristics, intraoperative findings, histopathological parameters, and prognostic outcomes were then compared between classical WT and TWT. To compare these variables, TWT and WT cases were matched at a 1:3 ratio.
Results: A total of 67 total WT cases, i.e., five diagnosed with TWT, were enrolled. While no significant differences in analyzed variables were detected between these groups, tumor volumes were notably larger in the TWT group relative to the classical WT group (203.30 ± 109.89 vs. 104.30 ± 66.97 cm3) despite similar tumor weight values in both groups (471.00 ± 80.65 vs. 432.67 ± 109.25 g). As for five patients diagnosed with TWT, all were alive during the follow-up, while one of them was diagnosed with pelvic metastasis.
Conclusions: This study is the first to our knowledge to have reported on the incidence of TWT among Chinese children, and our results preliminarily suggest that a combination of surgery and chemotherapy may be appropriate for the treatment of patients with WT, although prognostic outcomes varied substantially among patients with different stages of the disease. TWT tumor density may be lower than classical WT tumor density. Further research regarding the basic biological characteristics of TWT and relevant theranostic markers associated with this tumor type is warranted to better guide the development of individualized treatments for this rare cancer type.
Introduction
One of the most prevalent forms of renal tumors detected among pediatric patients is Wilms tumor (WT) (1), accounting for six and 95% of total pediatric cancers and pediatric kidney tumors, respectively (2). Both tumor stage and histologic subtype are key prognostic factors in patients with WT (3). However, WT clinical characteristics are often similar to those of other rare renal tumor subtypes, necessitating careful differentiation to guide appropriate patient diagnosis and treatment (4, 5).
One of the rare forms of heterologous nephroblastoma is teratoid WT (TWT), which was first explained in 1984 by Variend et al. (6). TWT tumors are generally triphasic, exhibiting blastemal, stromal, and epithelial cell types, with other cell and tissue types, such as mucinous epithelial tissue, neurogenic tissue, osteoid tissue, adipose tissue, and cartilage, also being present in some cases. In 1988, Fernandes et al. (7) suggested that TWT diagnosis should be dependent on a <50% heterogeneous component, while the Beckwith criteria state that renal teratomas must be fully contained within the renal capsule with evidence of both a renal component and other tissue types. Given the rarity of TWT, its pathogenesis remains the subject of debate, although these heterologous tissues most likely originate from a primitive metanephric blastema (8).
Up to now, just 62 TWT cases have been explained in the English language articles, with none having been reported in China. Herein, 67 cases with WT treated at our hospital from 2010 to 2020, i.e., five cases with TWT, were retrospectively evaluated, with a particular focus on individual TWT cases with respect to their clinical presentation, preoperative findings, chemotherapeutic treatment, histopathological characteristics, and postoperative treatment and associated outcomes, which may deepen our understanding of this rare condition.
Materials and Methods
Patient Enrollment
From December 2010 to December 2020, 67 total cases with WT were evaluated in the hospital and were incorporated in the present survey. The treatment method is mainly based on the Children's Oncology Group (COG) and the International Society of Pediatric Oncology (SIOP). A retrospective analysis of patient clinicopathological findings, pre- and postoperative chemotherapeutic treatment strategies, intraoperative achievements histopathological findings, and prognostic outcomes was conducted. Patient follow-up was conducted every 3 months, with abdominal ultrasound scans being performed every 6 months for the first 2 years.
WT and TWT Diagnosis and Assessment
Cases with TWT were diagnosed based upon histopathologic examination results demonstrating a >50% heterogeneous component. These tumors typically exhibit a prominent mature adipose tissue component composed of blastemal, epithelial, and stromal cell categories and may also exhibit other characteristics of other tissues, such as neurogenic tissue, cartilage, smooth muscle, osteoid tissue, squamous tissue, and mucinous epithelial tissue. Final diagnoses were established after a multi-disciplinary treatment discussion (Table 1).
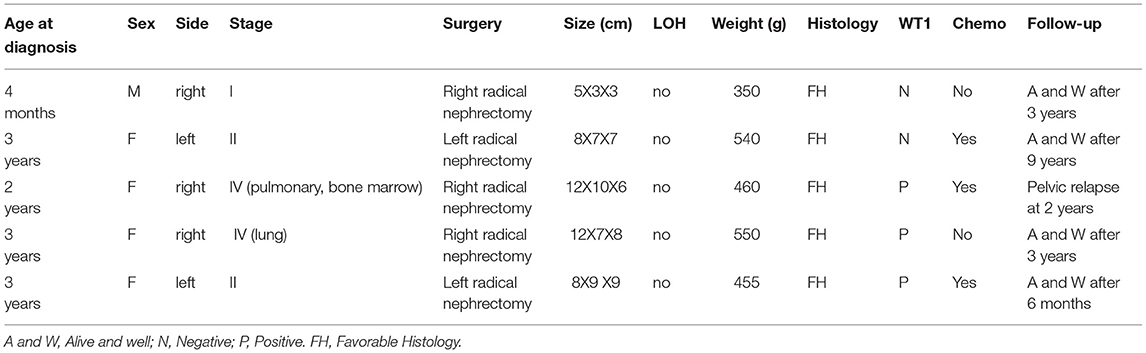
Table 1. Teratoid Wilms tumor patient information in our hospital.
