
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Astron. Space Sci. , 07 March 2024
Sec. Astrobiology
Volume 11 - 2024 | https://doi.org/10.3389/fspas.2024.1342694
 Chhandak Basu1,2*
Chhandak Basu1,2* Scott M. Perl2,3
Scott M. Perl2,3This opinion paper describes the potential (yet underutilized) use of a transcriptomic tool, RNA-seq, to study the cellular responses and adaptations of halophilic microbes to extreme environments. Halophilic microorganisms respond to environmental changes by differential expression of genes, and the end products of these gene expressions (proteins and other metabolites) help the organisms adapt to new environments and can serve as a biosignature of halophilic microbial communities. Transcriptomic-derived analyses can be used to understand such differential expressions of genes. Here we hypothesize that RNA-seq could give insights into commonalities in gene expression between ancient and modern-day microbes, and the gene expression data could be used to assess microbial evolution and adaptation by providing insights into what the microorganisms had to overcome and adapt to. Moreover, extremophilic microorganisms, including halophiles, are useful models for astrobiological investigations. If microbial life existed in the harsh UV-C, low water activity, chemically reactive, and anoxygenic early environments of the Martian surface, we hypothesize that they would have undergone a two-stage environmental-microbial response: 1) ecological stress, and 2) stress response mechanisms. This causal relationship would have hypothetically triggered differential gene expressions in microbial communities, if they existed, in ancient saline waters ∽3.5 Gyr ago during the late Noachian, when surface water started to lose its stability on Mars. We propose that available tools of transcriptomics, including RNA-seq, could be used for investigating this hypothetical ancient life on Mars. Our opinion paper's foundation is indeed built upon the concept of commonalities in gene expression between terrestrial life and hypothetical Martian life.
Transcriptomics gives us insights into genes and traits of adaptive significance (Voelckel et al., 2017). Zhou et al. (2019) used publicly available transcriptomics data, and using bioinformatics tools, they constructed a phylogenetic tree of Marine Benthic Group D (MBG-D) archaea (Order: Thermoprofundales). Hence, it is possible to use transcriptomics data to predict evolutionary lineages and geographic distribution of species. The ancient-to-modern Martian brine and lacustrine environmental processes, as shown by several articles (McLennan et al., 2005; Ming et al., 2006; McLennan and Grotzinger, 2008; Murchie et al., 2009; Ehlmann and Edwards, 2014; Vaniman et al., 2014) highlights the brine-to-evaporite mineral formation from ancient groundwater and dried lakebeds. The eventual mineral-microbial interactions from a drying sedimentary lake can induce hypothetical stress responses similar to the measured closed-basin lake systems responses (Jones and Baxter, 2017; Perl and Baxter, 2020) in terrestrial systems. Furthermore, these mineral-induced microbial stress responses can trigger expression of a hypothetical set of genes and their necessary activated stress-induced proteins that planetary instruments can measure as a potential life detection technique as Neveu et al. (2018) suggested.
DNA and RNA have already been established as valuable molecules used for life detection and biosignature studies (Neveu et al., 2018). One of the limitations in utilizing RNA-seq data from Earth-based microorganisms to predict potential Martian microorganisms (if they existed) could be that Martian microorganisms may have completely different genetic codes (equivalent to DNA, RNA, and proteins, as we know). Additionally, the biochemistry of Martian microorganisms might involve unknown molecules without any ‘Earthly equivalents.’ Modern transcriptomic techniques, including RNA-seq, Serial Analysis of Gene Expression (SAGE), Massively Parallel Signature Sequencing (MPSS), NanoString Technology, etc., could identify a series of up or downregulated desiccation-responsive genes in modern-day microbes. We propose these data could be extrapolated to understand hypothetical microbial life on Mars and how the microbes could have survived the loss of water on Mars, if life were ever present and had evolved to adapt to desiccation, high salinity, low water activity environments, and harsh gradients of UV-C. These are some of the strategies (Figure 1) describing how transcriptomic techniques, including RNA-seq, could be used to study microbial evolution on Mars or Mars-analog environments on Earth:
(1) Comparative transcriptomics: Comparative transcriptomics was used to study the evolution of cadmium stress-response among Escherichia coli, Saccharomyces cerevisiae, and Chlamydomonas reinhardtii by cross-kingdom comparison (Chen et al., 2021). Libourel et al. (2023) reconstructed the transcriptome of ancient nitrogen-fixing root nodule symbiosis (RNS) in Mimosa pudia and concluded that RNS could have originated more than 90 million years ago. Yoneda et al. (2016) studied the evolution of phenol tolerance in Rhodococcus opacus PD630. Blasdel et al. (2017) reported that gene expression patterns in two bacteriophage genera they studied were inherited from their common ancestor. Based on these reports, we propose that tools of comparative transcriptomics could be used to study the genomes of modern-day microbes and predict (stress-induced) gene expression as it occurred billions of years ago.
(2) Commonalities in gene expression: We can study the differential expression of genes in terrestrialmicrobes, and assuming that these genes evolved over time, our hypothesis is that we can predict commonalities in gene expression in ancient Mars (if life existed) or in ancient Earth. In fact, our entire paper is based on the assumption that commonalities in gene expression exist between ancient cells and modern-day cells in Mars (hypothetical) and Earth.
(3) Prediction of adaptive gene expression: The upregulated genes in extant microbes could provide a snapshot of the adaptive evolution of extinct microbes. For example, RNA-seq was used to study UV-tolerance mechanisms and adaptations to radiations by Deinococcus gobiensis (Yuan et al., 2012) and in a dinoflagellate, Karenia mikimotoi (Wang et al., 2019). By studying gene expression in modern-day microbes, we can predict adaptive strategies employed by their extinct counterparts in harsh environmental conditions. This type of information could be extrapolated to predict how life could have adapted in Mars-like or ancient Earth environments.
