 Corey T. Walker
Corey T. Walker Phillip A. Bonney
Phillip A. Bonney Nikolay L. Martirosyan
Nikolay L. Martirosyan Nicholas Theodore
Nicholas Theodore- Department of Neurosurgery, St. Joseph’s Hospital and Medical Center, Barrow Neurological Institute, Phoenix, AZ, USA
Adult spinal disorders are a significant cause of morbidity across the world and carry significant health and economic burdens. Genetic predispositions are increasingly considered for these conditions and are becoming understood. Advances in molecular technologies since the mid-1990s have made possible genetic characterizations of these diseases in many populations, and recent findings have provided insight into the underlying pathophysiologic mechanisms. These studies have made clear the genetic heterogeneity producing clinical phenotypes and suggest that individualized treatments are possible in the future. We review the genetics and heritability of cervical spondylotic myelopathy and ossification of the posterior longitudinal ligament and perform a systematic review of the genetics of adult lumbar degenerative scoliotic deformity, highlighting recent discoveries and the potential for personalized future therapeutics for these patients.
Introduction
Degenerative diseases of the spine affect the majority of individuals over a lifetime, causing pain and neurologic dysfunction, and presenting a significant challenge to physicians. These clinical entities develop from a complex interplay of genetic and environmental factors that are incompletely understood. Patients with a given disease may appear similar radiographically, yet outcomes may be disparate regarding disease progression and responses to medical, rehabilitative, or surgical interventions. Although there is a role for surgery in certain conditions, patient selection must be carefully considered, given the risks associated with surgery. Better stratification of patients is needed to guide treatments.
Advances in molecular technologies over the last 15 years have provided much insight into the genetics of numerous diseases, including those of the spine. Candidate gene approaches and genome-wide association studies have shed light on underlying pathophysiologic mechanisms at work in the development of such diseases. The potential application of these studies to patient care includes providing a clearer picture of who will benefit from surgical intervention. Furthermore, they offer an exciting opportunity for future therapies targeting specific genetic aberrations that predispose to disease.
Another article in this issue of Frontiers highlights the genetics of intervertebral disc (IVD) disease, which may play a partial role in almost all degenerative spine disorders and, therefore, is not discussed here. Alternatively, we review the genetics of several of the most common spinal disorders, including cervical spondylotic myelopathy (CSM), ossification of the posterior longitudinal ligament (OPLL), and adult scoliosis. Rather than enumerate specific single nucleotide polymorphisms (SNPs) and other genetic anomalies, we highlight the overarching mechanisms uncovered by recent studies and discuss the potential for development of personalized approaches to treating these diseases in the future.
Methods
Literature Review
We performed a systematic review of the literature to evaluate the contributions of current evidence on the genetics of adult degenerative scoliosis (DS). A recent systematic review that evaluated the genetic contributions for CSM and OPLL by Wilson et al. (1) was found and therefore was not performed in this study. The inclusion criteria included the studies comparing genetic variables in humans with this disease. Only studies in the English language were included. MEDLINE was queried with the terms “genetics” and “adult degenerative scoliosis” for articles published from 1966 to September 26, 2016. Studies focusing on other forms of scoliosis were excluded, as were those focusing solely on IVD degeneration. These queries returned 24 studies. The citation information for each result was examined by two of the authors for relevant studies. Six potentially relevant studies were identified, and the abstracts (and, if necessary, full manuscripts) of these studies were reviewed. The references of all reviewed manuscripts were also reviewed to identify other potential studies. Six studies met the inclusion criteria and were the focus of the present study. The studies were published between 2011 and 2015. All of the studies represented Level III evidence (small, non-randomized case–control studies). Results of the included studies were extracted and interpreted by the two reviewing authors.
Cervical Spondylotic Myelopathy
Cervical spondylosis is a nearly ubiquitous finding that occurs with aging as IVD degeneration, ligamentous laxity, facet hypertrophy, and osteophyte formation contribute to narrowing of the spinal canal (2). CSM occurs when neural elements of the spinal cord are compressed. Nevertheless, many patients incidentally show radiographic evidence of spinal cord compression but remain clinically asymptomatic (3). Although the exact reasons for this remain unknown, a potential explanation relates to the dynamic nature of the cervical spine and cord, and that static compression does not correlate exactly with the micropathological changes that occur in this disease (4).
Multiple human and animal studies have implicated various mechanisms in the acute and chronic pathophysiology of CSM (5). Direct mechanical forces result in static and dynamic injuries to neuronal and glial cells (6, 7). This injury is likely paralleled by ischemic changes seen in the disease caused by obstructed spinal cord perfusion and consequent microvascular changes (7–10). Several studies have also suggested a perpetuating cycle of ischemia related to blood–spinal cord barrier breakdown and dysregulation of the neurovascular unit (11, 12). Vascular permeability promotes edema through the release of inflammatory molecules and other potentially cytotoxic proteins into the cord parenchyma (13). This edema may potentiate neuronal damage and play an active role in the chronic degenerative component of the disease (14, 15). Additionally, glutamatergic toxicity (16), free radical-mediated cell injury (17, 18), and apoptosis (19) are also suggested as aggravating secondary injury pathways in the disease.
Heritability of CSM
An appreciation for the genetics of a disease is important for understanding how it is passed from one generation to the next. Although environmental factors undoubtedly play a role in the multifactorial pathogenesis, they allow providers and patients to assess the probability that the patient could develop the disease. Meaningful genealogy, however, is difficult to evaluate, and studies are limited (20). A study by Wilson et al. systematically reviewed the literature documenting the heritability of CSM and OPLL (1). Several authors have suggested genetic susceptibility of cervical spondylosis in twin–twin comparison studies (21–23). However, only one study has successfully used a population-based methodology to show inheritance patterns among non-twins (24). Patel et al. examined a database of 2 million Utah residents and found 486 patients with CSM and compared them with 1000 case controls (24). They used an index measuring genetic distance between pairs of patients to quantify familial clustering and found a statistically significant relationship related to the disease. Moreover, they identified a greater than five times relative risk of developing the disease among first-degree relatives. Studies corroborating these findings in other populations need to be done, but the data suggest heritability among the studied individuals.