Non-TWT cases in this study were considered to represent classical WT cases, and a definitive classical WT diagnosis was made by experienced pathologists. A 1:3 matching ratio was used when comparing data between TWT and classical WT cases to reduce the risk of bias (Tables 2, 3).
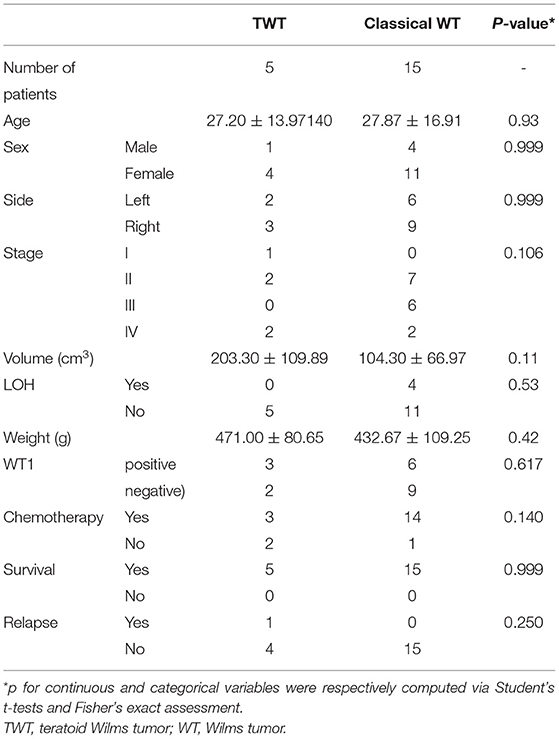
Table 2. Patient demographics, clinical characteristics, and prognostic outcomes in individual groups.
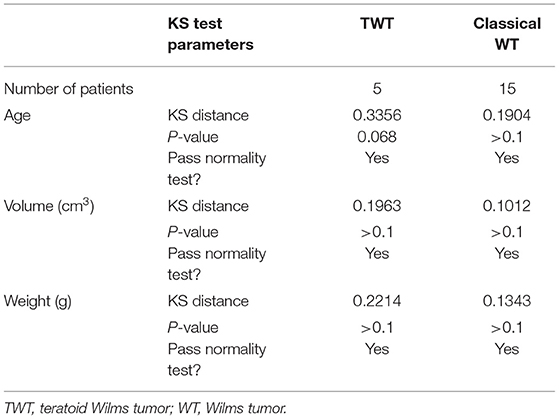
Table 3. Normality tests for quantitative data by Kolmogorov-Smirnov analysis.
Statistical Assessment
To analyze all outcomes in the present exploration, R v 3.6.3 was employed. Data are given as means with SD for continuous variables, while categorical numbers are given as numbers and percentages. Normality was assessed via the Kolmogorov-Smirnov assessment, with data being scrutinized via Fisher's exact assessment and Student's t-tests as appropriate. A two-sided p < 0.05 was the threshold of significance. Event-free survival (EFS) and overall survival (OS) curves were generated using the Kaplan-Meier method and compared using the log-rank test, using the R system for Windows (version 3.5.4). We selected all of the quantitative data from the manuscript, such as age, tumor volume, and weight, and finally found that these data pass the normality test.
Literature Review
The PubMed/NCBI database was employed to execute a literature review using the search terms “teratoid nephroblastoma” and “TWT”, leading to the identification of 62 cases reported previously (6–41) in addition to the five cases discussed in this article (Table 4). Overall, the ages and clinical findings reported for patients with TWT are similar to those for classical patients with WT. Both girls and boys exhibited an average age of 3.1 years (range: 3 months−11 years) at diagnosis, with initial presenting symptoms, i.e., abdominal pain and an abdominal mass. At the time of presentation, 30 cases demonstrated stage I/II disease, 10 cases demonstrated stage III disease with involvement of the local lymph node, seven exhibited stage IV disease, and 15 exhibited stage V bilateral disease. A total of five patients exhibited hypertension at the time of presentation, while eight exhibited congenital abnormalities, such as horseshoe kidneys, inguinal hernias, clubbed feet, Beckwith-Wiedemann syndrome, bilateral cryptorchidism, and an ectopic ureteropelvic system. In nearly whole cases, these TWT masses exhibited favorable histological findings (Table 5).