(4) Differential gene expression: We expect that a group of conserved genes would have differentially expressed over various stressful environments throughout evolutionary history and across a wide range of geographic locations. Hence, we can conclude that these adaptations (and eventually the genes) are essential for the microbes to survive in these hostile environments.
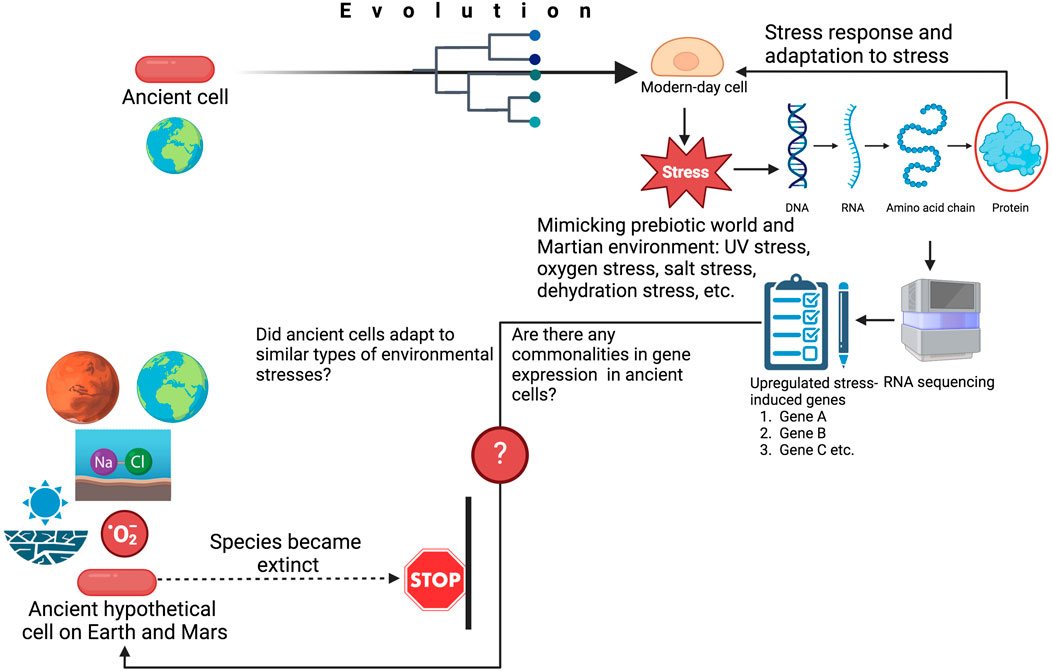
FIGURE 1. The utilization of RNA-seq to predict species habitability in ancient environments on both Earth and Mars. The strategies shown here involve study of modern-day cells, which evolved from ancient cells. The modern-day cell would be subjected to prebiotic world stress conditions or a simulated Martian environment. RNA-seq analysis will be performed on the stressed cell, generating a list of differentially expressed genes. If commonalities in gene expression existed between extant and extinct cells, these genes might have also been upregulated in extinct cells on Earth or, hypothetically, in extinct cells on Mars (if they existed). The gene expression data will help us to hypothesize whether the extinct cells on Earth or Mars (if they existed) adapted to similar types of stresses. This figure was generated from www.biorender.com.
The purpose of this paper aims to discuss the utility of RNA-seq, a transcriptomic analysis tool, to study the details of halophile adaptations within terrestrial evolution and for projected Martian ecological stresses and applications for transcriptomics for astrobiology research.
This paper discusses the potential of next-generation sequencing tools like RNA-seq to study gene expression in halophiles for astrobiology research. RNA-seq is used to quantify mRNA in a given sample at a particular time point. In this technique, mRNA is extracted from “control” and “treated” samples. Then, the mRNAs are converted to cDNAs (complementary DNAs or “photocopies of the genes”) using the enzyme reverse transcriptase. After that, cDNAs from various samples are “tagged” with short DNA sequences (barcodes) to distinguish among various samples. After that, the barcoded cDNAs are sequenced using a high-throughput DNA sequencer (like Illumina miSeq ™ or Illumina hiSeq ™). It is possible to obtain between 10 million and 100 million sequences per sample within 24 h or less. The sequenced cDNAs could be searched against public databases like GenBank to catalog a list of differentially upregulated or downregulated genes amongst various treatments or samples. We propose that RNA-seq data can unpack the gene expressions of halophilic microorganisms to show the links between the ecological settings of this planetary analog site and what gene expressions need to be activated in life elsewhere to survive.
RNA-seq is a powerful tool for studying microbial diversity without the need to conduct additional sequencing (Cox et al., 2017). RNA-seq can also be used to study microbes whose genome is not sequenced or when no reference genome is available (Cox et al., 2017); this is especially important in astrobiology research where the collected species samples might not have any background information available. Because of its ease of use, RNA-seq has already replaced microarray in transcriptomic studies and has become the most potent tool in bacterial gene regulation studies (Poulsen and Vinther, 2018). However, special care should be taken during RNA-seq experiments to minimize sampling and environmental variation (Harrison et al., 2012); for example, samples collected from the environment will have more environmental variance compared to lab-cultured samples. RNA-seq can also have other limitations, including gene length bias (Mandelboum et al., 2019), inability to detect less abundant transcripts (Mehta et al., 2016), technical variations (Williams et al., 2014), etc. Despite these limitations, RNA-seq is still widely used to study the entire transcriptome. A recently developed 3D spatial transcriptomics technique, also known as “Single-cell RNA-seq”, can potentially be used for cellular gene expression studies in space biology research (Gurazada et al., 2021). Although RNA-seq is a robust technique to detect changes in gene expression levels, it is evidently underutilized in astrobiology research. We searched the PubMed database for the number of publications that used these search terms. Although more than 29,000 and 21,000 publications (cumulatively from 1919) used the terms “RNA-seq” and “archaea” respectively, only 98 publications contained both the terms “RNA-seq and Archaea” together. And only two papers were found when we used both the terms “astrobiology” and “RNA-seq.”