Genetics of CSM
As methods for evaluating genomics have evolved, SNPs and proteomics have become easier to evaluate, and the literature regarding their contributions to CSM has grown. Nevertheless, identifying individual components of CSM is difficult as different genes spur degenerative changes that lead to spondylosis but not necessarily myelopathy. For example, Wang et al. have associated two different genetic polymorphisms with CSM (25). First, they identified two polymorphisms of the vitamin D receptor gene (VDR), ApaI and TaqI, which are related to the presence of CSM and the magnetic resonance imaging–based severity of disease in Chinese patients (25). They also found a strong link between CSM and the tryptophan allele (Trp2) of the collagen 9A2 gene, as well as smoking exposure (26). These polymorphisms promote IVD degeneration independently (27–32), a process that can cause central canal stenosis. Neither of these studies delineated how these genetic changes compared in patients with cervical stenosis with and without myelopathy. This also underlines the putative effects of environmental stressors on pathogenesis.
Another genetic relationship has been drawn between apolipoprotein E, a protein that plays a critical role in the repair and regeneration processes of multiple central nervous system diseases. Specifically, the ε4 allele of the apolipoprotein E gene is implicated in impairment of these repair mechanisms. In a study by Setzer and colleagues, 106 patients with radiographic cervical stenosis were collected prospectively, and the ε4 allele was strongly associated with the development of CSM, independent of imaging findings, and other confounders (the allele was not related to the degree of stenosis) (33). The group showed that the allele also had negative effects on treatment outcomes in 60 of the patients who underwent surgical decompression (34). These results suggest that this genetic link portends a worse prognosis both for developing the disease and recovering from it. Large-scale studies evaluating the clinical usefulness of this association are necessary before its application can become widespread. Still, this knowledge may provide clues to the pathogenesis of the disease and potential therapeutic targets.
Ossification of the Posterior Longitudinal Ligament
Ossification of the posterior longitudinal ligament is a condition of ectopic bone formation within the posterior longitudinal ligament, typically occurring at the cervical spine levels. It was first described as a disease of aging in Asian populations, with a prevalence of approximately 1–4%, though the prevalence is reported to be as high as 1.7% in Caucasian populations (35). About 17% of individuals with OPLL present with cervical myelopathy, while 29% of asymptomatic OPLL patients go on to develop myelopathy over the next three decades (36). Additionally, OPLL adds complexity to the treatment of cervical spondylosis and, ultimately, affects the surgical approach to treating symptomatic patients (37). Studies of the natural history of OPLL are clouded by the common presence of other coexisting degenerative spinal pathologies.
Little is known about the exact pathophysiologic mechanisms underlying OPLL. Multiple factors are suspected to play roles in the ectopic bone formation, including numerous biomechanically and metabolically mediated growth factors and cytokines (38). In vivo findings from human OPLL samples demonstrate degenerative elastic and cartilaginous fibers with metaplastic, hypertrophic cartilage cells (38, 39). Neovascularization, vascular endothelial growth factor-positive metaplastic chondrocytes, and abnormal collagen expression are thought to play a role in the spreading endochondral ossification front (38, 39). Additional studies are required to expand our understanding of OPLL and likely will be influenced by the wealth of genomic and proteomic results.
Heritability of OPLL
Several genetic studies have been performed to establish the heritability of OPLL. A study of 347 families of patients with OPLL found a 26% prevalence in parents and a 28% prevalence in siblings (40). In this study, the relative risk of first-degree relatives developing the disease was statistically significant and greater than five times that of the expected incidence in the general population. Another study looking at approximately 100 patients and relatives with OPLL found a prevalence of 27% with a relative risk of seven times that of the general population (41). Although a high segregation rate among siblings and a high prevalence of disease in parents suggest an autosomal dominant pattern of inheritance, neither study showed autosomal dominant (or recessive) inheritance on further analysis. Likewise, a polygene inheritance hypothesis was also rejected in these studies (40, 41). Altogether, these data suggest a high rate of heritability but not in a predictable fashion that would allow for practical genetic counseling.
Genetics of OPLL
Multiple genes have been targeted as possible contributors to the pathogenesis of OPLL. One of the first to be investigated was the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP1) gene, a transmembrane metalloenzyme that regulates soft-tissue calcification and bone mineralization via the production of inorganic pyrophosphate, a known inhibitor of calcification (42). ENPP1 was first implicated after studies in ttw mice showed altered gene expression causing tiptoe walking (43). The mice harbor a naturally recessive mutation that results in ectopic spinal ligament ossification and myelopathy that mirrors the disease traits in human OPLL (43). Several case–control studies in humans have examined SNPs in the ENPP1 gene, the main enzyme that controls inorganic pyrophosphate in osteoblasts and chondrocytes. The results have linked various polymorphisms to disease susceptibility, severity, and location, but the results have been inconsistent regarding which SNPs are involved (44–46). Nonetheless, these findings implicate ENPP1 as a possible therapeutic target. Further work needs to be done to elucidate the exact mechanisms by which it is modified in OPLL.
Collagen molecules have also received significant attention in the genetic research for OPLL. Mutations in type XI collagen within the COL11A2 gene are thought to affect the formation of fibril networks in the extracellular matrix and change the conformation of Type II collagen, which is responsible for bone and cartilage formation (38). Two large genome linkage studies found five different SNPs in COL11A2 that correlated with disease presence, and one was present in both reports (47, 48). Type VI collagen is also associated with multiple SNPs in chromosome 21, localizing to the COL6A1 gene (49). Other studies have shown similar findings and linked this SNP to ossification of the ligamentum flavum and diffuse idiopathic skeletal hyperostosis (50, 51). However, a study of these COL6A1 SNPs in a Korean population revealed conflicting results (52). Although significant data support the two collagen molecules as contributing to the pathogenesis of OPLL, lack of data congruency has made reliable conclusions difficult to make, likely due to disease heterogeneity.