Table 4. Published teratoid Wilms tumor cases.
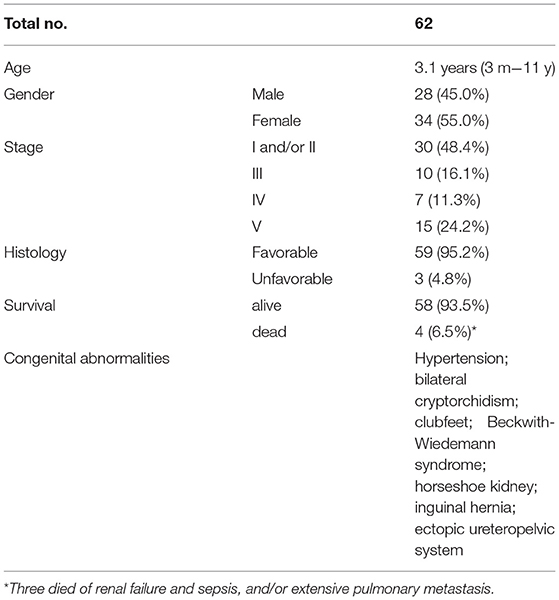
Table 5. Summary of teratoid Wilms tumor cases in the published literature.
Results
Patient Demographics, Presentation, and Prognosis
Of the 67 patients with WT in our hospital, five patients (7.64%) were diagnosed with TWT. A 1:3 matching ratio was used when comparing data between TWT and classical WT cases to reduce the risk of bias. There were no significant differences in clinicopathological variables when comparing patients with TWT and WT, although there was a clear trend toward an increase in tumor size for patients with TWT as compared to patients with WT (203.30 ± 109.89 vs. 104.30 ± 66.97 cm3), despite similar tumor weight values in both groups (471.00 ± 80.65 vs. 432.67 ± 109.25). Further details regarding patient demographics, clinical characteristics, and prognostic outcomes are given in Table 2.
As to EFS and OS (62 cases with non-TWT and five cases with TWT), EFS for TWT was 80% (4/5 patients; follow-up: 6–128 months, median: 97.5 months) vs. 95% (59/62 patients; follow-up: 5–161 months, median: 76.5 months) for non-TWT. The OS for TWT was 100% (5/5 patients; follow-up: 6–128 months, median: 97.5 months) vs. 98.4% (61/62, follow-up: 5–161 months, median: 76.5.5 months) for non-TWT (Figure 1).
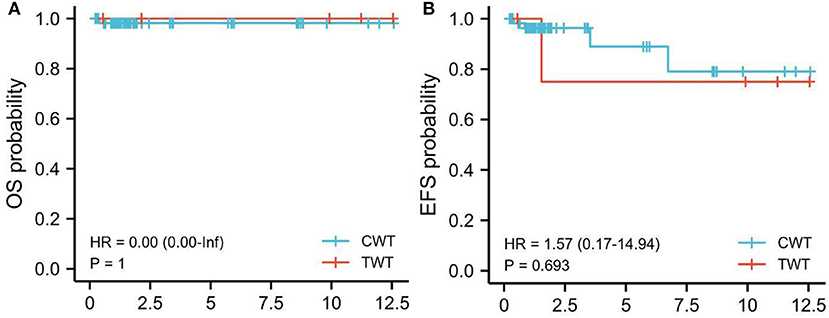
Figure 1. Kaplan-Meier survival analysis. EFS, TWT vs. non-TWT (CWT). Kaplan-Meier survival analysis. (A) OS, TWT vs. non-TWT (CWT). (B) EFS, TWT vs. non-TWT. Red line: TWT; Blue line: non-TWT. TWT, teratoid Wilms tumor; WT, Wilms tumor.
Patient With TWT Clinical Details
Case 1
The first case was a 4-month-old boy with a right-sided abdominal mass that had been evident for 1 month. Routine preoperative analyses of serum creatinine, urea, blood urea nitrogen, and alpha-fetoprotein levels were within normal ranges. He underwent nephrectomy, yielding a sample weighing <350 g (mass weight ≈100 g) that was histologically favorable and diagnosed as a stage I tumor. Immunohistochemical staining revealed the cells of the tumor to be negative for the WT1 tumor suppressor gene. The patient was discharged without undergoing preoperative or postoperative chemotherapeutic treatment, and 9 years after initial presentation remains alive and had no recurrent disease.
