
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Soil Sci. , 18 August 2021
Sec. Plant-Soil Interactions
Volume 1 - 2021 | https://doi.org/10.3389/fsoil.2021.659454
This article is part of the Research Topic Belowground Interactions and Implications for Nutrient Use Eco-Efficiency in Cropping Systems View all 6 articles
 Ridhdhi Rathore1,2
Ridhdhi Rathore1,2 David N. Dowling1
David N. Dowling1 Patrick D. Forristal2John Spink2
Patrick D. Forristal2John Spink2 Paul D. Cotter3,4
Paul D. Cotter3,4 Kieran J. Germaine1*
Kieran J. Germaine1*Microbes play vital roles in many soil ecosystem functions and services, which are crucial for agricultural productivity. Among different agricultural management practices, soil tillage methods can result in changes in a soil's physical, chemical and biological properties, including the soil microbiome. In addition, crop type and the plant developmental stage are important drivers of rhizosphere bacterial microbiota structure and composition. Here, we have used high-throughput, 16S amplicon sequencing to explore the rhizosphere bacterial structure and composition of Brassica napus (winter oilseed rape) in two contrasting tillage practices; conventional-plough based tillage and conservation strip tillage, over three different plant growth stages (vegetative, flowering and harvesting stage). This was the first year that conservation strip tillage was used in this field, as in previous years plough based tillage practices has been used. Our findings show that tillage and growth stages were important determinants of microbial community structure and composition, but the effect of tillage became stronger at plant maturity. The combined effect of conservation strip tillage and harvesting stage had a impact on the rhizosphere microbiota selection. The rhizosphere bacterial community of winter oilseed rape under conservation strip tillage was different to that under conventional tillage. Our data suggests that different tillage regimes created distinct ecological niches that selected different microbiota with potential consequences for the ecosystem services provided to the plants and the soil environment.
Soil is one of the richest, most diverse and wide-ranging ecosystems on earth. It provides many essential ecosystem services and products such as nutrient cycling, water filtration and a growth medium for food production (1). The provision of these ecosystem services are mostly driven by the complex interactions of soil biota and abiotic parameters (2). Soil microbes are sensitive and rapid indicators of perturbations and land use changes. Thus, the quantitative description of soil biota structure, composition and diversity is a potential tool for soil quality assessment (3).
Tillage systems are one of the most Important agricultural management practices which can alter the soil's physical, chemical and biological properties. These changes ultimately influence soil microorganism diversity, community structure and soil microbial processes (4–7). Conventional plough tillage is the most common tillage system utilized by farmers. It involves the inversion of the soil, to a depth of ~25–30 cm by mouldboard ploughing. This mechanical disruption of soil may adversely affect long-term soil productivity due to soil compaction, erosion and loss of soil organic matter (4). As a result, farmers are showing interest in alternative, conservation tillage systems which minimizes soil disturbance and helps to maintain soil productivity (8).
Several studies have reported the effect of agricultural management and tillage practices on soil microbial communities (6, 7, 9–13). However, for many crops, including Brassica napus (Oilseed rape), the impact of tillage practices on the rhizosphere microbiota are not well-understood. Rhizosphere microbes are critical in supporting the exchange of resources between the plant and its associated soil environment. Rhizosphere processes contribute to plant health and development, disease suppression and ultimately to crop productivity (14). However, soil microbes are affected by various abiotic factors such as soil texture, moisture, pH, organic matter content, oxygen and nutrient availability, soil aeration, temperature and biotic factors such as plant communities, and the presence of other soil organisms like fungi and soil fauna (10, 13, 15–18). Many researchers have reported that some of these parameters are altered, to various degrees, by the different tillage practices utilized by farmers (4, 10, 18–21). This disturbance may influence the soil microbial communities and associated ecosystem services.
In addition to tillage practices, plant developmental stages are also an important driver of bacterial community structure and composition (6, 22–24). The dynamics of bacterial community changes over the life-time of a plant are mainly brought about by root development and associated changes in rhizodeposition (25). Earlier studies have reported the impact of growth stages on soil and rhizosphere microbial community structure and dynamics, under field and glasshouse conditions (22, 24, 26, 27). However, these studies have not reported the interaction of tillage practices and plant growth stages on the rhizosphere microbiota of Brassica napus. In the current study we attempted to understand the effect that conservation strip tillage has on the rhizosphere microbiome associated with winter Oilseed rape (wOSR) at three different developmental stages. We used the microbiome of bulk and rhizosphere soil under conventional tillage as a reference to identify any potential changes in the microbiome for the bulk and rhizosphere soil as a result of conservation strip tillage.
The plant and soil samples for this study were taken from a field experiment evaluating the effect of crop establishment systems on the growth and development of wOSR. The establishment systems comprised of: (1) a conventional plough-based system (CT) and (2) a low-disturbance conservation strip tillage (ST) system. The conventional establishment system comprised of mouldboard ploughing which inverted the soil to a depth of 230 mm, 2 days prior to sowing. The ploughed soil then received secondary ploughing to 100 mm depth with a rotary power harrow and the wOSR was sown at 10 mm depth at row spacing of 125 mm using a conventional mechanical delivery seed drill operated in combination with the power harrow. The conservation strip tillage (ST) establishment system deployed was a non-inversion system, comprised of a single cultivation/seeding pass of a rigid leg cultivator with legs spaced at 600 mm apart which were operated at 200 mm depth. These forward-facing tines, with side “wings” giving additional soil disturbance, worked directly in the cereal residue of the previous crop, disturbing ~50% of the surface width between the legs. This was the first year that ST was used in this field, as in previous years plough based tillage practices has been used. Seeding was by metered pneumatic delivery of seed to a point behind the cultivator leg, giving a row spacing of 600 mm. For the microbiota studies, plant and soil samples were taken from these two establishment systems (CT and ST) in three replicated plots. The trial was a randomized block design with individual plot dimensions of 24 × 4.8 m and was located in an area known locally as the sawmill field at the Teagasc Crop Research Centre, Oak Park, Carlow, Ireland (52.857478°N, −6.922467°W). The previous crop was winter barley and cereal crops had been sown for more than 5 years previously. The wOSR variety “Compass” was sown at a seed-rate of 60 seeds/m2 on August 28th, 2013 in both establishment systems. Subsequent to seeding, the soil surface was rolled using a ring roller. The topsoil was a well-drained sandy loam overlying inter-bedded layers of sand, gravel and silt/clay. According to WRB classification (28), the site represented a Haplic Cambisol. The topsoil had a sand content of 50–70% with <20% clay. Physical and chemical characterization of the soil substrates used in this study described in Table 1. Crop management, other than crop establishment, followed standard practices for wOSR production in this region. A pre-emergence selective herbicide (quinmerac and metazachlor) was applied post-seeding for weed control. The crop received two fungicide applications (prothioconazole) in October and March for phoma stem canker (Leptosphaeria sp.) and light leaf spot (Pyrenopeziza brassicae) control. Phosphate (P) and potassium (K) fertilizers were applied on the basis of soil test results post-sowing according to Teagasc guidelines (29). Fertilizer N (a total of 225 kg N/ha) was applied in three equal applications at 2-week intervals starting in late February.
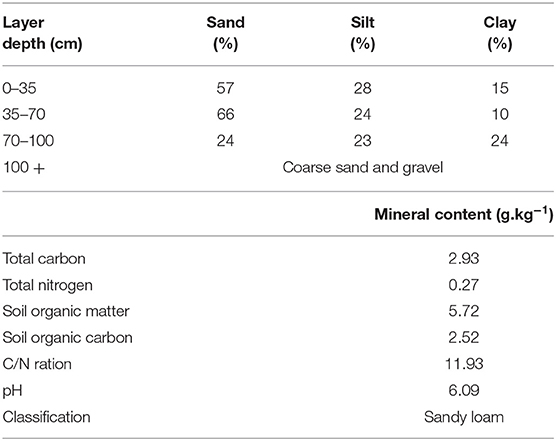
Table 1. Soil descriptions of the experimental site.