Bone morphogenetic proteins and transforming growth factor-β have been studied extensively due to their role in physiological and pathological pathways of bone formation and metabolism. Several SNPs are associated with both of these proteins, specifically bone morphogenetic protein-2, bone morphogenetic protein-4, and transforming growth factor-β1 (53–58). Although fewer studies have focused on these molecules and replication studies still are needed, they present attractive targets for future research. Likewise, multiple other candidate genes have been investigated independently (Table 1) (43–51, 53–66). A full list of SNP associations is well summarized in other studies (1, 67, 68).
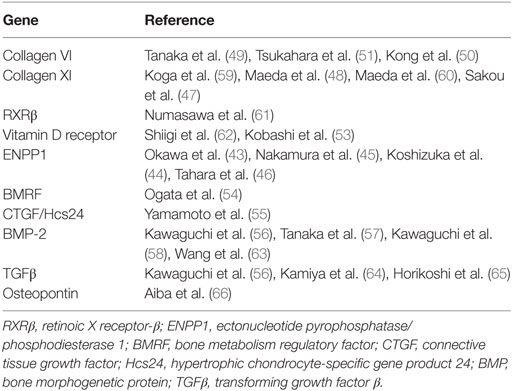
Table 1. Genetics of ossification of the posterior longitudinal ligament.
The first genome-wide association study for OPLL identified 26 SNPs on 3 chromosomes at 8p11.21, 8q23.1, 8q23.3, 12p11.22, 12p12.2, and 20p12.3 that are considered to be significantly associated with OPLL. Six of those SNPs were confirmed in a replication test as highly susceptible gene loci for OPLL (69). Interestingly, their comparison with previously reported gene loci from prior studies uncovered no significant associations. Two of the genes, radial spoke head 9 homolog, RSPH9 (coding for a protein that composes cilia and plays a role in the hedgehog pathway of skeletal development), and serine/threonine kinase 38 like, STK38L (a protein kinase that inhibits cell cycle progression), are believed to have a part in the pathobiology of OPLL through membranous ossification (69). Hydroxyacid oxidase 1, HAO1 (encodes hydroxyacid oxidase 1, which oxidizes 2-hydroxyacid), R-spondin 2, RSPO2 (encodes R-spondin 2 protein that contributes to osteoblastogenesis through Wnt/β-catenin signaling pathways), and coiled-coil domain containing 91, CCDC91 (encodes a trans-Golgi network protein) have putative roles in the endochondral ossification process (69). A follow-up study by the same group focused on RSPO2 and further implicated it by evaluating the putative SNP in vitro and how it affected the binding of a vital transcription factor, CCAAT-enhancer-binding protein β (C/EBPβ) (70). Macroscopically, these associations remain speculative at this time; nevertheless, the cumulative findings of these studies open the door for investigation of multiple new gene targets and provide insight into the novel mechanisms of OPLL.
Degenerative Lumbar Scoliosis
DS is a disease that occurs after skeletal maturity, typically after the third decade of life, and is a distinct entity from idiopathic scoliosis. It is associated with severe back and leg pain, which leads to spinal dysfunction and debilitation. Pain may result from asymmetric muscular loading, facet joint arthritis, or nerve root impingement/traction (71). A Cobb angle of greater than 10° in the coronal plane is considered diagnostic (72). Although it has been recognized for many decades as a significant cause of pain and disability, increasing clinical awareness has yielded a growing body of literature on treatment and greatly improved clinical outcomes.
Although interest in outcomes and surgical treatments has gained ground, little is known about the pathogenesis of DS. Multiple studies have implicated osteoporosis in DS. There is a high degree of overlap of these two pathologies; however, a causal relationship has not been established, and definitive correlations are lacking (73). Other studies suggest that asymmetric IVD degeneration is the cause, resulting in uneven loading forces that perpetuate the rate of asymmetric degeneration. Recent evidence suggests that cytokines and growth factors are differentially expressed within various locations of the IVD, likely creating regional discrepancies in the rates of cellular apoptosis, inflammation, and angiogenesis (74–76). Whether or not these differences are the result of other causative etiologies or are the instigating impetus behind the disease remains to be seen. Investigations into other sources of asymmetric spinal degeneration, such as myopathy and mechanical instability, are lacking and provide future directions for research (76, 77).
Heritability of DS
Few studies have been performed to examine the heritability of DS to date. Twin–twin and family-based comparison studies should be conducted to help identify patterns of inheritance. The paucity of data likely is due to the relatively recent attention to this disease since the mid-1990s. Unlike OPLL, which is found at particularly high rates in specific parts of the world, DS appears to show less geographic variation.
Genetics of DS
For adolescent idiopathic scoliosis (AIS), a number of investigations have been conducted to examine the genetic basis of disease. In 2010, Ward et al. performed a genome-wide association study that identified 53 SNPs that correlated with scoliotic curvature in Caucasian females (78). Using this genotype information and an initial Cobb angle, they devised an algorithm that calculated the risk of curvature progression in selected patients, which they commercialized under the name ScoliScore. This DNA-based predictive calculator theoretically enabled clinicians to forecast which individual patients were at low likelihood of curve progression (78). However, replication studies failed to demonstrate the same SNP associations in different geographically diverse populations (79–82). Nevertheless, this attempt at personalized, genome-based, clinical and outcome prediction exemplifies methods by which genetic outcomes could guide future treatments for a multitude of diseases.
Comparatively, in DS, there have been fewer genetic incongruities identified than in other degenerative spine diseases. It is thought that distinct genetic characteristics define these seemingly similar, but quite clinically different syndromes. Our systematic review identified six works in the literature (Table 2) (52, 83–87) that identified genetic contributions for DS. The quality of the data is relatively limited (Level III studies) but provides some insight into potential genetic mechanisms for disease pathogenesis that could potentially be used in future studies.
