
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Surg. , 18 June 2021
Sec. Thoracic Surgery
Volume 8 - 2021 | https://doi.org/10.3389/fsurg.2021.688236
This article is part of the Research Topic Case Reports in Thoracic Surgery: 2021 View all 11 articles
 Alessandra Mazzucco1*
Alessandra Mazzucco1* Eleonora Poirè1Andrea Leporati1Matteo Chiari1
Eleonora Poirè1Andrea Leporati1Matteo Chiari1 Laura Moneghini2Giorgio Ghilardi1
Laura Moneghini2Giorgio Ghilardi1 Alessandro Baisi1
Alessandro Baisi1Introduction: Primary pulmonary paraganglioma is a rare tumor with few cases reported in literature and unspecific clinical presentation.
Case Presentation: A 49-year-old woman presented to our department with an incidental finding of a pulmonary mass at chest X-ray and no associated clinical symptom. The CT scan and the FDG-PET showed mild uptake of contrast, but a definitive diagnosis was only possible after surgery through histopathological examination.
Conclusion: Paragangliomas originating in the pulmonary tissue are generally non-functioning masses discovered incidentally in otherwise asymptomatic patients. Surgery appears to be the best treatment option, with only radiologic follow-up necessary afterwards.
Primary pulmonary paragangliomas are rare neuroendocrine tumors with few cases reported in the literature. Generally, affected patients show no symptoms and usually discover the mass incidentally during unrelated medical examinations. We report a case of primary pulmonary paraganglioma in an asymptomatic 49-year-old woman.
A 49-year-old Caucasian woman, an active smoker (15 packs/year) with an otherwise silent past medical history, presented with a dry cough that worsened in the supine position. While the cough resolved with proton-pump inhibitor (PPI) therapy, suggesting a gastrointestinal nature of the symptom, the patient also underwent a routine chest radiograph. The imaging showed a nodule with a diameter of 3.5 cm in the right lower lobe. Thus, a chest CT scan was performed, which confirmed the presence of a solid lesion with well-defined margins, mild contrast enhancement, and a diameter of 34 × 26 mm in the anterior basal segment of the inferior right pulmonary lobe. The exam also revealed an enlarged axillary lymph node that was later confirmed to be inflammatory in nature. To further characterize the lesion, a PET-CT scan with an injection of 18F-fluorodeoxyglucose (FDG) was done; the images showed mild uptake at the level of the nodule in the right lower lobe (with a maximum SUV of 2.8) (Figures 1, 2), suggesting a possibly benign or locally invasive biological behavior. Fine-needle biopsy for typing of the lesion was attempted but ultimately not performed due to poor compliance of the patient during the CT-guided procedure. Therefore, wedge resection of the right inferior lobe and nodal sampling were performed with video-assisted thoracoscopic surgery. A frozen section procedure was performed during the operation, which gave unconclusive results. Hence, it was decided not to perform a completion lobectomy but to wait for the final histological results. The tissue specimen analyzed at histology showed richly vascularized intrapulmonary solid proliferation (Figure 3A) that comprised two types of cells: epitheliomorphic and spindle-shaped cells. The former type of cells had large eosinophilic cytoplasm and moderately atypical nuclei (nucleolates) partially dispersed in a loose stroma crossed by ill-defined septa (Figure 3B); the latter cells, also called “sub-tentacular cells,” were interposed with moderate infiltrate of lympho-plasma cells (Figure 3C). No necrosis was found in the sections examined, and the mitotic index was <1 mitosis for 10 high-magnification fields (10 HPF, 40X). At immunocytochemistry, all cellular elements showed a strong positive reaction for synaptophysin (Syn) and neuron-specific enolase (Figure 3D), while there were only areas of positivity to the S-100 protein in correspondence of the sub-tentacular elements. The dissected lymph nodes were negative. The final diagnosis was a primary pulmonary paraganglioma. The postoperative course was uneventful, with the thoracic drainage removed on the third postoperative day (POD) and no signs of pneumothorax on the chest radiograph performed afterward. The patient was discharged from the hospital on the fourth POD. At the latest checkup, 1 month after the hospital discharge, she showed no sign of relapse on the chest radiograph. After multidisciplinary discussion with the Oncology, Radiology, and Pneumology Departments, it was decided to proceed with radiologic follow-up at 3-month intervals for the first semester with a chest CT scan, then in 6 months for the following year, and later maintain a 1-year radiologic follow-up either with chest radiographs or a CT scan. The main limitation on deciding the timing of the follow-up was the scarcity of available literature on both the treatment and the recurrence rates; however, the available reports seem to suggest an indolent nature of this tumor with an unlikely tendency to recur, which is what informed about our decision on not performing completion of a lobectomy.