Case 2
A 26-month-old male with a 2-week history of a distended abdomen was associated with a large abdominal mass presented. Upon physical assessment, a hard mass was palpable in the right flank without any associated tenderness. Chest and cerebral CT and bone marrow biopsy did not reveal any conclusive findings. Intraoperatively, the right kidney was found to be almost fully replaced by a tumor enveloped by a smooth capsule. No pieces of evidence of vascular invasion or inferior vena cava involvement were detected. The size of the collected nephrectomy specimen was 9 cm × 7 cm × 5 cm and its weight was 540 g. Histological examination revealed this tumor to consist of many mature epithelial glandular elements and smooth muscle elements exhibiting squamous and goblet cell-like differentiation. In addition, admixed regions of classical embryonal epithelial, blastemal, and stromal elements were evident. These cells of the tumor exhibited WT1 positivity. The patient underwent four cycles of postoperative chemotherapeutic treatment with Ifosfamide, Etoposide, and Vincristine and remained alive and disease-free as of 9 years after the surgical treating.
Case 3
A 20-month-old female with anemia (hemoglobin: 95 gm/dl) and a right-sided renal tumor was admitted to the hospital following diagnosis at a separate hospital via needle biopsy and exhibited metastatic stage IV disease at the time of diagnosis with bone marrow and pulmonary metastases. The patient had undergone four cycles of preoperative chemotherapeutic treatment with Ifosfamide, Etoposide, and Vincristine and exhibited a 50% reduction in renal tumor size after 8 weeks as measured via abdominal CT. Four weeks later, nephrectomy was performed, and neither tumors were observed within intrarenal vessels nor were lymph node metastases detected. An ovoid cystic-solid mass (9 cm × 10 cm × 9 cm) with adherent renal tissue on either side was observed, with this tumor being primarily composed of heterologous tissues (cartilage, lack of keratin, muscle, adipose, or lace-like osteoid) together with regions that were histologically consistent with classic WT (blastemal, epithelial, and stromal). Immunohistochemical analyses revealed these tumor cells to be positive for WT1. After surgery, the patient underwent four cycles of treatment with Ifosfamide, Etoposide, and Vincristine. However, pelvic implantation metastasis was observed via abdominal CT at the 6-month follow-up time point. The patient remains alive 2 years after the surgical treating (Figure 2).
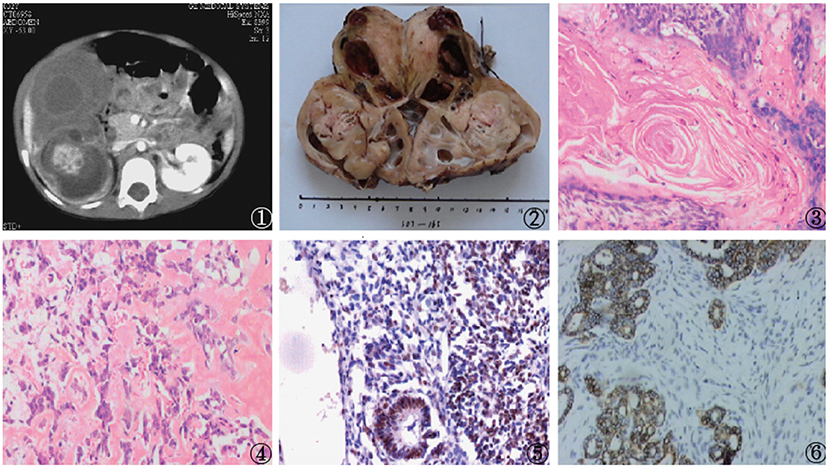
Figure 2. Findings from a 20-month-old girl with a right renal tumor presenting to our hospital with a background of anemia. (1) Abdominal CT scan exhibiting a large mass that had almost fully replaced the right kidney; Nephrectomy samples demonstrating the large mass that had replaced the kidney; (3–5) Histologic examination of the mass revealed a heterologous epithelium with squamous epithelial, adipose, and calcified bead characteristics (H and E, original magnification × 40); (6) Positive WT-1 immunostaining in intratumoral tubules.
Case 4
The next case was a 38-month-old female with a 1-week experience of abdominal pain, and abdominal ultrasonography revealed a solid right renal tumor (7 cm × 8 cm × 9 cm) exhibiting microcalcification. Abdominal CT scans demonstrated the presence of a large heterogeneous mass in the right renal posterior region (8 cm × 8 cm × 9 cm, Figure 3), with this tumor extending further on the cortex without crossing over the midline. The patient did not exhibit any evidence of bone marrow, pulmonary, or abdominal metastases and underwent right nephrectomy, yielding a 350 g mass. Histological analyses revealed this tumor to harbor regions with classical WT-like morphology admixed with regions of keratinized squamous cells in the form of discrete epidermoid cyst-like nodules composing >70% of the tumor volume. The patient underwent four cycles of postoperative chemotherapeutic treatment and was discharged from the hospital 10 years previously. The patient remains in good health without relapse (Figure 4).
