 Amy Bradshaw
Amy Bradshaw Agadha Wickremsekera
Agadha Wickremsekera Swee T. Tan
Swee T. Tan Lifeng Peng
Lifeng Peng Paul F. Davis
Paul F. Davis Tinte Itinteang
Tinte Itinteang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Surg., 15 April 2016
Sec. Neurosurgery
Volume 3 - 2016 | https://doi.org/10.3389/fsurg.2016.00021
Glioblastoma multiforme (GBM), an aggressive tumor that typically exhibits treatment failure with high mortality rates, is associated with the presence of cancer stem cells (CSCs) within the tumor. CSCs possess the ability for perpetual self-renewal and proliferation, producing downstream progenitor cells that drive tumor growth. Studies of many cancer types have identified CSCs using specific markers, but it is still unclear as to where in the stem cell hierarchy these markers fall. This is compounded further by the presence of multiple GBM and glioblastoma cancer stem cell subtypes, making investigation and establishment of a universal treatment difficult. This review examines the current knowledge on the CSC markers SALL4, OCT-4, SOX2, STAT3, NANOG, c-Myc, KLF4, CD133, CD44, nestin, and glial fibrillary acidic protein, specifically focusing on their use and validity in GBM research and how they may be utilized for investigations into GBM’s cancer biology.
Glioblastoma multiforme (GBM), a grade 4 astrocytoma, is the most aggressive form of glioma (1, 2) with the median survival of approximately 25 months following treatment (3). Despite advances in cancer research and treatment over several decades, there has only been a 2% improvement in 5-year survival (4). GBM has been shown to be resistant to radiotherapy and chemotherapy (5–7) and invariably recurs following surgical resection (8) and chemoradiation (9). GBM typically shows a space-occupying lesion with heterogeneous rim enhancement, causing mass effect with surrounding edema on computerized tomography (Figure 1A) and magnetic resonance imaging (Figure 1B).
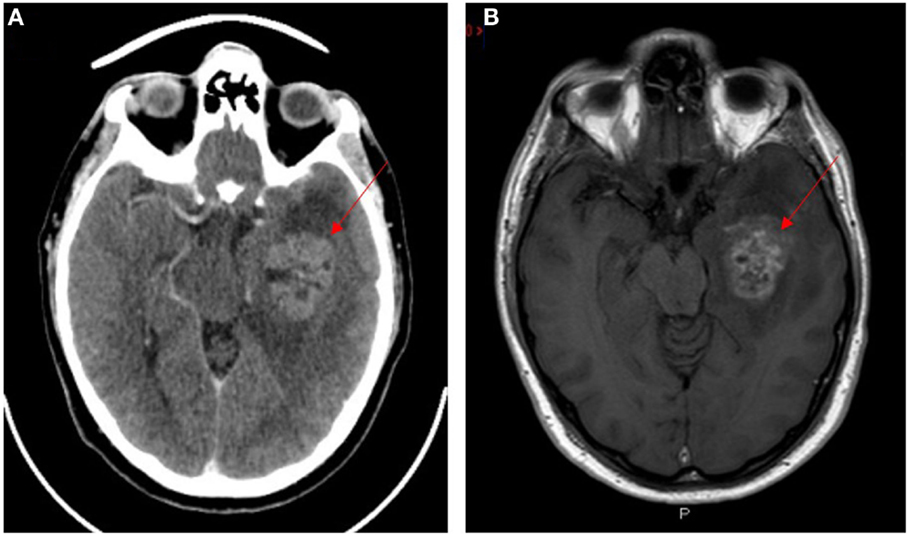
Figure 1. CT (A) and T1 weighted MRI (B) scans with contrast of a patient with glioblastoma multiforme. Lesion indicated by red arrows.
The presence of central necrosis (Figure 2A, arrows) and marginal proliferation of endothelial cells (microvascular hyperplasia) (Figure 2B, arrows) are hallmark histological features that separate GBM from lower grade glial tumors. Another characteristic feature of GBM is the presence of palisading cells around the area of necrosis (Figure 2C, arrows), which is widely regarded as a poor prognostic hallmark of GBM (10, 11). Increased mitosis, hypercellularity, atypical nuclei and cellular pleomorphism, and the development of lumina, reminiscent of kidney glomeruli (10, 12), are other histological features of GBM. Combinations of some or all of these features result in marked histological heterogeneity, indicating that GBM tumors can change and grow rapidly even while the central bulk of the tumor undergoes necrosis (13). While much is known about the histological features and chromosomal abnormalities (14, 15) in GBM, the molecular characteristics and the origin of the lesion are not fully elucidated.
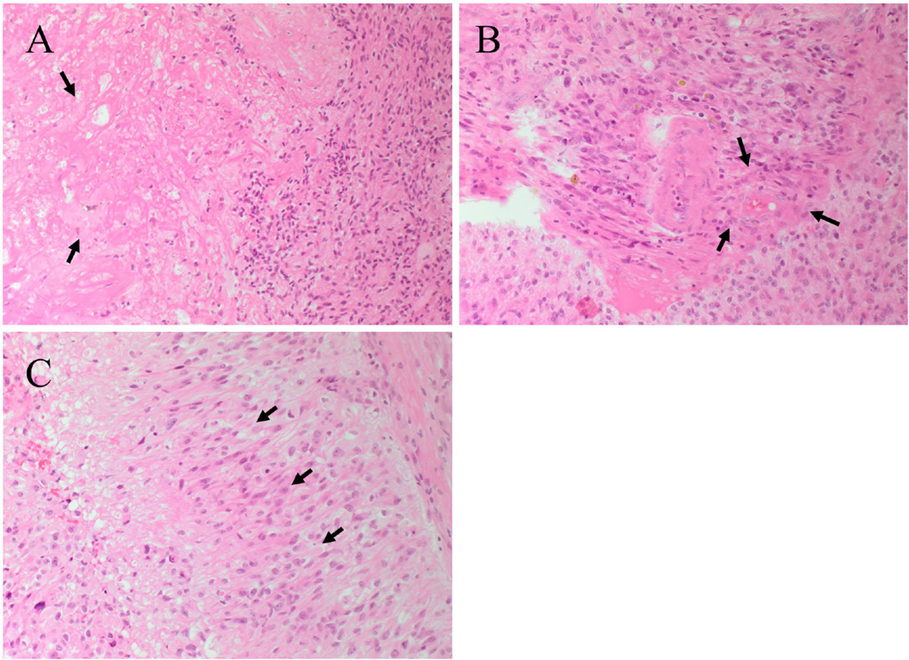
Figure 2. Hematoxylin and eosin staining of a glioblastoma multiforme. (A) The interface between tumor cells and the area of necrosis. The necrotic area (arrows) show greatly reduced nuclear staining. (B) Proliferation of the endothelial cells (arrows) within a microvessel. (C) Palisading cells (arrows) around the necrotic area. Original magnification: 200×.
This article reviews the data on cancer stem cell (CSC) markers currently used in GBM research and attempt to place them in the context of a hierarchical model of cancer.
The two current concepts on the origin of cancer and its continued propagation are: (1) the clonal evolution (or stochastic) model (Figure 3A) (16, 17), and (2) the hierarchical CSC model (Figure 3B) (16, 18).
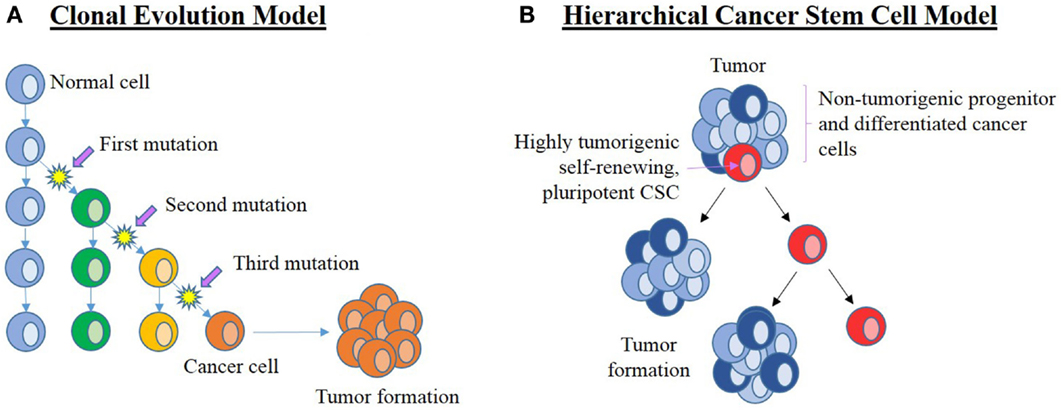
Figure 3. Current leading models of carcinogenesis. (A) The clonal evolution model hypothesizes that a normal cell (blue) within the organism undergoes a series of mutations to form a cancer cell (orange) that clonally expands and form the bulk of the tumor. Successful treatment must, therefore, eliminate all cancer cells. (B) The cancer stem cell (CSC) hierarchical model proposes that the origin of cancer being CSCs (red) that are pluripotent and self-renewing. They are highly tumorigenic with the ability to establish new tumors. CSCs divide asymmetrically to form new CSCs and progenitor (dark blue) cells that in turn give rise to differentiated cancer cells (light blue) that form the bulk of the tumor. These downstream cancer cells are low or non-tumorigenic. Adapted from Adams and Strasser (16).
The clonal evolution model of cancer proposes cumulative genetic mutations that occur over time in a normal cell, leading to the formation of a cancer cell that clonally expands to form identical copies, each with identical tumorigenic potential (16, 19). If these changes confer a selective advantage to a particular cell, then this allows the selected “clone” to outcompete other potential tumor forming clones (17). Propagation of this selected clone means that a substantial number of cells in the tumor are able to maintain tumor growth, so any effective treatment would require the elimination of all clonal cells, a theory that is inconsistent with the identification of CSCs in cancer (16).
Stem cells are cells that possess the capacity for self-renewal, proliferation, and differentiation (20–22). From a hierarchical viewpoint, embryonic stem cells (ESCs) are the most primitive cells within a biological system, and are considered pluripotent in that they are capable of differentiating into any type of cell in a particular organism (23). Downstream from ESCs, which are progenitor cells, a group that includes neural stem cells (NSCs) (24), mesenchymal (MES) stem cells (25), endothelial progenitor cells (26), and hematopoietic stem cells (HSCs) (27). These cells are multipotent, have more restricted lineage differentiation capacity, and, therefore, are no longer pluripotent (24). From here, the multipotent NSCs further differentiate, giving rise to more downstream progenitor cells with reducing differentiation, mitotic, and self-renewal potential, ultimately forming the majority of the organism (Figure 4) (24, 28).
