 Alexandria G. Watrous
Alexandria G. Watrous Megan C. Davis
Megan C. Davis Ryan C. Fortenberry
Ryan C. Fortenberry
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Astron. Space Sci., 09 March 2021
Sec. Astrochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fspas.2021.643348
This article is part of the Research TopicRefractory AstrochemistryView all 8 articles
Small, inorganic hydrides are likely hiding in plain sight, waiting to be detected toward various astronomical objects. AlH2OH can form in the gas phase via a downhill pathway, and the present, high-level quantum chemical study shows that this molecule exhibits bright infrared features for anharmonic fundamentals in regions above and below that associated with polycyclic aromatic hydrocarbons. AlH2OH along with HMgOH, HMgNH2, and AlH2NH2 are also polar with AlH2OH having a 1.22 D dipole moment. AlH2OH and likely HMgOH have nearly unhindered motion of the hydroxyl group but are still strongly bonded. This could assist in gas phase synthesis, where aluminum oxide and magnesium oxide minerals likely begin their formation stages with AlH2OH and HMgOH. This work provides the spectral data necessary to classify these molecules such that observations as to the buildup of nanoclusters from small molecules can possibly be confirmed.
Currently, Al–O bonds are known in evolved stars and one exoplanet atmosphere in the form of AlOH and AlO (Tenenbaum and Ziurys, 2009; Tenenbaum and Ziurys, 2010; Takigawa et al., 2017; Chubb et al., 2020). Silicon monoxide has been observed toward Sgr B2 (Wilson et al., 1971), but no other small molecules containing Si–O or even Mg–O bonds have yet to be reported in the astrochemical literature (McGuire, 2018). Differently, crystalline forms of minerals containing Si, Mg, and O have been observed spectroscopically. One of the most notable cases is the mineral enstatite (MgSiO3), whose infrared features have been noted toward NGC 6302 (Mölster et al., 2001, Mölster et al., 2002). Most of these enstatite features fall in the 17–50 μm region of the observered spectrum with those features from 2.4–12 μm attributed to carbon-rich materials like polycyclic aromatic hydrocarbons (PAHs). Hence, crystalline structures with Mg–O and Si–O bonds are perceived in at least one astronomical object. However, the region from 12–17 μm does not have a broad consensus as to the origins of these features, partially due to instrumental artifacts affecting the ISO spectrum. Additionally, O-rich evolved stars will not have PAH features in the 2–20 μm region, but only other silicate features. Therefore, any strong vibrational features in this region might aid in identification if they do not overlap with the strong Si–O–Si modes (Mölster et al., 2002).
Most of the anharmonic vibrational frequencies for the related and previously explored OAlOH molecule fall at 12.82 μm (the internal Al–O stretch) or longer directly in this “uncertain area of spectrum” (Mölster et al., 2001) from high-level quantum chemical computations (Fortenberry et al., 2020). This is also in line with (MO)2 molecules where M = Mg, Al, Si, P, S, Ca, and Ti and also where all of the spectral features of these molecules lie below 12 μm with many of these also above 17 μm (Westbrook and Fortenberry, 2020). Similar spectra behavior has also been noted in the enstatite monomer (Valencia et al., 2020). These fundamental vibrational frequencies also possess notable intensities making them more observable than molecules containing the more abundant carbon atoms that are often the central atom of astrochemical analyses.
Recent work has shown that the strongest bonds between atoms on the first three rows of the periodic table (i.e. a majority of the most common elements in the Universe) are between one part oxygen and another part from one of aluminum, silicon, and magnesium, respectively (Doerksen and Fortenberry, 2020). Granted, this is after the astrophysically-depleted Be, B, and F atoms are discounted. The strongest of the remaining bonds is between the aluminum and oxygen atoms in AlH2OH which is likely one of the first refractory chemical bonds. Further Al and O additions continue to build the material such that covalent network nanocrystals form followed by larger macromolecular solids in the form of common minerals. These mineral nanocrystals then form dust grains that eventually aggregate into rocky bodies like meteors or, ultimately, planets. Therefore, something more than a coincidence may be present between bond strength and elemental abundance in rocky bodies like the Earth, but determination of any such relationship is remains to be tested fully.
Previous, high-level quantum chemical computations show that water reacts with the simplest aluminum hydride, AlH3, to form AlH2OH and molecular hydrogen via an exothermic pathway producing a net of 30.5 kcal/mol with a submerged transition state lying 3.8 kcal/mol below the relative energy of the starting materials (Swinnen et al., 2009). Consequently, AlH2OH could readily form in the atmospheres of massive stars, in protoplanetary disks, or even in molecular clouds. This gas phase pathway for the creation of AlH2OH also produces omnipresent hydrogen molecules which would provide no further evidence for such a reaction potentially allowing such a pathway to remain hidden in plain sight. The work by Swinnen et al., (2009) is actually motivated by alternative energy applications in the production of H2 from common materials like water. Even so, the known presence of water in stellar atmospheres (McGuire, 2018) and perceived presence of aluminium trihydride (which is likely unobserved due to its lack of a permanent dipole moment rendering it rotationally dark) imply that such a reaction could be common under interstellar conditions. The regions where AlH2OH may be found will be dependent upon the abundance of the necessary starting materials, constraining the regions where it may be detected. Conversely, detection of this molecule would strongly imply the presence of AlH3 and H2O in the gas phase, and give an implication as to the presence of the rotationally-dark AlH3 molecule.