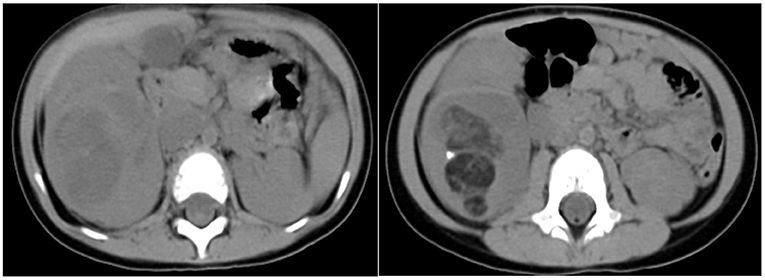
Figure 3. Findings in a 38-month-old girl presenting with abdominal pain for 1 week. Contrast-enhanced abdominal CT images revealed a heterogeneous mass in the middle and lower portions of the right kidney (8 cm × 8 cm × 9 cm) with imaging findings consistent with fat or hair.
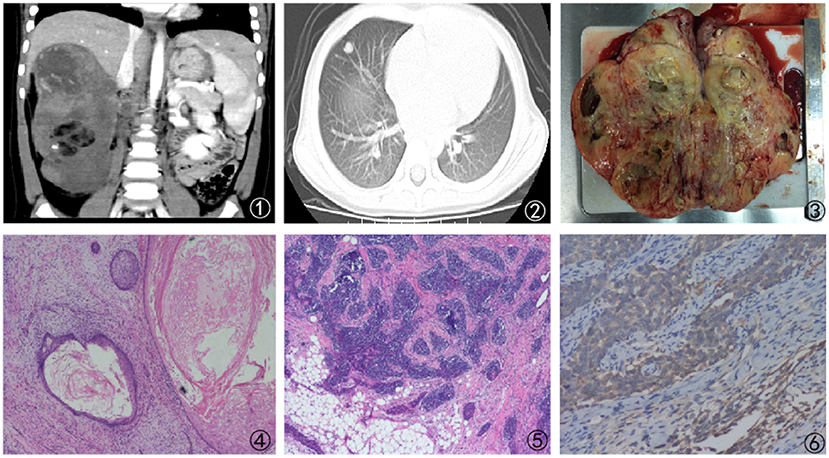
Figure 4. Findings from a 38-month-old girl presenting with abdominal pain for 1 week. (1) Abdominal CT scan revealing a large mass that had almost fully replaced the right kidney; (2) Lung metastases as revealed by the presents of a large lesion in the right lung; (3) Cross-sectional surface of the kidney, exhibiting a large mass that had largely replaced the normal tissue compartment; (4, 5) Imaging findings revealing prominent heterologous components that include squamous epithelial cells; (6) Positive WT-1 immunostaining in intratumoral tubules.
Case 5
A 3-year-old girl came to our hospital with a left-sided abdominal mass that had been present since 2 months of age and a low-grade fever that had been present for 2 weeks. The blood pressure was normal at the time of presentation. In the left lumbar region, an 8 cm3 × 9 cm3 × 9 cm3 mass was palpable. This mass did not cross the midline and was not correlated with any other abnormalities. Urinary analyses revealed evidence of microscopic hematuria and sterile culture findings. Hemoglobin levels of the patient were 8.7 gm/dl. Renal and chest radiographic findings were normal, as were liver function tests. Abdominal ultrasonography revealed a large heteroechoic mass (10 cm × 10 cm × 9 cm) extending from the lower pole of the left kidney. Right kidney of the patient was normal, and no blood vessels were observed. Contrast-enhanced abdominal CT scans confirmed the presence of a heterogeneous mass (10 cm × 10 cm × 9 cm) extending from the middle and lesser poles of the left kidney. Good excretion of contrast with splaying of pelvicalyceal system was seen. There was no enlargement of local lymph nodes. Laparotomy was performed via a supraumbilical transverse transperitoneal incision, and a left radical nephrectomy was conducted. Examination of the contralateral kidney revealed it to be normal. The patient was doing well as of a 6-month follow-up.
Discussion
One of the most prevalent forms of primary renal tumors in children is WT. These tumors exhibit characteristic features including efforts to recapitulate various stages of the nephrogenic process, and harbor blastemal, stromal, and epithelial cell types in a triphasic combination. In some cases, heterotopic mesodermal elements are also evident, representing a small proportion of the overall tumor. Atypical renal tumor times, i.e., TWT, clear cell kidney sarcomas, rhabdoid kidney tumors, multilocular cystic nephromas, and renal teratomas, are relatively rare (35). TWT tumors exhibit the characteristics of a diverse array of cell categories and tissues in addition to areas with classical nephroblastoma characteristics, and are diagnosed when >50% of the solid tumor exhibits the clear predominance of teratoid elements. In previously published reports, these TWTs have been reported to include a range of elements, such as neurological tissues, skeletal muscle, mucinous epithelial tissue, adipose tissue, squamous epithelial cysts, and cartilage (27, 28). Herein, differences in the volume of the tumor were not significant, in large part because of the very small size and n SD values in the TWT group. However, tumor volumes were substantially larger for patients with TWT relative to those in classical patients with WT, despite similar tumor weight values in both groups. This thus suggests that the density of TWT tumors may be lesser compared to classical WT tumors, potentially owing to the unique adipose tissue and squamous epithelial cysts present within TWTs.