Bulk soil and root samples were collected from the CT and ST treatments from three blocks per treatment at three different plant developmental stages: vegetative stage (~120 days after sowing), flowering stage (~240 days after sowing), and at harvesting (~330 days after sowing) stage. Bulk soil samples were collected from a depth of 0–25 cm, in triplicate from the edges of each plot, using a hand auger. For each plot, composite soil samples were prepared by thoroughly mixing the triplicate samples and a representative subsample of this was collected in sterile 50 mL Falcon tubes. Triplicate plants were sampled per plot. The excess soil was removed from the roots by manual shaking. The rhizosphere soil attached to the root was scraped off with a sterile forceps into sterile 50 mL Falcon tube and thoroughly mixed. All the samples were stored in −80°C until required for DNA extraction.
DNA extractions were performed on 3 bulk soil and 3 rhizosphere soil samples per plot with 3 plots per treatment (CT and ST). For DNA extraction, 0.25 g of soil was taken individually from each soil sample and processed according to the protocol from MoBio PowerSoil™ DNA isolation kit (Carlsbad, CA, United States). Total soil DNA was eluted in 50 μL of sterile water (Sigma–Aldrich). Concentration and purity of DNA was determined by Qubit 4 fluorimeter and Nanodrop spectrophotometry (Thermo Scientific, Wilmington, DE, United States), respectively. Post-quantification, all DNA samples were normalized to 10 ng/μL. The number of DNA samples per treatment was 3 (n = 3).
The amplicon library of bacterial DNA was generated using the PCR primers: 341F (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGN
GGCWGCAG-3′), 785R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHV
GGGTATCTAATCC-3′), with Illumina adapter overhang sequences (underlined) which covered ~464 bp of the hypervariable regions V3 and V4 of the 16S rRNA gene (30). Amplicons were generated, purified, indexed and sequenced with some modifications according to the Illumina MiSeq 16S Metagenomics Sequence Library Preparation protocol (16S-Metagenomic-library-prep, 2014). An initial PCR reaction contained 25 μL of 2 × KAPA HiFi Hotstart ReadyMix (KAPA Biosystems, Wilmington, MA, United States), 1 μL of forward primer (1 μM), 1 μL of reverse primer (1 μM), 2.5 μL of DNA (~10 ng/μL) and 20.5 μL of nuclease free H2O in a total volume of 50 μL. The PCR reaction was performed on a 96-well Thermocycler using the following program: 95°C for 3 min, followed by 25 cycles of 95, 55, and 72°C for 30 s and a final extension step at 72°C for 5 min. All amplicons were cleaned using Ampure DNA capture beads (Agencourt-Beckman Coulter; Inc.) following addition of Illumina sequencing adapters and dual-index barcodes to each amplicon with the Nextera-XT Index kit (Illumina Inc., San Diego, CA, United States) according to the manufacturer's instructions. The amplicon libraries were pooled in equimolar concentrations. The final library was paired end sequenced at 2 × 300 bp using a MiSeq Reagent Kit v3 on the Illumina MiSeq platform. Sequencing was performed on the Next Generation Sequencing Platform at Teagasc Moorepark Research Centre, Fermoy, Cork, Ireland.
16S rRNA gene sequences were analyzed using USEARCH v9 32 bit (31) and QIIME, v1.9.0 (Quantitative Insight into Microbial Ecology) (32), unless otherwise specified the default parameters were used. Paired-end reads were merged using the command fastq merge pairs in USEARCH by specifying a minimum overlap of 16 bp. Barcode sequences were removed from the merged paired-end sequences using the command extract_barcodes.py in QIIME. We used USEARCH to demultiplex the pre-processed sequencing reads and to generate a quality report. We used the fastq_filter function in USERACH to truncate all the reads to a length of 400 bp and discard sequences shorter than this length and sequences that contained more than four expected continuous base errors per read. The retained high-quality sequencing reads then clustered into operational taxonomic units (OTUs) at 97% sequence identity using the USERACH pipeline. Singletons were discarded from further analysis and the “Gold” reference database was used to identify and remove chimeras. Taxonomic classification of OTU-representative sequences was performed in QIIME using RDP (Ribosomal Database Project) classifier (33) trained against the Greengenes database (34), release 13_5. Likewise, we used OTU representative sequences to generate a phylogenetic tree in QIIME using “Phynast” as alignment method. The generated OTU table, taxonomy information and phylogenetic tree were used to implement the ecological and statistical analyses. The analysis generated 1,823,764 sequence reads of which 1,524,423 (~86.1% total sequence reads) were retained upon quality trimming, 1,400,091 were retained after removing de-replicate sequence reads and 1,049,731 sequence reads were retained upon singletons removal. These 1,049,731 sequence reads were clustered into 3,755 OTUs at > 97% sequence similarity. After removal of chimeric and a small fraction of plant derived (chloroplast/ mitochondria) OTUs, 3,594 OTUs were used for the downstream analysis.
Data analysis and visualization were performed using Phyloseq (35) package from R operated through R Studio v 0.99.893. All OTUs belonging to chloroplast and mitochondria were identified and removed from the data set prior the analysis. For alpha diversity analysis, observed OTUs, Chao1 and Shannon indexes, normal distribution of the data were checked with the Shapiro-Wilk test. Significant differences in the variance of parameters were evaluated, depending on the distribution of the estimated parameters, with analysis of variance (ANOVA) to identify significant differences between the plant developmental stages, two tillage systems and microhabitat zones. Post-hoc comparisons were conducted using the Tukey test. For such analysis, sequencing reads of soil samples were rarefied at an even sequencing depth of 5,025 reads per sample. To compare community diversity between the samples (beta-diversity), Principal Coordinate Analysis (PCoA) based on Bray-Curtis, sensitive to OTU abundances, and Weighted UniFrac (WUF), sensitive to OTU abundances and taxonomic affiliation, distances were calculated by using counts per million transformed OTU abundances. Permutational multivariate analysis of variance using distance matrices was performed in R using the -adonis function to define the strength and significance of tillage and/or microhabitat zone in determining variation of microbial abundances over the growth stages of wOSR. A differential analysis of the OTUs relative abundances using moderated shrinkage estimation for dispersions and fold changes as an input for a pair-wise Wald test was carried out in DESeq2 package from R version 1.14.1 (36). This test identifies the number of OTUs significantly enriched or depleted (by at least one order of magnitude) between different samples (i.e., between different developmental stages, microhabitats or tillage practices, with an adjusted P-value (False Discovery Rate, FDR P < 0.05). ANOVA analysis was carried out to see the effect of plant developmental stages, tillage and microhabitat zones on the top nine most abundant bacterial phyla. Relative abundance was calculated on the rarefied dataset for such analysis. For bacterial taxonomic classification at phylum and family level, the abundance data were transformed in percentage to calculate the percent relative abundance. In addition, ANOVA analysis was calculated of normally distributed bacterial sequence reads at phylum level, to see the effect of plant developmental stages, tillage practices and microhabitat zones of the top nine most abundant bacterial phyla (with an alpha level of 0.05). Phylum level abundance was calculated based on the number of sequence reads classified to phylum level.
The influence of tillage practices, [conventional tillage (CT) and conservation strip tillage (ST)] on the bacterial communities in the bulk soil and rhizosphere over three growth stages of wOSR was analyzed by amplicon (16S rRNA) next generation sequencing (NGS) Illumina MiSeq platform.
Our analysis showed that there were 386 OTUs shared among the 18 rhizosphere samples analyzed in our study (CT and ST plots). OTU1 (f_ Bradyrhizobiaceae) was the only OTU that was consistently found in all 18 rhizosphere samples, in abundances of >1% of the total reads per sample. It had an average abundance of 3.6% of the total reads/sample. Other OTUs that were strongly associated with the rhizosphere but only found in 13–17 out of the 18 samples included OTU5 (a member of the phylum Chloroflexi) with an average abundance of 2.8% per sample, OTU3 (f_Micrococcaceae) with an average abundance of 2.7% per sample, OTU33 (g_Mycobacterium) at 1.6%, OTU29 (f_Sphingomonadaceae) 1.7%, OTU8 (g_Candidatus) with an average of 2.2%, OTU7 and OTU4 (both f_Hyphomicrobiaceae) at 1.2 and 1.5%, respectively. Collectively these 8 OTUs made up 16.98 ± 4.1% of the total reads per sample. There were 582 OTUs shared among all 18 samples of bulk soil included in our study. OTU1 (f_Bradyrhizobiaceae, 3.5%), was the most abundant OTU consistently found in all bulk soil samples. OTU3 (f_Micrococcaceae, 1.6), OTU5 (a member of the phylum Chloroflexi, 2.7%) and OTU8 (g_Candidatus, 2.1%) were consistently found in all bulk soil samples. There was no significant difference in the number of OTUs found in the bulk soil (1,109 ± 72) compared to the rhizosphere soil under CT (1,061 ± 122) (P = 0.165). However, under ST there was a significantly greater number of OTUs in the bulk soil (1,146 ± 64) than in the rhizosphere soil (1,047 ± 115) (P = 0.012). Comparison of the 18 wOSR rhizophere samples and the 18 bulk soil samples showed that there was a significant reduction in OTU numbers (P < 0.05, average reduction = 219 OTUs) in the rhizosphere.
