 Taylor J. Santaloci
Taylor J. Santaloci  Marie E. Strauss 2
Marie E. Strauss 2 Ryan C. Fortenberry
Ryan C. Fortenberry - 1Department of Chemistry and Biochemistry, University of Mississippi, Oxford, MS, United States
- 2Department of Chemistry, Physics, and Engineering, Biola University, La Mirada, CA, United States
Functionalizing deprotonated polycyclic aromatic hydrocarbon (PAH) anion derivatives gives rise to electronically excited states in the resulting anions. While functionalization with −OH and −C2H, done presently, does not result in the richness of electronically excited states as it does with −CN done previously, the presence of dipole-bound excited states and even some valence excited states are predicted in this quantum chemical analysis. Most notably, the more electron withdrawing −C2H group leads to valence excited states once the number of rings in the molecule reaches three. Dipole-bound excited states arise when the dipole moment of the corresponding neutral radical is large enough (likely around 2.0 D), and this is most pronounced when the hydrogen atom is removed from the functional group itself regardless of whether functionalized by a hydroxyl or enthynyl group. Deprotonatation of the hydroxyl group in the PAH creates a ketone with a delocalized highest occupied molecular orbital (HOMO) unlike deprotonation of a hydrogen on the ring where a localized lone pair on one of the carbon atoms serves as the HOMO. As a result, hydroxyl functionlization and subsequent deprotonation of PAHs creates molecules that begin to exhibit structures akin to nucleic acids. However, the electron withdrawing −C2H has more excited states than the electron donating −OH functionalized PAH. This implies that the −C2H electron withdrawing group can absorb a larger energy range of photons, which signifies an increasing likelihood of being stabilized in the harsh conditions of the interstellar medium.
1 Introduction
The Cassini-Huygens mission investigated the atmospheric structure, surface morphology, composition, and meteorology of Saturn’s moon Titan (Coustenis, 2007). The Cassini plasma spectrometer electron spectrometer (CAPS-ELS) detected negatively charged materials with masses large enough such that polynucleotides could be one potential explanation for these detected species. The CAPS-ELS found a large breadth of possible anion sizes in Titan’s atmosphere with the least populated region of counts for atomic sizes of 10–100 amu/q.Ali et al. (2015) The most populous region reported was between 100 and 10,000 amu/q. Hence, the most abundant anions are the same size as nucleic acids, polycyclic aromatic hydrocarbons (PAHs), and polypeptides. This indicates that smaller anions react with molecules to form into larger anions. These anions may have lifetimes in extraterrestrial environments long enough for them to engage in novel astrobiochemistry (Schild et al., 1974; Snow, 1975; Hanel et al., 1981; Kunde et al., 1981; Maguire et al., 1981; Samuelson et al., 1981; Yung et al., 1984; Yung, 1987; Lepp and Dalgarno, 1988; Bradforth et al., 1993; Kawaguchi et al., 1995; Aoki et al., 1996; Clifford et al., 1997; Moustefaoui et al., 1998; Tulej et al., 1998; Aoki, 2000; Millar et al., 2000; Terzieva and Herbst, 2000; Molina-Cuberos et al., 2002; McCarthy et al., 2006; Brünken et al., 2007; Cernicharo et al., 2007; Coates et al., 2007; Remijan et al., 2007; Cernicharo et al., 2008; Ross et al., 2008; Thaddeus et al., 2008; Agúndez et al., 2010; Cordiner et al., 2011; López-Puertas et al., 2013).
Additionally, theoretical studies show that nitrogen containing PAHs, or polycyclic nitrogenated hydrocarbons (PANHs), likely also exist in Titan’s atmosphere (Kuiper, 1944; Allen et al., 1980; McKay, 1996; Clarke and Ferris, 1997; Ricca et al., 2001). The formation of PANHs starts with the addition of a cyano-containing group to a benzene ring. This has a small reaction barrier of 8 kcal/mol and forms aromatic nitriles. Additionally, these are known intermediates in the formation of purine, pyrimidine, and their derivatives (Ricca et al., 2001). Also, Titan’s atmosphere produces enough energy to surpass the 8 kcal/mol barrier (Walsh, 1995). However, the interplay between neutral PAHs or PANHs and anions of the same molecular mass has yet to be established for Titan or really anywhere. While PAHs like naphthalene can capture free electrons to form anions, PANHs or functionalized PAHs with electronegative functional groups like −CN, −OH or −C2H can capture free electrons more efficiently (Theis et al., 2015a). Hence, sprinkling a few different atomic species within the PAH structure enhances the anionic chemistry significantly. As a result, the foundations of nucleic acid molecular structure, purine and pyrimidine, may enhance the growth of molecular anions in astrophysical environments like Titan’s atmosphere. Additionally, nucleic acids are thought to be a first step for the creation of extraterrestrial life since they are one of the main constituents of DNA and RNA strands making such chemistry a fascinating venue for the production of biologically-relevent astromolecules.
The chemistry behind the formation of biologically-relevant molecular anions in Titans atmosphere is unknown. Puzzarini and coworkers suggestion follows suit with Olah’s non-classical carbocationic chemistry in that the formation of large anionic macromolecules proceeds through a series of carbocationic reactions, which eventually results in an open shell neutral radical. The open shell neutral radical would then undergo a radiative electron attachment process (Olah et al., 2016; Puzzarini et al., 2017). The radiative electron attachment process can form anions through dipole-bound states along pathway. McCarthy and coworkers provide the strongest support for the dipole-bound formation pathway by the observation of the interstellar C2nH radicals and their corresponding anions. The ground state for C4H is 2Σ+, and the excited state is 2Π. However, for larger C2nH radicals, the ground state becomes 2Π. Typically, the 2Σ+ states will have dipole moments of ≈0.8 D or smaller, and the 2Π state will have dipole moments of ≈4.0 D or larger(Maier, 1998; Pino et al., 2002; Agúndez et al., 2008). To follow the dipole-bound formation pathway, the dipole moment must be ≈2.0 D or larger. Thus, C4H must excite into the 2Π excited state to undergo the dipole-bound formation pathway. Whereas, the larger C2nHs will undergo the dipole-bound formation pathway at the 2Π ground state. The latter process is the energetically favorable reaction. Furthermore, experimental interstellar observations show that C4H− is less abundant in space than their corresponding radical, and C6H− is more abundant than their corresponding radical implying that excitation must take place from the 2Σ+ state of C4H. As such, the focus of this present work is to study open-shell radicals and determine if there are any anionic excited states as these likely play a vital role in interstellar anion chemistry as it is currently understood (Yamamoto et al., 1987; Shen et al., 1990; McCarthy et al., 1995; Woon, 1995; Hoshina et al., 1998; Taylor et al., 1998; van Hemert and van Dishoeck, 2008; Fortenberry et al., 2010; Fortenberry, 2015).
Computational analysis on isomeric anionic complexes of uracil shows evidence of dipole-bound anion formation (Hendricks et al., 1998) In order to explore such behavior, Gutowski and co-workers (Gutowski et al., 1996) previously utilized both photoelectron spectroscopy (PES) and theoretical computations in examining a uracil-glycine anion complex. The PES spectra show a broad feature with a maximum intensity at 1.8 eV implying removal of an electron from an anion state at this energy. In support of this, the theoretical work concludes that the excess electron in the uracil-glycine complex is a π* orbital localized on the uracil rings. Furthermore, later theoretical work by Adamowics and coworkers also predicts that uracil-glycine complexes have dipole moments of approximately 5 D. These items imply that uracil can form dipole-bound anions (Hendricks et al., 1996, 1998; Desfrancois et al., 1996; Jalbout et al., 2003).
Fermi and Teller, the first to study dipole-bound anions with the aid of quantum mechanics, found that the binding energy of an excess electron is dependent upon the corresponding neutral radical’s dipole moment (Fermi and Teller, 1947). Specifically, the dipole moment should be greater than 1.63 D in order to keep a free electron bound to the positive end of the dipole moment. Dipole-bound anions of the type described by Fermi and Teller are dominated by the excess electron residing in a highly-diffuse, Rydberg-like s orbital with higher dipole moments required for higher angular momentum states in these hydrogen-like, diffuse orbitals. In more recent studies, the neutral radical dipole moment has been shown to require closer to or even exceeding 2.0 D for the diffuse s orbitals to be populated in dipole-bound anions (Compton et al., 1996; Gutowski et al., 1996; Jordan and Wang, 2003; Simons, 2008). This suggests that when the magnitude of the dipole moment becomes larger, the probability also increases that a dipole-bound state of an anion will exist underneath the electron binding energy (eBE), the energy required to remove the electron from the molecular system) giving a proportional relationship between the magnitude of the dipole moment and the eBE. However, dipole-bound states do not have to solely be ground electronic states. If the anion is valence in nature for its ground electronic state, dipole-bound excited states (DBXSs) may be accessed via electronic transitions (Mead et al., 1984; Lykke et al., 1987; Mullin et al., 1992, 1993; Gutsev and Adamowicz, 1995; Fortenberry, 2015). Since Fermi and Teller’s equations (Fermi and Teller, 1947) dictate that only one dipole-bound state can be present for each angular momentum value, DBXSs are rare, but uracil appears to demonstrate such behavior. Hence, the analysis of DBXSs for molecular precursors to nucleotides will require specialized tools.