Furthermore, this same work by Swinnen et al., (2009) shows that H2O + 2AlH3 → Al2H6O + H2, where Al2H6O possesses a highly symmetric structure and an Al–O–Al motif, is also strongly exothermic producing 71 kcal/mol of energy. Curiously, the H2O + BH3 → BH2OH + H2 reaction has the transition state 11.8 kcal/mol above the starting materials (Swinnen et al., 2009). Such a barrier would stop any gas-phase astrochemical reactions before the reaction could progress, but this may be limited to second-row atoms due to the small charge density.
The present work utilizes similar quantum chemical computations as employed previously for OAlOH and AlOH (Fortenberry et al., 2020) to produce molecular structures, energies, and spectroscopic data for the four molecules at the nexus of strongly bound and containing highly abundant atoms as determined in earlier work (Doerksen and Fortenberry, 2020): AlH2OH, HMgOH, AlH2NH2, and HMgNH2. Additionally, these structures are known to challenge conventional thinking in that AlH2OH is planar, HMgOH is linear, and the two N-containing species are
Two QFFs are utilized in this work. Both are based on coupled cluster theory at the singles, doubles, and perturbative triples [CCSD(T)] level of theory (Raghavachari et al., 1989; Shavitt and Bartlett, 2009; Crawford and Schaefer III, 2000), but one utilizes the explicitly correlated F12b formalism (CCSD(T)-F12b) (Adler et al., 2007). This level of theory solely employs the cc-pVTZ-F12 basis set (Peterson et al., 2008; Yousaf and Peterson, 2008; Knizia et al., 2009) and will be referred to from here on as the F12-TZ approach. The other method is a composite scheme based on canonical CCSD(T) but with considerations for complete basis set (CBS) limit extrapolations (“C”), core electron correlation (“cC”), and relativity (“R”) to give the so-called CcCR QFF (Fortenberry et al., 2011). The F12-TZ QFF has been shown to produce fundamental anharmonic vibrational frequencies nearly coincident with the much more costly CcCR method (Agbaglo et al., 2019; Agbaglo and Fortenberry, 2019a; Agbaglo and Fortenberry, 2019b), but the composite method is still required for accurate rotational constants (Gardner et al., 2021). The F12-TZ QFF will be utilized on all four molecules of the present study. The CcCR will only be employed for HMgOH and AlH2OH due to the lower number of displacements required for HMgOH compared to the other molecules and also the prevalance of oxygen over nitrogen in most astrophysical media.
Both QFFs utilize the MOLPRO 2015.1 software package (Werner et al., 2015) for the electronic structure computations, and both begin with geometry optimizations. The F12-TZ approach simply but tightly optimizes the geometry at this level of theory, with gradients converged to less than 10–8 Å or radians. The CcCR QFF reference geometry is based on CCSD(T)/aug-cc-pV5Z, or aug-cc-pV(5 + d)Z for Al and Mg (Dunning, 1989; Kendall et al., 1992; Peterson and Dunning, 1995), structures corrected for shifts in the structure brought about by inclusion of core electron correlation. The Martin-Taylor (MT) core correlating basis set (Martin and Taylor, 1994) is utilized for CCSD(T) computations with the core electrons included and excluded. The difference in the MT structures is then added to the CCSD(T)/aug-cc-pV5Z geometry. Relativity has no marked effect on the reference geometries (Huang and Lee, 2008; Huang and Lee, 2009).
From this reference geometry, displacements are made to produce the actual QFF. Bond lengths are displaced by 0.005 Å per each step and angles/torsions by 0.005 radians. The coordinate system employed for
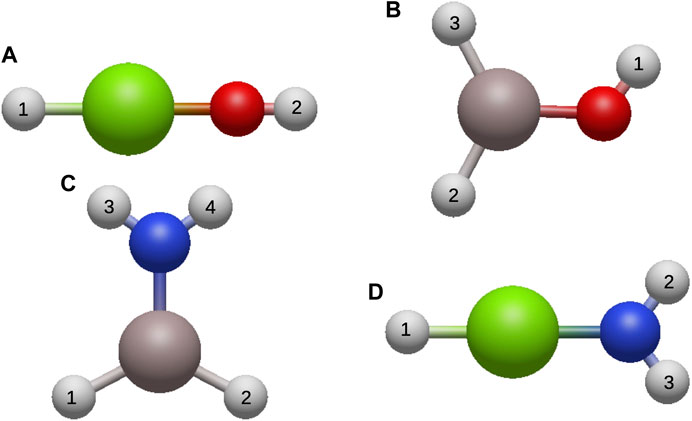
FIGURE 1. The Optimized Structures of (A) HMgOH, (B) AlH2OH, (C) AlH2NH2, and (D) HMgNH2 with H atoms in white, O in red, N in blue, Mg in green, and Al in gray.
The
The F12-TZ QFF only requires energies at each point at this level of theory. The CcCR QFF requires CCSD(T)/aug-cc-pVTZ, aug-cc-pVQZ, and aug-cc-pV5Z (with the tight d functions assumed for Mg and Al) energies computed at each point and extrapolated via a three-point formula to the CBS limit (Martin and Lee, 1996). Each point has CCSD(T)/MT computations with and without the core electrons included as well as scalar relativity (Douglas and Kroll, 1974) from CCSD(T)/cc-pVTZ-DK energies with and without the relativity turned on. For either QFF, the fitting of the points is done by means of a least squared procedure where the sum of squared residuals are on the order of 10–16 a.u.2 or less for all system including the F12-TZ QFF for AlH2NH2. The resulting force constants are transformed from simple-internal or symmetry-internal coordinates into more generic Cartesian coordinates using the INTDER program (Allen et al., 2005). Then, the SPECTRO program (Gaw et al., 1991) utilizes rotational and vibrational perturbation theory at second-order (VPT2) to produce the rotational constants and the fundamental vibrational frequencies (Mills, 1972; Watson, 1977; Papousek and Aliev, 1982). SPECTRO can not only treat Fermi resonances, but polyads of these resonances for more accurate descriptions of the frequencies (Martin and Taylor, 1997). The resonances included are: a
MP2/aug-cc-pVTZ double-harmonic intensities are computed with the Gaussian09 program (Møller and Plesset, 1934; Frisch et al., 2009) and have previously been shown to be reliable for semi-quantitative descriptions of the fundamental intensities (Yu et al., 2015; Finney et al., 2016; Westbrook and Fortenberry, 2020). These are also reported for the molecules of interest. The equilibrium dipole moments have all been computed within Molpro at the F12-TZ level at the corresponding reference geometries for this level of theory.