The alpha diversity within the bacterial communities in both bulk soil and rhizosphere soil at the vegetative, flowering and harvesting growth stages, was determined using the observed OTUs, Chao1 and Shannon diversity indices. In addition to this, we examined the effect of tillage practices (CT vs. ST) on alpha diversity within these samples (Figures 1A–C; Supplementary Material WS-1–4, respectively). The dataset was rarefied to 5,025 sequence reads per sample before calculating diversity indices. In the rhizosphere there was an increase in alpha diversity at the flowering stage, compared to the vegetative stage, but this diversity declined significantly at the harvesting stage. The alpha diversity of the rhizosphere was higher in the ST system compared to the CT tillage system only at the vegetative stage (Figure 1C). Overall, the alpha diversity in the bulk soil decreases over the lifetime of the crop. Alpha diversity in the bulk soil was similar under CT and ST tillage systems in both the vegetative and harvesting stages but was much greater in the ST tillage system at the flowering stage. Observed OTUs indices showed a statistically significant difference in alpha diversity as a result of plant growth stages (P < 0.01) and microhabitat zones (P < 0.01). The Chao1 index also showed a statistically significant difference due to growth stages (P < 0.05) but did not show a significant difference as a result of tillage practices or in the interactions of tillage, growth stages and microhabitat zones (P > 0.05). Similarly, the Shannon index showed a statistically significant difference in the diversity due to the plant growth stage, the microhabitat (rhizosphere vs. bulk soil) and the interaction of tillage*microhabitat zones (P < 0.05), but again did not show a significant difference due to tillage practices.
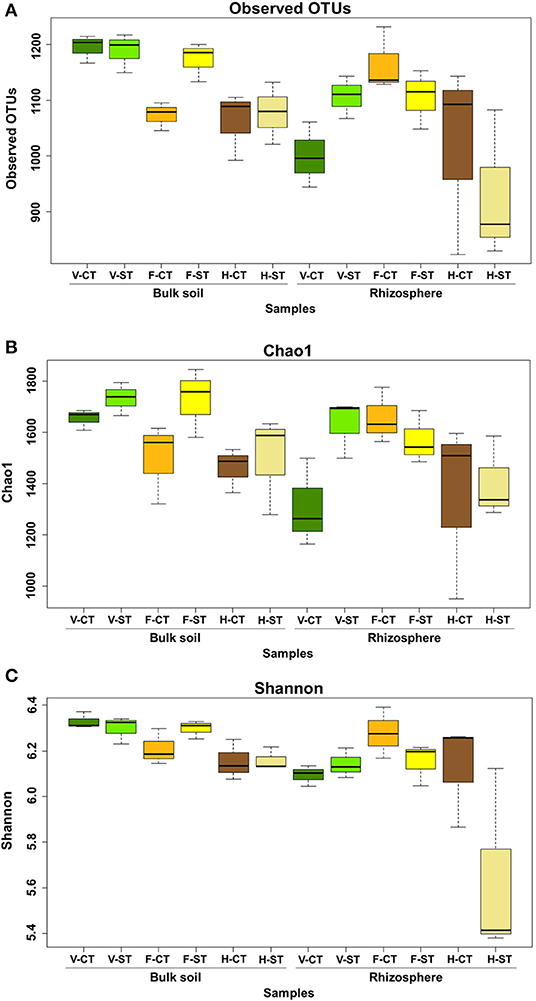
Figure 1. Variation patterns of alpha diversities of the bacterial communities associated with rhizosphere and bulk soil under two tillage practices; CT and ST at three plant growth stages. The alpha diversity estimates: Total number of observed OTUs, Chao1 estimator and Shannon's diversity are displayed in (A–C) respectively. Sequencing reads of soil samples were rarefied at an even sequencing depth 5,025 reads/sample prior the analysis. Letters in the caption stands for conventional tillage (CT), conservation strip tillage (ST), vegetative stage (V), flowering stage (F) and harvesting stage (H). Statistical analysis of alpha diversity is displayed in Supplementary Material WS-1–4. Data are represented by three replicates from each stage. Center line of boxes represent median of samples. The upper and lower sides of the boxes represent the third and first quartiles, respectively. Whiskers represent ± 1.5 times the interquartile range.
To visualize whether tillage regimes, growth stages and microhabitats were important factors driving microbial beta diversity, Bray-Curtis (BC) and Weighted UniFrac (WUF) dissimilarity matrices were used with Principal Coordinate Analysis (PCoA) an unconstrained ordination (Figure 2). At vegetative stage, PCoA analysis showed a clear separation between the two tillage practices in both BC (P < 0.05) and WUF (P < 0.01) matrices. In BC matrix, soil microhabitats also presented minor separation though it was not significant (P = 0.057; Figure 2A) whereas WUF matrix exhibited the clear clustering of bulk soil samples under CT (Figure 2B). At the Flowering stage there was no influence of tillage or microhabitat type on soil microbiota structure in both dissimilarity matrices (P > 0.05; Figures 2C,D). Whereas, at harvesting stage, rhizosphere microbiota under ST presented strong clustering compared to other soil samples and showed the significant influence of tillage and microhabitat type in both BC (P < 0.02) and WUF (P < 0.01) matrices. R2 and P-values of ADONIS based Bray-Curtis and Weighted Unifrac distance matrices are listed in Table 2. These results suggest that the niche separation of rhizosphere microbiota from the soil biota is influenced by the plant growth stages and tillage fine-tunes its composition.
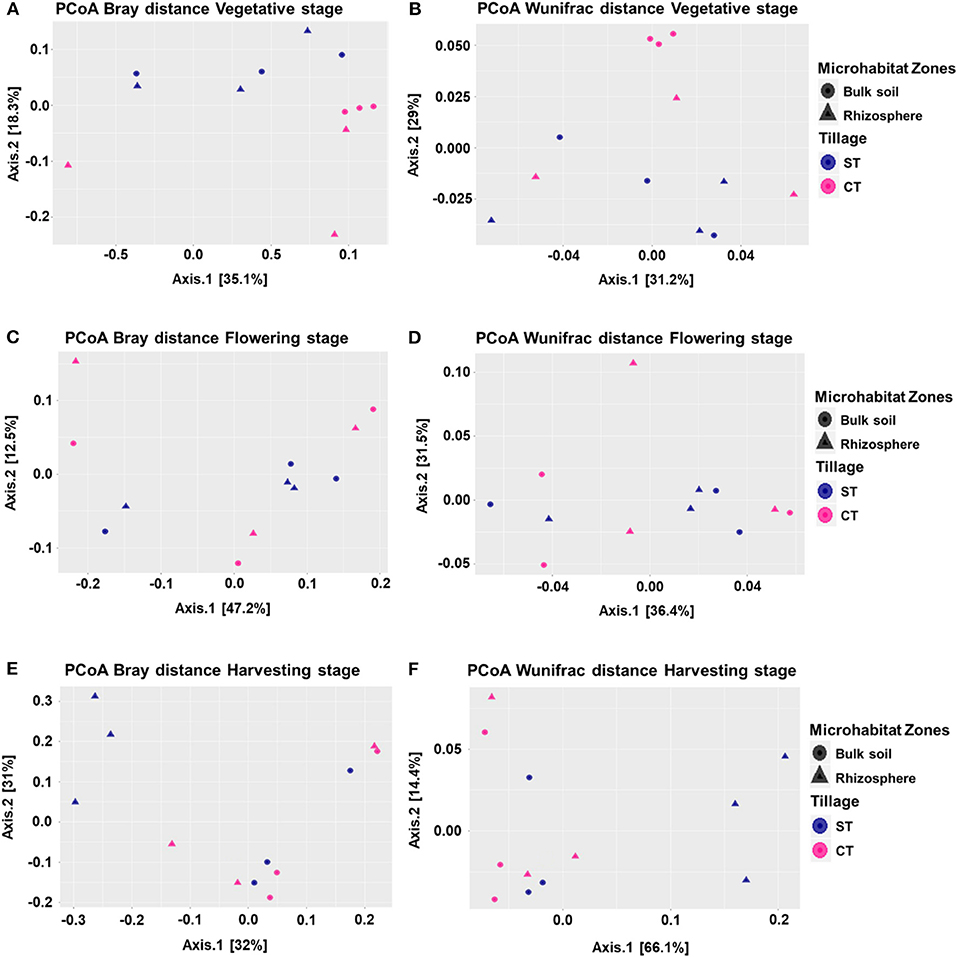
Figure 2. Beta diversity of microhabitats (bulk soil and rhizosphere) at wOSR plant developmental stages under CT and ST tillage regimes. Principle Coordinate Analysis (PCoA) based on Bray-Curtis (BC) and Weighted UniFrac (WUF) distances calculated using counts per million transformed OTU abundances. Comparison between the bulk soil and rhizosphere at vegetative stage (A) BC (B) WUF, flowering stage (C) BC (D) WUF, and harvesting stage (E) BC (F) WUF under CT and ST regimes. In both panels, colors define the tillage practices, while shapes depict the indicated microhabitats. Statistical results of beta diversity are displayed in supplementary excel file WS-5.