There are some challenges to the use of RNA-seq in haloarchaeal research. For example, a high rRNA: mRNA ratio (80%–90%: ∼10%) in archaea is a bottleneck for transcript enrichment, and hence rRNA depletion is necessary (Pastor et al., 2022). However, the unavailability of commercially available archaeal rRNA removal kits could be the reason for the sudden decrease in the publication of archaeal RNA-seq data in recent years (Pastor et al., 2022). We propose, there is also potential to use RNA-seq to understand how some halophilic species evolved and survived in so-called hostile environments (Figure 1). Our hypothesis is bolstered by these reports on using RNA-seq in halophiles research as described below: Chromohalobacter salexigens (Salvador et al., 2018), Cenococcum geophilum (Li J. et al., 2022), Halobacterium salinarum NRC-1 (Pastor et al., 2022; Lorenzetti et al., 2023; Ten-Caten et al. (2018), Haloferax mediterranei ATCC33500 (Artieri et al., 2017; Payá et al., 2020; Pastor et al., 2022), Haloarcula hispainca DF60 (Pastor et al., 2022), Halolamina sp. YKT1 (Kurt-Kızıldoğan et al., 2017), Halomonas bluephagenesis TD01 (Xu et al., 2022), Halomonas socia NY-011 (Peng et al., 2020), Haloferax volcanii (Ammar et al., 2012; Babski et al., 2016; Artieri et al., 2017; Gebetsberger et al., 2017; Laass et al., 2019; Gelsinger and DiRuggiero, 2022; Pastor et al., 2022; Pastor et al., 2023), Haloferax alexandrinus DSM 27206 (Buda et al., 2023), Dunaliella bardawil (Liang et al., 2020), Tetraselmis sp. (D'Adamo et al., 2014), Dunaliella salina (Gao et al., 2023), Euhalothece sp. Z-M001 (Yang et al., 2020), Jeotgalibacillus malaysiensis (Yaakop et al., 2016), Natrinema sp. J7-2 (Mei et al., 2017), Tetragenococcus halophilus (Liu et al., 2015; Yao et al., 2022), Vibrio parahaemolyticus (Al Kadi et al., 2021; Li et al., 2022; Zhang et al., 2023). Besides these, metagenomic analysis using RNA-seq was performed to study the genetic diversity of multiple halophilic microorganisms (Cycil et al., 2020).
Collaborative use of RNA-seq will significantly redefine our understanding and knowledge of species diversity and gene expression studies involving life in extreme environments for astrobiological investigations.
Halophiles, which grow in extreme saline conditions, have diverse metabolic pathways, including oxygenic and anoxygenic photosynthesis, electron acceptors including oxygen, nitrate, and sulfur, and can perform chemoautotrophy and fermentation to survive (Sundarasami et al., 2019). Some halophiles were reported to be excellent models for astrobiology research, for example, Halorubrum lacusprofundi (Laye and DasSarma, 2018), H. salinarum NRC-1 (DasSarma, 2006), Haloarcula marismortui (DasSarma et al., 2019), H. mediterranei (DasSarma et al., 2019).
Now, the question we ask: can genomic expressions within planetary analog environments be used as a sign of life adapting to ecological stresses? If potential Martian microbial life independently started outside of Earth, what environmental and geochemical stresses existed that a proposed microbial community on Mars had to adapt to in order to thrive? Fairén et al. (2023) studied successions of ecological communities in a planetary analog site in Spain following desiccation. They conclude this data could be used interpret and predict hypothetical Martian microbial community at the end of Hesperian (wet-to-dry transition, Figure 2). We postulate that since biological changes occur magnitudes faster than geological events, then life (if existed) would have ample time to adapt to planetary changes over geologic time (Figure 2 for ecological stresses on Mars). The efficiency of metabolic processes over geological time would yield a status quo of microbial communities in local extreme environments that would eventually become Mars’s typical soil and regolith microbial communities. These would be resistant and thrive in settings that are UV-C resistant, basaltic, Fe-rich, low water activity (aw), and desiccation tolerant (Figure 2). The activated and expressed genes to combat the stresses mentioned above within these conditions are generally shared among all extremophile species that inhabit these environments.
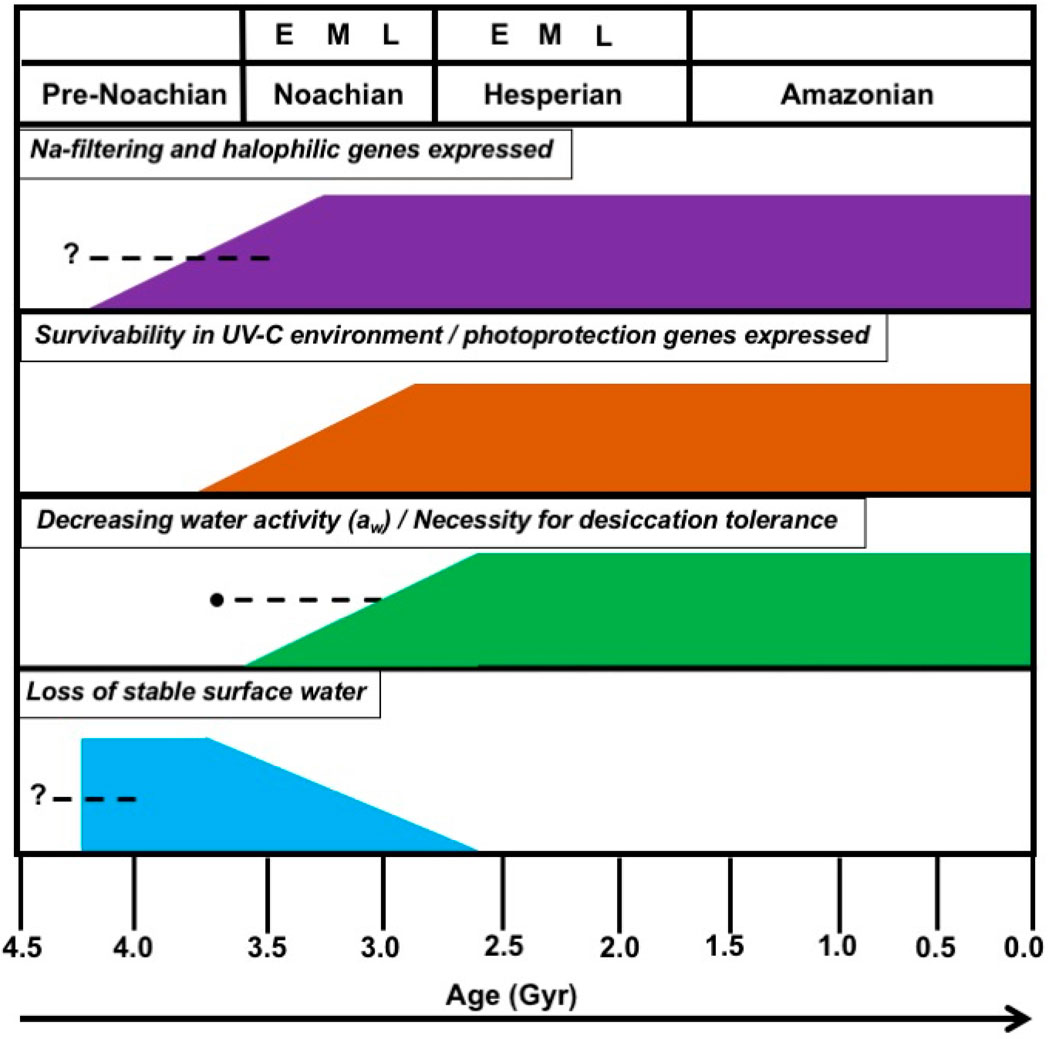
FIGURE 2. Martian epochs in-parallel with potential ecological events over geologic time. Since the late Noachian/early Hesperian boundary, the stability of Martian surface waters has decreased alongside the rise of UV-C and the cumulative loss of atmospheric integrity. While these changes are slow geologically, potential microbial life that would utilize such surface waters as a solvent within their nutrient cycling could have continued such processes with the deeper subsurface regolith further away from harmful UV-C. The approximate error bar for decreasing aw aligns with the approximate loss of surface water stability for variable porosities and permeabilities in the shallow subsurface (Perl et al., 2021). This does not apply to Martian subsurface environments.