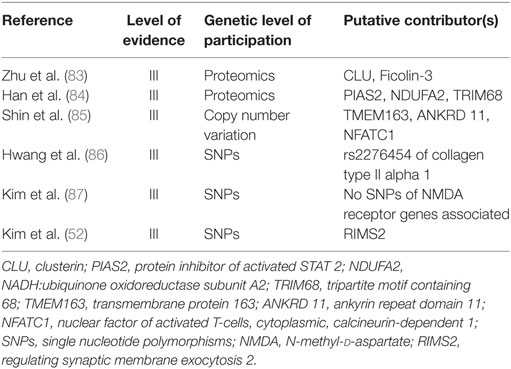
Table 2. Review of studies identifying genetic contributions to adult degenerative scoliosis.
Proteomic analyses of the sera of patients with DS have identified 11 proteins that are differentially expressed in such patients, 2 of which were secondarily confirmed with Western blot analyses, CLU (also known as apolipoprotein J, testosterone-repressed prostate message-2, SP 40-40, complement lysis inhibitor, gp80, glycoprotein III, and sulfate glycoprotein-2) and Ficolin-3 (also named Hakata antigen, thermolabile β-2 macroglycoprotein, thermolabile substance, and H-ficolin) (83). Both of these proteins have been suggested to have roles in autoimmunity, although they both likely have multiple roles in the human body and their association is non-specific. This protein expression analysis does not definitively implicate them in the disease pathogenesis but suggests that they may serve as potential future biomarkers and potentially raises the question of whether an autoimmune component of DS exists. The same group also compared proteomic expression in cultured mesenchymal stem cells of DS patients (84). This comparison revealed differential levels of three proteins, protein inhibitor of activated STAT 2 (PIAS2), NADH:ubiquinone oxidoreductase subunit A2 (NDUFA2), and tripartite motif containing 68 (TRIM68), none of which correspond to those elevated in the serum. All three of these proteins play various roles in biological processes and, therefore, pinpointing their roles may be difficult. Although the above proteomic analysis is helpful in identifying biomarkers of disease and drug targets, the proteins characterized in the analysis do not correlate exactly with genetic differences in the disease and are susceptible to environmental and other outside factors that change the genomic output via epigenetic influences. Consequently, much more work is required to tease out the meaning of these proteomic differences and reproduce these results with different patient populations.
One of the studies comparing genomic differences in DS examined copy number variations, which represent regional gene dosages of DNA segments 1 kb or larger (85). Of the 260 copy number variations identified by microarray analysis, quantitative polymerase chain reaction validation identified three genes with significant differences from the control group. These genes included transmembrane protein 163, TMEM163, a gene coding for a transmembrane protein of unknown function; ankyrin repeat domain 11, ANKRD11, an ankyrin repeating gene implicated in autism spectrum disorder and skeletal formation; and nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1, NFATC1, a gene reportedly involved in bone mineral density (85). This novel study provided evidence that DS could be predisposed by inherent genomic differences rather than resulting from external environmental forces causing asymmetric degeneration. Further work will be needed to expand on the biosignaling cascades by which these genes may affect disease pathogenesis.
Likewise, another area of gene-based research relates to SNPs associated with DS. Prior studies have given collagen molecules significant attention for similar diseases, including IVD degeneration and AIS (88). Collagen II has been investigated because of its structural role in stress-bearing of the spine (88). Investigators studied SNPs of COL2A1 and found a significant association of SNP (rs2276454) in COL2A1 to DS in Korean patients (86). Another study tested SNPs of glutamate receptors (N-methyl-d-aspartate receptors), given their role in controlling bone remodeling through stimulation, maturation, and differentiation of osteoblasts and osteoclasts (87). Interestingly, they found no association with any of the SNPs investigated. A similar study found that one of the SNPs in regulating synaptic membrane exocytosis 2, RIMS2, coding for a presynaptic active zone protein that regulates vesicle exocytosis of neurotransmitters, was significantly associated with DS (87). Therefore, glutamate, or other neurotransmitters, may still contribute to the disease progression of DS. The heterogeneity of these findings suggests that our insight into DS remains minimal; yet, these results lay the groundwork for further basic science in this area.
Conclusion
Adult degenerative spinal disease has tremendous health costs on a global level. CSM, OPLL, and DS have gained significant attention from medical providers and researchers as disease entities that merit further focus and investigation (Table 3). Studies of families and twins suggest that there may be a significant component of heritability to CSM and OPLL, although few genetic studies of DS have been published. Thus, DS presents an opportunity for further research. Advances in genomic analysis and biostatistics continue to open new doors to finding genetic linkages with these degenerative spinal diseases, which ultimately could guide the course of future pathophysiological studies, clinical diagnoses, and treatments.

Table 3. Summary of cervical spondylotic myelopathy (CSM), ossification of the posterior longitudinal ligament (OPLL), and lumbar degenerative scoliosis (DS).
Author Contributions
All the authors made substantial contributions to the conception or design of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Financial support was received from the Barrow Neurological Research Foundation.
Abbreviations
AIS, adolescent idiopathic scoliosis; CSM, cervical spondylotic myelopathy; DS, degenerative scoliosis; IVD, intervertebral disc; OPLL, ossification of posterior longitudinal ligament; SNP, single nucleotide polymorphism.
References
1. Wilson JR, Patel AA, Brodt ED, Dettori JR, Brodke DS, Fehlings MG. Genetics and heritability of cervical spondylotic myelopathy and ossification of the posterior longitudinal ligament: results of a systematic review. Spine (Phila Pa 1976) (2013) 38(22 Suppl 1):S123–46. doi:10.1097/BRS.0b013e3182a7f478
2. Gore DR, Sepic SB, Gardner GM. Roentgenographic findings of the cervical spine in asymptomatic people. Spine (Phila Pa 1976) (1986) 11(6):521–4. doi:10.1097/00007632-198607000-00003
3. Boden SD, Davis DO, Dina TS, Patronas NJ, Wiesel SW. Abnormal magnetic-resonance scans of the lumbar spine in asymptomatic subjects. A prospective investigation. J Bone Joint Surg Am (1990) 72(3):403–8.