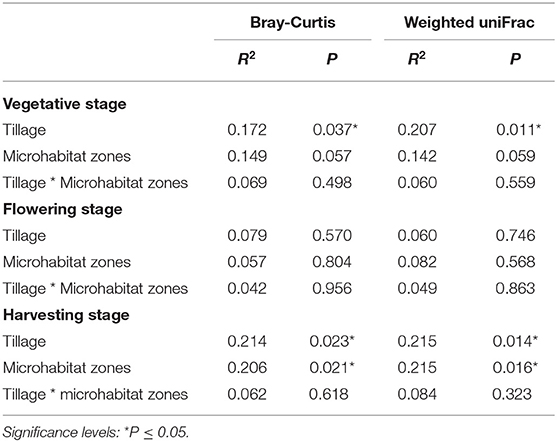
Table 2. Statistical analysis of beta diversity.
To identify OTUs that were responsible for significant differences in the communities with respect to plant growth stages, tillage practices and microhabitat zones, we performed a pair-wise comparison using a model based on a negative binomial distribution (Table 3; Supplementary Material WS-7–10; Walt-test P < 0.05, FDR corrected). Here, individual OTUs are identified which differentiate the growth stages, tillage practices and the microhabitat zones, respectively (Tables 3A–C, respectively). OTUs differentially enriched in the indicated pair-wise comparison are defined by the mean relative abundance and the logarithmic fold change in abundance. In all comparisons a positive fold change is associated with the enrichment of the OTU in a specific sample type.
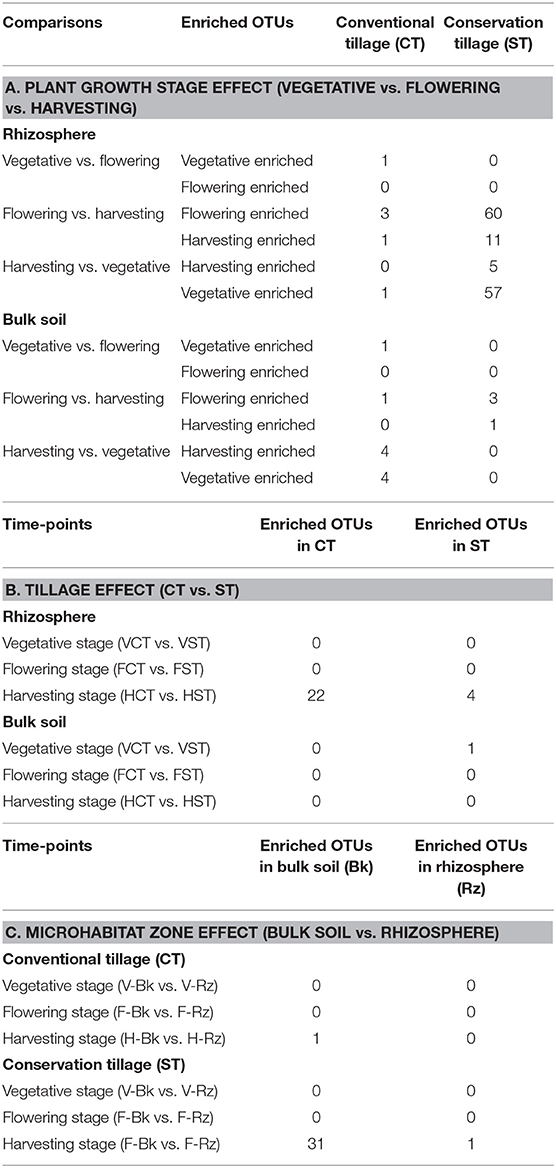
Table 3. Enriched OTUs retrieved from moderated estimation of fold change and pair-wise Wald test (FDR, P < 0.05) of bulk soil and WOSR rhizosphere prokaryotic microbiota profiles at plant developmental stages under tillage regimes.
To investigate the impact of growth stages on wOSR microbiota, the OTUs identified in each growth stage were progressively excluded from the other two growth stages. For example, OTUs identified in vegetative stage were excluded from the flowering stage (vegetative vs. flowering) and harvesting stage (vegetative vs. harvesting). Individual bacterial OTUs which were enriched or depleted in each growth stage contributed to differentiating the bacterial communities. Intriguingly, these OTUs represented just a minor fraction of the total wOSR microbiota. The results showed that at different time points in the rhizosphere under CT, very few OTUs were enriched suggesting that the rhizosphere community is relatively stable over the lifetime of the crop. Likewise in bulk soil under CT, at different growth stages, few OTUs were enriched significantly, again suggesting a relatively stable bacterial community. Under ST at the vegetative and flowering stage there was no significant OTU enrichment. However, when we compared the flowering stage with the harvesting stage, we observed 60 OTUs enriched in the flowering stage and 11 OTUs enriched in the harvesting stage. Likewise, when we compared the harvesting stage with the vegetative stage, we observed 5 OTUs enriched in the harvesting stage and 57 OTUs enriched in the vegetative stage (Table 3A, Supplementary Material WS-7, P < 0.05, FDR corrected). This suggests a dynamic and variable bacterial community structure driven by plant growth and development.
After aligning the OTUs with RDP against the Greengenes database, the wOSR soil microbial community was classified into the top ten most abundant bacterial phyla, which accounted for ~97 % of wOSR rhizosphere and bulk soil bacterial community (Figure 3; Supplementary WS-12). Among them were the phyla Actinobacteria, Proteobacteria, Bacteroidetes, and Chloroflexi, which occupied ~60% of wOSR microbiota. To elucidate whether these bacterial phyla showed any significant difference between the developmental growth stages, tillage regimes, and microhabitat zones, ANOVA analysis was carried out on relative abundance of rarefied dataset (Figure 4; Supplementary WS-11; P < 0.05). The phyla Proteobacteria (P < 0.001), Chloroflexi (P < 0.01) and Firmicutes (P < 0.05) showed significant difference in growth stages of wOSR microbiota (Figures 4A–C, respectively). The bacterial abundance of phylum Proteobacteria and Chloroflexi was similar in all samples at vegetative and flowering stages. However, at harvesting stage the phylum Proteobacteria abundance was remarkably higher while, the abundance of phylum Chloroflexi was lower, especially in rhizosphere under ST. The Firmicutes population showed a significant difference as a result of tillage method (P < 0.05) and growth stages. Overall Firmicutes abundance was higher in ST compared to CT, and at harvesting stage compared to other growth stages in both microhabitat zones.