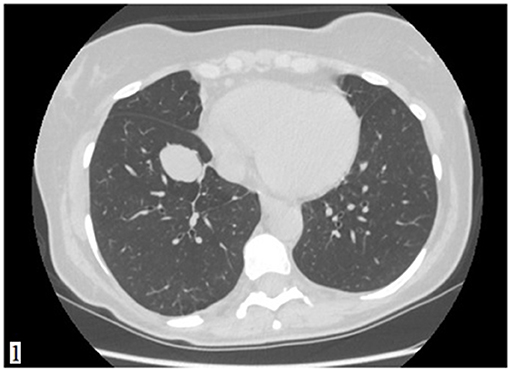
Figure 1. A CT scan showing a solid lesion with well-defined margins in the anterior segment of the inferior lobe of the right lung.
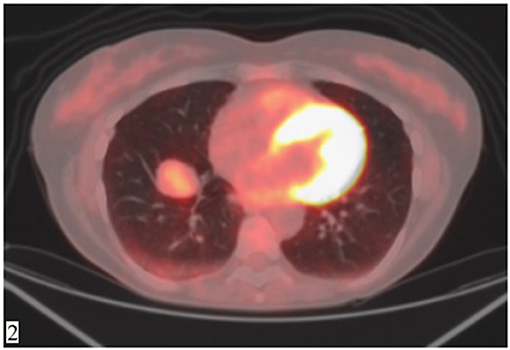
Figure 2. FDG-PET showing mild contrast uptake at the level of the nodule (SUV max 2.8).
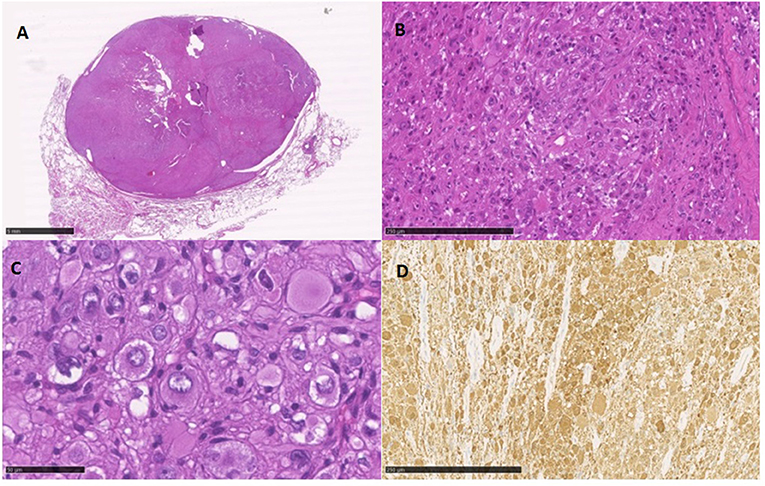
Figure 3. (A) low magnification view of the solid, well-defined intrapulmonary neoformation; (B) cellular growth is limited by the surrounding fibrous septae; (C) the epitheliomorphic ganglion elements are clearly evident with the interposition of some sub-tentacular cells and lymphocytes; (D) largely positive immunocytochemical staining for neuron-specific enolase.
Paragangliomas are rare neuroendocrine tumors that arise from the chromaffin cells of the extra-adrenal autonomic paraganglia (1). Their incidence is largely unknown, as they tend to be described in the literature mostly in association with pheochromocytomas; however, the incidence of these tumors combined has been estimated to be about two to eight cases/million a year (2, 3).
Most paragangliomas appear to be sporadic (4), with the majority of patients affected being females and middle-aged (5). Paragangliomas can be associated with either sympathetic or parasympathetic cells; the former are usually “functioning tumors,” as they secrete catecholamines, while the latter have a tendency to be “non-functioning.” Catecholamine-secreting paragangliomas often present with symptoms similar to those of pheochromocytomas (1) (i.e., diaphoresis, headache, and hypertension), whereas non-functioning tumors are silent and are discovered incidentally.