Wilms tumor can arise as a consequence of abnormal genetic changes in the 11p13 (WT1) and 11p15 (WT2) regions of the short arm of chromosome 11. In nine described TWT cases identified to date, monosomy 11 or 11p deletion has been detected (35). The WT1 gene encoded in the 11p13 region is composed of 10 exons spread over a 50 kb region that gives rise to a 3 kb mRNA. The carboxyl-terminal region of WT1 (exons 7–10) consists of four zinc finger motifs that give rise to a DNA-binding domain. This domain enables WT1 to bind to corresponding promoter sequences in downstream target genes, and mutations within this domain can alter this DNA-binding activity to adversely impact WT1 function (10). Ghamdi et al. reported on the presence of germline WT1 gene mutations in children that were predisposed to WT in a mutational analysis, although they specifically focused on tumor tissue samples. In line with reported WT1 mutation rates in sporadic WT cases, only one of the three cases exhibited a WT1 mutation. There have been few studies of WT1 gene mutations in TWT to date, and available evidence suggests that these findings are comparable to those in classical WT with mutations only being evident in a small subset of patients (36). Interestingly, three of our patients exhibited WT1 positivity in immunohistochemical staining assays, suggesting WT1 may offer potential value in this context, although further genetic analyses will be needed to further clarify this issue.
In general, TWT is considered to be a relatively non-aggressive tumor type to be relatively chemoresistant and radioresistant owing to its differentiated teratomatous composition. However, it is possible that these assumptions may be inaccurate. Treatment strategies for TWT have yet to be determined owing to the scarcity of this tumor type and its varied composition. To date, four patients with TWT who have died have been reported to date, with two dying of metastatic disease whereas the third died of renal failure and sepsis (7, 37). Regarding the considered cases, two exhibited metastases and one experienced postoperative pelvic recurrence potentially related to needle biopsy. These tumors may thus exhibit aggressive features, such as anaplasia and regional or distant metastasis, with anaplastic features contributing to the risk of such metastasis and associated mortality. To further establish the relative malignancy of TWT, further research is warranted in an effort to establish more appropriate and reliable treatment guidelines.
This study has several limitations. A relatively small sample size of children with TWT, patients originating from one hospital, and a retrospective study design constituted the limitations of this study. Despite these limitations, this was the first study to evaluate TWT in the context of WT, which may provide new insights in a previously unknown continent.
Conclusion
In summary, our patients with WT are the first reported cases of TWT in Chinese children to date. These preliminary results suggest that the tumors in patients with TWT may be less dense than those in patients with classical WT. Based on these findings, we recommend the treatment of this rare tumor type with specific protocols. There is additionally a clear need to conduct more basic research aimed at clarifying the biological nature of this tumor type and to identify appropriate theranostic markers that can be leveraged to optimize individualized treatment strategies for affected patients. Genetic studies also have the potential to clarify whether there are specific biomarkers associated with this cancer type. Owing to the highly differentiated and varied nature of these tumors, however, they are likely to respond poorly to chemotherapy, although we believe that surgical treatment alone is unsatisfactory as a means of treating this scarce tumor type.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Shanghai Children's Hospital, Shanghai Jiao Tong University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
WW and YW contributed to the study design. JL and WX contributed to data collection. WW and JL contributed to the analysis of data. WW and ZL contributed to manuscript writing. All authors approved the submitted version.
Funding
This study was funded by the Shanghai Health Committee (20214Y0476).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the Department of General Surgery, Shanghai Children's Hospital, and Shanghai Jiao Tong University for their support.
Abbreviations
WT, Wilms tumor; TWT, teratoid Wilms tumor.