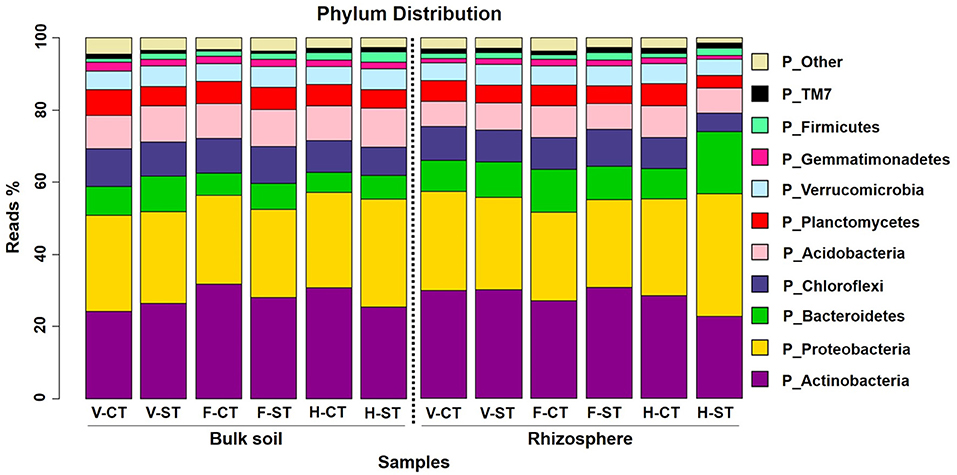
Figure 3. Phylum-level relative abundance of bacterial communities across growth stage and crop management practices. Average relative abundance (% of sequence reads) of top ten most abundant prokaryotic phyla associated with bulk soil and rhizosphere of wOSR under tillage practices at three different growth stages; vegetative stage, flowering stage and harvesting stage are displayed in different colors. Letters in the caption stands for conventional tillage (CT), conservation strip tillage (ST), vegetative stage (V), flowering stage (F) and harvesting stage (H). For each sample type, the number of replicates is n = 3.
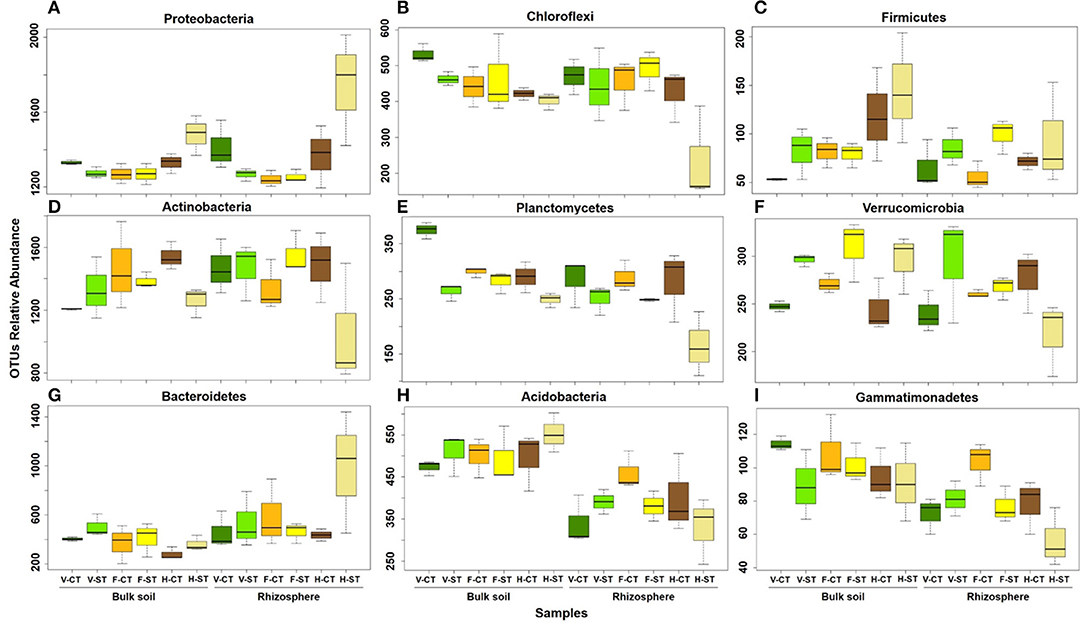
Figure 4. Number of sequencing reads classified to phylum level that significantly (ANOVA; P < 0.05) change with growth stages (A–C), tillage practices (D–F) and microhabitat zones (G–I) in rhizosphere and bulk soil under tillage practices at different plant growth stages. The colors of the box plots represent the three plant developmental stages of wOSR: green (vegetative stage), yellow (flowering stage), and brown (harvesting stage). The dark colors represent for CT and light colors represent for ST samples. Letters in the caption stands for conventional tillage (CT), conservation strip tillage (ST), vegetative stage (V), flowering stage (F) and harvesting stage (H). Data are represented by three replicates of each sample. Center line of boxes represents median of samples. The upper and lower sides of the boxes represent the third and first quartiles, respectively. Whiskers represent ± 1.5 times the interquartile range.
The phylum Actinobacteria displayed a significant difference in tillage*growth stage interaction (P < 0.05, Figure 4D). The soil samples of ST at harvesting stage showed significantly lower Actinobacteria abundance. While there was a significant difference in the Planctomycetes populations as a result of tillage practice (P < 0.001) and a minor contribution of the growth stages and microhabitat zones (P < 0.01). Bacterial abundance was significantly higher in CT compared to ST at all growth stages. The bulk soil of CT at vegetative stage and under ST at harvesting stage displayed higher bacterial abundance compared to rhizosphere. Moreover, the bulk soil of CT at the vegetative stage showed the highest abundance of Planctomycetes while the rhizosphere of ST at harvesting stage showed the lowest abundance (Figure 4E). A significant difference in the abundance of phylum Verrucomicrobia was observed between the tillage and tillage*microhabitat zones (P < 0.05). The bacterial abundance in both microhabitat zones was higher in ST compared to CT at vegetative and flowering stage. However, at harvesting stage in the rhizosphere, the Verrucomicrobia abundance was lower in ST compared to CT (Figure 4F).
Significant differences between the microhabitat zones were observed in abundance of phyla Bacteroidetes (P < 0.01) and Acidobacteria (P < 0.001). Bacteroidetes abundance in rhizosphere was higher compared to bulk soil, while the rhizosphere of ST at harvesting stage displayed the highest bacterial abundance (Figure 4G). Abundance of Acidobacteria was higher in bulk soil compared to rhizosphere under both tillage practices at all growth stages (Figure 4H). The phylum Gemmatimonadetes exhibited the significant difference between the microhabitat zones (P < 0.001), tillage (P < 0.05) and time point (P < 0.05). The bacterial abundance was higher in bulk soil compared to rhizosphere under both tillage regimes and at all growth stages except the vegetative stage of ST and flowering stage of CT where the abundance was similar.
We further analyzed the soil microbial communities at the family level to determine which families were affected by the tillage practice over the developmental stages, in both rhizosphere and bulk soil (Figure 5). We noticed that the relative abundance of families Bradyrhizobiaceae, Hyphomicrobiaceae, and Nocardiaceae were higher in CT whereas, Micrococcaceae, Sphingomonadaceae were higher in the ST rhizosphere samples. The abundance of families Pseudomonadaceae and Sphingobacteriaceae were markedly higher in the ST rhizosphere compared to the CT in rhizosphere samples. Families Streptomycetaceae, Rhizobiaceae, Microbacteriaceae, Nocardiaceae, and Pseudomonadaceae only observed at harvesting and Gaiellaceae at flowering stage in rhizosphere, whereas Streptomycetaceae and Gaiellaceae only present at harvesting and vegetative stage, respectively, in bulk soil. While some bacterial families such as Sphingobacteriaceae, Rhizobiaceae, Pseudomonadaceae, and Microbacteriaceae were only presented at the harvesting stage in rhizosphere and were not present in bulk soil. The bacterial family Nakamurellaceae was observed at vegetative and flowering stage but not at harvesting stage. On the other hand, the family Intrasporangiaceae was observed at vegetative stage, it was not observed at the flowering stage, then reappeared at harvesting stage in the rhizosphere. Both families were not observed in bulk soil. In the rhizosphere, Sphingomonadaceae and Micrococcaceae abundances were dramatically increased at harvesting stage compared to vegetative and flowering stage. These results further support our hypothesis that at different growth stages, Brassica napus can select the subset of microbes as per its requirement and their selection is influenced by tillage practice.