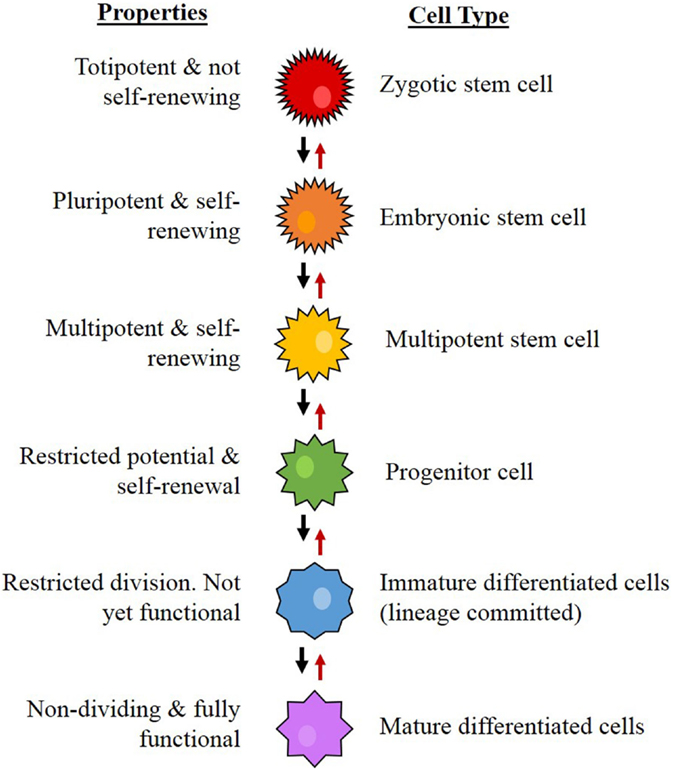
Figure 4. The proposed hierarchy for neural stem cell differentiation. The system begins with the most primitive and multipotent cell and moves through stages of differentiation to the most restricted cell. Concept from Gage (24).
Current literature uses the terms stem cell and progenitor cell interchangeably, essentially lumping these two cell types together (29–34). However, the validity of this practice has been questioned (35) as progenitor cells and stem cells differ in terms of hierarchy and biology and, therefore, should be regarded as distinct entities. Stem cells are multipotent with an unlimited capacity for self-renewal, whereas progenitor cells are most often unipotent with restricted capacity for self-renewal. Distinguishing between stem cells and progenitor cells in cancer is important in the understanding of the CSC concept for carcinogenesis. However, as they presumably belong to a spectral continuum distinguishing between the two populations remains a challenge.
The hierarchical CSC model of cancer proposes that a tumor arises from CSCs generated by mutations in either normal ESCs or progenitor cells, which may be present at birth or accumulated over time resulting in cells possessing the ability for uncontrolled growth and propagation (36–39). Recent studies have also observed the ability of non-CSCs to “de-differentiate” into CSCs due to epigenetic or environmental factors, which further increases the complexity of tumor biology and treatment (40). Cancer consists of a heterogeneous population of cells, proposed to arise from CSCs. Cells in a tumor are thought to be structured in a similar hierarchical manner to normal tissues, ranging from the most primitive cells to the most mature cells (Figure 4) (24, 41). Within a tumor, there may only be a small number of CSCs that are highly tumorigenic (Figure 3B) (16) and have the capacity to divide asymmetrically giving rise (1) to additional CSCs that migrate to form new tumors and (2) to downstream progenitor cells and differentiated cancer cells that possess no or low tumorigenic potential (42) and form the main bulk of the tumor (38, 41, 43).
It is important to note that these two different hypotheses may not be mutually exclusive, as clonal evolution has been shown to play a role in the formation of CSCs (44, 45).
A combination of clinical evaluation and genome-wide expression profiling has revealed that high-grade gliomas can be separated into four subtypes: proneural (PN), MES, neural, and proliferative (or classical) (15, 46). There remains some debate regarding the number and defining characteristics of these subtypes (46), but some criteria, such as chromosomal deletions and molecular markers (such as Notch and VEGF) have been proposed (47). The existence of multiple subtypes provides another explanation for therapy resistance in GBM, which needs to be taken into account when characterizing GBM cells (7). This adds another level of complexity to the study of GBM, as in addition to the known intra-tumoral cellular heterogeneity, there is also a degree of inter-tumor cellular heterogeneity.
In addition to the tumor subtypes, CSCs isolated from high-grade gliomas are also categorized into two distinct groups: PN and MES (48, 49). Several studies have adopted the term glioma stem cells to describe CSCs found in GBM (40, 49, 50), but for the purpose of clearly differentiating between stem cells in lower grade gliomas and those found in GBM, this review will use the term glioblastoma cancer stem cells (GBCSCs). GBCSCs are thought to originate from either neuronal stem cells or de-differentiate from normal brain cells, such as astrocytes and oligodendrocytes (18, 40), although this de-differentiation is not universally accepted (46). PN GBCSCs appear to share similarities with fetal NSCs, while MES GBCSCs more closely resemble adult NSCs (46, 51). MES GBCSCs are more aggressive, invasive, angiogenic, and resistant to radiotherapy than PN GBCSCs. MES GBCSCs are predominantly derived from primary GBMs that arise de novo, whereas PN GBCSCs reside in both Grade III gliomas and GBM (49, 52). Primary GBM can also contain multiple (polygenomic) or single (monogenomic) tumor cell clones and different genetic clones impact on tumorigenesis differently (53). However, even genetically diverse clones possess the stem cell markers CD133, CD15, A2B5, and CD44 (53), which suggests that despite the large amount of inter- and intra-tumor cell heterogeneity and the influence of the brain tumor microenvironment (54), at least some (if not all) stem cell markers remain consistent, thereby providing arguably one of the best targets for cancer therapy.
ESCs were originally identified and characterized from cells of the inner cell mass (ICM) in an embryonic blastocyst (55–57). ESCs and their more differentiated progeny all express a variety of markers, ranging from surface markers to transcription factors (57). As cells with stem cell properties isolated from cancers have been proposed to originate from ESCs, they also express these markers, making it possible to both identify and isolate CSCs using these markers (57, 58). Schoenhals et al. (58) showed that at least one of the ESC markers OCT-4, SOX2, KLF4, and c-Myc was expressed in 18 out of 40 cancer types investigated. Takahashi et al. (59) obtained induced pluripotent stem cells (iPSC) by transduction of these same markers into murine embryonic fibroblasts. iPSCs are made by reprograming adult somatic cells via transfection with specific markers, causing them to de-differentiate and regain ESC-like characteristics (60). Many more studies using additional ESC markers have resulted in a growing body of evidence for the presence of CSCs in many cancer types (61, 62). CSCs have been identified in lung (63), breast (64), head and neck (65), prostate (66, 67), pancreatic (68, 69), and colon (70, 71) cancers. GBCSCs were first identified by Ignatova et al. (72) and their presence has been confirmed in several other studies (20, 73–77). The list of proposed GBCSC markers includes CD133, nestin, NANOG, SALL4, STAT3, SOX2, c-Myc, Olig2, Bmi1, CD44, L1CAM, and KLF4 (1, 78–81).
The literature on normal ESCs describes a hierarchical differential expression pattern of stem cell markers, with ESC markers at the top of the hierarchy and progenitor cell markers more downstream (20, 28, 82–84). This is consistent with the observation that during formation of blood cells, HSCs rarely divide (85), making them slow in responding to any environmental changes and that they produce slightly more differentiated progenitor cells (Figure 5) (86). These cells are more numerous and proliferative, making them much more adaptable to change but still able to drive blood cell formation (87). Evidence for core stem cells and more malleable progenitor cells has also been found in some forms of cancer (38, 88, 89). In GBM, despite the known cellular heterogeneity present both within tumors and between patients (40, 49, 53) and the discovery of GBCSCs (52), characterization studies of the stem cell markers present on different GBCSC subtypes or at different tumor stages are relatively rare. The hierarchical model of cancer which proposes that a core group of stem cells exists at the top of the tumor hierarchy, from which other more differentiated cells are formed, descending from the most primitive cells to the most mature cells that make up the bulk of the tumor mass, remains relatively unexplored in GBM.
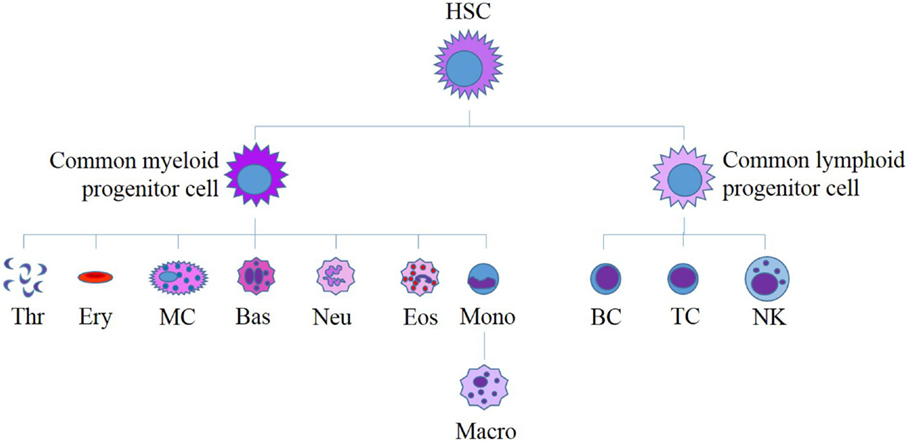
Figure 5. Current model for human hematopoiesis. All myeloid and lymphoid cells originate from a single hematopoietic stem cell (HSC). HSCs differentiate to form myeloid and lymphoid progenitor cells, which in turn differentiate to produce all of the diverse cells found in human blood. HSC, hematopoietic stem cell; Thr, thrombocytes; Ery, erythrocytes; MC, mast cell; Bas, basophil; Neu, neutrophil; Eos, eosinophil; Mono, monocyte; Macro, macrophage; B, B-cell; T, T-cell, NK, natural killer cell. Adapted from Bartis and Pongracz (86).
This review aimed to provide a perspective on CSC markers SALL4, OCT-4, SOX2, STAT3, NANOG, c-Myc, KLF4, CD133, CD44, nestin, and Glial fibrillary acidic protein (GFAP) reported in GBM, and attempt to place these markers in the context of the GBM CSC hierarchy, from the most primitive ESC markers to the more mature. These markers are presented in two categories: embryonic CSC markers and neural progenitor CSC markers.