As with the uracil anion complexes examined above (Hendricks et al., 1998, 1996), quantum chemistry provides a detailed means for determining the properties of dipole-bound anions and/or DBXSs. The diffuse, Rydberg-like s orbitals need an adequate quantum chemical basis set in order to be accurately described. One effective basis set is utilizing standard correlation consistent basis sets with large numbers of diffuse functions, such as the t-aug-cc-pVTZ (tapVTZ) set. However, such linear combinations of atomic orbitals to make molecular orbitals (LCAO-MO) approaches are too computationally expensive (Fortenberry and Crawford, 2011a). An alternate approach is to add even-tempered diffuse functions onto a dummy atom and keep the LCAO-MO approach for the valence orbital description (Mach et al., 2010; Fortenberry and Crawford, 2011b; Santaloci and Fortenberry, 2021). For the aug-cc-pVDZ basis set on the real atoms, this method lowers the number of basis functions by 48%, which, in return, lowers the computational cost significantly, without affecting the accuracy of the results (Fortenberry and Crawford, 2011b; Theis et al., 2015a; Morgan and Fortenberry, 2015).
Another important aspect of the basis set includes the placement and number of the employed diffuse functions. Past studies (Fortenberry and Crawford, 2011b; Theis et al., 2015b; Bassett and Fortenberry, 2017; Santaloci and Fortenberry, 2020, 2021) have placed the diffuse functions at either the center of charge (COC) or center of mass (COM). The COM is the mass weighted Cartesian origin, and the COC is at the positive pole of the corresponding neutral radical dipole moment computed, in this case, at the anion geometry. Placement of the diffuse functions at the COC lowers the energies of the excitations and is less likely to destabilize the occupied molecular orbitals (Santaloci and Fortenberry, 2020). Additionally, such a position is consistent with theory in that the binding of the excess, diffuse electron has its locus at the point with the most positive charge. As a result, the COC will be utilized for incorporating the additional diffuse functions.
The goal of this present work is to determine how functionalizing deprotonated PAHs will affect the anion photochemistry of such species. Previous work has shown that deprotonated anions will almost always exhibit valence ground states opening the possibility for DBXSs, if the corresponding neutral radical dipole moments are large enough (Fortenberry and Crawford, 2011a; Fortenberry and Crawford, 2011b; Hammonds et al., 2011; Fortenberry et al., 2014). Additionally, the presence of nitrogen heteroatoms promotes additional excited states of anions including DBXS (Theis et al., 2015b). Cyano-functionalization also promotes such behavior (Santaloci and Fortenberry, 2020, 2021), but other functional groups remain to be studied. Herein, the photochemical behavior of deprotonated PAH anions with hydroxyl and ethynyl functional groups are explored. Unlike −CN, the resulting molecular anions for both of these additions can be deprotonated on the functional group themselves likely shifting the behavior of the electronic structure.
Most notably, hydroxides can become ketones and vice versa in nucleobases under certain conditions. As this work will show, the most stable deprotonated anions for −OH and −C2H functionalized PAHs have the hydrogen atoms removed from these functional groups. In the case of the hydroxyl group, this actually forms a ketone and a delocalized π highest occupied molecule orbital (HOMO). Consequently, such a process could open the door for new photo physically-driven chemistry and faster kinetics for the formation of nucleic acids in astrophysical environments such as Titan’s atmosphere (Bevilacqua, 2003; Vay et al., 2019). Additionally, ethynyl-functionalized molecules have now been observed in the interstellar medium (Cernicharo et al., 2021), and the triatomic radical is one of the most abundant molecules in space (Heikkilä et al., 1999) likely helping in the buildup of larger PAHs (Tielens, 2008). This work will provide the isomerization energies, transition energies, and oscillator strengths for singly-deprotonated anions of benzene, naphthalene, and anthracene functionalized with a hydroxyl or ethynyl group. The goal of this work is to analyze the absorption properties of these singly deprotonated anionic PAHs, which will assist in understanding the type of anions that could potentially arise in the building of larger PAHs-like molecules including nulceotides.
2 Computational Methods
In this work, benzene, naphthalene, and anthracene singly deprotonated and functionalized with either a hydroxyl or ethynyl group and analyzed quantum chemically. The functional groups are placed at multiple positions for naphthalene and anthracene. Similar to past studies (Santaloci and Fortenberry, 2020, 2021), the closed shell anion and neutral radical geometries are optimized with B3LYP/aug-cc-pVDZ within Gaussian09 and Gaussian16 (Dunning, 1989; Becke, 1993; Frisch et al., 2016). The optimized geometries provide the relative energies for each radical or anion isomer of each molecule/functional group combination. The optimized geometries of the anion are then utilized to compute a neutral radical dipole moment and the coordinates for the closed-shell anion excited state computations.
The electronically excited states are computed with both CFOUR (Stanton et al., 2008) and Molpro (Werner et al., 2015) using equation of motion coupled cluster theory at the singles and doubles level (EOM-CCSD) (Stanton et al., 1993; Krylov, 2008; Shavitt and Bartlett, 2009) with a double-zeta correlation consistent basis set (aug-cc-pVDZ) expanded with six s-type, six p-type, and two d-type additional, even-tempered diffuse basis functions called the aug-cc-pVDZ+6s6p2d, or more simply the apVDZ+6s6p2d, basis set. The augmented basis functions are placed at the COC, and the anion geometries are utilized to mimic absorption behavior because the closed-shell anion is being excited. A past study on cyano functionalized PAHs shows that the EOM-CCSD computations with the apVDZ+6s6p2d basis approach the Hartree-Fock Limit, but this level of theory still has an error range of roughly 1 meV when comparing between computed values for the DBXS energy and the eBE (Bassett and Fortenberry, 2017; Santaloci and Fortenberry, 2021). The eBE is the maximum amount of energy than can be absorbed by the anion before the electron leaves the system entirely. Specifically here, the eBEs are computed using EOM-CCSD under the ionization potential formalism (EOMIP) (Stanton and Gauss, 1994) with the aug-cc-pVDZ basis set. The electrons in the system are of a valence nature and do not need additional diffuse functions to accurately portray the existence of the resulting neutral radical. The experimental eBE for CH2CN− is 1.543 eV, for example, and the benchmark theoretical value computed in the same way as that described above (EOMIP-CCSD/aug-cc-pVDZ) is 1.524 eV, a difference of less than 0.02 eV (Lykke et al., 1987; Gutsev and Adamowicz, 1995; Cordiner and Sarre, 2007; Fortenberry and Crawford, 2011a; Morgan and Fortenberry, 2015).
3 Results
The singly functionalized −C2H or −OH PAH anions are of either Cs or C2v symmetry. Correspondingly, the electronically excited state’s term will either be 1A′/1A1 or 1A″/1B1. The HOMO is typically isolated to a lone pair on the carbon where the hydrogen is removed. However, the HOMO is defined by the out of plane π orbitals when deprotonation takes place either on the hydroxyl or the ethynyl group. Consistent with past work, herein the dipole-bound excitation has an electron transition from the HOMO into a large, diffuse, Rydberg s-type orbital, and the defined +6s6p2d, additional diffuse functions mimic the diffuseness of the Rydberg-like orbitals. The dipole moment for the neutral radical computed at the optimized geometry of the anion also must be larger than 2.0 D in order to confidently determine whether the dipole-bound state exists in this simulation absorption. Furthermore, any possible valence excitation will naturally be well below the eBE, and the neutral radical dipole moment is independent of the excitation. As a result, the valence excitation does not need a neutral radical dipole moment of greater than 2.0 D.
3.1 Benzene
3.1.1 Relative Energies and Dipole Moments
Table 1 shows the relative energies and the radical dipole moments for the deprotonated phenol and ethynylbenzene classes of molecules. Furthermore, the deprotonation sites are labeled in Figure 1. The deprotonation sites correspond to the first column of the tables throughout the paper. For instance, in Table 1, phenol radical/anion 1 is the removal of the hydrogen on the oxygen atom.
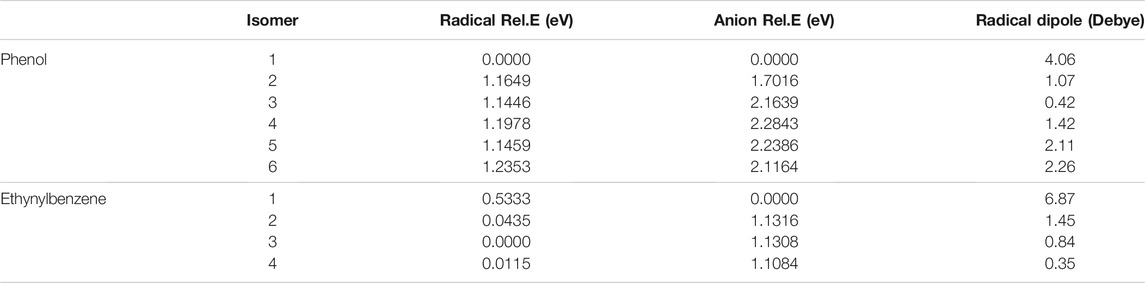
TABLE 1. The relative energies and neutral radical dipole moments of singly deprotonated phenol isomers.
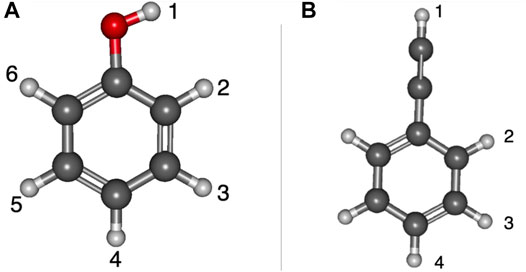
FIGURE 1. Optimized structures of phenol and ethynylbenzene with numbered positions of deprotonation: (A) phenol, (B) ethynylbenzene.