Of the four molecules examined in this work, AlH2OH is the most polar. Its rotational constants, given in Table 1, show a clear near-prolate character (κ = −0.97) and a dipole moment (Table 2) of 1.22 D. This is even higher than the 1.11 D dipole moment of AlOH (Fortenberry et al., 2020) and should help facilitate detection through rotational spectroscopy. There is little deviation of the dipole moment from the principle axis (
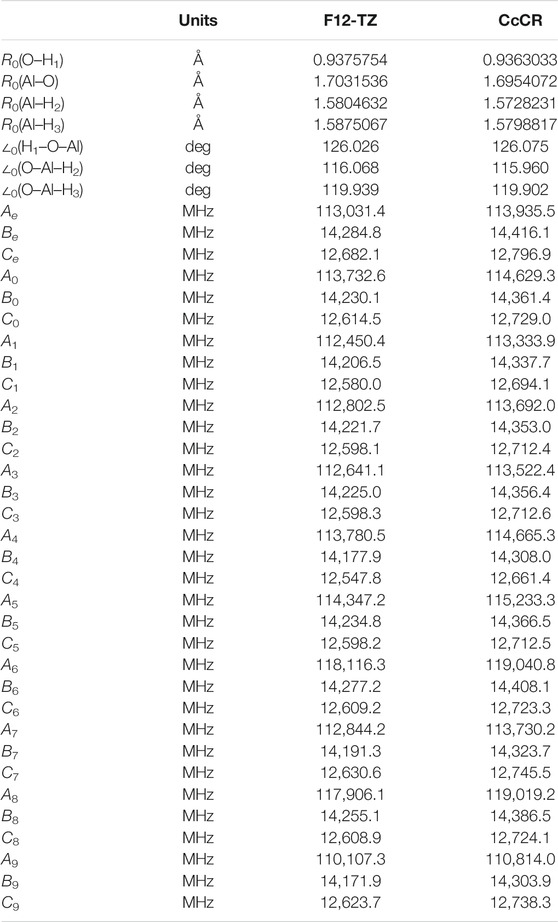
TABLE 1. Geometry and rotational constants for AlH2OH.
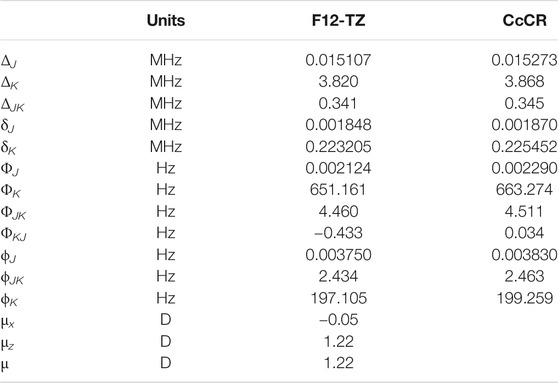
TABLE 2. Quartic and sextic constants for AlH2OH.
Most of the vibrational frequencies agree well between the CcCR and F12-TZ approaches. This is especially true of the harmonic frequencies, but, as has been observed previously (Fortenberry et al., 2020), the anharmonic frequencies diverge more. This is most pronounced in the
The vibrational frequencies imply that this molecule should be detectable in the infrared. All of the double-harmonic intensities from Table 3 are at least on the order of that for the antisymmetric stretch in water (∼70 km/mol) if not two or even three or more times greater. The strongly polarized Al−O bond is borne out in the 203 km/mol intensity for

TABLE 3. Vibrational Frequencies (in cm−1) of AlH2OH with MP2/aug-cc-pVTZ Intensities in Parentheses (in km/mol).
The HMgOH molecule is not nearly as polar as the AlH2OH, but its 0.57 D dipole moment (Table 4) should still support a potential detection through rotational spectroscopy or radioastronomical observation. The CcCR Mg−O bond length of 1.782 Å is notably longer than the 1.695 Al−O bond in the equivalent aluminum structure owing to the slightly weaker (but still quite strong) bonding present for the magnesium-bearing molecule. This also produces a slightly smaller
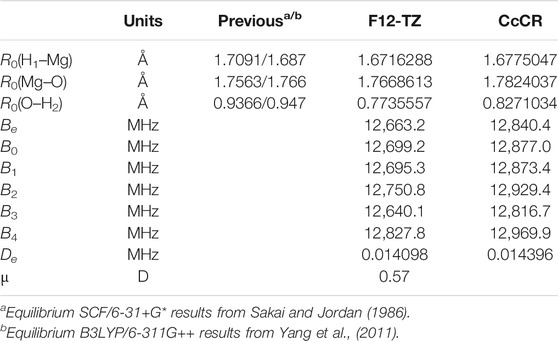
TABLE 4. Geometry and spectroscopic constants of HMgOH.