SALL4 is a spalt-like C2H2 zinc-finger transcription factor that is expressed on ESCs in a similar manner to OCT-4 and SOX2 (90, 91). SALL4 is mandatory for the development of the ICM to ensure zygotic survival and maintenance of ESC pluripotency (90, 92, 93). Interaction between SALL4 and NANOG has also been confirmed by co-immunoprecipitation experiments and it has been suggested that they work together in a similar pairwise manner to ESC markers OCT-4 and SOX2 to regulate transcription (94). However, other evidence suggests that, as well as NANOG, this regulatory function also has a greater involvement of OCT-4, SOX2, c-Myc, and KLF4. Yang et al. (91) report that when the level of the SALL4 protein is reduced, the levels of all four of the aforementioned ESC proteins also decrease, suggesting a relatively diverse role for SALL4. SALL4 plays a role in multiple types of cancers (95–97) and has been previously used as a CSC marker. It has also been demonstrated that SALL4 is expressed at a higher level in gliomas than in normal brain tissue and that increased levels correlate with a poor prognosis (80). Additionally, inhibition of SALL4 reduces cellular proliferation in gliomas and stimulates apoptosis (98). Di Tomaso et al. (99) find that CSCs in GBM express SALL4 and that these same cells also express NANOG. However, the use of SALL4 as a marker for CSCs in GBM is limited to a small number of reports (80, 98, 100, 101).
Together with NANOG, the transcription factor, OCT-4, is required for propagation of ESCs and they both work synergistically with SOX2 to achieve this regulation (102). OCT-4 is essential for pluripotency and mammalian embryonic development (103). It has also been associated with cancer, functioning as a driver for the self-renewal of CSCs (78, 104). OCT-4 is expressed by glioma cells, but not normal brain tissue, and is implicated in the pathogenesis of GBM (105, 106). Indeed, OCT-4 together with SOX2 and NANOG is expressed in most if not all gliomas and their expression correlates with tumor aggressiveness with GBM cells showing greater nuclear staining for OCT-4 and SOX2 (104). Furthermore, most cells expressing OCT-4 also express SOX2 and NANOG (104). Hence, OCT-4, SOX2, and NANOG are thought to be key players in the transcriptional regulation of CSCs.
SOX2 is a member of the family of transcriptional co-factors that are associated with various developmental milestones and is over-expressed in tumors (107, 108). It plays a role in maintaining pluripotency in several types of cancer, including rectal (109), breast (110), and lung (111) cancers. SOX2 is also over-expressed in GBM with little detected in normal brain tissue (1). Additionally, GBM demonstrates greater SOX2 mRNA expression than lower grade tumors (104). Along with OCT-4 and NANOG, SOX2 has been used in numerous studies to characterize iPSCs derived from somatic cells (59, 112, 113), demonstrating that SOX2 is critical for stem cell maintenance. Furthermore, SOX2 inhibition using shRNA halts tumor growth when GBM cells are transplanted into immunodeficient mice (114).
Although SOX2 has been implicated as a transcriptional regulator, it has also been proposed as a neural progenitor cell marker. Expression of SOX2 is essential for cells from the neural tube in chicks to maintain progenitor characteristics (115). There is also a body of evidence on co-expression of SOX2 and nestin and as such SOX2 has been used in some studies as a progenitor cell marker (116–119). Ellis et al. (120) show SOX2 as a persistent NSC marker that is present throughout the entire development of the mouse. The expression of SOX2, therefore, appears to be maintained even after stem cells have progressed through different stages of differentiation.
Signal transducers and activators of transcription (STAT) proteins are both activated by cytokines and regulate many cytokine and growth factor responses (121). STAT3 has more generalized functions than the rest of the STAT family and has been implicated in cell-cycle signaling, cell survival, and ESC self-renewal and pluripotency (122–124). The latter activity has been proposed to be maintained via the leukemia inhibitory factor (LIF) pathway, in which LIF binds to its receptor and produces phosphorylation of STAT3 that subsequently translocates to the nucleus, triggering the expression of other ESC-associated proteins, such as KLF4, SOX2, SALL4, and c-Myc (62, 125, 126). Loss of STAT3 expression has been shown to reduce ESC self-renewal, but enhance cellular differentiation, resulting in embryo lethality in mice (127). This would, therefore, indicate that ESC expression of STAT3 is essential, but that it also performs multiple other functions in adult tissues, such as cytokine release and cell signaling (124), suggesting STAT3 expression persists on further differentiated cells and, therefore, cannot be used purely as a primitive ESC marker.
Abnormal STAT3 signaling has also been associated with promoting cellular proliferation, weakening the immune system, and promoting angiogenesis and inflammation in cancer (123, 128). There is ample evidence for the role of STAT3 in cancer, with its activation contributing to cancers in the head and neck (129), breast (130), prostate (131), thyroid (132), skin (melanoma) (133), and GBM (134–136). In comparison to normal brain tissue and cells, particularly astrocytes, GBM expresses high levels of STAT3 and inhibition of this molecule results in the induction of apoptosis and cessation of tumor proliferation (81). Multiple subsequent studies of STAT3 in GBM have demonstrated its downregulation or inhibition leads to reduced tumor growth, suggesting a potential target for cancer treatment (137–141). However, as STAT3 is also required for non-cancer cell function, any form of inhibition will not be specific to the tumor and will likely result in major side effects for the patient (7, 142).
NANOG is an ESC transcription factor and its expression has been associated with multiple types of cancer, including those affecting the lung (143), oral cavity (144), breast (145, 146), and prostate (147). It has also been implicated in the regulation of GBM and has been found to be highly expressed in stem cells extracted from the cerebellum and medulloblastoma (104, 148–150). NANOG modulates GBM stem cell tumorigenicity, clonogenicity, and proliferation (151). Inhibition of NANOG in GBM prevents tumor proliferation and invasion (152). It is proposed that together with OCT-4 and SOX2, NANOG is responsible for ESCs’ capacity to maintain their pluripotency and self-renewal (7, 153). Deletion of NANOG from murine ESCs results in a loss of pluripotency (154) and NANOG has been used as a marker in the induction of pluripotent stem cell characteristics in normal human fibroblasts (112, 155). Current data implicate a role for NANOG in the regulation GBCSCs.
c-Myc is a member of the family of Myc genes, although only c-Myc, l-Myc, and N-Myc have been linked to tumor growth, and as such they have been termed nuclear oncogenes (156, 157). Upregulated c-Myc has been linked to cellular proliferation (158, 159). The deletion of c-Myc from rat fibroblast lines resulted in a prolonged cellular doubling time (160) and proved fatal to murine embryos, indicating its importance in embryonic development (161). Furthermore, c-Myc can be used to induce cellular de-differentiation, resulting in iPSCs (112).
c-Myc has been implicated in the pathogenesis of lung (162), pancreatic (163), prostate (164), and breast (165, 166) cancers as well as medulloblastoma (167) and GBM (168). Despite its experimental use in generating iPSCs, there is evidence indicating that c-Myc may be more of a marker for progenitor cells rather than ESCs. Successful generation of iPSCs without the expression of c-Myc implies that the oncogene is not essential for cellular de-differentiation (169). Additionally, in normal lung tissue c-Myc expression is strongest in hyperplastic alveolar type II pneumocytes, also known as bronchopulmonary progenitor cells (170). c-Myc also enhances the tumor forming capacity of nestin-expressing progenitor cells in medulloblastoma (171). This would suggest that c-Myc is expressed on progenitor cells, although its role as a neural progenitor cell marker is not fully established. Despite this, c-Myc has been strongly associated with GBM, CSC maintenance, and self-renewal, and its over-expression has been correlated with the poor prognosis of GBM (168, 171–173).
Krüppel-like factor 4 (KLF4) is a transcription factor involved in cell proliferation, differentiation, and apoptosis (174). It is a member of the KLF family characterized by the presence of Cys2/His2 zinc fingers (57, 175). KLF4 is essential for the maintenance of pluripotency and self-renewal of ESCs (176, 177) and is one of the factors, along with OCT-4 and SOX2, required to re-program fibroblasts to generate iPSCs (59, 112, 169). It is, therefore, not surprising that KLF4 over-expression is associated with cancer (58, 178). KLF4 was first identified as a potential oncogene in 1999 (179) and since then its over-expression has been shown to induce cellular dysplasia, similar to that found in squamous cell carcinomas (180). More recently, it has been shown that KLF4 is over-expressed in 70% of breast cancer specimens (178). However, there is growing evidence indicating that KLF4 actually inhibits tumor formation and metastasis in many types of cancer (181–185).
A possible explanation for these discrepancies has been proposed, suggesting that the cell-cycle inhibitor p21 can act as a “switch” between suppression and proliferation (186). It is hypothesized that KLF4 can activate p21-induced cell-cycle arrest and prevent tumor proliferation, but can also inhibit p53, blocking both cell senescence and apoptosis. These responses are also thought to be influenced by the cellular context. For example, it has been theorized that inhibition of p21 by additional pathways such as Ras or the adenoviral oncoprotein E1A can override the activation signals of KLF4. Therefore, inhibition of both apoptosis via p53 and cell-cycle arrest via p21 induces tumor formation. KLF4 expression in the first scenario can produce completely opposite outcomes for different pathways, yet in the second produces only one outcome no matter which pathway is activated. This observation may explain the aforementioned contrasting results for KLF4 and has been supported by further studies in the area of cell-cycle regulation (187–189), although the exact mechanism of “switching” remains unclear. Unfortunately, the apparent heavy reliance of KLF4 function on other proteins and inconsistencies in its expression make it difficult to use KLF4 as a CSC marker.
Information on KLF4 expression in GBM is limited. An analysis of gene expression data indicates that KLF4 is over-expressed in brain tumors, with no specific data on GBM (58). A more recent study shows that micro-RNA targeting of KLF4 suppresses tumor growth in GBM cells (190) but the role of KLF4 in GBM remains undetermined.
The nestin gene (previously known as Rat. 401), the neuroepithelial stem cell gene, encodes a novel intermediate filament that does not fit into one of the five classes of intermediate filaments that have already been defined (191). Nestin is expressed in several types of cancer (191–193) and it is strongly associated with GBM (20, 21, 73, 194–196). Increased nestin expression has been associated with higher grade gliomas and lower patient survival rates (197). Additionally, inducing differentiation of GBM cells leads to downregulation of nestin (198). It also binds to a large percentage of cells in the mammalian embryonic brain and its presence is correlated with cellular propagation during the development of the central nervous system (199, 200). These data, plus the observation that nestin-expressing cells have the ability to differentiate into multiple cell types (72), implicate nestin as an effective stem cell marker. However, existing evidence indicates that nestin is more of a neural progenitor cell marker as it is found on immediate neuron precursor cells (199, 201) and is downregulated when precursor cells differentiate into glial cells or neurons (201). Nestin is currently used as a marker for cells immediately preceding the dedication of CNS cells to a restricted lineage (202).