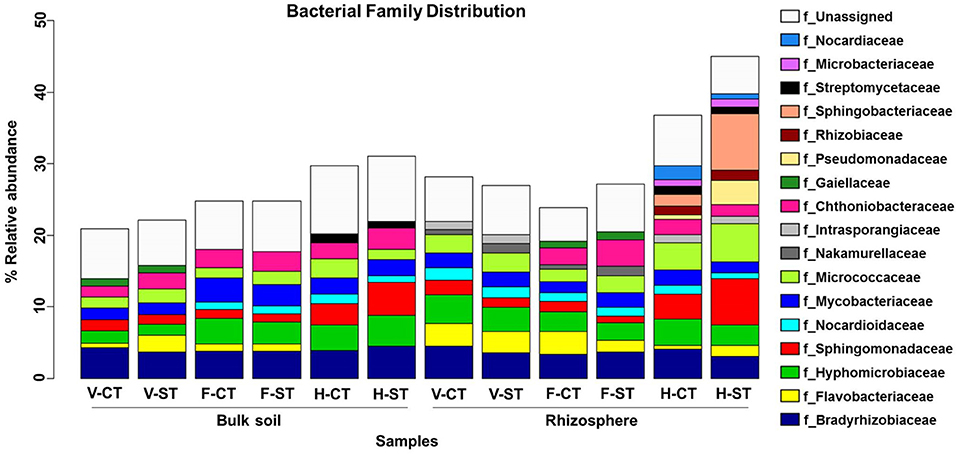
Figure 5. Family-level relative abundance of bacterial communities across growth stage and crop management practices. The bacterial families that present > 1% average relative abundance of sequence reads in bulk soil and rhizosphere of wOSR under conventional tillage (CT) and conservation strip tillage (ST) practices at three plant growth stages; vegetative (V), flowering (F), harvesting (H) are displayed in different colors. The sequencing reads of samples were rarefied at an even sequencing depth 5,025 reads/sample prior to analysis.
The composition and abundances of microorganisms in the bulk soil and rhizosphere are likely to be determined by many different abiotic and biotic factors, such as soil structure (6), microclimate (37), plant age (23) or plant species (38).
Here, we investigated the effects of tillage and plant growth stage on the rhizosphere microbial community in wOSR, in comparison to the bulk soil bacterial community under either conventional or conservation strip tillage practices. At all stages (from DNA extraction to sequencing) of the process there is bias introduced that can influence the results e.g., different DNA extraction protocols and 16S rRNA PCR primer sets used can dramatically affect the observed microbial diversity. Even the presence of relic DNA (DNA from dead cells in the sample) can lead to overestimation of microbial diversity and abundance (39). A major limitation of our study is the low sample number used (3 technical replicates). However, we are confident that each replicate is representative of each trial plot as they were generated from a composite DNA sample create by combining DNA extractions from 3 separate samples within the plot. The limited data does point to some interesting findings.
Our beta diversity plots confirmed that strip tillage practices did result in distinct microbial communities at all growth stages investigated, compared to conventionally tilled samples. PCoA plots of either weighted UniFrac or Bray-Curtis distance matrices both showed a clear separation of rhizosphere microbiomes as a result of the tillage practice utilized. Likewise soil microbiomes were found to separate out based on tillage practice. Bziuk et al. (13) also reported that tillage practices are one of the key factors in shaping soil and rhizosphere microbiome of barley. The beta diversity plots also showed that the rhizosphere and bulk soil microbiota were influenced by the developmental stage of the plant. This result is in agreement with the findings of (24, 40, 41).
In most plants species the release of vast quantities of photosynthates into the root zone is known to stimulate bacterial growth in, and attraction to, the rhizosphere. This leads to the so called “rhizosphere effect,” where the microbial population size and diversity is much greater in the rhizosphere than in the bulk soil (18, 24). We did not observe this rhizosphere effect in the 18 rhizosphere samples from wOSR. There was either no significant difference between the rhizosphere and the bulk soil or there was a significantly greater number of OTUs in the bulk soil. Although we have no data on population sizes, in terms of bacterial diversity we can conclude that wOSR has a negative effect on bacterial diversity in its rhizosphere. Brassicas do not form mycorrhizal associations, presumably by inhibiting the growth of mycorrhizal fungi. Brassicas are known to exude glucoinolates many of which are known to have antimicrobial activity against Gram-positive and Gram-negative bacteria (42). Brassica napus was found to produce progoitrin, glucoalyssin, gluconapin and glucobrassicapin (43). The release of these compounds into the rhizosphere might explain the reduced bacterial biodiversity that we observed in the rhizosphere compared to the bulk soil. Another possibility is that plants select the microorganisms from the available pool in the surrounding environment. While the presence of nutrients and secreted exudates helps increase microbial abundance, the plant's selective pressure increases from bulk soil to rhizoplane and hence decreases bacterial diversity. This concept is in agreement with the study reported by Steinauer et al. (44) that root exudate diversity did not increase the number of microbial species in grassland rhizosphere, but induced alterations in species-specific abundances and compositional shift toward fast-growing copiotrophic soil microorganisms due to a nutrient-enriched environment. The number of species and their relative abundance within specific root locations are a particularly debated aspect of rhizosphere microbial ecology.
Strip tillage practice did not result in any statistically significant difference in the alpha diversity in either the bulk soil or rhizosphere at the vegetative or flowering stages compared to conventional tillage. Wasserman et al. (45) found no significant difference in the alpha diversity among 14 different Brassica species. Maintaining microbial diversity in the soil and rhizosphere is important in order to ensure functional redundancy in carrying out essential ecosystem functions. However, we did observe an overall decrease in diversity over the lifetime of the crop. A similar result was observed by Copeland et al. (46) who found a decrease in bacterial diversity in canola. This is in contrast to other studies on cereals, which found that there was a general increase in microbial diversity over the growing season (47). We observed a statistically significant drop in the alpha diversity in the rhizosphere at the harvesting stage under strip tillage compared with conventional tillage. These findings are further supported by our percent phylum distribution data showing significantly higher relative abundance of bacterial phyla Proteobacteria and Bacteroidetes, and remarkably lower relative abundance of Actinobacteria and Chloroflexi only observed in rhizosphere under ST at the harvesting stage compared to all other samples.
Dorr de Quadros et al. (48) demonstrated through 16S amplicon metagenomic sequencing that soil microbial diversity (in particular the anaerobes as Verrucomicrobia, Firmicutes, Crenarchaeota, Chlamydiae, Euryarchaeota, and Chlorobi), and soil nutrients (phosphate, magnesium, total organic carbon, and nitrogen) were significantly higher in no-tillage compared to conventionally tillage plots. Navarro-Noya et al. (49) observed similar results, in that under reduced tillage, the microbial biomass, plant organic matter and nutrient content were higher. These changes in the physical-chemical properties of the soil as a result of different tillage practices create different ecological niches that select for different microbial lifestyles and which can lead to the development of very different microbial community structures. Yin et al. (50) used a metagenomic approach to study the effect of conventional and no-till practices on the microbiome of wheat crops. They found that copiotrophic bacterial families such as Oxalobacteriaceae, Pseudomonadaceae, and Cytophagaceae were more abundant in reduced tillage and oligotrophic bacterial families were more abundant in conventional tillage. However, Degrune et al. (10) observed higher abundance of copiotrophic bacteria under conventional tillage and oligotrophic bacteria under reduced tillage. While tillage practices alter soil microbial community structure, the response of individual microbial groups appears to be very context-specific and cannot be generalized across various agroecosystems. The response is largely dependent on the soil physical-chemical conditions, soil types, climatic conditions and sampling depths.
Hale et al. (51) reported that root exudation patterns can be affected by agriculture practices, soil water availability, stress, soil temperature, light intensity, degree of anaerobiosis, application of agro-chemicals, plant age, plant species, mineral nutrition, and soil microbes. Several studies demonstrated that root exudates and soil environmental changes can strongly affect the rhizosphere microbial community structure and composition over the life period of plants (15, 23, 44, 52–55).
A number of bacterial families were consistently found in the rhizosphere microbiome regardless of the tillage method and stage of plant growth. Bradyrhizobiaceae, Flavobacteriacecea, Hyphomicrobiaceae, Micrococcaceae and Mycobacteriaceae were all dominant members of the bacterial community. Many of are involved in nutrient cycling Bradyrhizobiaceae, Flavobacteriaceae and Hyphomicrobacteriaceae have all been associated with nitrogen cycling either through nitrogen fixation or nitrifying/denitrification activities (56). These groups have been consistently found in wet European soils (57). Many of their members are oligotrophic/oligocarbophilic and their ubiquitous presence has been linked to stable ecological niches, such as moist soils rich in humic substances or in the plant rhizosphere. Many of the bacterial species within these families have been reported to increase nutrient obtainability; subdue disease-causing fungi; reduce stress from non-living environmental factors and promote the growth of the plants. Flavobacteriacecea represent a significant fraction of root- and leaf-associated microbiomes in a broad range of plant species. Several of these studies have shown that the relative abundance of members of this genus increases substantially along the soil, rhizosphere, and rhizoplane continuum, indicating a specialized capacity to proliferate in plant environments and suggesting a role in plant functioning (56).