Geochemical similarities exist between geological features in terrestrial salty brine systems and their hydrated minerals to those same minerals observed globally on Mars by the CRISM (Compact Reconnaissance Imaging Spectrometer for Mars) instrument onboard the Mars Reconnaissance Orbiter (Viviano-Beck et al., 2014; Murchie et al., 2007). When surface water was stable, the saline and hypersaline conditions on Mars would have presented a challenge for cellular life, should it have existed at all, and would have presented confined evolutionary pathways for the earliest forms of life. We assume ancient Martian fluids were stable (resistant to freezing), although the temperature was well below 273 K (Fairén et al., 2009) and possibly could support life (Rivkina et al., 2000) because the accumulation of solutes possibly generated saline aqueous solutions. Terrestrially, we study halophilic microorganisms and compare their adaptive processes and gene expressions to understand their habitability in brine settings. However, on Mars, microbial life would have had to evolve and incorporate these genomic adaptations early in evolution to pump harmful sodium ions and accumulate osmoprotectant molecules like glycine betaine (Deole and Hoff, 2020). Martian microbes (if they existed) also must encounter (detoxify) toxic perchlorate ions due to the prevalence of perchlorate ions on the Martian surface. In fact, using RNA-seq, Harris et al. (2021) reported that the archaea Methanosarcina barkeri metabolized perchlorate in Martian simulated environments by upregulating methylamine methanogenesis genes. Heinz et al. (2022) grew halotolerant yeast Debaryomyces hansenii on perchlorates, and after a thorough proteomic study, they reported the yeast cells went through protein glycosylation and cell wall remodulations in response to perchlorate stress. Hence we postulate modern tools of transcriptomics (e.g., RNA-seq) could have the potential to identify extinct and extant life on Earth and beyond.
Astrobiological investigations involving halophiles often encounter various limitations and challenges. RNA-seq has the promise to address some of those challenges and assist in astrobiological research (Table 1).

TABLE 1. Notable challenges for usage of RNA-seq in astrobiological data interpretation.
Life detection typically refers to measurements of potential ancient biosignatures from long-extinct microorganisms in in-situ planetary environments. The science mission objectives for the in-situ rover for current Martian exploration have focused on preserving organics as a sign of potential ancient microbial life. This opinion paper directly relates to the preservation medium and relies on the highest probability that geologically old regolith and lacustrine environments have preserved signs of ancient biology (Summons et al., 2011). As we move into an extant life-driven set of science mission objectives, our criteria and burden of proof needed for distinguishing between abiotic and biogenic features in the mineral and rock record are of the utmost importance (Perl et al., 2021; Perl et al., 2021). Utilizing extant life experiments to detect and compare adaptations between microorganisms that have thrived in planetary conditions, we would expect the differences in gene expressions due to adaptive processes; it would be critical to quantify how extant microbial life can survive under these conditions.
Available tools of transcriptomics, including RNA-seq, could be used for the study of the origins of life, including genetic adaptations. Unfortunately, these tools are underutilized in astrobiology research. Proper applications of these tools can also provide us insights into species diversity and origins of life, including halophilic evolution in extreme environments. For example, an RNA-seq analysis of Serratia liquefaciens cells grown under a simulated Martian environment revealed differential expression of genes, including ABC transporters, motility proteins, and transcription factors (Fajardo-Cavazos et al., 2018). Nepal and Kumar (2020) showed E. coli could survive up to 1.25 M MgSO4 salt, accompanied by differential expression of osmotically inducible gene (osmC) and sulfate transport (cysP) genes. This finding has an important astrobiological aspect, as the Martian surface can have a 1.25 M MgSO4 salt concentration during subzero temperatures (Nepal and Kumar, 2020).