4. Toledano M, Bartleson JD. Cervical spondylotic myelopathy. Neurol Clin (2013) 31(1):287–305. doi:10.1016/j.ncl.2012.09.003
5. Karadimas SK, Gatzounis G, Fehlings MG. Pathobiology of cervical spondylotic myelopathy. Eur Spine J (2015) 24(Suppl 2):132–8. doi:10.1007/s00586-014-3264-4
6. Henderson FC, Geddes JF, Vaccaro AR, Woodard E, Berry KJ, Benzel EC. Stretch-associated injury in cervical spondylotic myelopathy: new concept and review. Neurosurgery (2005) 56(5):1101–13; discussion 1101–13. doi:10.1227/01.NEU.0000157929.85251.7C
7. Baptiste DC, Fehlings MG. Pathophysiology of cervical myelopathy. Spine J (2006) 6(6 Suppl):190S–7S. doi:10.1016/j.spinee.2006.04.024
8. Breig A, Turnbull I, Hassler O. Effects of mechanical stresses on the spinal cord in cervical spondylosis: a study on fresh cadaver material. J Neurosurg (1966) 25(1):45–56. doi:10.3171/jns.1966.25.1.0045
9. Gooding MR, Wilson CB, Hoff JT. Experimental cervical myelopathy: autoradiographic studies of spinal cord blood flow patterns. Surg Neurol (1976) 5(4):233–9.
10. Doppman JL. The mechanism of ischemia in anteroposterior compression of the spinal cord. Invest Radiol (1975) 10(6):543–51. doi:10.1097/00004424-197511000-00001
11. Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A (2014) 111(11):E1035–42. doi:10.1073/pnas.1401595111
12. Figley SA, Khosravi R, Legasto JM, Tseng YF, Fehlings MG. Characterization of vascular disruption and blood-spinal cord barrier permeability following traumatic spinal cord injury. J Neurotrauma (2014) 31(6):541–52. doi:10.1089/neu.2013.3034
13. Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci (2002) 22(17):7526–35.
14. Karadimas SK, Gialeli CH, Klironomos G, Tzanakakis GN, Panagiotopoulos E, Karamanos NK, et al. The role of oligodendrocytes in the molecular pathobiology and potential molecular treatment of cervical spondylotic myelopathy. Curr Med Chem (2010) 17(11):1048–58. doi:10.2174/092986710790820598
15. Karadimas SK, Erwin WM, Ely CG, Dettori JR, Fehlings MG. Pathophysiology and natural history of cervical spondylotic myelopathy. Spine (Phila Pa 1976) (2013) 38(22 Suppl 1):S21–36. doi:10.1097/BRS.0b013e3182a7f2c3
16. Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci (1996) 16(2):545–52.
17. Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury: results of the Second National Acute Spinal Cord Injury Study. N Engl J Med (1990) 322(20):1405–11. doi:10.1056/NEJM199005173222001
18. Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury: results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA (1997) 277(20):1597–604.
19. Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med (1997) 3(1):73–6. doi:10.1038/nm0197-73
20. Singh A, Tetreault L, Fehlings MG, Fischer DJ, Skelly AC. Risk factors for development of cervical spondylotic myelopathy: results of a systematic review. Evid Based Spine Care J (2012) 3(3):35–42. doi:10.1055/s-0032-1327808
21. Bull J, el Gammal T, Popham M. A possible genetic factor in cervical spondylosis. Br J Radiol (1969) 42(493):9–16. doi:10.1259/0007-1285-42-493-9
22. Sambrook PN, MacGregor AJ, Spector TD. Genetic influences on cervical and lumbar disc degeneration: a magnetic resonance imaging study in twins. Arthritis Rheum (1999) 42(2):366–72. doi:10.1002/1529-0131(199902)42:2<366:AID-ANR20>3.0.CO;2-6
23. Mukerji N, Sinar EJ. Identical twins with cervical myelopathy: a case for hereditary cervical spondylosis? Report of two cases and review of the literature. J Neurosurg Spine (2007) 6(4):344–9. doi:10.3171/spi.2007.6.4.10
24. Patel AA, Spiker WR, Daubs M, Brodke DS, Cannon-Albright LA. Evidence of an inherited predisposition for cervical spondylotic myelopathy. Spine (Phila Pa 1976) (2012) 37(1):26–9. doi:10.1097/BRS.0b013e3182102ede
25. Wang ZC, Chen XS, Wang da W, Shi JG, Jia LS, Xu GH, et al. The genetic association of vitamin D receptor polymorphisms and cervical spondylotic myelopathy in Chinese subjects. Clin Chim Acta (2010) 411(11–12):794–7. doi:10.1016/j.cca.2010.01.031
26. Wang ZC, Shi JG, Chen XS, Xu GH, Li LJ, Jia LS. The role of smoking status and collagen IX polymorphisms in the susceptibility to cervical spondylotic myelopathy. Genet Mol Res (2012) 11(2):1238–44. doi:10.4238/2012.May.9.2
27. Annunen S, Paassilta P, Lohiniva J, Perala M, Pihlajamaa T, Karppinen J, et al. An allele of COL9A2 associated with intervertebral disc disease. Science (1999) 285(5426):409–12. doi:10.1126/science.285.5426.409
28. Kales SN, Linos A, Chatzis C, Sai Y, Halla M, Nasioulas G, et al. The role of collagen IX tryptophan polymorphisms in symptomatic intervertebral disc disease in Southern European patients. Spine (Phila Pa 1976) (2004) 29(11):1266–70. doi:10.1097/00007632-200406010-00017
29. Jim JJ, Noponen-Hietala N, Cheung KM, Ott J, Karppinen J, Sahraravand A, et al. The TRP2 allele of COL9A2 is an age-dependent risk factor for the development and severity of intervertebral disc degeneration. Spine (Phila Pa 1976) (2005) 30(24):2735–42. doi:10.1097/01.brs.0000190828.85331.ef
30. Kawaguchi S, Yamashita T, Katahira G, Yokozawa H, Torigoe T, Sato N. Chemokine profile of herniated intervertebral discs infiltrated with monocytes and macrophages. Spine (Phila Pa 1976) (2002) 27(14):1511–6. doi:10.1097/00007632-200207150-00006