On the other hand, there are families (Pseudomonadaceae and Sphingbacteriaceaea) which only became dominant members of the bacteria communities at certain stages in the plant life cycle and which were affected by the tillage practice. These families only became dominant members of the community at the harvesting stage and the relative abundance of both were significantly increased as a result of strip tillage (ST). The Pseudomonadaceae is a family of Gram-negative bacteria that includes four genera Frateuria, Pseudomonas, Xanthomonas, and Zoogloea. While some members of these groups are known plant and animal pathogens the majority are linked with beneficial soil ecosystem functions such as nitrogen cycling (denitrification), plant growth promotion through phosphate solubilisation, ACC deaminase activity, and biocontrol of insects and plant pathogenic fungi. One of the benefits of conservation tillage is the retention of soil organic matter which in turn leads to a greater soil biodiversity, including insects and fungi. Chitin is a component with makes up a significant proportion of the biomass of these organisms. Both Pseudomonadeaceae and Sphingbacteriaceaea are known to have significant chitinase activity (58) and perhaps the enrichment of these two families is linked to the possible increased level of chitin in the soil by the harvesting stage. Mendes et al. (59) and Fang et al. (60) reported that as a plant ages, it releases specific antimicrobial substances which can select for specific groups of resistant microbes. Gkarmiri et al. (61) also reported these bacterial families in rhizosphere of oilseed rape though stable isotope high throughput sequence analysis.
Our results show that plant developmental stage and the rhizosphere effect led to dynamic changes in the bacterial community structure in the root zone and that different tillage practices lead to lasting differences in bacterial communities both in the rhizosphere and in the bulk soil. Further investigations are required to understand, what consequences, if any, these changes in microbial community have on plant health/development and important soil ecosystem functions.
The sequences generated in this study were deposited in the European Nucleotide Archive (ENA) under the accession numbers ERS2166256–ERS2166291 under the ENA Bioproject PRJEB24119. The scripts used to analyse the sequencing data and generate the figures of this study are available at https://github.com/KieranGermaine-Lab/Brassica-napus-rhizosphere-microbiota.
RR, KG, DD, PF, and JS conceived of and designed the experiments. RR performed the experimental work, conceived, and executed the analysis of the 16S rRNA sequencing dataset. PC carried out the sequencing work. RR, KG, and DD wrote the paper. All authors contributed to the article and approved the submitted version.
This work was supported through the Teagasc Walsh Scholarship Scheme which funded to RR and through funding from the Institutes of Technology, Carlow Presidents post-graduate Award Scheme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Dr. Fiona Crispie for her assistance in the library preparation and sequencing work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsoil.2021.659454/full#supplementary-material
1. Altieri MA. The ecological role of biodiversity in agroecosystems. Agricu Ecosyst Environ. (1999) 74:19–31. doi: 10.1016/S0167-8809(99)00028-6
2. Kibblewhite MG, Ritz K, Swift MJ. Soil health in agricultural systems. Philos Trans R Soc B Biol Sci. (2008) 363:685–701. doi: 10.1098/rstb.2007.2178
3. Zelles L. Fatty acid patterns of phospholipids and lipopolysaccharides in the characterisation of microbial communities in soil: a review. Biol Fert Soils. (1999) 29:111–29. doi: 10.1007/s003740050533
4. Vian JF, Peigne J, Chaussod R, Roger-Estrade J. Effects of four tillage systems on soil structure and soil microbial biomass in organic farming. Soil Use Manag. (2009) 25:1–10. doi: 10.1111/j.1475-2743.2008.00176.x
5. Jangid K, Williams MA, Franzluebbers AJ, Sanderlin JS, Reeves JH, Jenkins MB, et al. Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol Biochem. (2008) 40:2843–53. doi: 10.1016/j.soilbio.2008.07.030
6. Kraut-Cohen JZA, Shaltiel-Harpaz L, Argaman E, Rabinovich R, Green SJ, Minz D. Effects of tillage practices on soil microbiome and agricultural parameters. Sci Total Environ. (2020) 705:791. doi: 10.1016/j.scitotenv.2019.135791
7. Hartman K, Van der Heijden MGA, Wittwer RA, Banerjee S, Walser J-C, Schlaeppi K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome. (2018) 6:14. doi: 10.1186/s40168-018-0456-x
8. Lahmar R. Adoption of conservation agriculture in Europe. Land Use Policy. (2010) 27:4–10. doi: 10.1016/j.landusepol.2008.02.001
9. Rathore R, Dowling DN, Forristal PD, Spink J, Cotter PD, Bulgarelli D, et al. Crop establishment practices are a driver of the plant microbiota in winter oilseed rape (Brassica napus L.). Front Microbiol. (2017) 8:e01489. doi: 10.3389/fmicb.2017.01489
10. Degrune F, Theodorakopoulos N, Colinet G, Hiel M-P, Bodson B, Taminiau B, et al. Temporal dynamics of soil microbial communities below the seedbed under two contrasting tillage regimes. Front Microbiol. (2017) 8:1127. doi: 10.3389/fmicb.2017.01127
11. Smith CR, Blair PL, Boyd C, Cody B, Hazel A, Hedrick A, et al. Microbial community responses to soil tillage and crop rotation in a corn/soybean agroecosystem. Ecol Evol. (2016) 6:8075–84. doi: 10.1002/ece3.2553
12. Carbonetto B, Rascovan N, Álvarez R, Mentaberry A, Vázquez MP. Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land use and tillage systems in argentine pampas. PLoS ONE. (2014) 9:e99949. doi: 10.1371/journal.pone.0099949
13. Bziuk N, Maccario L, Douchkov D, Lueck S, Babin D, Sørensen SJ, et al. Tillage shapes the soil and rhizosphere microbiome of barley-but not its susceptibility towards Blumeria graminis f. sp. hordei. FEMS Microbiol Ecol. (2021) 97:fiab018. doi: 10.1093/femsec/fiab018
14. Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, et al. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci. (2013) 110:6548–53. doi: 10.1073/pnas.1302837110
15. Garbeva P, Van Veen JA, Van Elsas JD. Microbial diversity in soil: selection microbial populations by plant and soil type and implications for disease suppressiveness. Annu Rev Phytopathol. (2004) 42:243–70. doi: 10.1146/annurev.phyto.42.012604.135455
16. Xu Y, Wang G, Jin J, Liu J, Zhang Q, Liu X. Bacterial communities in soybean rhizosphere in response to soil type, soybean genotype, and their growth stage. Soil Biol Biochem. (2009) 41:919–25. doi: 10.1016/j.soilbio.2008.10.02
17. Hery M, Singer AC, Kumaresan D, Bodrossy L, Stralis-Pavese N, Prosser JI, et al. Effect of earthworms on the community structure of active methanotrophic bacteria in a landfill cover soil. ISME J. (2007) 2:92–104. doi: 10.1038/ismej.2007.66
18. Praeg N, Pauli H, Illmer P. Microbial diversity in bulk and rhizosphere soil of ranunculus glacialis along a high-alpine altitudinal gradient. Front Microbiol. (2019) 10:e01429. doi: 10.3389/fmicb.2019.01429
19. Six J, Elliott ET, Paustian K. Aggregate and soil organic matter dynamics under conventional and no-tillage systems. Soil Sci Soc Am J. (1999) 63:1350–8. doi: 10.2136/sssaj1999.6351350x
20. Lipiec J, Kuś J, Słowińska-Jurkiewicz A, Nosalewicz A. Soil porosity and water infiltration as influenced by tillage methods. Soil Tillage Res. (2006) 89:210–20. doi: 10.1016/j.still.2005.07.012
21. Schmidt R, Gravuer K, Bossange AV, Mitchell J, Scow K. Long-term use of cover crops and no-till shift soil microbial community life strategies in agricultural soil. PLoS ONE. (2018) 13:e0192953. doi: 10.1371/journal.pone.0192953
22. Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. (2012) 488:91–5. doi: 10.1038/nature11336
23. Chaparro JM, Badri DV, Vivanco JM. Rhizosphere microbiome assemblage is affected by plant development. Isme J. (2014) 8:790–803. doi: 10.1038/ismej.2013.196
24. Wang Z, Liu L, Chen Q, Wen X, Liu Y, Han J, et al. Conservation tillage enhances the stability of the rhizosphere bacterial community responding to plant growth. Agron Sust Dev. (2017) 37:44. doi: 10.1007/s13593-017-0454-6
25. Philippot L, Raaijmakers JM, Lemanceau P, Van der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Micro. (2013) 11:789–99. doi: 10.1038/nrmicro3109
26. Shi SJ, Richardson AE, O'Callaghan M, Deangelis KM, Jones EE, Stewart A, et al. Effects of selected root exudate components on soil bacterial communities. FEMS Microbiol Ecol. (2011) 77:600–10. doi: 10.1111/j.1574-6941.2011.01150.x
27. Zhang B, He H, Ding X, Zhang X, Zhang X, Yang X, et al. Soil microbial community dynamics over a maize (Zea mays L.) growing season under conventional- and no-tillage practices in a rainfed agroecosystem. Soil Tillage Res. (2012) 124:153–60. doi: 10.1016/j.still.2012.05.011
28. FAO. World Reference Base for Soil Resources 2014, International Soil Classification System for Naming Soils and Creating Legends for Soil Maps. World Soil Resource Reports 106. Rome (2015).