For phenol, radical/anion 1 is the most stable isomer of each respective group and has the largest radical dipole moment of 4.06 D. Furthermore, radical/anion 1 is lower in energy than the rest by more than 1 eV, which is a result of the hydroxyl group becoming a ketone functional group stabilizes the system. Radical 6 and anion 4 are the least stable of their sets, but most of the relative energies are self-similar because the electron donating nature of the hydroxyl group does not greatly affect the stability of the electrons in the HOMO. Radical 6 is 1.2353 eV higher in energy than radical 1, and anion 4 is 2.2843 eV less stable than anion 1. Additionally, anion 2 is the second most stable. This is likely due to the hydrogen on the oxygen constructively interfering with the adjacent lone pair stabilizing the anionic charge. This trend is not as clear for the radical section, but the difference between the least stable (radical 6) to radical 2 is only 0.07 eV. Besides radical 1, only radicals 5 and 6 have dipole moments large enough to have a dipole-bound excitation in their corresponding anions. At the opposite end of the spectrum, the radical 2 dipole moment is only 1.07 D.
3.1.2 Vertical Excitation Energies
Conversely for the other functional group and due to the electron withdrawing nature of ethynyl, the singly-occupied HOMO is destabilized to a much greater extent in the radical form when the hydrogen removal site on the ethynyl group. Radical 3, where the singly-occupied orbital is in the meta position, is the most stable, but there is littler isomeric energy difference between it and radicals 2 and 4. This is similar to the previous benzonitrile anion study where the least stable radical has the largest dipole moment at 5.18 D and is also the most stable anion for the benzonitrile isomer group (Santaloci and Fortenberry, 2020, 2021). Hence, functionalization with −C2H and −CN show similarities.
Table 2 lists the electronically excited state transition energies and eBEs of the phenol and ethynylbenzene anion isomers. Figure 2 shows the orbitals involved in electronic excitations for ethynylbenzene anion 1 as an example. The HOMO, in this case, is the out-of-plane π orbital (Figure 2B). In the other isomers, when the deprotonation is on the ring and the symmetry of the anion becomes Cs, the HOMO looks more like Figure 2A. Most of the anions throughout the paper are Cs. When this C2v to Cs shift occurs, the deprotonation moves to the ring, and the in-plane lone pair is the least stable orbital.
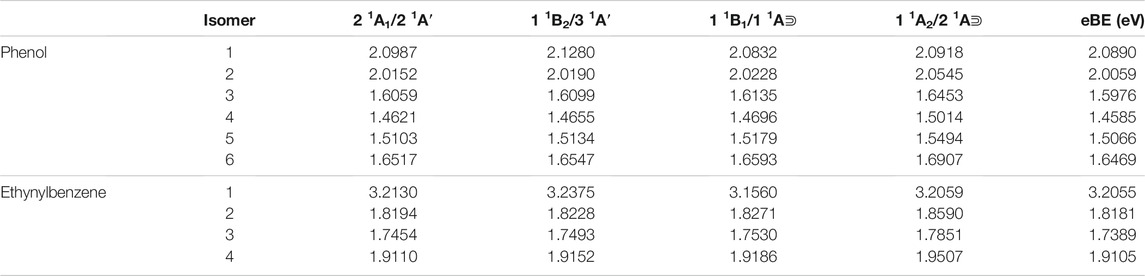
TABLE 2. The singly deprotonated anion derivatives eBEs and excited state transition energies.
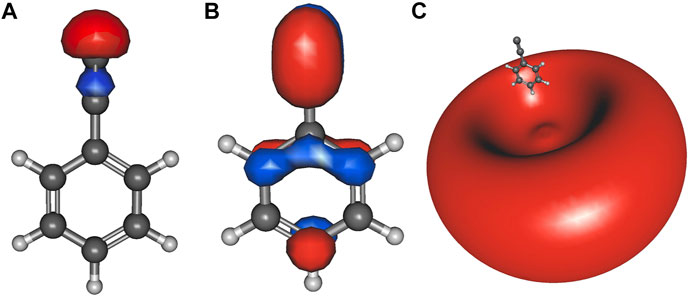
FIGURE 2. Ethynylbenzene anion 1 a1 HOMO-2 (A); b1 HOMO (B), and the a1 LUMO (C) dipole-bound orbital.
When the deprotonation occurs on the hydroxyl or ethynyl group, a valid dipole-bound excitation occurs, but such is not present for any of the other isomers where the deprotonation occurs on the ring even though phenol radicals 5 and 6 showed dipole moments of greater than 2.0 D. Regardless, the phenol anion 1 dipole-bound transition is 5.8 meV and ethynylbenzene anion 1 is 49 meV underneath their respective eBEs. Furthermore, the bound phenol absorption wavelength is 595 nm, and the ethynylbenzene absorption wavelength is 392 nm. The phenol DBXS transition is capable of capturing much lower energy photons, whereas the ethynylbenzene absorption is approaching the violet region but barely within the UV region. Lastly, both phenol and ethynyl derivatives do not exhibit any valence excited states.
3.2 Naphthalene
3.2.1 Relative Energies and Dipole Moments
Figure 3 displays the deprotonation positions of naphthalene isomers functionalized with the hydroxyl groups. The ethynyl group naphthalene isomer numbering is much the same. The two isomers of naphthalene have the functional groups placed either at position 1 or 2. Position 1 has functional groups placed on the inner portion of the ring, and position 2 is when functionalization takes place on the outer part of the naphthalene like with quinoline and isoquinoline. Additionally, the deprotonation sites are labeled to correspond with the subsequent tables.
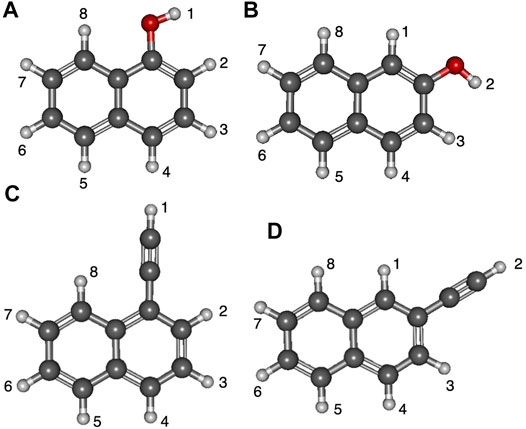
FIGURE 3. Optimized structures of the four naphthalene derivatives with numbered positions of deprotonation: (A) 1-hydroxynaph, (B) 2-hydroxynaph, (C) 1-ethynylnaph, (D) 2-ethynylnaph.
Tables 3, 4 portray the relative energies of the radicals and anions for the 1-naphthalene isomers. 1-hydroxynaphthalene has 10 and 1-ethynylnaphthalene has 8 radical/anion isomers. The hydroxynaphthalene class has an additional two isomers because the hydrogen on the oxygen points towards either side of the naphthalene. When the hydrogen is rotated to the opposite side an “R” is added to the label. For example, 1-hydroxynaphthalene 2 has the hydrogen facing toward the deprotonated carbon and 2R has the hydrogen rotated away. All other arrangements of “R” produce insignificant changes in the electronic structure and will not be explored.
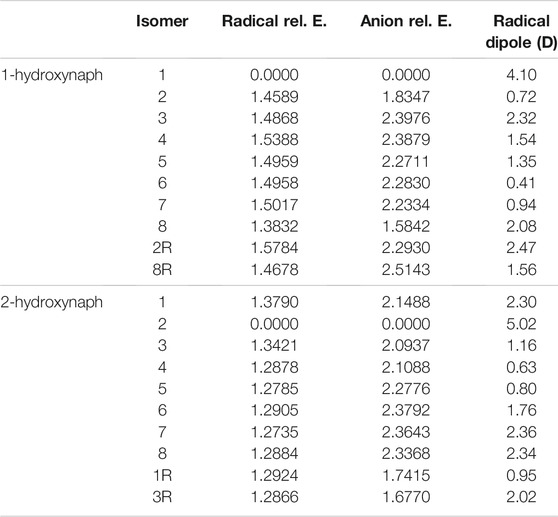
TABLE 3. The relative energies and neutral radical dipole moments of hydroxynaphthalene derivatives.
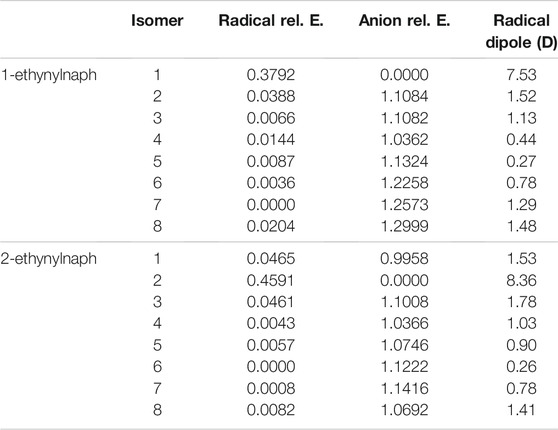
TABLE 4. The relative energies and neutral radical dipole moments of ethynylnaphthalene derivatives.
The relative energies of both ethynyl- and hydroxy-functionalization follow the same trend as the benzene section. For the hydroxynaphthalene, the radical/anion energies are the most stable when the ketone group forms. The hydroxynaphthalene isomers have a total of 9 radicals with a dipole moment above 2.0 D. Specifically, 1-hydroxynaphthalene neutral radicals 1, 2R, 3, and 8 along with 2-hydroxynaphthalene radicals 1, 2, 7, 8, and 3R have dipole moments greater than 2.0 D. 2-hydroxynaphthalene radical 2 has the largest dipole moment at 5.0 D, but the next highest is the 1-hydroxynaphthalene radical 1 at 4.1 D. Additionally, the dipole moments of 1-hydroxynaphthalene 2R and 2-hydroxynaphthalene 3R are significantly larger than their unrotated counterparts. The 1-hydroxynaphthalene radical 2 and 2-hydroxynaphthalene radical 3 both possess dipole moments below 1.2 D. This is a result of the hydrogen on the oxygen, which distorts the dipole moment and stabilizes the partial negative charge.