Extra-adrenal paragangliomas are more commonly silent, and if they do present symptoms, these are mostly related to the mass effect at the site of the tumor. Primary pulmonary paragangliomas are rare even among the extra-adrenal paragangliomas, with <30 cases reported in literature (5). The majority of reported patients were asymptomatic and had a nodule discovered during imaging studies of the chest (6). In a minority of cases, there were symptoms consistent with the localization of the mass, such as cough or chest pain (5).
The differential diagnosis of primary pulmonary paraganglioma should include many different conditions, including pulmonary carcinoma, pulmonary tuberculosis, pulmonary mycosis, either round or organizing pneumonia, inflammatory pseudo-tumors, and metastatic lung cancer (5).
Paragangliomas are best evaluated at a contrast-enhanced CT scan, where they show enhancement in the arterial phase (7). However, poor enhancement at a CT scan does not necessarily exclude the diagnosis, but it may be suggestive of a more benign lesion, as it has been reported that the degree of vascularization in non-functioning paragangliomas is not always as rich (5). Silent paragangliomas tend to show mild FDG uptake at a PET scan (5, 7, 8).
These tumors are slow growing and usually benign; however, because they do have a tendency for expansive growth and there are cases reported of low-grade malignant lesions (5, 9), surgical excision, when possible, is the preferred treatment.
In regard to the primary pulmonary paraganglioma, patients have mostly been treated surgically, with either local excision or a lobectomy, depending on the extension of the lesion. No recurrence or metastatic disease has been reported in any of the known cases even though no chemo or radiotherapy was performed afterwards (5, 7). It appears that only 10% of pheochromocytomas and paragangliomas are malignant (4), but there are no biochemical or histological examinations that can reliably predict the tendency of these tumors, and, therefore, surgery remains the only approach to treatment combined with radiologic follow-up.
This report presents a case of a primary pulmonary paraganglioma, a rare tumor with only a few cases reported in the literature (5, 7, 8). As the patients are mostly asymptomatic, these tumors are generally discovered incidentally. Surgical excision seems to be the best approach to treatment, followed by radiologic follow-up and no further pharmacological therapy.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
All authors participated equally in the case and in the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. (2019) 381:552–65. doi: 10.1056/NEJMra1806651
2. Stenström G, Svärdsudd K. Pheochromocytoma in Sweden 1958–1981. An analysis of the National Cancer Registry Data. Acta Med Scand. (1986) 220:225–32. doi: 10.1111/j.0954-6820.1986.tb02755.x
3. Santos P, Pimenta T, Taveira-Gomes A. Hereditary Pheochromocytoma. Int J Surg Pathol. (2014) 22:393–400. doi: 10.1177/1066896914537683
4. Brink I, Hoegerle S, Klisch J, Bley TA. Imaging of pheochromocytoma and paraganglioma. Fam Cancer. (2005) 4:61–8. doi: 10.1007/s10689-004-2155-y
5. Zhang JJ, Liu T, Peng F. Primary paraganglioma of the lung: a case report and literature review. J Int Med Res. (2012) 40:1617–26. doi: 10.1177/147323001204000442
6. Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. (2001) 86:5210–6. doi: 10.1210/jcem.86.11.8034
7. Fiorentino G, Annunziata A, De Rosa N. Primary paraganglioma of the lung: a case report. J Med Case Rep. (2015) 9:166. doi: 10.1186/s13256-015-0639-z
8. Huang X, Liang QL, Jiang L, Liu QL, Ou WT, Li DH, et al. Primary pulmonary paraganglioma: a case report and review of literature. Medicine. (2015) 94:e1271. doi: 10.1097/MD.0000000000001271
Keywords: paraganglioma, thoracic surgery, VATS, lung nodule, rare tumor, case report
Citation: Mazzucco A, Poirè E, Leporati A, Chiari M, Moneghini L, Ghilardi G and Baisi A (2021) An Unusual Histology for a Lung Nodule: A Case Report of Primary Pulmonary Paraganglioma. Front. Surg. 8:688236. doi: 10.3389/fsurg.2021.688236
Received: 30 March 2021; Accepted: 27 May 2021;
Published: 18 June 2021.
Edited by:
Nuria Maria Novoa, University of Salamanca Health Care Complex, SpainReviewed by:
Savvas Lampridis, 424 General Military Hospital, GreeceCopyright © 2021 Mazzucco, Poirè, Leporati, Chiari, Moneghini, Ghilardi and Baisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Mazzucco, YWxlc3NhbmRyYS5tYXp6dWNjb0Bob3RtYWlsLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.