31. Cheung KM, Chan D, Karppinen J, Chen Y, Jim JJ, Yip SP, et al. Association of the Taq I allele in vitamin D receptor with degenerative disc disease and disc bulge in a Chinese population. Spine (Phila Pa 1976) (2006) 31(10):1143–8. doi:10.1097/01.brs.0000216530.41838.d3
32. Akmal M, Kesani A, Anand B, Singh A, Wiseman M, Goodship A. Effect of nicotine on spinal disc cells: a cellular mechanism for disc degeneration. Spine (Phila Pa 1976) (2004) 29(5):568–75. doi:10.1097/01.BRS.0000101422.36419.D8
33. Setzer M, Hermann E, Seifert V, Marquardt G. Apolipoprotein E gene polymorphism and the risk of cervical myelopathy in patients with chronic spinal cord compression. Spine (Phila Pa 1976) (2008) 33(5):497–502. doi:10.1097/BRS.0b013e3181657cf7
34. Setzer M, Vrionis FD, Hermann EJ, Seifert V, Marquardt G. Effect of apolipoprotein E genotype on the outcome after anterior cervical decompression and fusion in patients with cervical spondylotic myelopathy. J Neurosurg Spine (2009) 11(6):659–66. doi:10.3171/2009.7.SPINE08667
35. Matsunaga S, Sakou T. Ossification of the posterior longitudinal ligament of the cervical spine: etiology and natural history. Spine (Phila Pa 1976) (2012) 37(5):E309–14. doi:10.1097/BRS.0b013e318241ad33
36. Matsunaga S, Sakou T, Taketomi E, Komiya S. Clinical course of patients with ossification of the posterior longitudinal ligament: a minimum 10-year cohort study. J Neurosurg (2004) 100(3 Suppl Spine):245–8.
37. Lawrence BD, Jacobs WB, Norvell DC, Hermsmeyer JT, Chapman JR, Brodke DS. Anterior versus posterior approach for treatment of cervical spondylotic myelopathy: a systematic review. Spine (Phila Pa 1976) (2013) 38(22 Suppl 1):S173–82. doi:10.1097/BRS.0b013e3182a7eaaf
38. Inamasu J, Guiot BH, Sachs DC. Ossification of the posterior longitudinal ligament: an update on its biology, epidemiology, and natural history. Neurosurgery (2006) 58(6):1027–39; discussion 1027–39. doi:10.1227/01.NEU.0000215867.87770.73
39. Sato R, Uchida K, Kobayashi S, Yayama T, Kokubo Y, Nakajima H, et al. Ossification of the posterior longitudinal ligament of the cervical spine: histopathological findings around the calcification and ossification front. J Neurosurg Spine (2007) 7(2):174–83. doi:10.3171/SPI-07/08/174
40. Terayama K. Genetic studies on ossification of the posterior longitudinal ligament of the spine. Spine (Phila Pa 1976) (1989) 14(11):1184–91. doi:10.1097/00007632-198911000-00009
41. Tanikawa E, Furuya K, Nakajima H. Genetic study on ossification of posterior longitudinal ligament. Bull Tokyo Med Dent Univ (1986) 33(3):117–28.
42. Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta (2003) 1638(1):1–19. doi:10.1016/S0925-4439(03)00058-9
43. Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet (1998) 19(3):271–3. doi:10.1038/956
44. Koshizuka Y, Kawaguchi H, Ogata N, Ikeda T, Mabuchi A, Seichi A, et al. Nucleotide pyrophosphatase gene polymorphism associated with ossification of the posterior longitudinal ligament of the spine. J Bone Miner Res (2002) 17(1):138–44. doi:10.1359/jbmr.2002.17.1.138
45. Nakamura I, Ikegawa S, Okawa A, Okuda S, Koshizuka Y, Kawaguchi H, et al. Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL). Hum Genet (1999) 104(6):492–7. doi:10.1007/s004390050993
46. Tahara M, Aiba A, Yamazaki M, Ikeda Y, Goto S, Moriya H, et al. The extent of ossification of posterior longitudinal ligament of the spine associated with nucleotide pyrophosphatase gene and leptin receptor gene polymorphisms. Spine (Phila Pa 1976) (2005) 30(8):877–80; discussion 881. doi:10.1097/01.brs.0000160686.18321.ad
47. Sakou T, Taketomi E, Matsunaga S, Yamaguchi M, Sonoda S, Yashiki S. Genetic study of ossification of the posterior longitudinal ligament in the cervical spine with human leukocyte antigen haplotype. Spine (Phila Pa 1976) (1991) 16(11):1249–52. doi:10.1097/00007632-199111000-00001
48. Maeda S, Ishidou Y, Koga H, Taketomi E, Ikari K, Komiya S, et al. Functional impact of human collagen alpha2(XI) gene polymorphism in pathogenesis of ossification of the posterior longitudinal ligament of the spine. J Bone Miner Res (2001) 16(5):948–57. doi:10.1359/jbmr.2001.16.5.948
49. Tanaka T, Ikari K, Furushima K, Okada A, Tanaka H, Furukawa K, et al. Genomewide linkage and linkage disequilibrium analyses identify COL6A1, on chromosome 21, as the locus for ossification of the posterior longitudinal ligament of the spine. Am J Hum Genet (2003) 73(4):812–22. doi:10.1086/378593
50. Kong Q, Ma X, Li F, Guo Z, Qi Q, Li W, et al. COL6A1 polymorphisms associated with ossification of the ligamentum flavum and ossification of the posterior longitudinal ligament. Spine (Phila Pa 1976) (2007) 32(25):2834–8. doi:10.1097/BRS.0b013e31815b761c
51. Tsukahara S, Miyazawa N, Akagawa H, Forejtova S, Pavelka K, Tanaka T, et al. COL6A1, the candidate gene for ossification of the posterior longitudinal ligament, is associated with diffuse idiopathic skeletal hyperostosis in Japanese. Spine (Phila Pa 1976) (2005) 30(20):2321–4. doi:10.1097/01.brs.0000182318.47343.6d