References
1. Hol JA, Jewell R, Chowdhury T, Duncan C, Nakata K, Oue T, et al. Wilms tumour surveillance in at-risk children: Literature review and recommendations from the SIOP-Europe Host Genome Working Group and SIOP Renal Tumour Study Group. Eur J Cancer. (2021) 153:51–63. doi: 10.1016/j.ejca.2021.05.014
2. Cone EB, Dalton SS, Van Noord M, Tracy ET, Rice HE, Routh JC, et al. Biomarkers for Wilms tumor: a systematic review. J Urol. (2016) 196:1530–5. doi: 10.1016/j.juro.2016.05.100
4. Dome JS, Perlman EJ, Graf N. Risk stratification for Wilms tumor: current approach and future directions. Am Soc Clin Oncol Educ Book. 2014, 215–23. doi: 10.14694/EdBook_AM.2014.34.215
5. Treger TD, Chowdhury T, Pritchard-Jones K, Behjati ST. genetic changes of Wilms tumour. Nat Rev Nephrol. (2019) 15:240–51. doi: 10.1038/s41581-019-0112-0
6. Variend S, Spicer RD, Mackinnon AE. Teratoid WT. Cancer. (1984) 53:1936–42. Available online at: 10.1002/1097-0142(19840501)53:9<1936::aid-cncr2820530922>3.0.co;2-w
7. Fernandes ET, Parham DM, Ribeiro RC, Douglass EC, Kumar AM, Wilimas JT. Teratoid Wilms tumor: the St Jude experience. J Pediatr Surg. (1988) 23:1131–4. doi: 10.1016/S0022-3468(88)80328-2
8. Beckwith JB. Wilms' tumor and other renal tumors of childhood: a selective review from the national Wilms' tumor study pathology center. Hum Pathol. (1983) 14:481–92. doi: 10.1016/S0046-8177(83)80003-3
9. Vujanic GM. Renal tumours of childhood: an overview. Diagn Histopathol. (2009) 15:5019. doi: 10.1016/j.mpdhp.2009.08.002
10. Al-Hussain T, Ali A, Akhtar M. Wilms tumor: an update. Adv Anat Pathol. (2014) 21:166–73. doi: 10.1097/PAP.0000000000000017
11. Vujanic GM. Teratoid WT: report of a unilateral case. Pediatr Pathol. (1991) 11:303–9. doi: 10.3109/15513819109064767
12. Magee JF, Ansari S, McFadden DE, Dimmick J. Teratoid Wilms' tumour: a report of two cases. Histopathology. (1992) 20:427–31. doi: 10.1111/j.1365-2559.1992.tb01014.x
13. Kotiloglu E, Kale G, Sevinir B, Akçören ZT. Teratoid WT. A unilateral case. Tumori. (1994) 80:61–3. doi: 10.1177/030089169408000112
14. Williams MA, Schropp KP, Noe HN. Fat containing renal mass in childhood: a case report of teratoid Wilms tumor. J Uro. (1994) 151:1662–3. doi: 10.1016/S0022-5347(17)35339-9
15. Ashworth MT, Pizer BL, Oakhill A, Berry PJ. A teratoid WT with raised serum alpha-fetoprotein level. Pediatr Pathol Lab Med. (1996) 16:853–9. doi: 10.1080/107710496175462
16. Paterson A, Sweeney LE. Teratoid Wilms' tumour occurring synchronously with classical Wilms' tumour in Beckwith Wiedemann syndrome. Pediatr Radiol. (2000) 30:656–7. doi: 10.1007/s002470000249
17. Karaca I, Sencan A, Ortaç R, Mir ET. Teratoid WT: A case report. Turk J Pediatr. (2000) 42:242–5.
18. Bakshi N, Mansoor I, Venkataramu NK, Katariya S. An unusual renal malignancy of childhood: Unilateral teratoid WT. Pediatr Pathol Mol Med. (2003) 22:435–41. doi: 10.1080/pdp.22.5.435.441
19. Inoue M, Uchida K, Kohei O, Nashida Y, Deguchi T, Komada Y, et al. Teratoid WT: A case report with literature review. J Pediatr Surg. (2006) 41:1759–63. doi: 10.1016/j.jpedsurg.2006.05.045
20. Cecchetto G, Alaggio R, Scarzello G, Dall'Igna P, Martino A, Bisogno G, et al. Teratoid WT: Report of a unilateral case. J Pediatr Surg. (2003) 38:259–61. doi: 10.1053/jpsu.2003.50059
21. Myers JB, Dall'Era J, Odom LF, McGavran L, Lovell MA, Furness P III. Teratoid WT, an important variant of nephroblastoma. J Pediatr Urol. (2007) 3:282–6. doi: 10.1016/j.jpurol.2006.11.004
22. Koksal Y, Varan A, Akyuz C, Kale G, Büyükpamukçu N, Büyükpamukçu M. Teratoid WT in a child. Pediatr Int. (2007) 49:414–7. doi: 10.1111/j.1442-200X.2007.02358.x
23. Gupta DK, Sharma S, Agarwal S, Carachi R. Saga of Wilms' tomor: lessons learnt from the past. J Indian Assoc Pediatr Surg. (2005) 10:217–28. doi: 10.4103/0971-9261.19271
24. Ibrahim OOK, Abdur-Rahman LO, Nasir AA, Folaranmi OO, Abdulmajeed AA, Nwosu DC, et al. Bilateral teratoid Wilms tumour: a report of a 16-month old female child in Ilorin. AMR. (2016) 5:63.