Likewise, the ethynylnaphthalene anion isomers are the most stable when the hydrogen is deprotonated from the ethynyl group. The ethynylnaphthalene radicals act differently than the hydroxynaphthalene isomers. The least stable ethynyl radicals are the most stable ethynylnaphthalene anions, again like with the −CN functionalization (Santaloci and Fortenberry, 2021), implying a trend for when the functional group is a strong electron withdrawing group. Furthermore, 1-ethynylnaphthalene radical 7 and 2-ethynylnaphthalene radical 6 are the most stable. Both of these have the lone pair on the opposite side of the ring from the functional group. Interestingly, the other radicals that surround the ethynyl group demonstrate relative energies within at most 0.046 eV of each other. This relatively small difference results in only two ethynylnaphthalene neutral radicals dipole moments being larger than 2.0 D. Both of these radicals are the most stable anions and have the highest corresponding neutral radical dipole moments seen so far: 7.5 D for 1-ethynylnaphthalene anion 1 and 8.4 D for 2-ethynylnaphthalene anion 2. This indicates only two anions in the ethynylnaphthalene class can bind a free electron to form a dipole-bound state.
When comparing the 1-naphthalene and 2-naphthalene classes, the most stable anions are found in the 2-naphthalene regardless of the functional group at position 2. Specifically, the 2-hydroxynaphthalene anion 2 is 0.16 eV lower in energy than the 1-hydroxynaphthalene anion 1, and the 2-ethynylnaphthalene anion 2 is 0.01 eV is lower in energy than 1-ethynylnaphthalene anion 1. This is, again, consistent with the previous work on cyanonaphthalenes (Santaloci and Fortenberry, 2021). The reason for such behavior is that the molecule is slightly enlongated in the 2 position which increases the total spatial extent for the orbitals. From a particle-in-a-box (PIB) argument, as the box becomes longer the wave function stabilizes the lower energy.
3.2.2 Vertical Excitation Energies
Table 5 shows that all of the possible valence excited state transition energies are above the eBEs for each isomer. The hydroxynaphthalene DBXS transition energies for 1-hydroxynaphthalene anion 1 and 2-hydroxynaphthalene anion 2 are clearly underneath the eBE. Specifically, 1-hydroxynaphthalene anion 1 is 6.9 meV and 2-hydroxynaphthalene anions 2 is 10.9 meV, respectively, below their eBEs. Other DBXS candidates, like 1-hydroxynaphthalene anions 8 and 2R, may exist since their excitation energies are within 1 meV of the eBE. Both of these anions are next to the oxygen when the hydrogen is facing away from it. The destabilization of the oxygen lone pair to their radical lone pair results in the dipole moment being above 2.0 D, but the excitation energies fall within the error range and can not be confidently concluded as a DBXS through the present quantum chemical analysis.
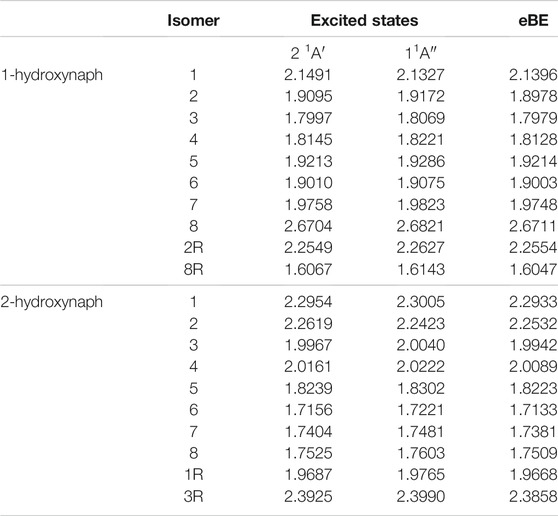
TABLE 5. The singly deprotonated hydroxynaphthalene anion derivatives eBEs and transition energies.
Table 6 shows the electronically excited state energies for the deprotonated ethynylnaphthalene anion isomers. Similar to ethynylbenzene, the only conclusive DBXS transition originates from the lone pair on the ethynyl group. However, there are a couple of transition energies that are below the eBE. These are 1-ethynylnaphthalene anion 4 and 8 as well as 2-ethynylnaphthalene anion 5 and 8. Since, the radicals’ dipole moments are not large enough to stabilize the electron in a diffuse orbital, the dipole-bound state likely cannot be present. Analysis of the MOs does not support these being valence excited states. Future work will analyze the behavior of these possibly bound electronically excited states. Furthermore, the DBXS transition energies for 1-ethynylnaphthalene anion 1 and 2-ethynylnaphthalene anion 2 are largely underneath the eBE. Specifically, 1-ethynylnaphthalene anion 1 is 41.3 meV and 2-ethynylnaphthalene anion 2 is 55.3 meV below the eBE. This is a result of the large radical dipole moments. Figure 4 shows the orbitals involved in the DBXS excitation of 2-ethynylnaphthalene anion 2, and the hydroxynaphthalene orbitals, not shown, appear similar. The left side of Figure 4 is the HOMO, and the right is the diffuse s-type Rydberg LUMO. Similar to the benzene class of anions discussed previously, the hydroxynaphthalene excitations are between the 550–581 nm range, and the ethynylnaphthalene excitations are clearly lower between 386 and 390 nm. Again, the ethynylnaphthalene can capture UV light and hydroxynaphthalene is in the greenish-yellow region.
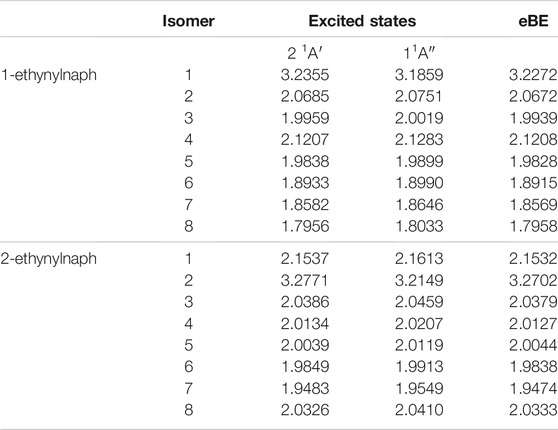
TABLE 6. The singly deprotonated ethynylnaphthalene anion derivatives eBEs and transition energies.
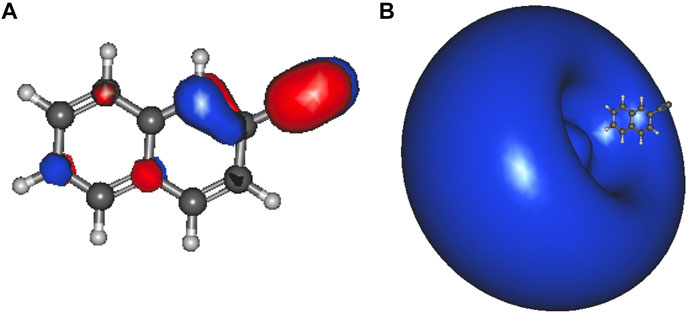
FIGURE 4. 2-ethynylnaphthalene anion 2 a′ HOMO (A);and the a′ LUMO (B) dipole-bound orbital.
3.3 Anthracene
3.3.1 Relative Energies and Dipole Moments
Supplementary Figures S1, S2 represent the three derivatives of deprotonated ethynyl- and hydroxyanthracene. There are ten isomers for 1-hydroxyanthracene and six isomers for 1-ethynylanthracene. Similar to past studies, only six of the 1-ethynylanthracene isomers are unique due to the symmetry. For both hydroxyl- and ethynylanthracene, the other two derivatives have ten isomers and are Cs symmetry.
Tables 7, 8 show the relative and excitation energies for all the hydroxyanthracene derivatives. In line with phenol and hydroxynaphthalene, the most stable radical and anion isomers exist when the hydrogen is deprotonated off of the oxygen. For all three hydroxyanthracene derivative sets, the second most stable radical and anion occur when deprotonation takes place on the adjacent carbon where the hydrogen on the oxygen is facing the resulting lone pair. The least stable radicals and anions differ for each hydroxyanthracene. However, there is no significant difference in energy between the least stable and third most stable radicals and anions for all three hydroxyanthracenes, which range from 0.01 to 0.06 eV from one another. An example is 1-hydroxyanthracene radical 6, which is the least stable compared to radical 8, which is the third most stable. These two radicals differ in relative energy by 0.06 eV.
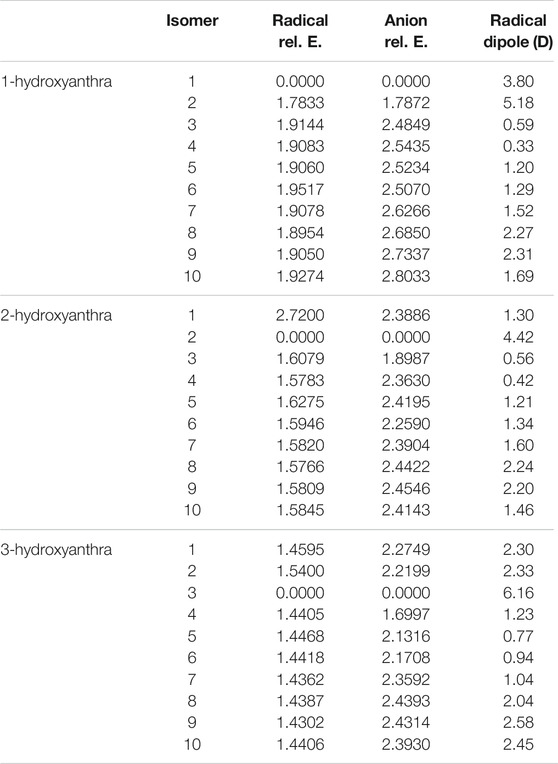
TABLE 7. The relative energies and neutral radical dipole moments of hydroxyanthracene derivatives.