Glial fibrillary acidic protein is an astrocyte maturation marker commonly used as a histological marker for tumors of glial origin known to be involved in normal astrocyte functions (203, 204). GFAP has been used previously to identify differentiated cells (20, 73), but some evidence indicates that astrocytes found in the subventricular zone (SVZ) of the mammalian adult brain are actually NSCs and are precursors to neurons (205). NSCs from the postnatal and adult brain have been found to express GFAP, but not NSCs from the early embryonic brain (206), indicating that GFAP is a marker of more mature glial cells. This would, therefore, suggest that GFAP is a progenitor rather than an ESC marker. GFAP has been previously shown to be co-expressed with nestin in GBM cells (207) and is over-expressed in the serum and peripheral blood of GBM patients in comparison to healthy controls (208, 209). However, the proportion of GBM patients with GFAP positivity varied greatly between these two studies. The study on serum found GFAP over-expression in 80% of GBM cases (208) [a finding that has been replicated recently (210)], whereas the peripheral blood study found that GFAP was over-expressed in only 20.6% of patients. It is possible that this discrepancy is due to the heterogeneous nature of GBM and that GFAP may be preferentially expressed in certain GBM subtypes or even subtypes of GBCSCs. Nevertheless, both studies indicate the migration of GBM cells outside of the CNS, and this is unexpected given that clinically recognizable hematogenous metastasis from GBM is extremely rare (211). While the relationship between GFAP and GBM metastasis clearly requires further elucidation, GFAP staining is considered a standard diagnostic marker for GBM for samples taken within the CNS (208, 212–214).
CD133, also known as prominin-1, is a protein found on plasma membrane projections and is one of the cluster of differentiation (CD) antigens (215). CD133 is expressed on HSCs (216) and was also found on NSCs in 2003 (20). Singh et al. (20) have identified stem-like cells lacking the expression of neural differentiation markers in pediatric brain tumors that express CD133, and showed that CD133+ human GBM cells can initiate tumor formation in the brains of immunodeficient mice (21). Interestingly, these cells also express nestin, indicating the possibility of CD133 expression on progenitor cells. CD133 expression has since been implicated in other cancers, including prostate and colorectal cancer, and an increased proportion of CD133+ cells in a tumor correlates with poorer survival (30, 47, 217). GBM tumors that have recurred after radiotherapy or chemotherapy contain an increased percentage of cells that are CD133+ compared with the original tumor, presumably due to increased progenitor cell activation (42). Similarly, the CD133+ gene transcription signal can distinguish GBM from low-grade tumors and its expression has been attributed to the aggressiveness of the tumor (218). These suggest a key role for CD133 in tumor recurrence and invasion.
However, not all stem cells express CD133. Subsequent studies have shown that tumors grow successfully from CD133− stem-like cells in xenograft models (74, 217, 219) and so identification of CSC cannot be solely based on CD133 expression. As CD133 is not essential for tumor formation, this implies that it is not present on all ESCs. Therefore, CD133 cannot be considered an ESC marker, but is further along on the stem cell hierarchy and can be considered as a marker for progenitor cells.
CD44 is a transmembrane glycoprotein and the receptor for the glycosaminoglycan hyaluronan (HA) (220, 221). It is found in a variety of tissues and is expressed on embryonic epithelia during development (222). Multiple isoforms of CD44 exist, altered through splicing and post-translational modifications. CD44s is the most common isoform, but other variants (CD44v) also exist (223). The potential for hundreds of variations on the CD44 receptor may contribute to its involvements in various pathways, including lymphocyte activation, angiogenesis, cytokine release, and cellular adhesion (222). Additionally, CD44 has been implicated in colorectal (224, 225), prostate (67, 226, 227), breast (64, 228–230), head and neck (65, 231), and non-small-cell lung (232) cancers. CD44+ cells can generate new tumors similar to the original tumor when xenografted onto mice, but CD44− cells cannot achieve this. (65). These findings are consistent with those indicating that CD44+ cells from prostate cancer are more proliferative than CD44− cells and that they also possess some progenitor cell properties (67).
CD44 activates NANOG in breast and ovarian cancers (145) demonstrating a role for CD44 in the regulation of ESCs, supported by the finding that in cancer CD44+ cells also express Bmi1 (65). Moreover, tumor aggressiveness and growth can be inhibited by preventing the HA-CD44 interaction (233). This observation suggests a key role for CD44 and its ligand in the development of cancer, although opposing views exist.
Increased CD44 expression is associated with a better outcome in thyroid cancer (234). Similar findings have also been demonstrated in ovarian cancer (235), non-small-cell lung cancer (236), and soft tissue sarcomas (237) although high expression of CD44 is also correlated with increased risks of recurrence (237). This variation is likely due to the CD44 isoform examined, as each study uses a different variant or epitope or CD44 as a whole. Caution is needed when interpreting the data in the attempt to elucidate the precise role of CD44 in cancer.
Li et al. (238) show that CD44 variants are not expressed by GBM, but only in metastases originating from the brain. However, a subsequent study shows that CD44 variants are expressed in 100% of all GBM cell lines and tumors (239). This latter finding has been supported by a more comprehensive study using immunohistochemical staining that demonstrates cells from GBM express CD44s and several other variants of CD44 (240). Furthermore, inhibition of CD44 prevents progression of GBM, indicating a definite role in tumorigenesis (241). However, different GBM cell lines have varying expression of CD44 (242). These results show changeable expression of CD44 in cancer, with some studies finding high expression while others show low expression in the same cancer type (67), indicating CD44 is not essential for tumor formation. Consistent with this observation is the current hypothesis that CD44 is a progenitor cell marker as opposed to an ESC marker. An extensive study of the expression of CD44 in mouse cerebellum show this cell surface marker to be co-expressed with nestin, SOX2, astrocyte specific glutamate transporter and brain lipid binding protein (BLBP), all of which are specific to neural stem/progenitor cells (243). CD44 is also co-expressed with the oligodendrocyte progenitor marker Olig2. This evidence would infer that CD44 is a progenitor cell marker, as it is present on partially differentiated cells.
While there is growing evidence supporting the CSC model of cancer, the field of CSCs in glial tumors remains relatively understudied, as evidenced by the difficulties in identification and characterization of this primitive population. In this article, we review a number of markers published in the recent literature and evaluated their usefulness in CSC research, in the context of GBM. While some markers are key to the identification of CSCs, others have a less defined association, requiring more study to define their precise role in carcinogenesis. It is becoming increasingly apparent that the hierarchical system observed in normal stem cells, such as HSCs, and in other forms of cancer, also applies to GBM.
Cells in GBM express OCT-4 (104, 105), SOX2 (1, 104, 114), pSTAT3 (81, 134), NANOG (104, 152), SALL4 (80, 99), c-Myc (78, 168, 172), KLF4 (58, 190), nestin (20, 21), CD44 (239, 240), CD133 (21, 218), and GFAP (207, 208), highlighting an overlapping hierarchical and heterogeneous population of stem and progenitor cells within GBM. This paper attempts to categorize each of the markers into one of two categories, (1) ESC markers and (2) progenitor cell markers, based on current evidence. NANOG, SALL4, OCT-4, KLF4, SOX2, and pSTAT3 all have essential roles in embryonic development, indicating that they must be expressed on more primitive cells. However, SOX2 and pSTAT3 are also expressed in cells that are more differentiated than ESCs, indicating that expression of a particular marker is not restricted to one cell type or developmental stage.
Current evidence suggests that GFAP, nestin, CD44, and CD133 are found further down in the stem cell hierarchy as they are expressed on more differentiated cells. Although involved in the modulation of tumor aggressiveness, both CD133 (74, 217, 219) and CD44 (67, 243) do not appear to be essential for cancer formation, leading to the inference that these markers represent markers of more differentiated progenitor cells in the hierarchy although they may be co-expressed with nestin and GFAP in “higher up” progenitors. The presence of the same markers on multiple cell types highlights the importance of using multiple markers to properly define and distinguish the most primitive CSCs from their specific but varying downstream lineage and progenitor cells. Such models have already been proposed for non-cancerous stem cells (Figure 4) (24); and in the case of HSCs, a number of markers have been assigned to specific stages of blood cell development (244). A similar paradigm for markers for GBM is currently missing from the literature and establishing a properly defined model of the hierarchy will improve the understanding CSCs in GBM. Additionally, it is important to remember that the stages of stem cell development and maturation are likely not static or strictly defined, but more of a free flowing continuum upon which multiple variations of stem cells can be found. This is particularly important given the high degree of intra- and inter-tumor cellular heterogeneity that is already known to exist within GBM.
Although it has been shown that stem cell markers such as CD133 and CD44 persist on genetically diverse clones (53), the presence of more primitive markers, such as OCT-4, SALL4, NANOG, SOX2, c-Myc, KLF4, and pSTAT3, on different GBM or GBCSC subtypes has not been defined. The ability to identify these extremely primitive CSCs may be key to developing novel and effective treatments for GBM. Finding markers that are consistently expressed by CSC populations within different GBM subtypes may enable effective targeting of CSCs by destroying the “roots” of the cancer. Properly defining the CSC markers will underscore precise identification and characterization of the CSC population in GBM.
TI and STT initiated and conceptualized the review. AB drafted the manuscript. AW, ST, LP, PD, and TI critically examined and commented on the manuscript. All authors revised and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Schmitz M, Temme A, Senner V, Ebner R, Schwind S, Stevanovic S, et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br J Cancer (2007) 96(8):1293–301. doi: 10.1038/sj.bjc.6603802
2. Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci U S A (2000) 97(12):6242–4. doi:10.1073/pnas.97.12.6242
3. Lonser RR, Walbridge S, Vortmeyer AO, Pack SD, Nguyen TT, Gogate N, et al. Induction of glioblastoma multiforme in nonhuman primates after therapeutic doses of fractionated whole-brain radiation therapy. J Neurosurg (2002) 97(6):1378–89. doi:10.3171/jns.2002.97.6.1378
4. Surawicz TS, Davis F, Freels S, Laws ER, Menck HR. Brain tumor survival: results from the National Cancer Data Base. J Neurooncol (1998) 40(2):151–60. doi:10.1023/A:1006091608586
5. Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res (2009) 7(7):989–99. doi:10.1158/1541-7786.MCR-09-0030
6. Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res (2002) 62(1):200–7.