25. Seo J, Yeon-Lim Suh YL, Choi HY. Adult teratoid WT with prominent neuroepithelial, differentiation. Pathol Int. (2009) 59:44–8. doi: 10.1111/j.1440-1827.2008.02323.x
26. Kajbafzadeh A, Tourchi A, Elmi A, Sadeghi Z, Ramyar A, Mahjoob FT. Teratoid WT with hypertension treated with partial nephrectomy: case report with literature review. Eur J Pediatr Surg. (2010) 20:270–2. doi: 10.1055/s-0029-1242730
27. Parikh B, Trivedi P, Shukla K. A unilateral teratoid WT with raised serum alpha-fetoprotein level. Indian J Pathol Microbiol. (2007) 50:317–9.
28. Sultan I, Ajlouni F, Al-Jumaily U, Al-Ashhab M, Hashem H, Ghandour K, et al. Distinct features of teratoid Wilms tumor. J Pediatr Surg. (2010) 45:e13–9. doi: 10.1016/j.jpedsurg.2010.06.035
29. Mukhopadhyay B, Shukla RM, Mukhopadhyay M, Mandi S, Roy D, Bhattacharya MK. Teratoid Wilms'tumor-A rare renaltumor. Urol Ann. (2011) 3:155–7. doi: 10.4103/0974-7796.84959
30. Treetipsatit J, Raveesunthornkiet M, Ruangtrakool R, Sanpakit K, Thorner PST. Wilms'tumor: case report of a rare variant that can mimic aggressive biology during chemotherapy. J Pediatr Surg. (2011) 46:e1–6. doi: 10.1016/j.jpedsurg.2011.09.049
31. Bardesi JH, Al-Sayyad AJ. Teratoid Wilms tumor in a child: a case report. Uro Today International Journal. (2012) 5:5–7. doi: 10.3834/uij.1944-5784.2012.02.08
32. Yadav YK, Sharma U, Gupta K, Arora R. Squamous predominant Wilms'tumor. J Lab Physicians. (2012) 4:50–2. doi: 10.4103/0974-2727.98675
33. Ramani M, Geetha K, Ramesh Reddy K, ahoor AR, Rani CS. A rare Wilms'tumor with teratoid differentiation in a 3 month old male child -a case report. J Evol Med Denta Sci. (2013) 2:4161–5. doi: 10.14260/jemds/816
34. Pawel BR, Chadsrevian JP, Smergel EM, Weintraub WHT. Teratoid Wilms tumor arising as a botryoid growth within a supernumerary ureteropelvic structure. Arch Pathol Lab Med. (1998) 122:925–8.
35. Chandra MK, Madhumita M, Shibsankar B, Halder P, Mukhopadhyay B, Kumar R. Uncommon renal tumors in children: A single center experience. J Indian Assoc Pediatr Surg, 21, 61–5. doi: 10.4103/0971-9261.176940
36. Ghamdi DA, Bakshi N, Akhtar M. Teratoid Wilms tumor: report of three cases and review of the literature. Turk Patoloji Derg. (2019) 35:61–8. doi: 10.5146/tjpath.2016.01363
37. Ellen D'H, William M, Vujanić GM. “Teratoid” Wilms tumor: the extreme end of heterologous element differentiation, not a separate entity. Am J Surg Pathol. (2019) 43:1583–90. doi: 10.1097/PAS.0000000000001335
38. Garje MU, Sakthivel V, Dhaimbekar PP, Rathod SG. Teratoid WT of kidney with neural tissue predominant: case report with review of literature. J Family Med Prim Care. (2019) 8:3447–9. doi: 10.4103/jfmpc.jfmpc_558_19
39. Green DM. The treatment of stages I-IV favorable histology WT. J Clin Oncol. (2004) 22:1366–72. doi: 10.1200/JCO.2004.08.008
40. Irtan S, Ehrlich PF, Pritchard-Jones K. Wilms tumor: “State-of-the-art” update, 2016. Semin Pediatr Surg. (2016) 25:250–6. doi: 10.1053/j.sempedsurg.2016.09.003
Keywords: Wilms tumor, teratoid, nephroblastoma, prognosis, histopathology
Citation: Wu W, Wu Y, Xu W, Liu J and Lv Z (2022) Teratoid Wilms Tumor and Classical Wilms Tumor: A Retrospective 10-Year Single-Center Study and Literature Review. Front. Surg. 8:781060. doi: 10.3389/fsurg.2021.781060
Received: 22 September 2021; Accepted: 24 December 2021;
Published: 02 February 2022.
Edited by:
Ezekiel E. Young, University at Buffalo, United StatesReviewed by:
Roberto Iglesias Lopes, Hospital for Sick Children, CanadaMatthieu Peycelon, Hôpital Robert Debré, France
Copyright © 2022 Wu, Wu, Xu, Liu and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhibao Lv, bHZ6aGliYW8mI3gwMDA0MDt5ZWFoLm5ldA==