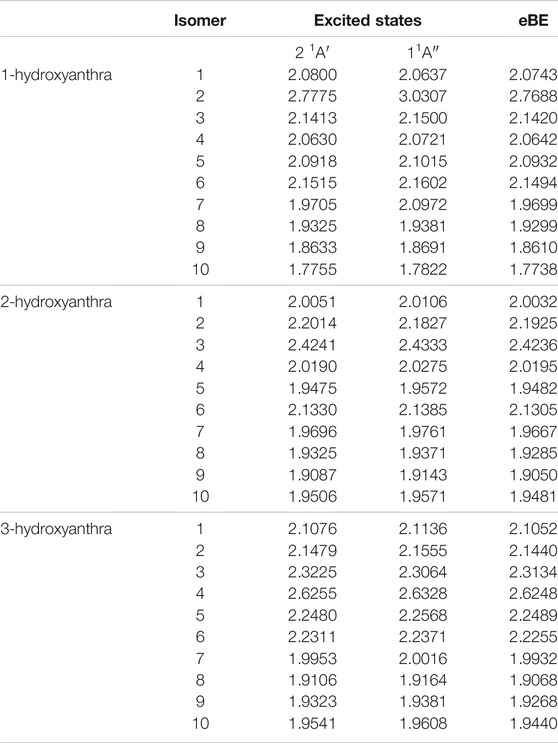
TABLE 8. The singly deprotonated hydroxyanthracene anion derivatives eBEs and transition energies.
Comparable to the hydroxynaphthalene derivatives, the most stable anion and radical isomer for the larger ring class is 1-hydroxyanthracene, and the least stable is 3-hydroxyanthracene. The radical dipole moment once more seemingly increases with radical isomeric instability; the largest radical dipole moments are in the 3-hydroxyanthracene. Specifically, the 3-hydroxyanthracene radical 3 dipole moment is 6.2 D, 2-hydroxyanthracene radical 2 is 4.4 D, and 1-hydroxyanthracene radical 1 is 3.8 D. There are other notable radicals with dipole moments larger than 2.0 D. For 1-hydroxyanthracene, radicals 8 and 9 are 2.2 and 2.1 D. 2-hydroxyanthracene radicals 8, and 9 are 2.2, and 2.0 D. 3-hydroxyanthracene radicals 1, 2, 8, 9 and 10 are 2.3, 2.3, 2.0, 2.6 and 2.5 D. A past study on cyanoanthracene reveals that the dipole moments of the radicals become larger when the ring length becomes larger (Santaloci and Fortenberry, 2020).
Tables 9, 10 display the relative and excitations energies for the ethynylanthracene derivatives. The relative energies follow the same type of trend reported in the previous cyanoanthracene study (Santaloci and Fortenberry, 2021) The least stable anion is the most stable radical and vice versa. Unlike hydroxyanthracene, the radical and anion do not have a significant energy difference between their isomers. This is a result of no hydrogen bonding effect and no formation of a different functional group. The largest energy difference for the radicals is 2-ethynylanthracene radical 4 with relative energy of 0.0396 eV compared to the most stable isomer, radical 9. For the anions, their largest relative energy is from 1-ethynylanthracene anion 2 at 1.4 eV compared to anion 1.
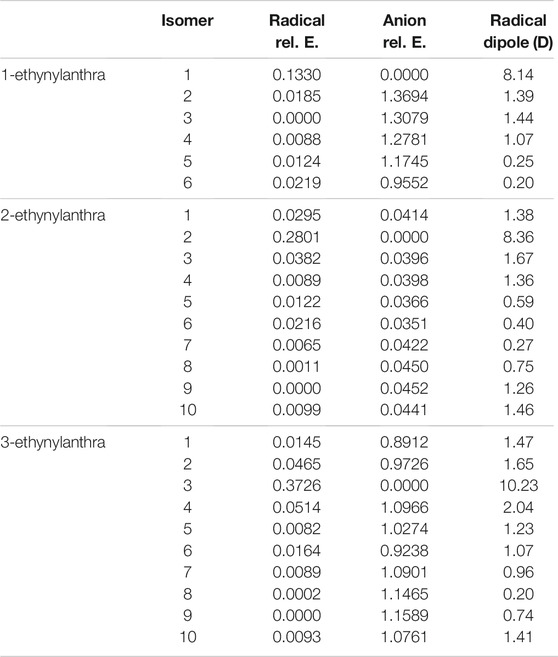
TABLE 9. The relative energies and neutral radical dipole moments of singly deprotonated ethynylanthracene derivatives.
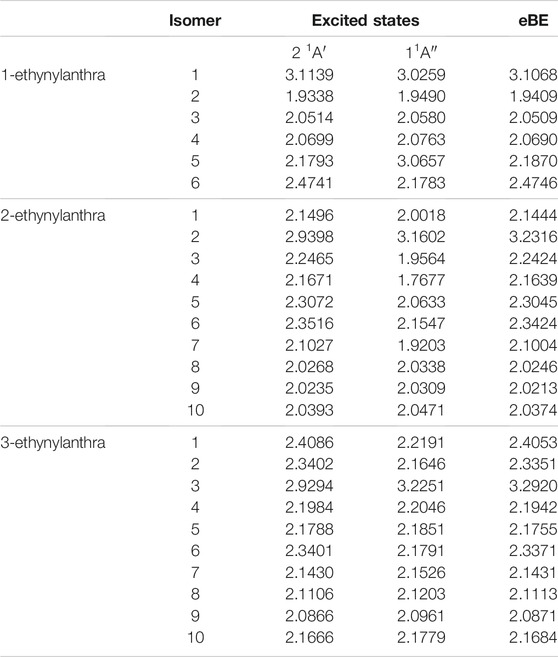
TABLE 10. The singly deprotonated ethynylanthracene anion derivatives eBEs and transition energies.
The largest radical dipole for the ethynylanthracene derivatives is, once more, for the isomer resulting from deprotonation on the ethynyl group. Specifically, the 1-ethynylanthracene radical 1 has a dipole moment of 8.1 D; 2-ethynylanthracene radical 2 has a dipole moment of 8.4 D; 3-ethynylanthracene radical 3 has a dipole moment of 10.2 D. 3-ethynylanthracene is the only ethynylanthracene isomer that has more than one radical with a dipole moment larger than 2.0 D. Namely, radical 4 has a dipole moment of 2.0 D. For the ethynylanthracene class, only 4 radicals have dipole moments greater than 2.0 D.
3.3.2 Vertical Excitation Energies
Tables 8, 10 display the electronically excited states of the deprotonated hydroxyl and ethynylanthracene anions. Comparable to ethynylbenzene, the carbon on the 1-ethynylanthracene functional group and adjacent to the ethynyl group is C2v symmetry with the 2 1A1 state being likely a DBXS, and the 1 1B1 state would be valence. Additionally, similar to phenol, the 1-hydroxyanthracene is C2v symmetry when the deprotonation site is on the oxygen. Furthermore, the excitation into the 2 1A1 state is potentially a DBXS, and 1 1B1 would be valence. Lastly, similar to the naphthalenes, all of the remaining derivatives will be Cs symmetry, which indicates the dipole-bound states will be 2 1A’, and any possible valence states will be 1 1A″.
In the hydroxy-functionalized isomers, the dipole-bound excitation only exists when the ketone group forms upon deprotonation from the oxygen atom. For 1-hydroxyanthracene, excitations from anions 1, 4, and 5 are outside the 1 meV error range of the eBE. However, the only neutral radical dipole moment greater than 2.0 D is anion 1. 2-hydroxyanthracene has a dipole-bound excitation for anion 2. Similar to 1-hydroxyanthracene, 2-hydroxyanthracene anions 4 and 5 do not have large enough dipole moments but have excitations that are only 0.5 and 0.7 meV underneath the eBE. Congruent to other hydroxyanthracenes, 3-hydroxyanthracene anion 5 has an excitation 0.9 meV below the eBE. These excitations may be more exotic types of excitation that go beyond the present study. Lastly, none of the hydroxyanthracene derivatives have any valence excitation characteristics.
When ethynyl functionalization on anthracene occurs, more types of excitations appear. For the ethynylanthracene class, there are both valence and dipole bound excitations. All three ethynylanthracene exhibit both dipole-bound and valence excitations. Figure 4 shows the HOMO to valence LUMO and dipole-bound LUMO orbitals for 3-ethynylanthracene anion 3. The dipole-bound orbital is closely related for all of the valid hydroxyanthracene and ethynylanthracene dipole-bound excitations. Interestingly, there are only four neutral radical dipole moments large enough to stabilize a free electron. Three of those four isomers are deprotonated on the ethynyl group. Specifically, anion 1 of 1-ethynylanthracene is 81 meV, anion 2 of 2-ethynylanthracene is 29 meV, and anion 3 of 3-ethynylanthracene is 66 meV underneath the eBE. 3-ethynylanthracene anion 4 is the remaining isomer with a large corresponding neutral radical dipole moment, but it does not exhibit an excited state below the eBE. Also, 1-ethynylanthracene anion 6, 2-ethynylanthracene anion 1-7, and 3-ethynylanthracene anion 1, 2, 3, and 6 have valence excitations. 1-ethynylanthracene anion 2 and 5, 3-ethynylanthracene anion 8, 9, and 10 do not have corresponding neutral radicals that possess large enough dipole moments. However, these isomers have excitation energies underneath the eBE, which again alludes to a more complex type of quantum electronics.