To study hypothetical cells (if they existed) on the Martian surface, we can simulate Martian environments on Earth as reported by other researchers, including de la Vega et al. (2007), Rettberg et al. (2004), and Osman et al. (2008). We then need to establish the growth conditions of halophiles (or another suitable species) in these simulated Martian environments, followed by RNA extraction from these cells and ultimately perform RNA-sequencing (Figure 1). The RNA-seq data will give us the following physiological insights on hypothetical halophiles that might haveexisted on Martian surface: i) hypothetical gene expression in Martian climate, and ii) adaptation and evolution in harsh Martian environment. We propose the establishment of publicly available databases generated from RNA-seq data from halophiles grown on Martian simulated environments, similar to NASA’s space-related omics database GeneLab (https://genelab.nasa.gov/). We believe that available halophilic RNA-seq data could be applied to predict the biochemistry and cell types of microorganisms that might have existed (and are now extinct) on Earth millions of years ago. The ideal approach to predicting the physiology of potential Martian microorganisms (if they existed) would involve a combination of Earth-based halophilic RNA-seq data and a comparison with samples from Mars (if possible). Readers are encouraged to explore NASA’s Mars Sample Return webpage for more information: https://mars.nasa.gov/msr/.
We conclude that RNA-seq and available tools of transcriptomics within molecular biology can significantly assist and support us in astrobiology and exobiology research.
CB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing–review and editing. SP: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by NASA’s Research and Technology Development Fund Spontaneous Concept Task (Grant no. 01STCR/R.20.021.266).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al Kadi, M., Ishii, E., Truong, D. T., Motooka, D., Matsuda, S., Iida, T., et al. (2021). Direct RNA sequencing unfolds the complex transcriptome of Vibrio parahaemolyticus. mSystems 6 (6), e0099621. doi:10.1128/mSystems.00996-21
Ammar, R., Torti, D., Tsui, K., Gebbia, M., Durbic, T., Bader, G. D., et al. (2012). Chromatin is an ancient innovation conserved between Archaea and Eukarya. Elife 1, e00078. doi:10.7554/eLife.00078
Artieri, C. G., Naor, A., Turgeman-Grott, I., Zhou, Y., York, R., Gophna, U., et al. (2017). Cis-regulatory evolution in prokaryotes revealed by interspecific archaeal hybrids. Sci. Rep. 7 (1), 3986. doi:10.1038/s41598-017-04278-4
Babski, J., Haas, K. A., Näther-Schindler, D., Pfeiffer, F., Förstner, K. U., Hammelmann, M., et al. (2016). Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq). BMC Genomics 17 (1), 629. doi:10.1186/s12864-016-2920-y
Blasdel, B., Chevallereau, A., Monot, M., Lavigne, R., and Debarbieux, L. (2017). Comparative transcriptomics analyses reveal the conservation of an ancestral infectious strategy in two bacteriophage genera. ISME J. 11, 1988–1996. doi:10.1038/ismej.2017.63
Buda, D. M., Szekeres, E., Tudoran, L. B., Esclapez, J., and Banciu, H. L. (2023). Genome-wide transcriptional response to silver stress in extremely halophilic archaeon Haloferax alexandrinus DSM 27206 T. BMC Microbiol. 23, 381. doi:10.1186/s12866-023-03133-z
Chen, M., Wang, L., Zheng, X., Cohen, M., and Li, X. (2021). Cross-kingdom comparative transcriptomics reveals conserved genetic modules in response to cadmium stress. mSystems 6 (6), e0118921. doi:10.1128/msystems.01189-21
Cox, J. W., Ballweg, R. A., Taft, D. H., Velayutham, P., Haslam, D. B., and Porollo, A. (2017). A fast and robust protocol for metataxonomic analysis using RNA-seq data. Microbiome 5 (1), 7. doi:10.1186/s40168-016-0219-5
Cycil, L. M., DasSarma, S., Pecher, W., McDonald, R., AbdulSalam, M., and Hasan, F. (2020). Metagenomic insights into the diversity of halophilic microorganisms indigenous to the karak salt mine, Pakistan. Front. Microbiol. 11, 1567. doi:10.3389/fmicb.2020.01567
D'Adamo, S., Jinkerson, R. E., Boyd, E. S., Brown, S. L., Baxter, B. K., Peters, J. W., et al. (2014). Evolutionary and biotechnological implications of robust hydrogenase activity in halophilic strains of Tetraselmis. PLoS ONE 9 (1), e85812. doi:10.1371/journal.pone.0085812
DasSarma, S. (2006). Extreme halophiles are models for astrobiology. Microbe 1, 120–126. doi:10.1128/microbe.1.120.1
DasSarma, S., Fomenkov, A., DasSarma, S. L., Vincze, T., DasSarma, P., and Roberts, R. J. (2019). Methylomes of two extremely halophilic archaea species, Haloarcula marismortui and Haloferax mediterranei. Microbiol. Resour. Announc 8 (27), e00577-19. doi:10.1128/MRA.00577-19
de la Vega, U. P., Rettberg, P., and Reitz, G. (2007). Simulation of the environmental climate conditions on martian surface and its effect on Deinococcus radiodurans. Adv. Space Res. 40, 1672–1677. doi:10.1016/j.asr.2007.05.022
Deole, R., and Hoff, W. D. (2020). A potassium chloride to glycine betaine osmoprotectant switch in the extreme halophile Halorhodospira halophila. Sci. Rep. 10 (1), 3383. doi:10.1038/s41598-020-59231-9
Ehlmann, B. L., and Edwards, C. S. (2014). Mineralogy of the martian surface. Annu. Rev. Earth Planet. Sci. 42 (1), 291–315. doi:10.1146/annurev-earth-060313-055024
Fairén, A., Davila, A., Gago-Duport, L., Amils, R., and McKay, C. P. (2009). Stability against freezing of aqueous solutions on early Mars. Nature 459, 401–404. doi:10.1038/nature07978
Fairén, A. G., Rodríguez, N., Sánchez-García, L., Rojas, P., Uceda, E. R., Carrizo, D., et al. (2023). Ecological successions throughout the desiccation of Tirez lagoon (Spain) as an astrobiological time-analog for wet-to-dry transitions on Mars. Sci. Rep. 13, 1423. doi:10.1038/s41598-023-28327-3
Fajardo-Cavazos, P., Morrison, M. D., Miller, K. M., Schuerger, A. C., and Nicholson, W. L. (2018). Transcriptomic responses of Serratia liquefaciens cells grown under simulated Martian conditions of low temperature, low pressure, and CO2-enriched anoxic atmosphere. Sci. Rep. 8, 14938. doi:10.1038/s41598-018-33140-4
Gao, F., Nan, F., Feng, J., Lü, J., Liu, Q., Liu, X., et al. (2023). Identification of novel salt stress-responsive microRNAs through sequencing and bioinformatic analysis in a unique halophilic Dunaliella salina strain. J. Ocean. Limnol. 41, 1558–1574. doi:10.1007/s00343-022-2130-1
Gebetsberger, J., Wyss, L., Mleczko, A. M., Reuther, J., and Polacek, N. (2017). A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol. 14 (10), 1364–1373. doi:10.1080/15476286.2016.1257470
Gelsinger, D. R., and DiRuggiero, J. (2022). “Small RNA-sequencing library preparation for the halophilic archaeon Haloferax volcanii,”. Archaea. Methods in molecular biology. Editor S. Ferreira-Cerca (New York, NY: Humana), 2522.