Again, F12-TZ and CcCR are mostly in agreement with one another for HMgOH save for the
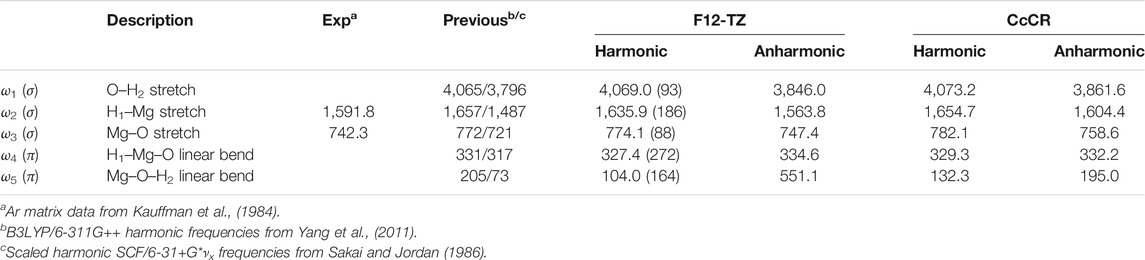
TABLE 5. Vibrational frequencies (in cm−1) of HMgOH with MP2/aug-cc-pVTZ intensities in parentheses (in km/mol).
HMgNH2 is even more prolate than AlH2OH with the A rotational constant a factor of more than 30 larger than B or C (Table 6) and κ = −0.998, but is less polar at 0.62 D (Table 7). The
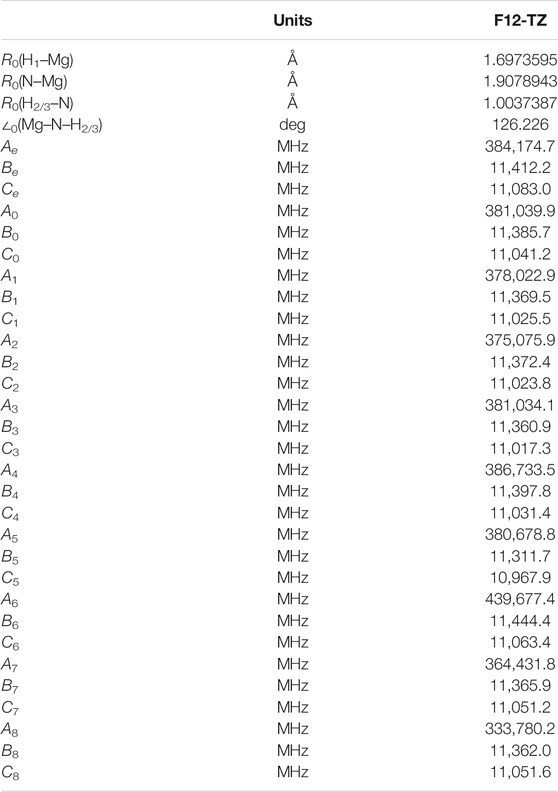
TABLE 6. Geometry and rotational constants for HMgNH2.
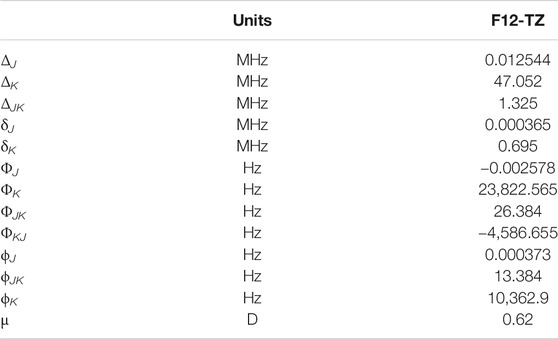
TABLE 7. Quartic and sextic constants for HMgNH2.
The CcCR QFF is not utilized for this molecule due to time constraints in the publication process. However, the F12-TZ QFF vibrational frequencies have been shown above to be comparable to those from CcCR and potentially more reliable for the low-frequency modes. The
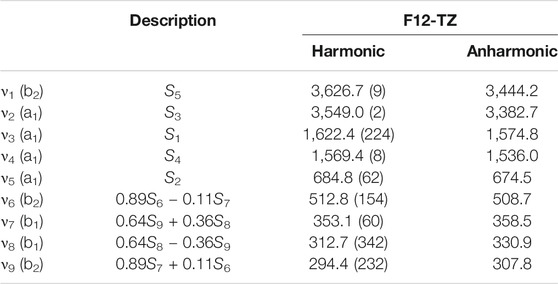
TABLE 8. Vibrational Frequencies (in cm−1) of HMgNH2 with MP2/aug-cc-pVTZ intensities in parentheses (in km/mol).
The number of points necessary to define the QFF increases dramatically for AlH2NH2 making F12-TZ the most practical choice of QFF for analysis of this molecule. AlH2NH2 has rotational constants (Table 9) similar to those present in the related AlH2OH molecule but with a slightly lower

TABLE 9. Geometry and rotational constants of AlH2NH2.
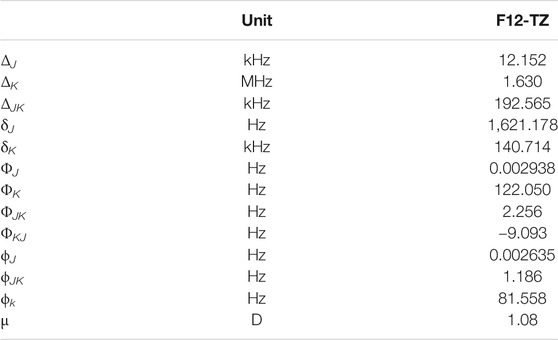
TABLE 10. Quartic and sextic constants of AlH2NH2.