Lastly, similar to the past classes, the hydroxyanthracene excitations are higher in energy than the ethynylanthracene. For hydroxyanthracene, the dipole bound transitions are 537–600 nm, and ethynylanthracene dipole-bound transitions are between the range of 384–409 nm. Also, ethynylanthracene has the first instance of valence excitations examined in this work, and the absorption range is 558–700 nm, which is in the same region as the hydroxyanthracene dipole-bound transitions.
4 Oscillator Strengths
The oscillator strengths are given in the Supplementary Material. Similar to past PAH and PANH studies, the oscillator strengths are on the order of 1 × 10–4 if the dipole-bound state exists (Theis et al., 2015a,b; Fortenberry et al., 2016; Santaloci and Fortenberry, 2021). Furthermore, the oscillator strengths become larger the farther away the HOMO electron density is away from the COC. Additionally, when the oscillator strength for the dipole-bound states computed here are not large but large enough to contribute to absorption spectra when found over large enough path lengths as likely takes place under interstellar conditions. Additionally, when the excited state transition energy is above the eBE, the resulting oscillator strengths tendency to be below 1 × 10–6, which shows that those states above the eBE would not contribute to any spectra even if they could somehow be stabilized.
5 Conclusion
When closed shell PAH anion single deprotonation occurs on the functional group, a DBXS will exist. However, when the deprotonation site is on the ring, the type of excited state for excitation becomes obscure. To be clear, if the excitation energy is greater than 1 meV underneath the eBE and the neutral radical dipole moment is larger than 2.0 D, then such a state is most likely a DBXS. On the other hand, if the excitation energy is within 1 meV underneath the eBE and the neutral radical dipole moment is less than 2.0 D, the state likely cannot be classified as dipole-bound. This implies a possibility of a different type of anion excitation. An example is 3-ethynylanthracene anions 6–10, which have neutral radical dipole moments of less than 2.0 D, but the DBXS excitation energies are within 1 meV underneath the eBE. When the dipole moment is less than 2.0 D, some other property of the PAH must keep the electron bound to result in the excitation being underneath the eBE.
Differently, the number of valence excitations depends on whether the functional group is electron donating or electron withdrawing. The electron donating hydroxyl group stabilizes the ring structures, which produces a larger gap between the HOMO and LUMO. As a result, the hydroxyl functionalized PAHs examined herein do not have any valence excited state anions. Typically, the stronger the electron donating group the better the ring stabilization. Electron withdrawing functionalization appears to produce the opposite effect. Past studies on singly deprotonated cyano functionalized PAHs show that valence excitations begin to appear when the number of rings is two (Santaloci and Fortenberry, 2021). When the ring number is three, then every deprotonated structure has a valence excited state. Interestingly, when the electron withdrawing group is a weaker ethynyl group (compared to −CN), valence excitations only begin to appear once three rings are present. Specifically, only 11 out of 30 ethynylanthracene anions possess valence excitations. The ethynyl group destabilizes the HOMO less than the cyano group but is still a destabilizing group. The valence excited state oscillator strengths are approximately <1 × 10−4, which is large enough to contribute to absorption spectra, but only when there is a sufficient population of molecules.
The energy of the photon that an anion absorbs to form a possible DBXS is higher in energy than the valence states. When the functional group is an electron donating group, like hydroxyl, the excitation energies become higher when the number of rings increases. Notably, for the electron withdrawing ethynyl group, the excitation energies lower when the ring number increases, but the excited states are well below the eBE. This suggests the hydroxyl groups are able to stabilize the HOMOs of larger PAH anions better than the ethynyl group. This supports the idea that deprotonation of hydroxyl groups may be an intermediate in the formation of nucleobases in gas-phase, astrophysical environments. However, broadly speaking, deprotonated PAH anions functionalized with electron donating groups will be less likely to have electronicallly excited anions than those with electron withdrawing groups.
Hence, from a stability perspective, the hydroxyl, electron donating group produces a more stable structure, especially for deprotonation on the functional group itself (thus creating the ketone like those found in nucleotides), which will better allow these anions to persist in collision-dominated environments. In regions with a higher photon flux, however, the less stable PAHs functionalized with electron withdrawing groups will be able to absorb the incident photons much more effeciently likely producing longer lifetimes for this opposite class of molecules. As a result, if anions are involved in the formation of nucleic acids, both of these seemingly counteracting processes may be taking place at different times in their evolution in order to assist in nucleic acid formation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
TS performed the computational work. RF concieved of the idea, managed the project, and secured the funding. All three authors were involved in analyzing the results as well as writing and editing the manuscript.
Funding
This work is supported by NASA grant NNX17AH15G, NSF grant OIA-1757220, NSF REU grant CHE-1757888, the College of Liberal Arts at the University of Mississippi, and a graduate resesearch fellowship from the Mississippi Space Grant Consortium.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The depictions of the molecules are provided by Gabedit.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2021.777107/full#supplementary-material
References
Agúndez, M., Cernicharo, J., Guélin, M., Gerin, M., McCarthy, M. C., and Thaddeus, P. (2008). Search for Anions in Molecular Sources: C4H-Detection in L1527. A&A 478, L19–L22. doi:10.1051/0004-6361:20078985
Agúndez, M., Cernicharo, J., Guélin, M., Kahane, C., Roueff, E., Kłos, J., et al. (2010). Astronomical Identification of CN-, the Smallest Observed Molecular Anion. A&A 517, L2. doi:10.1051/0004-6361/201015186
Ali, A. E. C. S., Sittler, E. C., Chornay, D., Rowe, B. R., and Puzzarini, C. (2015). Organic Chemistry in Titan׳s Upper Atmosphere and its Astrobiological Consequences: I. Views towards Cassini Plasma Spectrometer (CAPS) and Ion Neutral Mass Spectrometer (INMS) Experiments in Space. Planet. Space Sci. 109-110, 46–63. doi:10.1016/j.pss.2015.01.015
Allen, M., Yung, Y. L., and Pinto, J. P. (1980). Titan - Aerosol Photochemistry and Variations Related to the sunspot Cycle. ApJ 242, L125–L128. doi:10.1086/183416
Aoki, K. (2000). Candidates for U-Lines at 1377 and 1394 MHz in IRC+10216: Ab Initio Molecular Orbital Study. Chem. Phys. Lett. 323, 55–58. doi:10.1016/s0009-2614(00)00469-3
Aoki, K., Ikuta, S., and Murakami, A. (1996). Equilibrium Geometries and Stabilities of the C3H Radical: Ab Initio MO Study. J. Mol. Struct. THEOCHEM 365, 103–110. doi:10.1016/0166-1280(96)04513-7
Bassett, M. K., and Fortenberry, R. C. (2017). Symmetry Breaking and Spectral Considerations of the Surprisingly Floppyc-C3h Radical and the Related Dipole-Bound Excited State Ofc-C3h−. J. Chem. Phys. 146, 224303. doi:10.1063/1.4985095
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Bevilacqua, P. C. (2003). Mechanistic Considerations for General Acid−Base Catalysis by RNA: Revisiting the Mechanism of the Hairpin Ribozyme. Biochemistry 42, 2259–2265. doi:10.1021/bi027273m
Bradforth, S. E., Kim, E. H., Arnold, D. W., and Neumark, D. M. (1993). Photoelectron Spectroscopy of CN−, NCO−, and NCS−. J. Chem. Phys. 98, 800–810. doi:10.1063/1.464244