Gurazada, S. G. R., Cox, K. L., Czymmek, K. J., and Meyers, B. C. (2021). Space: the final frontier — achieving single-cell, spatially resolved transcriptomics in plants. Emerg. Top. Life Sci. 5 (2), 179–188. doi:10.1042/etls20200274
Harris, R. L., Schuerger, A. C., Wang, W., Tamama, Y., Garvin, Z. K., and Onstott, T. C. (2021). Transcriptional response to prolonged perchlorate exposure in the methanogen Methanosarcina barkeri and implications for Martian habitability. Sci. Rep. 11, 12336. doi:10.1038/s41598-021-91882-0
Harrison, P. W., Wright, A. E., and Mank, J. E. (2012). The evolution of gene expression and the transcriptome-phenotype relationship. Semin. Cell Dev. Biol. 23 (2), 222–229. doi:10.1016/j.semcdb.2011.12.004
Heinz, J., Doellinger, J., Maus, D., Schneider, A., Lasch, P., Grossart, H.-P., et al. (2022). Perchlorate-specific proteomic stress responses of Debaryomyces hansenii could enable microbial survival in Martian brines. Environ. Microbiol. 24 (11), 5051–5065. doi:10.1111/1462-2920.16152
Jones, D. L., and Baxter, B. K. (2017). DNA repair and photoprotection: mechanisms of overcoming environmental ultraviolet radiation exposure in halophilic archaea. Front. Microbiol. 8, 1882. doi:10.3389/fmicb.2017.01882
Kurt-Kızıldoğan, A., Abanoz, B., and Okay, S. (2017). Global transcriptome analysis of Halolamina sp. to decipher the salt tolerance in extremely halophilic archaea. Gene 601, 56–64. doi:10.1016/j.gene.2016.11.042
Laass, S., Monzon, V. A., Kliemt, J., Hammelmann, M., Pfeiffer, F., Förstner, K. U., et al. (2019). Characterization of the transcriptome of Haloferax volcanii, grown under four different conditions, with mixed RNA-Seq. PLoS ONE 14 (4), e0215986. doi:10.1371/journal.pone.0215986
Laye, V. J., and DasSarma, S. (2018). An antarctic extreme halophile and its polyextremophilic enzyme: effects of perchlorate salts. Astrobiology 18 (4), 412–418. doi:10.1089/ast.2017.1766
Li, J., Li, C., Tsuruta, M., Matsushita, N., Goto, S., Shen, Z., et al. (2022a). Physiological and transcriptional responses of the ectomycorrhizal fungus Cenococcum geophilum to salt stress. Mycorrhiza 32 (3-4), 327–340. doi:10.1007/s00572-022-01078-1
Li, Y., Sun, W., Wang, Q., Yu, Y., Wan, Y., Zhou, K., et al. (2022b). The GntR-like transcriptional regulator HutC involved in motility, biofilm-forming ability, and virulence in Vibrio parahaemolyticus. Microb. Pathog. 167, 105546. doi:10.1016/j.micpath.2022.105546
Liang, M. H., Jiang, J. G., Wang, L., and Zhu, J. (2020). Transcriptomic insights into the heat stress response of Dunaliella bardawil. Enzyme Microb. Technol. 132, 109436. doi:10.1016/j.enzmictec.2019.109436
Libourel, C., Keller, J., Brichet, L., Cazalé, A. C., Carrère, S., Vernié, T., et al. (2023). Comparative phylotranscriptomics reveals ancestral and derived root nodule symbiosis programmes. Nat. Plants 9, 1067–1080. doi:10.1038/s41477-023-01441-w
Liu, L., Si, L., Meng, X., and Luo, L. (2015). Comparative transcriptomic analysis reveals novel genes and regulatory mechanisms of Tetragenococcus halophilus in response to salt stress. J. Ind. Microbiol. Biotechnol. 42 (4), 601–616. doi:10.1007/s10295-014-1579-0
Lorenzetti, A. P. R., Kusebauch, U., Zaramela, L. S., Wu, W. J., de Almeida Jpp, , Turkarslan, S., et al. (2023). A genome-scale atlas reveals complex interplay of transcription and translation in an archaeon. mSystems 8 (2), e0081622. doi:10.1128/msystems.00816-22
Mandelboum, S., Manber, Z., Elroy-Stein, O., and Elkon, R. (2019). Recurrent functional misinterpretation of RNA-seq data caused by sample-specific gene length bias. PLoS Biol. 17 (11), e3000481. doi:10.1371/journal.pbio.3000481
Mclennan, S. M., Bell, J. F., Calvin, W. M., Christensen, P. R., Clark, B. C., De Souza, P. A., et al. (2005). Provenance and diagenesis of the evaporite-bearing burns formation, meridiani planum, Mars. Earth Planet. Sci. Lett. 240, 95–121. doi:10.1016/j.epsl.2005.09.041
Mclennan, S. M., and Grotzinger, J. P. (2008). “The sedimentary rock cycle of Mars,” in The martian surface: composition, mineralogy, and physical properties. Editor J. F. Bell III (Cambridge: Cambridge University Press), 541–577.