Significantly more previous experimental and high-level theoretical data exist for the vibrational frequencies of AlH2NH2 than any of the other four molecules examined in this work. Both of the previous (Davy and Jaffrey, 1994; Grant and Dixon, 2005) coupled cluster harmonic frequency sets corroborate well the present F12-TZ results shown in Table 11 implying that the harmonic foundation for the subsequent QFF computations is grounded. The Ar matrix data (Himmel et al., 2000) perform a similar benchmark for the F12-TZ QFF anharmonic vibrational frequencies. Most frequencies agree between present theory and this previous experiment to within 10 cm−1 or so. The notable exceptions are
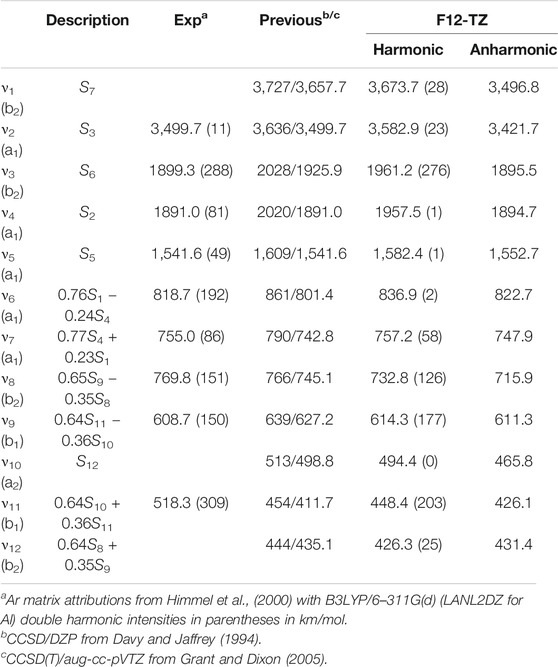
TABLE 11. Vibrational Frequencies (in cm−1) of AlH2NH2 with MP2/aug-cc-pVTZ Intensities in Parentheses (in km/mol).
Beyond such benchmarks, the most notable item for the vibrational spectrum of AlH2NH2 is that the Al−H stretches, especially the
AlH2OH is a detectable and likely interstellar molecule waiting for observation. Previous work has shown its kinetic and thermodynamic formation are favorable in the gas phase from AlH3, which is likely to be present, and water, which is abundant. The notable dipole moment and infrared intensities for AlH2OH should allow for detection in simulated astrophysical contexts utilizing the spectral data produced here. Furthermore, the largely unhindered rotation of the hydroxyl group is typical of what recent work has shown for similar molecules with Al−O−H motifs. This does not appear to be negatively affecting the predicted anharmonic frequencies here as much as has been reported previously.
AlH2OH along with HMgOH, HMgNH2, and AlH2NH2 share many similarities in that the O−H and N−H stretches have smaller intensities but have frequencies above those found in molecules not containing third-row atoms. Most frequencies for these molecules come in roughly three sets: 1) the second-row hydride stretches in the 2.5–3.0 μm range above that typically associated with PAHs; 2) the third-row hydride stretches and second-row hydride bends between 5.0–6.7 μm in regions typically associated with bending in PAHs; and 3) the >12 μm range for the heavy atom stretches and other bend/dihedral fundamental frequencies. The first and last of these three imply that such frequencies rest in infrared spectral windows less clouded with seemingly ubiquitous PAHs. Hence, these molecules may prove to be novel candidates for understanding the provenance for many currently unidentified interstellar infrared emission and absorption features, and may viable candidates for detection with the upcoming James Webb Space Telescope. Additionally, all four molecules are near-prolate (if not linear) and polar allowing for rotational and radioastronomical observation. The data provided herein should aid in growing the census of simple inorganic molecules observed in space.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
AW gathered the data. AW and MD analyzed and formatted the data. RF provided management and funding. All three wrote and edited the paper.
All funding sources have been declared.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work has been supported by NSF Grant OIA-1757220, NASA Grant NNX17AH15G, and the University of Mississippi. Additionally, computational resources have been provided in part by the Mississippi Center for Supercomputing Research funded through NSF Grant CHE-1338056.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2021.643348/full#supplementary-material.
Adler, T. B., Knizia, G., and Werner, H. J. (2007). A simple and efficient CCSD(T)-F12 approximation. J. Chem. Phys. 127, 221106. doi:10.1063/1.2817618
Agbaglo, D., and Fortenberry, R. C. (2019a). The performance of CCSD(T)-F12/aug-cc-pVTZ for the computation of anharmonic fundamental vibrational frequencies. Int. J. Quantum Chem. 119, e25899. doi:10.1002/qua.25899
Agbaglo, D., and Fortenberry, R. C. (2019b). The performance of explicitly correlated wavefunctions [CCSD(T)-F12b] in the computation of anharmonic vibrational frequencies. Chem. Phys. Lett. 734, 136720. doi:10.1016/j.cplett.2019.136720
Agbaglo, D., Lee, T. J., Thackston, R., and Fortenberry, R. C. (2019). A small molecule with pah vibrational properties and a detectable rotational spectrum: (C)C3H2, cyclopropenylidenyl carbene. Astrophys. J. 871, 236. doi:10.3847/1538-4357/aaf85a
Alabugin, I. V., Bresch, S., and Manoharan, M. (2014). Hybridization trends for main group elements and expandingthe bentâs rule beyond carbon: more than electronegativity. J. Phys. Chem. A. 118, 3663–3677. doi:10.1021/jp502472u
Allen, W. D., et al. (2005). INTDER 2005 is a General Program Written by W. D. Allen and Coworkers, which performs vibrational analysis and higher-order non-linear transformations.