Brünken, S., Gupta, H., Gottlieb, C. A., McCarthy, M. C., and Thaddeus, P. (2007). Detection of the Carbon Chain Negative Ion C 8 H - in TMC-1. ApJ 664, L43–L46. doi:10.1086/520703
Cernicharo, J., Agúndez, M., Cabezas, C., Tercero, B., Marcelino, N., Pardo, J. R., et al. (2021). Pure Hydrocarbon Cycles in Tmc-1: Discovery of Ethynyl Cyclopropenylidene, Cyclopentadiene, and Indene. A&A 649, L15. doi:10.1051/0004-6361/202141156
Cernicharo, J., Guélin, M., Agúndez, M., Kawaguchi, K., McCarthy, M., and Thaddeus, P. (2007). Astronomical Detection of C$\mathsf{_{4}H^-}$, the Second Interstellar Anion. A&A 467, L37–L40. doi:10.1051/0004-6361:20077415
Cernicharo, J., Guélin, M., Agúndez, M., McCarthy, M. C., and Thaddeus, P. (2008). Detection of C 5 N - and Vibrationally Excited C 6 H in IRC +10216. ApJ 688, L83–L86. doi:10.1086/595583
Clarke, D. W., and Ferris, J. P. (1997). Titan Haze: Structure and Properties of Cyanoacetylene and Cyanoacetylene-Acetylene Photopolymers. Icarus 127, 158–172. doi:10.1006/icar.1996.5667
Clifford, E. P., Wenthold, P. G., Lineberger, W. C., Petersson, G. A., and Ellison, G. B. (1997). Photoelectron Spectroscopy of the NCN- and HNCN- Ions. J. Phys. Chem. A. 101, 4338–4345. doi:10.1021/jp964067d
Coates, A. J., Crary, F. J., Lewis, G. R., Young, D. T., Waite, J. H., and Sittler, E. C. (2007). Discovery of Heavy Negative Ions in Titan’s Ionosphere. Geophys. Res. Lett. 34. doi:10.1029/2007gl030978
Compton, R. N., Carman, H. S., Abdoul‐Carime, H., Schermann, J. P., Hendricks, J. H., Lyapustina, S. A., et al. (1996). On the Binding of Electrons to Nitromethane: Dipole and Valence Bound Anions. J. Chem. Phys. 105, 3472–3478. doi:10.1063/1.472993
Cordiner, M. A., and Sarre, P. J. (2007). The CH$_\mathsf{2}$CN- Molecule: Carrier of the $\mathsf{\lambda}$8037 Diffuse Interstellar Band. A&A 472, 537–545. doi:10.1051/0004-6361:20077358
Cordiner, M., Charnley, S., Buckle, J., Walsh, C., and Millar, T. (2011). Discovery of Interstellar Anions in Cepheus and Auriga. Astrophys. J. Lett. 730, 1–5. doi:10.1088/2041-8205/730/2/l18
Coustenis, A. (2007). What Cassini-Huygens Has Revealed about Titan. Astron. Geophys. 48, 2.14–2.20. doi:10.1111/j.1468-4004.2007.48214.x
Desfrancois, C., Abdoul-Carime, H., and Schermann, J. (1996). Electron Attachment to Isolated Nucleic Acid Bases. J. Chem. Phys. 104, 7762. doi:10.1063/1.471484
Dunning, T. H. (1989). Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms boron through Neon and Hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
Fermi, E., and Teller, E. (1947). The Capture of Negative Mesotrons in Matter. Phys. Rev. 72, 399–408. doi:10.1103/physrev.72.399
Fortenberry, R. C., and Crawford, T. D. (2011a). Singlet Excited States of Silicon-Containing Anions Relevant to Interstellar Chemistry. J. Phys. Chem. A. 115, 8119–8124. doi:10.1021/jp204844j
Fortenberry, R. C., and Crawford, T. D. (2011b). Theoretical Prediction of New Dipole-Bound Singlet States for Anions of Interstellar Interest. J. Chem. Phys. 134, 154304. doi:10.1063/1.3576053
Fortenberry, R. C. (2015). Interstellar Anions: The Role of Quantum Chemistry. J. Phys. Chem. A. 119, 9941–9953. doi:10.1021/acs.jpca.5b05056
Fortenberry, R. C., King, R. A., Stanton, J. F., and Crawford, T. D. (2010). A Benchmark Study of the Vertical Electronic Spectra of the Linear Chain Radicals C2H and C4H. J. Chem. Phys. 132, 144303. doi:10.1063/1.3376073
Fortenberry, R. C., Moore, M. M., and Lee, T. J. (2016). Excited State Trends in Bidirectionally Expanded Closed-Shell PAH and PANH Anions. J. Phys. Chem. A. 120, 7327–7334. doi:10.1021/acs.jpca.6b06654
Fortenberry, R. C., Morgan, W. J., and Enyard, J. D. (2014). Predictable Valence Excited States of Anions. J. Phys. Chem. A. 118, 10763–10769. doi:10.1021/jp509512u
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Revision C.01. Wallingford CT [Dataset]: Gaussian Inc.
Gutowski, M., Skurski, P., Boldyrev, A. I., Simons, J., and Jordan, K. D. (1996). Contribution of Electron Correlation to the Stability of Dipole-Bound Anionic States. Phys. Rev. A. 54, 1906–1909. doi:10.1103/physreva.54.1906
Gutsev, G. L., and Adamowicz, L. (1995). The Valence and Dipole-Bound States of the Cyanomethide Ion, CH2CN−. Chem. Phys. Lett. 246, 245–250. doi:10.1016/0009-2614(95)01097-s
Hammonds, M., Pathak, A., Candian, A., and Sarre, P. (2011). Spectroscopy of Protonated and Deprontonated PAHs. EAS Publ. Ser. 46, 373–379.
Hanel, R., Conrath, B., Flasar, F. M., Kunde, V., Maguire, W., Pearl, J., et al. (1981). Infrared Observations of the Saturnian System from Voyager 1. Science 212, 192–200. doi:10.1126/science.212.4491.192
Heikkilä, A., Johansson, L. E. B., and Olofsson, H. (1999). Molecular Abundance Variations in the Magellanic Clouds. Astron. Astrophys. 344, 817–847.
Hendricks, J. H., Lyapustina, S. A., de Clercq, H. L., Snodgrass, J. T., and Bowen, K. H. (1996). Dipole Bound, Nucleic Acid Base Anions Studied via Negative Ion Photoelectron Spectroscopy. J. Chem. Phys. 104, 7788–7791. doi:10.1063/1.471482
Hendricks, J., Lyapustine, A., de Clercq, H., and Bowen, K. (1998). The Dipole Bound-To-Covalent Anion Transformation in Uracil. J. Chem. Phys. 8, 108. doi:10.1063/1.475360
Hoshina, K., Kohguchi, H., Ohshima, Y., and Endo, Y. (1998). Laser-induced Fluorescence Spectroscopy of the C4H and C4D Radicals in a Supersonic Jet. J. Chem. Phys. 108, 3465–3478. doi:10.1063/1.475746
Jalbout, A. F., Pichugin, K. Y., and Adamowicz, L. (2003). An Excess Electron Connects Uracil to glycine. Eur. Phys. J. D - At. Mol. Opt. Phys. 26, 197–200. doi:10.1140/epjd/e2003-00224-4
Jordan, K. D., and Wang, F. (2003). Theory of Dipole-Bound Anions. Annu. Rev. Phys. Chem. 54, 367–396. doi:10.1146/annurev.physchem.54.011002.103851
Kawaguchi, K., Kasai, Y., Ishikawa, S., and Kaifu, N. (1995). A Spectral-Line Survey Observation of IRC+10216 between 28 and 50 GHz. Publ. Astron. Soc.-Japan 47, 853–876.
Krylov, A. I. (2008). Equation-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker's Guide to Fock Space. Annu. Rev. Phys. Chem. 59, 433–462. doi:10.1146/annurev.physchem.59.032607.093602
Kunde, V. G., Aikin, A. C., Hanel, R. A., Jennings, D. E., Maguire, W. C., and Samuelson, R. E. (1981). C4H2, HC3N and C2N2 in Titan's Atmosphere. Nature 292, 686–688. doi:10.1038/292686a0
Lepp, S., and Dalgarno, A. (1988). Polycyclic Aromatic Hydrocarbons in Interstellar Chemistry. Astrophys. J. 324, 533. doi:10.1086/165915
López-Puertas, M., Dinelli, B. M., Adriani, A., Funke, B., García-Comas, M., Moriconi, M. L., et al. (2013). Large Abundances of Polycyclic Aromatic Hydrocarbons in Titan's Upper Atmosphere. ApJ 770, 132. doi:10.1088/0004-637x/770/2/132
Lykke, K. R., Neumark, D. M., Andersen, T., Trapa, V. J., and Lineberger, W. C. (1987). Autodetachment Spectroscopy and Dynamics of CH2CN− and CD2CN−. J. Chem. Phys. 87, 6842–6853. doi:10.1063/1.453379
Mach, T. J., King, R. A., and Crawford, T. D. (2010). A Coupled Cluster Benchmark Study of the Electronic Spectrum of the Allyl Radical†. J. Phys. Chem. A. 114, 8852–8857. doi:10.1021/jp102292x
Maguire, W. C., Hanel, R. A., Jennings, D. E., Kunde, V. G., and Samuelson, R. E. (1981). C3H8 and C3H4 in Titan's Atmosphere. Nature 292, 683–686. doi:10.1038/292683a0
Maier, J. P. (1998). Electronic Spectroscopy of Carbon Chains. J. Phys. Chem. A. 102, 3462–3469. doi:10.1021/jp9807219
McCarthy, M. C., Gottlieb, C. A., Gupta, H., and Thaddeus, P. (2006). Laboratory and Astronomical Identification of the Negative Molecular Ion C 6 H -. ApJ 652, L141–L144. doi:10.1086/510238
McCarthy, M. C., Gottlieb, C. A., Thaddeus, P., Horn, M., and Botschwina, P. (1995). Structure of the Cccn and Cccch Radicals: Isotopic Substitution and Ab Initio Theory. J. Chem. Phys. 103, 7820–7827. doi:10.1063/1.470198
McKay, C. P. (1996). Elemental Composition, Solubility, and Optical Properties of Titan's Organic Haze. Planet. Space Sci. 44, 741–747. doi:10.1016/0032-0633(96)00009-8
Mead, R. D., Lykke, K. R., Lineberger, W. C., Marks, J., and Brauman, J. I. (1984). Spectroscopy and Dynamics of the Dipole‐bound State of Acetaldehyde Enolate. J. Chem. Phys. 81, 4883–4892. doi:10.1063/1.447515