Mehta, S., Tsai, P., Lasham, A., Campbell, H., Reddel, R., Braithwaite, A., et al. (2016). A study of TP53 RNA splicing illustrates pitfalls of RNA-seq methodology. Cancer Res. 76 (24), 7151–7159. doi:10.1158/0008-5472.CAN-16-1624
Mei, Y., Liu, H., Zhang, S., Yang, M., Hu, C., Zhang, J., et al. (2017). Effects of salinity on the cellular physiological responses of Natrinema sp. J7-2. PLoS One 12 (9), e0184974. doi:10.1371/journal.pone.0184974
Ming, D. W., Mittlefehldt, D. W., Morris, R. V., Golden, D. C., Gellert, R., Yen, A., et al. (2006). Geochemical and mineralogical indicators for aqueous processes in the Columbia Hills of Gusev crater, Mars. J. Geophys. Res. 111, E02S12. doi:10.1029/2005JE002560
Murchie, S., Arvidson, R., Bedini, P., Beisser, K., Bibring, J.-P., Bishop, J., et al. (2007). Compact reconnaissance imaging spectrometer for Mars (CRISM) on Mars reconnaissance orbiter (MRO). J. Geophys. Res. Planets 112 (E5). doi:10.1029/2006je002682
Murchie, S. L., Mustard, J. F., Ehlmann, B. L., Milliken, R. E., Bishop, J. L., McKeown, N. K., et al. (2009). A synthesis of Martian aqueous mineralogy after 1 Mars year of observations from the Mars Reconnaissance Orbiter. J. Geophys. Res. Planets 114 (E2). doi:10.1029/2009je003342
Nepal, S., and Kumar, P. (2020). Growth, cell division, and gene expression of Escherichia coli at elevated concentrations of magnesium sulfate: implications for habitability of europa and Mars. Microorganisms 8 (5), 637. doi:10.3390/microorganisms8050637
Neveu, M., Hays, L. E., Voytek, M. A., New, M. H., and Schulte, M. D. (2018). The ladder of life detection. Astrobiology 18, 1375–1402. doi:10.1089/ast.2017.1773
Osman, S., Peeters, Z., La Duc, M. T., Mancinelli, R., Ehrenfreund, P., and Venkateswaran, K. (2008). Effect of shadowing on survival of bacteria under conditions simulating the Martian atmosphere and UV radiation. Appl. Environ. Microbiol. 74 (4), 959–970. doi:10.1128/aem.01973-07
Pastor, M., Sakrikar, S., Hwang, S., Hackley, R., Soborowski, A., Maupin-Furlow, J., et al. (2023). TroR is the primary regulator of the iron homeostasis transcription network in the halophilic archaeon Haloferax volcanii. doi:10.1101/2023.08.16.553580
Pastor, M. M., Sakrikar, S., Rodriguez, D. N., and Schmid, A. K. (2022). Comparative analysis of rRNA removal methods for RNA-seq differential expression in halophilic archaea. Biomolecules 12 (5), 682. doi:10.3390/biom12050682
Payá, G., Bautista, V., Camacho, M., Bonete, M. J., and Esclapez, J. (2020). New proposal of nitrogen metabolism regulation by small RNAs in the extreme halophilic archaeon Haloferax mediterranei. Mol. Genet. Genomics 295 (3), 775–785. doi:10.1007/s00438-020-01659-9
Peng, S., Kai, M., Yang, X., Luo, Y., and Bai, L. (2020). Study on the osmoregulation of "Halomonas socia" NY-011 and the degradation of organic pollutants in the saline environment. Extremophiles 24 (6), 843–861. doi:10.1007/s00792-020-01199-5
Perl, S., Adeli, S., Basu, C., Baxter, B. K., Bowman, J., Boyd, E., et al. (2021b). Salty Environments: the importance of evaporites and brine environments as habitats and preservers of biosignatures. Bull. Am. Astronomical Soc. 53 (4). doi:10.3847/25c2cfeb.d0fffbba
Perl, S. M., and Baxter, B. K. (2020). “Great salt lake as an astrobiology analogue for ancient martian hypersaline aqueous systems,” in Great salt lake biology: a terminal lake in a time of change. Editors B. K. Baxter, and J. K. Butler (Cham: Springer International Publishing), 487–514.