Bassett, M. K., and Fortenberry, R. C. (2017). Symmetry breaking and spectral considerations of the surprisingly floppy C3H radical and the related dipole-bound excited state of C3H−. J. Chem. Phys. 146, 224303. doi:10.1063/1.4985095
Bizzocchi, L., Lattanzi, V., Laas, J., Spezzano, S., Giuliano, B. M., Prudenzano, D., et al. (2017). Accurate sub-millimetre rest frequencies for HOCO+ and DOCO+ ions. Astronom. Astrophys. 602, A34. doi:10.1051/0004-6361/201730638
Burton, M. A., Russ, B. T., Bucchino, M. P., Sheridan, P. M., and Ziurys, L. M. (2019). Quadrupole coupling in alkali metal amides MNH2(
Chubb, K. L., Min, M., Kawashima, Y., Helling, C., and Waldmann, I. (2020). Aluminium oxide in the atmosphere of hot Jupiter WASP-43b. Astronom. Astrophys. 639, A3. doi:10.1051/0004-6361/201937267
Crawford, T. D., and Schaefer, H. F. (2000). “An introduction to coupled cluster theory for computational chemists,” in Reviews in computational chemistry. Editors K. B. Lipkowitz, and D. B. Boyd (New York: Wiley), 14, 33–136.
Davy, R. D., and Jaffrey, K. L. (1994). Aluminum-nitrogen multiple bonds in small a1nh molecules: structures and vibrational frequencies of AlNH2, AlNH3, and AlNH4. J. Phys. Chem. 98, 8930–8936. doi:10.1021/j100087a019
Doerksen, E. S., and Fortenberry, R. C. (2020). A coincidence between bond strength, atomic abundance, and the composition of rocky materials. ACS Earth Sp. Chem. 4, 812–817. doi:10.1021/acsearthspacechem.0c00029
Douglas, M., and Kroll, N. M. (1974). Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. 82, 89–155. doi:10.1016/0003-4916(74)90333-9
Dunning, T. H. (1989). Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
Finney, B., Fortenberry, R. C., Francisco, J. S., and Peterson, K. A. (2016). A spectroscopic case for SPSi detection: the third-row in a single molecule. J. Chem. Phys. 145, 124311. doi:10.1063/1.4963337
Fortenberry, R. C., Huang, X., Francisco, J. S., Crawford, T. D., and Lee, T. J. (2012a). Fundamental vibrational frequencies and spectroscopic constants of HOCS+, HSCO+, and isotopologues via quartic force fields. J. Phys. Chem. A. 116, 9582–9590. doi:10.1021/jp3073206
Fortenberry, R. C., Huang, X., Francisco, J. S., Crawford, T. D., and Lee, T. J. (2012b). Quartic force field predictions of the fundamental vibrational frequencies and spectroscopic constants of the cations HOCO+ and DOCO+. J. Chem. Phys. 136, 234309. doi:10.1063/1.4729309
Fortenberry, R. C., Trabelsi, T., and Francisco, J. S. (2020). Anharmonic frequencies and spectroscopic constants of OAlOH and AlOH: strong bonding but unhindered motion. J. Phys. Chem. A. 124, 8834–8841. doi:10.1021/acs.jpca.0c07945
Fortenberry, R. C., and Francisco, J. S. (2017). On the detectability of the
Fortenberry, R. C., Huang, X., Francisco, J. S., Crawford, T. D., and Lee, T. J. (2011). The trans-HOCO radical: fundamental vibrational frequencies, quartic force fields, and spectroscopic constants. J. Chem. Phys. 135, 134301. doi:10.1063/1.3643336
Fortenberry, R. C., and Lee, T. J. (2019). Computational vibrational spectroscopy for the detection of molecules in space. Annu. Rep. Comput. Chem. 15, 173–202. doi:10.1016/bs.arcc.2019.08.006
Fortenberry, R. C., and Lukemire, J. A. (2015). Electronic and rovibrational quantum chemical analysis of C3P−: the next interstellar anion? Mon. Not. R. Astron. Soc. 453, 2824–2829. doi:10.1093/mnras/stv1844
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09 Revision D.01. Wallingford CT: Gaussian Inc.
Fuente, A., Goicoechea, J. R., Pety, J., Gal, R. L., Martín-Doménech, R., Gratier, P., et al. (2017). First detection of interstellar S2H. Astrophys. J. Lett. 851, 49. doi:10.3847/2041-8213/aaa01b
Fugel, M., Beckmann, J., Jayatilaka, D., Gibbs, G. V., and Grabowsky, S. (2018). A variety of bond analysis methods, one answer? an investigation of the element-oxygen bond of hydroxides HnXOH. Chem. Eur. J. 24, 6248–6261. doi:10.1002/chem.201800453
Gardner, M. B., Westbrook, B. R., Fortenberry, R. C., and Lee, T. J. (2021). Highly-accurate quartic force fields for the prediction of anharmonic rotational constants and fundamental vibrational frequencies. Spectrochim. Acta A: Mol. Biomol. Spectrosc., 248, 119184. doi:10.1016/j.saa.2020.119184 (in press).
Gaw, J. F., Willets, A., Green, W. H., and Handy, N. C. (1991). “SPECTRO: a program for the derivation of spectrscopic constants from provided quartic force fields and cubic dipole fields,” in Advances in molecular vibrations and collision dynamics. Editors J. M. Bowman, and M. A. Ratner (Greenwich, Connecticut: JAI Press, Inc.), 170–185.