7. Seymour T, Nowak A, Kakulas F. Targeting aggressive cancer stem cells in glioblastoma. Front Oncol (2015) 5:159. doi:10.3389/fonc.2015.00159
8. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi:10.1016/S1470-2045(09)70025-7
9. Milano MT, Okunieff P, Donatello RS, Mohile NA, Sul J, Walter KA, et al. Patterns and timing of recurrence after temozolomide-based chemoradiation for glioblastoma. Int J Radiat Oncol Biol Phys (2010) 78(4):1147–55. doi:10.1016/j.ijrobp.2009.09.018
10. Outlines P. CNS Tumor Astrocytic Tumors Glioblastoma Multiforme. (2013). Available from: http://www.pathologyoutlines.com/topic/cnstumorglioblastoma.html
11. Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol (2006) 65(6):529–39. doi:10.1097/00005072-200606000-00001
12. Moore K, Kim L. Primary brain tumours: characteristics, practical diagnostic and treatment approaches. In: Ray SK, editor. Glioblastoma: Molecular Mechanisms of Pathogenesis and Current Therapeutic Strategies. New York: Springer Science + Business Media, LLC (2009). p. 43–75.
13. Liu Q, Liu Y, Li W, Wang X, Sawaya R, Lang FF, et al. Genetic, epigenetic, and molecular landscapes of multifocal and multicentric glioblastoma. Acta Neuropathol (2015) 130(4):587–97. doi:10.1007/s00401-015-1470-8
14. Kim DH, Mohapatka G, Bollen A, Waldman FM, Feuerstein BG. Chromosomal abnormalities in glioblastoma multiforme tumors and glioma cell lines detected by comparative genomic hybridization. Int J Cancer (1995) 60(6):812–9. doi:10.1002/ijc.2910600615
15. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi:10.1016/j.ccr.2009.12.020
16. Adams JM, Strasser A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res (2008) 68(11):4018–21. doi:10.1158/0008-5472.CAN-07-6334
17. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell (2009) 138(5):822–9. doi:10.1016/j.cell.2009.08.017
18. Schneider M, Ströbele S, Nonnenmacher L, Siegelin MD, Tepper M, Stroh S, et al. A paired comparison between glioblastoma “stem cells” and differentiated cells. Int J Cancer (2016) 138(7):1709–18. doi:10.1002/ijc.29908
19. Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol (2010) 5:51. doi:10.1146/annurev-pathol-121808-102113
20. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res (2003) 63(18):5821–8.
21. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432(7015):396–401. doi:10.1038/nature03128
22. Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell (2011) 8(5):482–5. doi:10.1016/j.stem.2011.04.013
23. Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, et al. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest (2007) 117(5):1305. doi:10.1172/JCI30740
24. Gage FH. Mammalian neural stem cells. Science (2000) 287(5457):1433–8. doi:10.1126/science.287.5457.1433
25. Billing AM, Dib SS, Hamidane HB, Goswami N, Al-mismar R, Cotton R, et al., editors. Comprehensive characterization of the differentiation of human embryonic stem cells into mesenchymal stem cells. Qatar Foundation Annual Research Conference. Doha, Qatar (2014).
26. Rafii S, Kloss CC, Butler JM, Ginsberg M, Gars E, Lis R, et al. Human ESC-derived hemogenic endothelial cells undergo distinct waves of endothelial to hematopoietic transition. Blood (2013) 121(5):770–80. doi:10.1182/blood-2012-07-444208
27. Doulatov S, Vo LT, Chou SS, Kim PG, Arora N, Li H, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell (2013) 13(4):459–70. doi:10.1016/j.stem.2013.09.002
28. Dietrich J, Imitola J, Kesari S. Mechanisms of disease: the role of stem cells in the biology and treatment of gliomas. Nat Clin Pract Oncol (2008) 5(7):393–404. doi:10.1038/ncponc1132
29. Zhao X, Das AV, Thoreson WB, James J, Wattnem TE, Rodriguez-Sierra J, et al. Adult corneal limbal epithelium: a model for studying neural potential of non-neural stem cells/progenitors. Dev Biol (2002) 250(2):317–31. doi:10.1006/dbio.2002.0793
30. Zeppernick F, Ahmadi R, Campos B, Dictus C, Helmke BM, Becker N, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res (2008) 14(1):123–9. doi:10.1158/1078-0432.CCR-07-0932
31. Suga H, Matsumoto D, Eto H, Inoue K, Aoi N, Kato H, et al. Functional implications of CD34 expression in human adipose-derived stem/progenitor cells. Stem Cells Dev (2009) 18(8):1201–10. doi:10.1089/scd.2009.0003
32. Horky LL, Galimi F, Gage FH, Horner PJ. Fate of endogenous stem/progenitor cells following spinal cord injury. J Comp Neurol (2006) 498(4):525–38. doi:10.1002/cne.21065
33. Tanaka M, Itoh T, Tanimizu N, Miyajima A. Liver stem/progenitor cells: their characteristics and regulatory mechanisms. J Biochem (2011) 149(3):231–9. doi:10.1093/jb/mvr001
34. Vankelecom H. Pituitary stem/progenitor cells: embryonic players in the adult gland? Eur J Neurosci (2010) 32(12):2063–81. doi:10.1111/j.1460-9568.2010.07523.x
35. Seaberg RM, van der Kooy D. Stem and progenitor cells: the premature desertion of rigorous definitions. Trends Neurosci (2003) 26(3):125–31. doi:10.1016/S0166-2236(03)00031-6
36. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367(6464):645–8. doi:10.1038/367645a0
38. Shipitsin M, Polyak K. The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab Invest (2008) 88(5):459–63. doi:10.1038/labinvest.2008.14
39. Kim EJ, Khan GN, Griffith K, Greenson J, Takebe N, Zalupski M, et al. Abstract A40: Cancer stem cells (CSC) and inhibition of hedgehog (Hh) pathway signaling in advanced pancreatic cancer: GDC-0449 in combination with gemcitabine (Gem). Cancer Res (2012) 72(Suppl 14):A40. doi:10.1158/1538-7445.panca2012-a40
40. Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis (2015) 2:152–63. doi:10.1016/j.gendis.2015.02.001
41. Foreman KE, Rizzo P, Osipo C, Miele L. The cancer stem cell hypothesis. In: Bagley RG, Teicher BA, editors. Stem Cells and Cancer. New York: Springer Science + Business Media, LLC (2009). p. 3–14.
42. Tamura K, Aoyagi M, Ando N, Ogishima T, Wakimoto H, Yamamoto M, et al. Expansion of CD133-positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy: laboratory investigation. J Neurosurg (2013) 119(5):1145–55. doi:10.3171/2013.7.JNS122417
43. Diehn M, Cho RW, Clarke MF. Therapeutic implications of the cancer stem cell hypothesis. Semin Radiat Oncol (2009) 19(2):78–86. doi:10.1016/j.semradonc.2008.11.002
44. Barabé F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science (2007) 316(5824):600–4. doi:10.1126/science.1139851
45. Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest (2007) 117(9):2562–9. doi:10.1172/JCI30890
46. Morokoff A, Ng W, Gogos A, Kaye A. Molecular subtypes, stem cells and heterogeneity: implications for personalised therapy in glioma. J Clin Neurosci (2015) 22:1219–26. doi:10.1016/j.jocn.2015.02.008
47. Jhanwar-Uniyal M, Labagnara M, Friedman M, Kwasnicki A, Murali R. Glioblastoma: molecular pathways, stem cells and therapeutic targets. Cancers (2015) 7(2):538–55. doi:10.3390/cancers7020538
48. Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell (2013) 24(3):331–46. doi:10.1016/j.ccr.2013.08.001
49. Mao P, Joshi K, Li J, Kim S-H, Li P, Santana-Santos L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A (2013) 110(21):8644–9. doi:10.1073/pnas.1221478110
50. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell (2009) 15(6):501–13. doi:10.1016/j.ccr.2009.03.018
51. Ricci-Vitiani L, Pallini R, Larocca LM, Lombardi DG, Signore M, Pierconti F, et al. Mesenchymal differentiation of glioblastoma stem cells. Cell Death Differ (2008) 15(9):1491–8. doi:10.1038/cdd.2008.72
52. Nakano I. Stem cell signature in glioblastoma: therapeutic development for a moving target. J Neurosurg (2015) 122(2):324–30. doi:10.3171/2014.9.JNS132253
53. Stieber D, Golebiewska A, Evers L, Lenkiewicz E, Brons NH, Nicot N, et al. Glioblastomas are composed of genetically divergent clones with distinct tumourigenic potential and variable stem cell-associated phenotypes. Acta Neuropathol (2014) 127(2):203–19. doi:10.1007/s00401-013-1196-4
54. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia (2012) 60(3):502–14. doi:10.1002/glia.21264
55. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature (1981) 292(5819):154–6. doi:10.1038/292154a0
56. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science (1998) 282(5391):1145–7. doi:10.1126/science.282.5391.1145
57. Zhao W, Ji X, Zhang F, Li L, Ma L. Embryonic stem cell markers. Molecules (2012) 17(6):6196–236. doi:10.3390/molecules17066196
58. Schoenhals M, Kassambara A, De Vos J, Hose D, Moreaux J, Klein B. Embryonic stem cell markers expression in cancers. Biochem Biophys Res Commun (2009) 383(2):157–62. doi:10.1016/j.bbrc.2009.02.156
59. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
60. Kim D, Kim C-H, Moon J-I, Chung Y-G, Chang M-Y, Han B-S, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell (2009) 4(6):472. doi:10.1016/j.stem.2009.05.005
61. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell (2014) 14(3):275–91. doi:10.1016/j.stem.2014.02.006
62. Zhao X, Sun X, Li XL. Expression and clinical significance of STAT3, P-STAT3, and VEGF-C in small cell lung cancer. Asian Pac J Cancer Prev (2012) 13(6):2873–7. doi:10.7314/APJCP.2012.13.6.2873
63. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ (2008) 15(3):504–14. doi:10.1038/sj.cdd.4402283
64. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100(7):3983–8. doi:10.1073/pnas.0530291100
65. Prince M, Sivanandan R, Kaczorowski A, Wolf G, Kaplan M, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A (2007) 104(3):973–8. doi:10.1073/pnas.0610117104
66. Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol (2008) 26(17):2862–70. doi:10.1200/JCO.2007.15.1472
67. Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene (2006) 25(12):1696–708. doi:10.1038/sj.onc.1209327
68. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell (2007) 1(3):313–23. doi:10.1016/j.stem.2007.06.002
69. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res (2007) 67(3):1030–7. doi:10.1158/0008-5472.CAN-06-2030
70. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature (2007) 445(7123):106–10. doi:10.1038/nature05372
71. Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. J Mol Med (2009) 87(11):1097–104. doi:10.1007/s00109-009-0518-4
72. Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia (2002) 39(3):193–206. doi:10.1002/glia.10094
73. Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann-Hogiu S, Farkas DL, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene (2004) 23(58):9392–400. doi:10.1038/sj.onc.1208311
74. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, et al. CD133+ and CD133- glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res (2007) 67(9):4010–5. doi:10.1158/0008-5472.CAN-06-4180
75. Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells (2009) 27(8):1722–33. doi:10.1002/stem.98
76. Piccirillo SG, Binda E, Fiocco R, Vescovi AL, Shah K. Brain cancer stem cells. J Mol Med (2009) 87(11):1087–95. doi:10.1007/s00109-009-0535-3
77. Field M, Alvarez A, Bushnev S, Sugaya K. Embryonic stem cell markers distinguishing cancer stem cells from normal human neuronal stem cell populations in malignant glioma patients. Clin Neurosurg (2010) 57:151–9.