Millar, T. J., Herbst, E., and Bettens, A. R. P. (2000). Large Molecules in the Envelope Surroundings IRC+10216. Mon. Not. R. Astron. Soc. 316, 195–200. doi:10.1046/j.1365-8711.2000.03560.x
Molina-Cuberos, G. J., Schwingenschuh, K., López-Moreno, J. J., Rodrigo, R., Lara, L. M., and Anicich, V. (2002). Nitriles Produced by Ion Chemistry in the Lower Ionosphere of Titan. J. Geophys. Res. Planet. 107, 9–11. doi:10.1029/2000je001480
Morgan, W. J., and Fortenberry, R. C. (2015). Additional Diffuse Functions in Basis Sets for Dipole-Bound Excited States of Anions. Theor. Chem. Acc. 134, 47. doi:10.1007/s00214-015-1647-1
Moustefaoui, T., Rebrion-Rowe, C., Le Garrec, J.-L., Rowe, R. C., and Mitchell, Brian. A. J. (1998). Low Temperature Electron Attachment to Polycyclic Aromatic Hydrocarbons. Faraday Discuss. 109, 71–82. doi:10.1039/a800490k
Mullin, A. S., Murray, K. K., Schulz, C. P., and Lineberger, W. C. (1993). Autodetachment Dynamics of Acetaldhyde Enolate Anion. Ch2cho−. J. Phys. Chem. 97, 10281–10286. doi:10.1021/j100142a005
Mullin, A. S., Murray, K. K., Schulz, C., Szaflarski, D. M., and Lineberger, W. (1992). Autodetachment Spectroscopy of Vibrationally Excited Acetaldehyde Enolate Anion. Ch2cho−. Chem. Phys. 166, 207–213. doi:10.1016/0301-0104(92)87019-6
Olah, G., Mathew, T., Prakesh, S., and Rusul, G. (2016). Chemical Aspects of Astrophysically Observed Extraterrestrial Methonal, Hydrocarbon Derivatives, and Ions. J. Am. Chem. Soc. 138, 1717–1722. doi:10.1021/jacs.6b00343
Pino, T., Tulej, M., Güthe, F., Pachkov, M., and Maier, J. P. (2002). Photodetachment Spectroscopy of the C2nH− (N=2–4) Anions in the Vicinity of Their Electron Detachment Threshold. J. Chem. Phys. 116, 6126–6131. doi:10.1063/1.1451248
Puzzarini, C., Baiardi, A., Bloine, J., Barone, V., Murphy, T., Drew, D., et al. (2017). Spectroscopic Characterization of Key Aromatic and Heterocyclic Molecules: A Route toward the Origin of Life. Astron. J. 154, 82. doi:10.3847/1538-3881/aa7d54
Remijan, A. J., Hollis, J. M., Lovas, F. J., Cordiner, M. A., Millar, T. J., Markwick-Kemper, A. J., et al. (2007). Detection of C8H− and Comparison with C8H toward IRC +10216. Astrophys. J. 664, L47–L50. doi:10.1086/520704
Ricca, A., Bauschlicher, C. W., and Bakes, E. L. O. (2001). A Computational Study of the Mechanisms for the Incorporation of a Nitrogen Atom into Polycyclic Aromatic Hydrocarbons in the Titanhaze. Icarus 154, 516–521. doi:10.1006/icar.2001.6694
Ross, T., Baker, E., Snow, T., Destree, J., and Rachford, B. (2008). The Search for H− in Astrophysical Environments. Astrophys. J. 684, 358–363. doi:10.1086/590242
Samuelson, R., Hanel, R., Kunde, V., and Maguire, W. (1981). Mean Molecular Weight and Hydrogen Abundance of Titan’s Atmosphere. Nature 292, 688–693. doi:10.1038/292688a0
Santaloci, T., and Fortenberry, R. (2021). Electronically Excited States of Closed-Shell, Cyano-Functionalized Polycyclic Aromatic Hydrocarbon Anions. Chem 3, 296–313. doi:10.3390/chemistry3010022
Santaloci, T. J., and Fortenberry, R. C. (2020). On the Possibilities of Electronically Excited States in Stable Amine Anions: Dicyanoamine, Cyanoethynylamine, and Diethynylamine. M. Astrophy. 19, 100070. doi:10.1016/j.molap.2020.100070
Schild, R., Chaffee, F., Frogel, J., and Persson, S. (1974). The Nature of Infrared Excesses in Extreme Be Stars. Astrophys. J. 190, 73–83. doi:10.1086/152848
Shavitt, I., and Bartlett, R. (2009). MBPT and Coupled-Cluster Theory of Many-Body Methods in Chemistry and Physics, 59. Cambridge: Cambridge University Press.
Shen, L. N., Doyle, T. J., and Graham, W. R. M. (1990). Fourier Transform Spectroscopy of C4H (Butadiynyl) in Ar at 10 K C-H and C ≡C Stretching Modes. J. Chem. Phys. 93, 1597–1603. doi:10.1063/1.459138
Snow, T. (1975). A Search for H− in the Shell Surrounding Chi Ophiuchi. Astrophys. J. 198, 361–367. doi:10.1086/153611
Stanton, J. F., and Gauss, J. (1994). Analytic Energy Derivatives for Ionized States Described by the Equation-Of-Motion Coupled Cluster Method. J. Phys. Chem. 101, 8938–8944. doi:10.1063/1.468022
Stanton, J. F., Gauss, J., Cheng, L., Harding, M. E., Matthews, D. A., and Szalay, P. G. (2008). CFOUR, Coupled-Cluster Techniques for Computational Chemistry, a Quantum-Chemical Program Package. With Contributions from A. J. Chem. Phys. 152 (21), 214108. Watts and the integral packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. vDataset. doi:10.1063/5.0004837
Stanton, J. F., and Bartlett, R. J. (1993). The Equation of Motion Coupled-Cluster Method: A Systematic Biorthogonal Approach to Molecular Excitation Energies, Transition-Probabilities, and Excited-State Properties. J. Chem. Phys. 98, 7029–7039. doi:10.1063/1.464746
Taylor, T. R., Xu, C., and Neumark, D. M. (1998). Photoelectron Spectra of the C2nH− (N=1–4) and C2nD− (N=1–3) Anions. J. Chem. Phys. 108, 10018–10026. doi:10.1063/1.476462
Terzieva, R., and Herbst, E. (2000). Radiative Electron Attachment to Small Linear Carbon Cluster and its Significance for the Chemistry of Diffuse Interstellar Clouds. Int. J. Mass. Spectrom. 201, 135–142. doi:10.1016/s1387-3806(00)00212-8
Thaddeus, P., Gottlieb, C. A., Gupta, H., Brünken, S., McCarthy, M. C., Agúndez, M., et al. (2008). Laboratory and Astronomical Detection of the Negative Molecular Ion C3N−. J. Astrophys. Astron. 677, 1132–1139. doi:10.1086/528947
Theis, M. L., Candian, A., Tielens, A. G. G. M., Lee, T. J., and Fortenberry, R. C. (2015b). Electronically Excited States of Anisotropically Extended Singly-Deprotonated PAH Anions. J. Phys. Chem. A. 119, 13048–13054. doi:10.1021/acs.jpca.5b10421
Theis, M. L., Candian, A., Tielens, A. G. G. M., Lee, T. J., and Fortenberry, R. (2015a). Electronically Excited States of PANH Anions. Phys. Chem. Chem. Phys. 17, 14761. doi:10.1039/c5cp01354b
Tielens, A. G. G. M. (2008). Interstellar Polycyclic Aromatic Hydrocarbon Molecules. Annu. Rev. Astron. Astrophys. 46, 289–337. doi:10.1146/annurev.astro.46.060407.145211
Tulej, M., Kirkwoods, D., Pachkov, M., and Maier, J. (1998). Gas-phase Electronic Transitions of Carbon Chain Anions Coinciding with Diffuse Interstellar Bands. Astrophys. J. Lett. 506, L69–L73. doi:10.1086/311637
van Hemert, M. C., and van Dishoeck, E. F. (2008). Photodissociation of Small Carbonaceous Molecules of Astrophysical Interest. Chem. Phys. 343, 292–302. doi:10.1016/j.chemphys.2007.08.011
Vay, K. L., Salibi, E., Song, E. Y., and Mutschler, H. (2019). Nucleic Acid Catalysis under Potential Prebiotic Conditions. Chem-asian J. 15, 214–230. doi:10.1002/asia.201901205
Walsh, S. P. (1995). Characterization of the Minimum Energy Paths for the Ring Closure Reactions of C4H3 with Acetylene. J. Chem. Phys. 103.
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., et al. (2015). Molpro, Version 2015.1, a Package of Ab Initio Programs. Dataset.
Woon, D. E. (1995). A Correlated Ab Initio Study of Linear Carbon-Chain Radicals CnH (N=2-7). Chem. Phys. Lett. 244, 45–52. doi:10.1016/0009-2614(95)00906-k
Yamamoto, S., Saito, S., Guelin, M., Cernicharo, J., Suzuki, H., and Ohishi, M. (1987). Laboratory Microwave Spectroscopy of the Vibrational Satellites for the ν 7 and 2 ν 7 States of C4H and Their Astronomical Identification. Astrophys. J. Lett. 323, L149. doi:10.1086/185076
Yung, Y. (1987). An Update of Nitrile Photochemistry on Titan. Icarus 72, 468–472. doi:10.1016/0019-1035(87)90186-2
Keywords: dipole bound anion, dipole bound states, quantum chemistry, basis sets, PAHs
Citation: Santaloci TJ, Strauss ME and Fortenberry RC (2021) Electronically Excited States of Potential Interstellar, Anionic Building Blocks for Astrobiological Nucleic Acids. Front. Astron. Space Sci. 8:777107. doi: 10.3389/fspas.2021.777107
Received: 14 September 2021; Accepted: 08 November 2021;
Published: 30 November 2021.
Edited by:
Ashraf - Ali, University of Maryland, United StatesReviewed by:
Jack Simons, The University of Utah, United StatesSankar Prasad Bhattacharyya, Indian Association for the Cultivation of Science, India
Copyright © 2021 Santaloci , Strauss and Fortenberry . This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan C. Fortenberry , cjQxMEBvbGVtaXNzLmVkdQ==