Grant, D. J., and Dixon, D. A. (2005). Thermodynamic properties of molecular borane phosphines, alane amines, and phosphine alanes and the [BH4+][PH4+], [AlH4−][NH4+], and [AlH4−][PH4+] salts for chemical hydrogen storage systems from ab initio electronic structure theory. J. Phys. Chem. A. 109, 10138–10147. doi:10.1021/jp054152y
Grotjahn, D. B., Sheridan, P. M., Al Jihad, I., and Ziurys, L. M. (2001). First synthesis and structural determination of a monomeric, unsolvated lithium amide. J. Am. Chem. Soc. 123, 5489–5494. doi:10.1021/ja003422h
Himmel, H.-J., Downs, A. J., and Greene, T. M. (2000). Thermal and photochemical reactions of aluminum, gallium, and indium atoms (M) in the presence of ammonia: generation and characterization of the species MNH3, HMNH2, MNH2, and H2MNH2. J. Am. Chem. Soc. 122, 9793–9807. doi:10.1021/ja001313x
Huang, X., and Lee, T. J. (2008). A procedure for computing accurate ab initio quartic force fields: application to HO2+ and H2O. J. Chem. Phys. 129, 044312. doi:10.1063/1.2957488
Huang, X., and Lee, T. J. (2009). Accurate ab initio quartic force fields for NH2− and CCH− and rovibrational spectroscopic constants for their isotopologs. J. Chem. Phys. 131, 104301. doi:10.1063/1.3212560
Huang, X., Taylor, P. R., and Lee, T. J. (2011). Highly accurate quartic force field, vibrational frequencies, and spectroscopic constants for cyclic and linear C3H3+. J. Phys. Chem. A. 115, 5005–5016. doi:10.1021/jp2019704
Kauffman, J. W., Hauge, R. H., and Margrave, J. L. (1984). Infrared matrix isolation studies of the interactions of magnesium, calcium, strontium, and barium atoms and small clusters with water. High Temp. Sci. 18, 97.
Kendall, R. A., Dunning, T. H., and Harrison, R. J. (1992). Electron affinities of the first-row atoms revisited. systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806. doi:10.1063/1.462569
Kitchens, M. J. R., and Fortenberry, R. C. (2016). The rovibrational nature of closed-shell third-row triatomics: HOX and HXO, X = Si+, P, S+, and Cl. Chem. Phys. 472, 119–127. doi:10.1016/j.chemphys.2016.03.018
Knizia, G., Adler, T. B., and Werner, H. J. (2009). Simplified CCSD(T)-F12 methods: theory and benchmarks. J. Chem. Phys. 130, 054104. doi:10.1063/1.3054300
Martin, J. M. L., and Lee, T. J. (1996). The atomization energy and proton affinity of nh3. an ab initio calibration study. Chem. Phys. Lett. 258, 136–143. doi:10.1016/0009-2614(96)00658-6
Martin, J. M. L., and Taylor, P. R. (1997). Accurate ab initio quartic force field for trans-HNNH and treatment of resonance polyads. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 53, 1039–1050. doi:10.1016/s1386-1425(96)01869-0
Martin, J. M. L., and Taylor, P. R. (1994). Basis set convergence for geometry and harmonic frequencies. are ℎ functions enough?. Chem. Phys. Lett. 225, 473–479. doi:10.1016/0009-2614(94)87114-0
McGuire, B. A. (2018). 2018 census of interstellar, circumstellar, extragalactic, protoplanetary disk, and exoplanetary molecules. Astrophys. J. Suppl. Ser. 239 (17), 48. doi:10.3847/1538-4365/aae5d2
Mills, I. M. (1972). “Vibration-rotation structure in asymmetric- and symmetric-top molecules,” in Molecular spectroscopy - modern research. Editors K. N. Rao, and C. W. Mathews (New York: Academic Press), 115–140.
Møller, C., and Plesset, M. S. (1934). Note on an approximation treatment for many-electron systems. Phys. Rev. 46, 618–622.
Mölster, F. J., Lim, T. L., Sylvester, R. J., Waters, L. B. F. M., Barlow, M. J., Beintema, D. A., et al. (2001). The complete ISO spectrum of NGC 6302. Astronom. Astrophys. 372, 165–172. doi:10.1051/0004-6361:20010465
Mölster, F. J., Waters, L. B. F. M., and Tielens, A. G. G. M. (2002). Crystalline silicate dust around evolved stars II. The crystalline silicate complexes. Astronom. Astrophys. 382, 222–240. doi:10.1051/0004-6361:20011551
Morgan, W. J., and Fortenberry, R. C. (2015). Theoretical rovibronic treatment of the
Novak, C. M., and Fortenberry, R. C. (2017). Vibrational frequencies and spectroscopic constants of three, stable noble gas molecules: NeCCH+, ArCCH+, and ArCN+. Phys. Chem. Chem. Phys. 19, 5230–5238. doi:10.1039/c6cp08140a
Peterson, K. A., Adler, T. B., and Werner, H. J. (2008). Systematically convergent basis sets for explicitly correlated wavefunctions: the atoms H, He, B-Ne, and Al-Ar. J. Chem. Phys. 128, 084102, doi:10.1063/1.2831537
Peterson, K. A., and Dunning, T. H. (1995). Benchmark calculations with correlated molecular wave functions. vii. binding energy and structure of the hf dimer. J. Chem. Phys. 102, 2032–2041. doi:10.1063/1.468725
Raghavachari, K., Trucks, G. W., Pople, J. A., and Head-Gordon, M. (1989). A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 157, 479–483. doi:10.1016/s0009-2614(89)87395-6
Sakai, S., and Jordan, K. D. (1986). Squared and vibrational frequencies of HBeOH, HBOH, HCOH, HMgOH, HAlOH, and HSiOH. Chem. Phys. Lett. 130, 103–110. doi:10.1016/0009-2614(86)80434-1
Shavitt, I., and Bartlett, R. J. (2009). Many-body methods in chemistry and physics: MBPT and coupled-cluster theory. Cambridge: Cambridge University Press.