Perl, S. M., Celestian, A. J., Cockell, C. S., Corsetti, F. A., Barge, L. M., Bottjer, D., et al. (2021a). A proposed geobiology-driven nomenclature for AstrobiologicalIn SituObservations and sample analyses. Astrobiology 21 (8), 954–967. doi:10.1089/ast.2020.2318
Poulsen, L. D., and Vinther, J. (2018). RNA-seq for bacterial gene expression. Curr. Protoc. Nucleic Acid. Chem. 73 (1), e55. doi:10.1002/cpnc.55
Rettberg, P., Rabbow, E., Panitz, C., and Horneck, G. (2004). Biological space experiments for the simulation of Martian conditions: UV radiation and Martian soil analogues. Adv. Space Res. 33 (8), 1294–1301. doi:10.1016/j.asr.2003.09.050
Rivkina, E. M., Friedmann, E. I., McKay, C. P., and Gilichinsky, D. (2000). Metabolic activity of permafrost bacteria below the freezing point. Appl. Environ. Microbiol. 66, 3230–3233. doi:10.1128/aem.66.8.3230-3233.2000
Salvador, M., Argandoña, M., Naranjo, E., Piubeli, F., Nieto, J. J., Csonka, L. N., et al. (2018). Quantitative RNA-seq analysis unveils osmotic and thermal adaptation mechanisms relevant for ectoine production in chromohalobacter salexigens. Front. Microbiol. 9, 1845. doi:10.3389/fmicb.2018.01845
Summons, R. E., Amend, J. P., Bish, D., Buick, R., Cody, G. D., Des Marais, D. J., et al. (2011). Preservation of martian organic and environmental records: final report of the Mars biosignature working group. Astrobiology 11 (2), 157–181. doi:10.1089/ast.2010.0506
Sundarasami, A., Sridhar, A., and Mani, K. (2019). “Halophilic archaea as beacon for exobiology: recent advances and future challenges,” in Advances in biological science research. Editors S. N. Meena, and M. M. Naik (Academic Press), 197–214.
Ten-Caten, F., Vêncio, R. Z. N., Lorenzetti, A. P. R., Zaramela, L. S., Santana, A. C., and Koide, T. (2018). Internal RNAs overlapping coding sequences can drive the production of alternative proteins in archaea. RNA Biol. 15 (8), 1119–1132. doi:10.1080/15476286.2018.1509661
Vaniman, D. T., Bish, D. L., Ming, D. W., Bristow, T. F., Morris, R. V., Blake, D. F., et al. (2014). Mineralogy of a mudstone at yellowknife bay, gale crater, Mars. Science 343 (6169), 1243480. doi:10.1126/science.1243480
Viviano-Beck, C. E., Seelos, F. P., Murchie, S. L., Kahn, E. G., Seelos, K. D., Taylor, H. W., et al. (2014). Revised CRISM spectral parameters and summary products based on the currently detected mineral diversity on Mars. J. Geophys. Res. Planets 119 (6), 1403–1431.
Voelckel, C., Gruenheit, N., and Lockhart, P. (2017). Evolutionary transcriptomics and proteomics: insight into plant adaptation. Trends Plant Sci. 22 (6), 462–471. doi:10.1016/j.tplants.2017.03.001
Wang, X., Niu, X., Chen, Y., Sun, Z., Han, A., Lou, X., et al. (2019). Transcriptome sequencing of a toxic dinoflagellate, Karenia mikimotoi subjected to stress from solar ultraviolet radiation. Harmful Algae 88, 101640. doi:10.1016/j.hal.2019.101640
Williams, A. G., Thomas, S., Wyman, S. K., and Holloway, A. K. (2014). RNA-Seq data: challenges in and recommendations for experimental design and analysis. Curr. Protoc. Hum. Genet. 83, 11–20. doi:10.1002/0471142905.hg1113s83
Xu, M., Chang, Y., Zhang, Y., Wang, W., Hong, J., Zhao, J., et al. (2022). Development and application of transcription terminators for polyhydroxylkanoates production in halophilic Halomonas bluephagenesis TD01. Front. Microbiol. 13, 941306. doi:10.3389/fmicb.2022.941306
Yaakop, A. S., Chan, K. G., Ee, R., Lim, Y. L., Lee, S. K., Manan, F. A., et al. (2016). Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci. Rep. 6, 33660. doi:10.1038/srep33660
Yang, H. W., Song, J. Y., Cho, S. M., Kwon, H. C., Pan, C. H., and Park, Y. I. (2020). Genomic survey of salt acclimation-related genes in the halophilic cyanobacterium Euhalothece sp. Z-m001. Z-M001. Sci. Rep. 10 (1), 676. doi:10.1038/s41598-020-57546-1
Yao, S., Zhou, R., Jin, Y., Huang, J., Qin, J., and Wu, C. (2022). Formation of biofilm changed the responses of Tetragenococcus halophilus to ethanol stress revealed by transcriptomic and proteomic analyses. Food Res. Int. 161, 111817. doi:10.1016/j.foodres.2022.111817
Yoneda, A., Henson, W. R., Goldner, N. K., Joo Park, K., Forsberg, K. J., Kim, S.Ji, et al. (2016). Comparative transcriptomics elucidates adaptive phenol tolerance and utilization in lipid-accumulating Rhodococcus opacus PD630. Nucleic Acids Res. 44 (Issue 5), 2240–2254. doi:10.1093/nar/gkw055
Yuan, M., Chen, M., Zhang, W., Lu, W., Wang, J., Yang, M., et al. (2012). Genome sequence and transcriptome analysis of the radioresistant bacterium Deinococcus gobiensis: insights into the extreme environmental adaptations. PLoS ONE 7 (3), e34458. doi:10.1371/journal.pone.0034458
Zhang, Y., Tan, X., Li, M., Liu, P., Jiao, X., and Gu, D. (2023). Transcriptome analysis reveals the effect of low NaCl concentration on osmotic stress and type III secretion system in Vibrio parahaemolyticus. Int. J. Mol. Sci. 24 (3), 2621. doi:10.3390/ijms24032621
Keywords: astrobiology, halophiles, RNA-seq, gene expression, evolution
Citation: Basu C and Perl SM (2024) The use of RNA-seq for the study of physiological adaptations of halophiles in extreme environments for astrobiological data interpretation. Front. Astron. Space Sci. 11:1342694. doi: 10.3389/fspas.2024.1342694
Received: 22 November 2023; Accepted: 15 February 2024;
Published: 07 March 2024.
Edited by:
Alberto Fairén, Spanish National Research Council (CSIC), SpainCopyright © 2024 Basu and Perl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Chhandak Basu, Y2hoYW5kYWsuYmFzdUBjc3VuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.