52. Kim KH, Kuh SU, Park JY, Lee SJ, Park HS, Chin DK, et al. Association between BMP-2 and COL6A1 gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the cervical spine in Korean patients and family members. Genet Mol Res (2014) 13(1):2240–7. doi:10.4238/2014.March.31.4
53. Kobashi G, Ohta K, Washio M, Okamoto K, Sasaki S, Yokoyama T, et al. FokI variant of vitamin D receptor gene and factors related to atherosclerosis associated with ossification of the posterior longitudinal ligament of the spine: a multi-hospital case-control study. Spine (Phila Pa 1976) (2008) 33(16):E553–8. doi:10.1097/BRS.0b013e31817e9de2
54. Ogata N, Koshizuka Y, Miura T, Iwasaki M, Hosoi T, Shiraki M, et al. Association of bone metabolism regulatory factor gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the spine and its severity. Spine (Phila Pa 1976) (2002) 27(16):1765–71. doi:10.1097/00007632-200208150-00015
55. Yamamoto Y, Furukawa K, Ueyama K, Nakanishi T, Takigawa M, Harata S. Possible roles of CTGF/Hcs24 in the initiation and development of ossification of the posterior longitudinal ligament. Spine (Phila Pa 1976) (2002) 27(17):1852–7. doi:10.1097/00007632-200209010-00009
56. Kawaguchi H, Kurokawa T, Hoshino Y, Kawahara H, Ogata E, Matsumoto T. Immunohistochemical demonstration of bone morphogenetic protein-2 and transforming growth factor-beta in the ossification of the posterior longitudinal ligament of the cervical spine. Spine (Phila Pa 1976) (1992) 17(3 Suppl):S33–6. doi:10.1097/00007632-199203001-00007
57. Tanaka H, Nagai E, Murata H, Tsubone T, Shirakura Y, Sugiyama T, et al. Involvement of bone morphogenic protein-2 (BMP-2) in the pathological ossification process of the spinal ligament. Rheumatology (Oxford) (2001) 40(10):1163–8. doi:10.1093/rheumatology/40.10.1163
58. Kawaguchi Y, Furushima K, Sugimori K, Inoue I, Kimura T. Association between polymorphism of the transforming growth factor-beta1 gene with the radiologic characteristic and ossification of the posterior longitudinal ligament. Spine (Phila Pa 1976) (2003) 28(13):1424–6. doi:10.1097/00007632-200307010-00013
59. Koga H, Sakou T, Taketomi E, Hayashi K, Numasawa T, Harata S, et al. Genetic mapping of ossification of the posterior longitudinal ligament of the spine. Am J Hum Genet (1998) 62(6):1460–7. doi:10.1086/301868
60. Maeda S, Koga H, Matsunaga S, Numasawa T, Ikari K, Furushima K, et al. Gender-specific haplotype association of collagen alpha2 (XI) gene in ossification of the posterior longitudinal ligament of the spine. J Hum Genet (2001) 46(1):1–4. doi:10.1007/s100380170117
61. Numasawa T, Koga H, Ueyama K, Maeda S, Sakou T, Harata S, et al. Human retinoic X receptor beta: complete genomic sequence and mutation search for ossification of posterior longitudinal ligament of the spine. J Bone Miner Res (1999) 14(4):500–8. doi:10.1359/jbmr.1999.14.4.500
62. Shiigi E, Sugiyama T, Tanaka H, Murata H, Shirakura Y, Kawai S. Possible involvement of vitamin D receptor gene polymorphism in male patients with ossification of spinal ligaments. J Bone Miner Metab (2001) 19(5):308–11. doi:10.1007/s007740170015
63. Wang H, Liu D, Yang Z, Tian B, Li J, Meng X, et al. Association of bone morphogenetic protein-2 gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the spine and its severity in Chinese patients. Eur Spine J (2008) 17(7):956–64. doi:10.1007/s00586-008-0651-8
64. Kamiya M, Harada A, Mizuno M, Iwata H, Yamada Y. Association between a polymorphism of the transforming growth factor-beta1 gene and genetic susceptibility to ossification of the posterior longitudinal ligament in Japanese patients. Spine (Phila Pa 1976) (2001) 26(11):1264–6; discussion 6–7. doi:10.1097/00007632-200106010-00017
65. Horikoshi T, Maeda K, Kawaguchi Y, Chiba K, Mori K, Koshizuka Y, et al. A large-scale genetic association study of ossification of the posterior longitudinal ligament of the spine. Hum Genet (2006) 119(6):611–6. doi:10.1007/s00439-006-0170-9
66. Aiba A, Nakajima A, Okawa A, Koda M, Yamazaki M. Evidence of enhanced expression of osteopontin in spinal hyperostosis of the twy mouse. Spine (Phila Pa 1976) (2009) 34(16):1644–9. doi:10.1097/BRS.0b013e3181aa01fc
67. Stapleton CJ, Pham MH, Attenello FJ, Hsieh PC. Ossification of the posterior longitudinal ligament: genetics and pathophysiology. Neurosurg Focus (2011) 30(3):E6. doi:10.3171/2010.12.FOCUS10271
68. Stetler WR, La Marca F, Park P. The genetics of ossification of the posterior longitudinal ligament. Neurosurg Focus (2011) 30(3):E7. doi:10.3171/2010.12.FOCUS10275
69. Nakajima M, Takahashi A, Tsuji T, Karasugi T, Baba H, Uchida K, et al. A genome-wide association study identifies susceptibility loci for ossification of the posterior longitudinal ligament of the spine. Nat Genet (2014) 46(9):1012–6. doi:10.1038/ng.3045
70. Nakajima M, Kou I, Ohashi H; Genetic Study Group of the Investigation Committee on the Ossification of Spinal Ligaments, Ikegawa S. Identification and functional characterization of RSPO2 as a susceptibility gene for ossification of the posterior longitudinal ligament of the spine. Am J Hum Genet (2016) 99(1):202–7. doi:10.1016/j.ajhg.2016.05.018
71. Daffner SD, Vaccaro AR. Adult degenerative lumbar scoliosis. Am J Orthop (Belle Mead NJ) (2003) 32(2):77–82; discussion 82.