Sheridan, P. M., and Ziurys, L. M. (2000). Laboratory detection of the MgNH2 radical (
Shimanouchi, T. (1972). Tables of molecular vibrational frequencies consolidated volume I. National Bureau of Standards, 1–160.
Swinnen, S., Nguyen, V. S., Sakai, S., and Nguyen, M. T. (2009). Calculations suggest facile hydrogen release from water using boranes and alanes as catalysts. Chem. Phys. Lett. 472, 175–180. doi:10.1016/j.cplett.2009.02.078
Takigawa, A., Kamizuka, T., Tachibana, S., and Yamamura, I. (2017). Dust formation and wind acceleration around the aluminum oxide-rich AGB star W Hydrae. Sci. Adv. 3, eaao2149. doi:10.1126/sciadv.aao2149
Tenenbaum, E. D., and Ziurys, L. M. (2010). Exotic metal molecules in oxygen-rich envelopes: detection of AlOH (
Tenenbaum, E. D., and Ziurys, L. M. (2009). Millimeter detection of AlO (
Thackston, R., and Fortenberry, R. C. (2018). Quantum chemical spectral characterization of CH2NH2+ for remote sensing of titan’s atmosphere. Icarus 299, 187–193. doi:10.1016/j.icarus.2017.07.029
Theis, R. A., and Fortenberry, R. C. (2016). Potential interstellar noble gas molecules: ArOH+ and NeOH+ rovibrational analysis from quantum chemical quartic force fields. Mol. Astrophys. 2, 18–24. doi:10.1021/acsomega.6b00249
Trabelsi, T., Davis, M. C., Fortenberry, R. C., and Francisco, J. S. (2019). Spectroscopic investigation of [Al,N,C,O] refractory molecules. J. Chem. Phys. 151, 244303. doi:10.1063/1.5125268
Valencia, E. M., Worth, C. J., and Fortenberry, R. C. (2020). Enstatite (MgSiO3) and forsterite (Mg2SiO4) monomers and dimers: highly-detectable infrared and radioastronomical molecular building blocks. Mon. Not. R. Astron. Soc. 492, 276–282. doi:10.1093/mnras/stz3209
Wagner, J. P., McDonald, D. C., and Duncan, M. A. (2018). An argon oxygen covalent bond in the ArOH+ molecular ion. Angew. Chem. Int. Ed. Engl. 57, 5081–5085. doi:10.1002/anie.201802093
Watson, J. K. G. (1977). “Aspects of quartic and sextic centrifugal effects on rotational energy levels,” in Vibrational spectra and structure. Editor J. R. During (Amsterdam: Elsevier), 1–89.
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., et al. (2015). Molpro, version 2015.1, a package of ab initio programs. http://www.molpro.net.
Westbrook, B. R., and Fortenberry, R. C. (2020). Anharmonic frequencies of (MO)2 & related hydrides for M = Mg, Al, Si, P, S, Ca, & Ti and heuristics for predicting anharmonic corrections of inorganic oxides. J. Phys. Chem. A. 124, 3191–3204. doi:10.1021/acs.jpca.0c01609
Wilson, R. W., Penzias, A. A., Jefferts, K. B., Kutner, M., and Thaddeus, P. (1971). Discovery of interstellar silicon monoxide. Astrophys. J. 167, L97–L100. doi:10.1086/180769
Xin, J., Brewster, M. A., and Ziurys, L. M. (2000). The pure rotational spectrum of gas-phase NaNH2 (
Yang, W., Han, Z., Zhou, J., Liu, J., and Cen, K. (2011). Theoretical study on the reaction of magnesium with water in the gas-phase. Int. J. Hydrogen Energy 36, 10608–10613. doi:10.1016/j.ijhydene.2011.05.112
Yousaf, K. E., and Peterson, K. A. (2008). Optimized auxiliary basis sets for explicitly correlated methods. J. Chem. Phys. 129, 184108. doi:10.1063/1.3009271
Yu, Q., Bowman, J. M., Fortenberry, R. C., Mancini, J. S., Lee, T. J., Crawford, T. D., et al. (2015). The structure, anharmonic vibrational frequencies, and intensities of NNHNN+. J. Phys. Chem. A. 119, 11623–11631. doi:10.1021/acs.jpca.5b09682
Keywords: astrochemistry, quantum chemistry, computational spectroscopy, coupled cluster theory, geochemistry
Citation: Watrous AG, Davis MC and Fortenberry RC (2021) Pathways to Detection of Strongly-Bound Inorganic Species: The Vibrational and Rotational Spectral Data of AlH2OH, HMgOH, AlH2NH2, and HMgNH2. Front. Astron. Space Sci. 8:643348. doi: 10.3389/fspas.2021.643348
Received: 17 December 2020; Accepted: 28 January 2021;
Published: 09 March 2021.
Edited by:
Majdi Hochlaf, Université Paris Est Marne la Vallée, FranceReviewed by:
Alessandra Candian, University of Amsterdam, NetherlandsCopyright © 2021 Watrous, Davis and Fortenberry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan C. Fortenberry, cjQxMEBvbGVtaXNzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.