78. Li W, Wei W, Zhu S, Zhu J, Shi Y, Lin T, et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell (2009) 4(1):16–9. doi:10.1016/j.stem.2008.11.014
79. Olmez I, Shen W, McDonald H, Ozpolat B. Dedifferentiation of patient-derived glioblastoma multiforme cell lines results in a cancer stem cell-like state with mitogen-independent growth. J Cell Mol Med (2015) 19:1262–72. doi:10.1111/jcmm.12479
80. Zhang L, Yan Y, Jiang Y, Cui Y, Zou Y, Qian J, et al. The expression of SALL4 in patients with gliomas: high level of SALL4 expression is correlated with poor outcome. J Neurooncol (2015) 121(2):261–8. doi:10.1007/s11060-014-1646-4
81. Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene (2002) 21(55):8404–13. doi:10.1038/sj.onc.1206047
82. Shin S, Sun Y, Liu Y, Khaner H, Svant S, Cai J, et al. Whole genome analysis of human neural stem cells derived from embryonic stem cells and stem and progenitor cells isolated from fetal tissue. Stem Cells (2007) 25(5):1298–306. doi:10.1634/stemcells.2006-0660
83. Ratajczak MZ, Kucia M, Majka M, Reca R, Ratajczak J. Heterogeneous populations of bone marrow stem cells – are we spotting on the same cells from the different angles? Folia Histochem Cytobiol (2004) 42(3):139–46.
84. Cai C, Grabel L. Directing the differentiation of embryonic stem cells to neural stem cells. Dev Dyn (2007) 236(12):3255–66. doi:10.1002/dvdy.21306
85. Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci U S A (1999) 96(6):3120–5. doi:10.1073/pnas.96.6.3120
86. Bartis D, Pongrácz J. Three Dimensional Tissue Cultures and Tissue Engineering. Hungary: University of Pécs (2011). Retrieved from Digitalis Tankonyvtar.
87. Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med (2005) 202(11):1599–611. doi:10.1084/jem.20050967
88. Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat Rev Cancer (2003) 3(6):434–43. doi:10.1038/nrc1095
89. Villadsen R, Fridriksdottir AJ, Rønnov-Jessen L, Gudjonsson T, Rank F, LaBarge MA, et al. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol (2007) 177(1):87–101. doi:10.1083/jcb.200611114
90. Zhang J, Tam W-L, Tong GQ, Wu Q, Chan H-Y, Soh B-S, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol (2006) 8(10):1114–23. doi:10.1038/ncb1481
91. Yang J, Chai L, Fowles TC, Alipio Z, Xu D, Fink LM, et al. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc Natl Acad Sci U S A (2008) 105(50):19756–61. doi:10.1073/pnas.0809321105
92. Elling U, Klasen C, Eisenberger T, Anlag K, Treier M. Murine inner cell mass-derived lineages depend on Sall4 function. Proc Natl Acad Sci U S A (2006) 103(44):16319–24. doi:10.1073/pnas.0607884103
93. Lim CY, Tam W-L, Zhang J, Ang HS, Jia H, Lipovich L, et al. Sall4 regulates distinct transcription circuitries in different blastocyst-derived stem cell lineages. Cell Stem Cell (2008) 3(5):543–54. doi:10.1016/j.stem.2008.08.004
94. Wu Q, Chen X, Zhang J, Loh Y-H, Low T-Y, Zhang W, et al. Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J Biol Chem (2006) 281(34):24090–4. doi:10.1074/jbc.C600122200
95. Kobayashi D, Kuribayshi K, Tanaka M, Watanabe N. SALL4 is essential for cancer cell proliferation and is overexpressed at early clinical stages in breast cancer. Int J Oncol (2011) 38(4):933–9. doi:10.3892/ijo.2011.929
96. Kobayashi D, Kuribayashi K, Tanaka M, Watanabe N. Overexpression of SALL4 in lung cancer and its importance in cell proliferation. Oncol Rep (2011) 26(4):965–70. doi:10.3892/or.2011.1374
97. Oikawa T, Kamiya A, Zeniya M, Chikada H, Hyuck AD, Yamazaki Y, et al. Sal-like protein 4 (SALL4), a stem cell biomarker in liver cancers. Hepatology (2013) 57(4):1469–83. doi:10.1002/hep.26159
98. He J, Zhang W, Zhou Q, Zhao T, Song Y, Chai L, et al. Low-expression of microRNA-107 inhibits cell apoptosis in glioma by upregulation of SALL4. Int J Biochem Cell Biol (2013) 45(9):1962–73. doi:10.1016/j.biocel.2013.06.008
99. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res (2010) 16(3):800–13. doi:10.1158/1078-0432.CCR-09-2730
100. Jha P, Patric IRP, Shukla S, Pathak P, Pal J, Sharma V, et al. Genome-wide methylation profiling identifies an essential role of reactive oxygen species in pediatric glioblastoma multiforme and validates a methylome specific for H3 histone family 3A with absence of G-CIMP/isocitrate dehydrogenase 1 mutation. Neuro Oncol (2014) 16(12):1607–17. doi:10.1093/neuonc/nou113
101. Mei K, Liu A, Allan RW, Wang P, Lane Z, Abel TW, et al. Diagnostic utility of SALL4 in primary germ cell tumors of the central nervous system: a study of 77 cases. Mod Pathol (2009) 22(12):1628–36. doi:10.1038/modpathol.2009.148
102. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell (2005) 122(6):947–56. doi:10.1016/j.cell.2005.08.020
103. Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell (1998) 95(3):379–91. doi:10.1016/S0092-8674(00)81769-9
104. Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, et al. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology (2011) 59(4):763–75. doi:10.1111/j.1365-2559.2011.03993.x
105. Talsma CE, Flack CG, Zhu T, He X, Soules M, Heth JA, et al., editors. Oct4 regulates GBM neurosphere growth and its expression is associated with poor survival in GBM patients. Neuro-Oncology. Cary, NC: Oxford University Press Inc. Journals Dept. (2011). p. 147–157.
106. Du Z, Jia D, Liu S, Wang F, Li G, Zhang Y, et al. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia (2009) 57(7):724–33. doi:10.1002/glia.20800
107. Wegner M. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res (1999) 27(6):1409–20. doi:10.1093/nar/27.6.1409
108. Dong C, Wilhelm D, Koopman P. Sox genes and cancer. Cytogenet Genome Res (2004) 105(2–4):442–7. doi:10.1159/000078217
109. Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Ioue Y, et al. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann Surg Oncol (2009) 16(12):3488–98. doi:10.1245/s10434-009-0617-z
110. Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem (2008) 283(26):17969–78. doi:10.1074/jbc.M802917200
111. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet (2012) 44(10):1111–6. doi:10.1038/ng.2405
112. Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc (2007) 2(12):3081–9. doi:10.1038/nprot.2007.418
113. Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol (2008) 26(11):1269–75. doi:10.1038/nbt.1502
114. Gangemi RMR, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells (2009) 27(1):40–8. doi:10.1634/stemcells.2008-0493
115. Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron (2003) 39(5):749–65. doi:10.1016/S0896-6273(03)00497-5
116. Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev (2008) 22(23):3320–34. doi:10.1101/gad.1726608
117. Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci U S A (2008) 105(8):2907–12. doi:10.1073/pnas.0707886105
118. Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A (2011) 108(24):9951–6. doi:10.1073/pnas.1016912108
119. Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature (2012) 482(7384):216–20. doi:10.1038/nature10821
120. Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci (2004) 26(2–4):148–65. doi:10.1159/000082134
121. Takeda K, Akira S. Multi-functional roles of Stat3 revealed by conditional gene targeting. Arch Immunol Ther Exp (2001) 49(4):279–83.
122. Raz R, Lee C-K, Cannizzaro LA, d’Eustachio P, Levy DE. Essential role of STAT3 for embryonic stem cell pluripotency. Proc Natl Acad Sci U S A (1999) 96(6):2846–51. doi:10.1073/pnas.96.6.2846
123. Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. New and old functions of STAT3: a pivitol target for individualized treatment of cancer. Cell Cycle (2005) 4(9):1131–3. doi:10.4161/cc.4.9.1985
124. Levy DE, Lee CK. What does Stat3 do? J Clin Invest (2002) 109(9):1143–8. doi:10.1172/JCI0215650
125. Niwa H, Ogawa K, Shimosato D, Adachi K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature (2009) 460(7251):118–22. doi:10.1038/nature08113
126. Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev (1998) 12(13):2048–60. doi:10.1101/gad.12.13.2048
127. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A (1997) 94(8):3801–4. doi:10.1073/pnas.94.8.3801
128. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi:10.1038/nrc2734
129. Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci U S A (2003) 100(7):4138–43. doi:10.1073/pnas.0534764100
130. Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res (2006) 12(1):11–9. doi:10.1158/1078-0432.CCR-04-1752
131. Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, et al. Constitutive activation of STAT3 in human prostate tumors and cell lines direct inhibition of STAT3 signaling induces apoptosis of prostate cancer cells. Cancer Res (2002) 62(22):6659–66.
132. Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simões M, et al. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci U S A (2012) 109(35):E2361–70. doi:10.1073/pnas.1201232109
133. Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev (2005) 24(2):315–27. doi:10.1007/s10555-005-1580-1
134. Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells (2009) 27(10):2383–92. doi:10.1002/stem.185
135. de la Iglesia N, Konopka G, Lim K-L, Nutt CL, Bromberg JF, Frank DA, et al. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci (2008) 28(23):5870–8. doi:10.1523/JNEUROSCI.5385-07.2008
136. de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev (2008) 22(4):449–62. doi:10.1101/gad.1606508
137. Senft C, Priester M, Polacin M, Schröder K, Seifert V, Kögel D, et al. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J Neurooncol (2011) 101(3):393–403. doi:10.1007/s11060-010-0273-y
138. Yang YP, Chang YL, Huang PI, Chiou GY, Tseng LM, Chiou SH, et al. Resveratrol suppresses tumorigenicity and enhances radiosensitivity in primary glioblastoma tumor initiating cells by inhibiting the STAT3 axis. J Cell Physiol (2012) 227(3):976–93. doi:10.1002/jcp.22806
139. Gariboldi MB, Ravizza R, Monti E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem Pharmacol (2010) 80(4):455–62. doi:10.1016/j.bcp.2010.05.011
140. Villalva C, Martin-Lannerée S, Cortes U, Dkhissi F, Wager M, Le Corf A, et al. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int J Cancer (2011) 128(4):826–38. doi:10.1002/ijc.25416
141. Fuh B, Sobo M, Cen L, Josiah D, Hutzen B, Cisek K, et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br J Cancer (2009) 100(1):106–12. doi:10.1038/sj.bjc.6604793
142. Cao F, Hata R, Zhu P, Nakashiro K-i, Sakanaka M. Conditional deletion of Stat3 promotes neurogenesis and inhibits astrogliogenesis in neural stem cells. Biochem Biophys Res Commun (2010) 394(3):843–7. doi:10.1016/j.bbrc.2010.03.092
143. Chiou S-H, Wang M-L, Chou Y-T, Chen C-J, Hong C-F, Hsieh W-J, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res (2010) 70(24):10433–44. doi:10.1158/0008-5472.CAN-10-2638
144. Chiou S-H, Yu C-C, Huang C-Y, Lin S-C, Liu C-J, Tsai T-H, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res (2008) 14(13):4085–95. doi:10.1158/1078-0432.CCR-07-4404
145. Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem (2008) 283(25):17635–51. doi:10.1074/jbc.M800109200
146. Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with protein kinase Cϵ promotes oncogenic signaling by the stem cell marker Nanog and the production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J Biol Chem (2009) 284(39):26533–46. doi:10.1074/jbc.M109.027466
147. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene (2011) 30(36):3833–45. doi:10.1038/onc.2011.114
148. Clement V, Sanchez P, De Tribolet N, Radovanovic I, i Altaba AR. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17(2):165–72. doi:10.1016/j.cub.2007.01.024
149. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle (2009) 8(20):3274–84. doi:10.4161/cc.8.20.9701
150. Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J (2010) 29(15):2646–58. doi:10.1038/emboj.2010.131
151. Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt SN, Borges I, i Altaba AR. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J (2010) 29(15):2659–74. doi:10.1038/emboj.2010.137
152. Niu CS, Yang Y, Cheng C-D. MiR-134 regulates the proliferation and invasion of glioblastoma cells by reducing Nanog expression. Int J Oncol (2013) 42(5):1533–40. doi:10.3892/ijo.2013.1844
153. Loh Y-H, Wu Q, Chew J-L, Vega VB, Zhang W, Chen X, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet (2006) 38(4):431–40. doi:10.1038/ng1760
154. Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell (2003) 113(5):631–42. doi:10.1016/S0092-8674(03)00393-3
155. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (2007) 318(5858):1917–20. doi:10.1126/science.1151526
156. Möröy T, Fisher P, Guidos C, Ma A, Zimmerman K, Tesfaye A, et al. IgH enhancer deregulated expression of L-myc: abnormal T lymphocyte development and T cell lymphomagenesis. EMBO J (1990) 9(11):3659.
157. Dang CV, Resar LM, Emison E, Kim S, Li Q, Prescott JE, et al. Function of the c-Myc oncogenic transcription factor. Exp Cell Res (1999) 253(1):63–77. doi:10.1006/excr.1999.4686
158. Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A (2000) 97(7):3260–5. doi:10.1073/pnas.97.7.3260
159. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A (2006) 103(48):18261–6. doi:10.1073/pnas.0606108103
160. Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ (1997) 8(10):1039–48.
161. Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev (1993) 7(4):671–82. doi:10.1101/gad.7.4.671
162. Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature (1983) 306:194–6. doi:10.1038/306194a0
163. Lewis BC, Klimstra DS, Varmus HE. The c-myc and PyMT oncogenes induce different tumor types in a somatic mouse model for pancreatic cancer. Genes Dev (2003) 17(24):3127–38. doi:10.1101/gad.1140403
164. Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol (2008) 21(9):1156–67. doi:10.1038/modpathol.2008.111
165. Dubik D, Dembinski TC, Shiu RP. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res (1987) 47(24 Pt 1):6517–21.
166. Chen C-R, Kang Y, Massagué J. Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor β growth arrest program. Proc Natl Acad Sci U S A (2001) 98(3):992–9. doi:10.1073/pnas.98.3.992
167. Bigner SH, Friedman HS, Vogelstein B, Oakes WJ, Bigner DD. Amplification of the c-myc gene in human medulloblastoma cell lines and xenografts. Cancer Res (1990) 50(8):2347–50.
168. Zheng H, Ying H, Yan H, Kimmelman A, Hiller D, Chen A-J, et al. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harbor Symposia on Quantitative Biology. Cold Spring Harbor Laboratory Press (2008).
169. Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol (2008) 26(1):101–6. doi:10.1038/nbt1374
170. Broers JL, Viallet J, Jensen SM, Pass H, Travis WD, Minna JD, et al. Expression of c-myc in progenitor cells of the bronchopulmonary epithelium and in a large number of non-small cell lung cancers. Am J Respir Cell Mol Biol (1993) 9:33–43. doi:10.1165/ajrcmb/9.1.33
171. Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia (2003) 5(3):198–204. doi:10.1016/S1476-5586(03)80052-0
172. Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One (2008) 3(11):e3769. doi:10.1371/journal.pone.0003769
173. Herms JW, von Loewenich FD, Behnke J, Markakis E, Kretzschmar HA. c-Myc oncogene family expression in glioblastoma and survival. Surg Neurol (1999) 51(5):536–42. doi:10.1016/S0090-3019(98)00028-7
174. Dang DT, Pevsner J, Yang VW. The biology of the mammalian Krüppel-like family of transcription factors. Int J Biochem Cell Biol (2000) 32(11):1103–21. doi:10.1016/S1357-2725(00)00059-5
175. Bieker JJ. Krüppel-like factors: three fingers in many pies. J Biol Chem (2001) 276(37):34355–8. doi:10.1074/jbc.R100043200
176. Ivey KN, Muth A, Arnold J, King FW, Yeh R-F, Fish JE, et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell (2008) 2(3):219–29. doi:10.1016/j.stem.2008.01.016
177. Nakatake Y, Fukui N, Iwamatsu Y, Masui S, Takahashi K, Yagi R, et al. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol (2006) 26(20):7772–82. doi:10.1128/MCB.00468-06
178. Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene (2011) 30(18):2161–72. doi:10.1038/onc.2010.591
179. Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, et al. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ (1999) 10(6):423–34.
180. Foster KW, Liu Z, Nail CD, Li X, Fitzgerald TJ, Bailey SK, et al. Induction of KLF4 in basal keratinocytes blocks the proliferation–differentiation switch and initiates squamous epithelial dysplasia. Oncogene (2005) 24(9):1491–500. doi:10.1038/sj.onc.1208307
181. Wei D, Kanai M, Huang S, Xie K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis (2005) 27(1):23–31. doi:10.1093/carcin/bgi243
182. Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE, et al. Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res (2010) 70(24):10182–91. doi:10.1158/0008-5472.CAN-10-2414
183. Chen H-Y, Lin Y-M, Chung H-C, Lang Y-D, Lin C-J, Huang J, et al. miR-103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res (2012) 72(14):3631–41. doi:10.1158/0008-5472.CAN-12-0667
184. Akaogi K, Nakajima Y, Ito I, Kawasaki S, Oie S, Murayama A, et al. KLF4 suppresses estrogen-dependent breast cancer growth by inhibiting the transcriptional activity of ERα. Oncogene (2009) 28(32):2894–902. doi:10.1038/onc.2009.151
185. Zhou Y, Hofstetter WL, He Y, Hu W, Pataer A, Wang L, et al. KLF4 inhibition of lung cancer cell invasion by suppression of SPARC expression. Cancer Biol Ther (2010) 9(7):507–13. doi:10.4161/cbt.9.7.11106
186. Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer (2006) 6(1):11–23. doi:10.1038/nrc1780
187. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Krüppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene (2004) 23(2):395–402. doi:10.1038/sj.onc.1207067
188. Yoon HS, Chen X, Yang VW. Krüppel-like factor 4 mediates p53-dependent G1/S cell cycle arrest in response to DNA damage. J Biol Chem (2003) 278(4):2101–5. doi:10.1074/jbc.M211027200
189. Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol (2005) 7(11):1074–82. doi:10.1038/ncb1314
190. Ma J, Yao Y, Wang P, Liu Y, Zhao L, Li Z, et al. MiR-152 functions as a tumor suppressor in glioblastoma stem cells by targeting Krüppel-like factor 4. Cancer Lett (2014) 355(1):85–95. doi:10.1016/j.canlet.2014.09.012
191. Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Mod Pathol (2007) 20(1):102–7. doi:10.1038/modpathol.3800720
192. Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res (2007) 67(10):4807–15. doi:10.1158/0008-5472.CAN-06-4608
193. Su H-T, Weng C-C, Hsiao P-J, Chen L-H, Kuo T-L, Chen Y-W, et al. Stem cell marker nestin is critical for TGF-β1-mediated tumor progression in pancreatic cancer. Mol Cancer Res (2013) 11(7):768–79. doi:10.1158/1541-7786.MCR-12-0511
194. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444(7120):756–60. doi:10.1038/nature05236
195. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer (2006) 5(1):67. doi:10.1186/1476-4598-5-67
196. Hatanpaa KJ, Hu T, Vemireddy V, Foong C, Raisanen JM, Oliver D, et al. High expression of the stem cell marker nestin is an adverse prognostic factor in WHO grade II–III astrocytomas and oligoastrocytomas. J Neurooncol (2014) 117(1):183–9. doi:10.1007/s11060-014-1376-7
197. Zhang M, Song T, Yang L, Chen R, Wu L, Yang Z, et al. Nestin and CD133: valuable stem cell-specific markers for determining clinical outcome of glioma patients. J Exp Clin Cancer Res (2008) 27(1):1–7. doi:10.1186/1756-9966-27-85
198. Staberg M, Villingshøj M, Stockhausen M, Poulsen H. P01.20epigenetic treatment and induction of differentiation in glioblastoma multiforme neurosphere cells leads to downregulation of EGFR, EGFRvIII and nestin together with reduced colony formation in vitro. Neurooncology (2014) 16(Suppl 2):ii31–ii. doi:10.1093/neuonc/nou174.113
199. Frederiksen K, McKay R. Proliferation and differentiation of rat neuroepithelial precursor cells in vivo. J Neurosci (1988) 8(4):1144–51.
200. Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell (1990) 60(4):585–95. doi:10.1016/0092-8674(90)90662-X
201. Dahlstrand J, Lardelli M, Lendahl U. Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Dev Brain Res (1995) 84(1):109–29. doi:10.1016/0165-3806(94)00162-S
202. Suzuki S, Namiki J, Shibata S, Mastuzaki Y, Okano H. The neural stem/progenitor cell marker nestin is expressed in proliferative endothelial cells, but not in mature vasculature. J Histochem Cytochem (2010) 58(8):721–30. doi:10.1369/jhc.2010.955609
203. Gomes FC, Paulin D, Moura Neto V. Glial fibrillary acidic protein (GFAP): modulation by growth factors and its implication in astrocyte differentiation. Braz J Med Biol Res (1999) 32(5):619–31. doi:10.1590/S0100-879X1999000500016
204. Middeldorp J, Hol E. GFAP in health and disease. Prog Neurobiol (2011) 93(3):421–43. doi:10.1016/j.pneurobio.2011.01.005
205. Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell (1999) 97(6):703–16. doi:10.1016/S0092-8674(00)80783-7
206. Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J Neurosci (2003) 23(7):2824–32.
207. Glass R, Synowitz M, Kronenberg G, Walzlein J-H, Markovic DS, Wang L-P, et al. Glioblastoma-induced attraction of endogenous neural precursor cells is associated with improved survival. J Neurosci (2005) 25(10):2637–46. doi:10.1523/JNEUROSCI.5118-04.2005
208. Jung C, Foerch C, Schänzer A, Heck A, Plate K, Seifert V, et al. Serum GFAP is a diagnostic marker for glioblastoma multiforme. Brain (2007) 130(12):3336–41. doi:10.1093/brain/awm263
209. Müller C, Holtschmidt J, Auer M, Heitzer E, Lamszus K, Schulte A, et al. Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med (2014) 6(247):ra101–247. doi:10.1126/scitranslmed.3009095
210. Tichy J, Spechtmeyer S, Mittelbronn M, Hattingen E, Rieger J, Senft C, et al. Prospective evaluation of serum glial fibrillary acidic protein (GFAP) as a diagnostic marker for glioblastoma. J Neurooncol (2015) 126(2):361–9. doi:10.1007/s11060-015-1978-8
211. Hamilton JD, Rapp M, Schneiderhan TM, Sabel M, Hayman A, Scherer A, et al. Glioblastoma multiforme metastasis outside the CNS: three case reports and possible mechanisms of escape. J Clin Oncol (2014) 32(22):e80–4. doi:10.1200/JCO.2013.48.7546
212. Hamaya K, Doi K, Tanaka T, Nishimoto A. The determination of glial fibrillary acidic protein for the diagnosis and histogenetic study of central nervous system tumors: a study of 152 cases. Acta Med Okayama (1985) 39(6):453–62.
213. Abaza MSI, Narayan RK, Atassi M. In vitro efficacy of anti-glial fibrillary acidic protein monoclonal antibodies against human malignant glioma cell lines. Cancer Sci (1997) 88(11):1094–9.
214. Jung C, Unterberg A, Hartmann C. Diagnostic markers for glioblastoma. Histol Histopathol (2011) 26(10):1327–41.
215. Fargeas CA, Corbeil D, Huttner WB. AC133 antigen, CD133, prominin-1, prominin-2, etc.: prominin family gene products in need of a rational nomenclature. Stem Cells (2003) 21(4):506–8. doi:10.1634/stemcells.21-4-506
216. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood (1997) 90(12):5002–12.
217. Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest (2008) 118(6):2111. doi:10.1172/JCI34401
218. Yan X, Ma L, Yi D, Yoon J-g, Diercks A, Foltz G, et al. A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc Natl Acad Sci U S A (2011) 108(4):1591–6. doi:10.1073/pnas.1018696108
219. Wang J, Sakariassen PØ, Tsinkalovsky O, Immervoll H, Bøe SO, Svendsen A, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer (2008) 122(4):761–8. doi:10.1002/ijc.23130
220. Stamenkovic I, Amiot M, Pesando JM, Seed B. A lymphocyte molecule implicated in lymph node homing is a member of the cartilage link protein family. Cell (1989) 56(6):1057–62. doi:10.1016/0092-8674(89)90638-7
221. Lesley J, English N, Perschl A, Gregoroff J, Hyman R. Variant cell lines selected for alterations in the function of the hyaluronan receptor CD44 show differences in glycosylation. J Exp Med (1995) 182(2):431–7. doi:10.1084/jem.182.2.431
222. Sneath R, Mangham D. The normal structure and function of CD44 and its role in neoplasia. Mol Pathol (1998) 51(4):191. doi:10.1136/mp.51.4.191
223. Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. CD44 in cancer. Crit Rev Clin Lab Sci (2002) 39(6):527–79. doi:10.1080/10408360290795574
224. Du L, Wang H, He L, Zhang J, Ni B, Wang X, et al. CD44 is of functional importance for colorectal cancer stem cells. Clin Cancer Res (2008) 14(21):6751–60. doi:10.1158/1078-0432.CCR-08-1034
225. Wielenga VJ, Heider K-H, Johan G, Offerhaus A, Adolf GR, van den Berg FM, et al. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res (1993) 53(20):4754–6.
226. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med (2011) 17(2):211–5. doi:10.1038/nm.2284
227. Kito H, Suzuki H, Ichikawa T, Sekita N, Kamiya N, Akakura K, et al. Hypermethylation of the CD44 gene is associated with progression and metastasis of human prostate cancer. Prostate (2001) 49(2):110–5. doi:10.1002/pros.1124
228. Kaufmann M, von Minckwitz G, Heider K, Ponta H, Herrlich P, Sinn H. CD44 variant exon epitopes in primary breast cancer and length of survival. Lancet (1995) 345(8950):615–9. doi:10.1016/S0140-6736(95)90521-9
229. Götte M, Yip GW. Heparanase, hyaluronan, and CD44 in cancers: a breast carcinoma perspective. Cancer Res (2006) 66(21):10233–7. doi:10.1158/0008-5472.CAN-06-1464
230. Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J Exp Med (1997) 186(12):1985–96. doi:10.1084/jem.186.12.1985
231. Joshua B, Kaplan MJ, Doweck I, Pai R, Weissman IL, Prince ME, et al. Frequency of cells expressing CD44, a head and neck cancer stem cell marker: correlation with tumor aggressiveness. Head Neck (2012) 34(1):42–9. doi:10.1002/hed.21699
232. Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty GJ, Sharma S, et al. Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem (2001) 276(24):20809–12. doi:10.1074/jbc.C100140200
233. Toole BP, Slomiany MG. Hyaluronan, CD44 and Emmprin: partners in cancer cell chemoresistance. Drug Resist Updat (2008) 11(3):110–21. doi:10.1016/j.drup.2008.04.002
234. Gasbarri A, Martegani MP, Del Prete F, Lucante T, Natali PG, Bartolazzi A. Galectin-3 and CD44v6 isoforms in the preoperative evaluation of thyroid nodules. J Clin Oncol (1999) 17(11):3494–502.
235. Saegusa M, Machida D, Hashimura M, Okayasu I. CD44 expression in benign, premalignant, and malignant ovarian neoplasms: relation to tumour development and progression. J Pathol (1999) 189(3):326–37. doi:10.1002/(SICI)1096-9896(199911)189:3<326::AID-PATH425>3.0.CO;2-6
236. Pirinen R, Hirvikoski P, Böhm J, Kellokoski J, Moisio K, Virén M, et al. Reduced expression of CD44v3 variant isoform is associated with unfavorable outcome in non-small cell lung carcinoma. Hum Pathol (2000) 31(9):1088–95. doi:10.1053/hupa.2000.16277
237. Maula S, Huuhtanen R, Blomqvist C, Wiklund T, Laurila P, Ristamäki R. The adhesion molecule CD44v6 is associated with a high risk for local recurrence in adult soft tissue sarcomas. Br J Cancer (2001) 84(2):244. doi:10.1054/bjoc.2000.1590
238. Li H, Hamou M-F, de Tribolet N, Jaufeerally R, Hofmann M, Diserens A-C, et al. Variant CD44 adhesion molecules are expressed in human brain metastases but not in glioblastomas. Cancer Res (1993) 53(22):5345–9.
239. Eibl RH, Pietsch T, Moll J, Skroch-Angel P, Heider K-H, von Ammon K, et al. Expression of variant CD44 epitopes in human astrocytic brain tumors. J Neurooncol (1995) 26(3):165–70. doi:10.1007/BF01052619
240. Kaaijk P, Troost D, Morsink F, Keehnen RM, Leenstra S, Bosch DA, et al. Expression of CD44 splice variants in human primary brain tumors. J Neurooncol (1995) 26(3):185–90. doi:10.1007/BF01052621
241. Breyer R, Hussein S, Radu DL, Pütz K-M, Gunia S, Hecker H, et al. Disruption of intracerebral progression of rat C6 glioblastoma by in vivo treatment with anti-CD44 monoclonal antibody. J Neurosurg (2000) 92(1):140–9. doi:10.3171/jns.2000.92.1.0140
242. Asher R, Bignami A. Hyaluronate binding and CD44 expression in human glioblastoma cells and astrocytes. Exp Cell Res (1992) 203(1):80–90. doi:10.1016/0014-4827(92)90042-7
243. Naruse M, Shibasaki K, Yokoyama S, Kurachi M, Ishizaki Y. Dynamic changes of CD44 expression from progenitors to subpopulations of astrocytes and neurons in developing cerebellum. PLoS One (2013) 8(1):e53109. doi:10.1371/journal.pone.0053109
Keywords: glioblastoma multiforme, cancer, cancer stem cell, markers, hierarchy
Citation: Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF and Itinteang T (2016) Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 3:21. doi: 10.3389/fsurg.2016.00021
Received: 17 February 2016; Accepted: 29 March 2016;
Published: 15 April 2016
Edited by:
Eberval Figueiredo, University of São Paulo, BrazilReviewed by:
Kareem Zaghloul, National Institute of Neurological Disorders and Stroke, USACopyright: © 2016 Bradshaw, Wickremsekera, Tan, Peng, Davis and Itinteang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swee T. Tan, c3dlZS50YW5AZ21yaS5vcmcubno=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.