73. Pappou IP, Girardi FP, Sandhu HS, Parvataneni HK, Cammisa FP Jr, Schneider R, et al. Discordantly high spinal bone mineral density values in patients with adult lumbar scoliosis. Spine (Phila Pa 1976) (2006) 31(14):1614–20. doi:10.1097/01.brs.0000222030.32171.5f
74. Koerner JD, Markova DZ, Yadla S, Mendelis J, Hilibrand A, Vaccaro AR, et al. Differential gene expression in anterior and posterior annulus fibrosus. Spine (Phila Pa 1976) (2014) 39(23):1917–23. doi:10.1097/BRS.0000000000000590
75. Chen B, Fellenberg J, Wang H, Carstens C, Richter W. Occurrence and regional distribution of apoptosis in scoliotic discs. Spine (Phila Pa 1976) (2005) 30(5):519–24. doi:10.1097/01.brs.0000154652.96975.1f
76. Bertram H, Steck E, Zimmerman G, Chen B, Carstens C, Nerlich A, et al. Accelerated intervertebral disc degeneration in scoliosis versus physiological ageing develops against a background of enhanced anabolic gene expression. Biochem Biophys Res Commun (2006) 342(3):963–72. doi:10.1016/j.bbrc.2006.02.048
77. Hiniker A, Wong LJ, Berven S, Truong CK, Adesina AM, Margeta M. Axial mitochondrial myopathy in a patient with rapidly progressive adult-onset scoliosis. Acta Neuropathol Commun (2014) 2:137. doi:10.1186/s40478-014-0137-3
78. Ward K, Ogilvie JW, Singleton MV, Chettier R, Engler G, Nelson LM. Validation of DNA-based prognostic testing to predict spinal curve progression in adolescent idiopathic scoliosis. Spine (Phila Pa 1976) (2010) 35(25):E1455–64. doi:10.1097/BRS.0b013e3181ed2de1
79. Tang QL, Julien C, Eveleigh R, Bourque G, Franco A, Labelle H, et al. A replication study for association of 53 single nucleotide polymorphisms in ScoliScore test with adolescent idiopathic scoliosis in French-Canadian population. Spine (Phila Pa 1976) (2015) 40(8):537–43. doi:10.1097/BRS.0000000000000807
80. Ogura Y, Takahashi Y, Kou I, Nakajima M, Kono K, Kawakami N, et al. A replication study for association of 53 single nucleotide polymorphisms in a scoliosis prognostic test with progression of adolescent idiopathic scoliosis in Japanese. Spine (Phila Pa 1976) (2013) 38(16):1375–9. doi:10.1097/BRS.0b013e3182947d21
81. Ogura Y, Takahashi Y, Kou I, Nakajima M, Kono K, Kawakami N, et al. A replication study for association of 5 single nucleotide polymorphisms with curve progression of adolescent idiopathic scoliosis in Japanese patients. Spine (Phila Pa 1976) (2013) 38(7):571–5. doi:10.1097/BRS.0b013e3182761535
82. Xu L, Qin X, Sun W, Qiao J, Qiu Y, Zhu Z. Replication of association between 53 single-nucleotide polymorphisms in a DNA-based diagnostic test and AIS progression in Chinese Han population. Spine (Phila Pa 1976) (2016) 41(4):306–10. doi:10.1097/BRS.0000000000001203
83. Zhu Y, Han S, Zhao H, Liang J, Zhai J, Wu Z, et al. Comparative analysis of serum proteomes of degenerative scoliosis. J Orthop Res (2011) 29(12):1896–903. doi:10.1002/jor.21466
84. Han S, Zhu Y, Wu Z, Zhang J, Qiu G. The differently expressed proteins in MSCs of degenerative scoliosis. J Orthop Sci (2013) 18(6):885–92. doi:10.1007/s00776-013-0444-8
85. Shin JH, Ha KY, Jung SH, Chung YJ. Genetic predisposition in degenerative lumbar scoliosis due to the copy number variation. Spine (Phila Pa 1976) (2011) 36(21):1782–93. doi:10.1097/BRS.0b013e318221a65f
86. Hwang DW, Kim KT, Lee SH, Kim JY, Kim DH. Association of COL2A1 gene polymorphism with degenerative lumbar scoliosis. Clin Orthop Surg (2014) 6(4):379–84. doi:10.4055/cios.2014.6.4.379
87. Kim KT, Kim J, Han YJ, Kim JH, Lee JS, Chung JH. Assessment of NMDA receptor genes (GRIN2A, GRIN2B and GRIN2C) as candidate genes in the development of degenerative lumbar scoliosis. Exp Ther Med (2013) 5(3):977–81. doi:10.3892/etm.2013.910
Keywords: cervical spondylotic myelopathy, genetics, genome-wide association study, heritability, intervertebral disc degeneration, ossification of posterior longitudinal ligament, proteomics
Citation: Walker CT, Bonney PA, Martirosyan NL and Theodore N (2016) Genetics Underlying an Individualized Approach to Adult Spinal Disorders. Front. Surg. 3:61. doi: 10.3389/fsurg.2016.00061
Received: 01 September 2016; Accepted: 26 October 2016;
Published: 22 November 2016
Edited by:
Eberval Figueiredo, University of Sao Paulo, BrazilCopyright: © 2016 Barrow Neurological Institute. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution, or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas Theodore, bmV1cm9wdWJAZGlnbml0eWhlYWx0aC5vcmc=