29. Coulter B, Lalor S. Major and Micronutrient Advice for Productive Agricultural Crops, 3rd Edn. (2008).
30. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. (2013) 41:e1. doi: 10.1093/nar/gks808
31. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Meth. (2013) 10:996–8. doi: 10.1038/nmeth.2604
32. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. (2010). 7:335–6. doi: 10.1038/nmeth.f.303
33. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. (2007) 73:5261–7. doi: 10.1128/AEM.00062-07
34. Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. (2006) 72:5069–72. doi: 10.1128/AEM.03006-05
35. Mcmurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. (2013) 8:e61217. doi: 10.1371/journal.pone.0061217
36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
37. Jansson JK, Hofmockel KS. Soil microbiomes and climate change. Nat Rev Microbiol. (2020) 18:35–46. doi: 10.1038/s41579-019-0265-7
38. Schneijderberg M, Cheng X, Franken CDE, Hollander M, Van Velzen R, et al. Quantitative comparison between the rhizosphere effect of Arabidopsis thaliana and co-occurring plant species with a longer life history. ISME J. (2020) 14:2433–48. doi: 10.1038/s41396-020-0695-2
39. Carini P, Marsden PJ, Leff JW, Morgan EE, Strickland M, Fierer N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. bioRxiv. (2016). doi: 10.1101/043372
40. Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. (2013) 64:807–38. doi: 10.1146/annurev-arplant-050312-120106
41. Li X, Rui J, Mao Y, Yannarell A, Mackie R. Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol Biochem. (2014) 68:392–401. doi: 10.1016/j.soilbio.2013.10.017
42. Guil-Guerrero JL, Ramos L, Moreno C, Zúñiga-Paredes JC, Carlosama-Yepez M, Ruales P. Antimicrobial activity of plant-food by-products: a review focusing on the tropics. Livestock Sci. (2016) 189:32–49. doi: 10.1016/j.livsci.2016.04.021
43. Velasco P, Soengas P, Vilar M, Cartea ME, del Rio M. Comparison of glucosinolate profiles in leaf and seed tissues of different Brassica napus crops. J Am Soc Hortic Sci. (2008) 133:551–8. doi: 10.21273/JASHS.133.4.551
44. Steinauer K, Chatzinotas A, Eisenhauer N. Root exudate cocktails: the link between plant diversity and soil microorganisms? Ecol Evol. (2016) 6:7387–96. doi: 10.1002/ece3.2454
45. Wassermann B, Rybakova D, Müller C, Berg G. Harnessing the microbiomes of Brassica vegetables for health issues. Sci Rep. (2017) 7:17649. doi: 10.1038/s41598-017-17949-z
46. Copeland JK, Yuan L, Layeghifard M, Wang PW, Guttman DS. Seasonal community succession of the phyllosphere microbiome. Mol Plant Microbe Interact. (2015) 28:274–85. doi: 10.1094/MPMI-10-14-0331-FI
47. Gdanetz K, Trail F. The Wheat Microbiome Under Four Management Strategies, and Potential for Endophytes in Disease Protection. (2017). doi: 10.1094/PBIOMES-05-17-0023-R
48. Dorr de Quadros P, Zhalnina K, Davis-Richardson A, Fagen JR, Drew J, Bayer C, et al. The effect of tillage system and crop rotation on soil microbial diversity and composition in a subtropical acrisol. Diversity. (2012) 4:375. doi: 10.3390/d4040375
49. Navarro-Noya YE, Gómez-Acata S, Montoya-Ciriaco N, Rojas-Valdez A, Suárez-Arriaga MC, Valenzuela-Encinas C, et al. Relative impacts of tillage, residue management and crop-rotation on soil bacterial communities in a semi-arid agroecosystem. Soil Biol Biochem. (2013) 65:86–95. doi: 10.1016/j.soilbio.2013.05.009
50. Yin C, Mueth N, Hulbert S, Schlatter D, Paulitz TC, Schroeder K, et al. Bacterial communities on wheat grown under long-term conventional tillage and no-till in the Pacific Northwest of the United States. Phytobiomes. (2017) 1:83–90. doi: 10.1094/PBIOMES-09-16-0008-R
51. Hale MG, Foy CL, Shay FJ. Factors affecting root exudation. In: Brady NC, editor. Advances in Agronomy. Cambridge, MA: Academic Press (1971).
52. Classen AT, Sundqvist MK, Henning JA, Newman GS, Moore JAM, Cregger MA, et al. Direct and indirect effects of climate change on soil microbial and soil microbial-plant interactions: what lies ahead? Ecosphere. (2015) 6:1–21. doi: 10.1890/ES15-00217.1
53. Van Horn DJ, Okie JG, Buelow HN, Gooseff MN, Barrett JE, Takacs-Vesbach CD. Soil microbial responses to increased moisture and organic resources along a salinity gradient in a polar desert. Appl Environ Microbiol. (2014) 80:3034–43. doi: 10.1128/AEM.03414-13
54. Cruz-Martínez K, Rosling A, Zhang Y, Song M, Andersen GL, Banfield JF. Effect of rainfall-induced soil geochemistry dynamics on grassland soil microbial communities. Appl Environ Microbiol. (2012) 78:7587–95. doi: 10.1128/AEM.00203-12
55. Marschner P, Marino W, Lieberei R. Seasonal effects on microorganisms in the rhizosphere of two tropical plants in a polyculture agroforestry system in Central Amazonia, Brazil. Biol Fertil Soils. (2002) 35:68–71. doi: 10.1007/s00374-001-0435-3
56. Fang X, Zheng R, Guo X, Fu Q, Fan F, Liu S. Yak excreta-induced changes in soil microbial communities increased the denitrification rate of marsh soil under warming conditions. Appl Soil Ecol. (2021) 165:103935. doi: 10.1016/j.apsoil.2021.103935
57. Pershina EV, Ivanova EA, Korvigo IO, Chirak EL, Sergaliev NH, Abakumov EV, et al. Investigation of the core microbiome in main soil types from the East European plain. Sci Total Environ. (2018) 631:1421–30. doi: 10.1016/j.scitotenv.2018.03.136
58. Wieczorek AS, Schmidt O, Chatzinotas A, Von Bergen M, Gorissen A, Kolb S. Ecological functions of agricultural soil bacteria and microeukaryotes in chitin degradation: a case study. Front Microbiol. (2019) 10:1293. doi: 10.3389/fmicb.2019.01293
59. Mendes R, Kruijt M, De Bruijn I, Dekkers E, Van der Voort M, Schneider JH, et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. (2011) 332:123980. doi: 10.1126/science.1203980
60. Fang Y, Zhang L, Jiao Y, Liao J, Luo L, Ji S, et al. Tobacco rotated with rapeseed for soil-borne phytophthora pathogen biocontrol: mediated by rapeseed root exudates. Front Microbiol. (2016) 7:894. doi: 10.3389/fmicb.2016.00894
Keywords: tillage, oilseed rape, rhizosphere, microbiota, plant growth stages, next generation sequencing, 16S rRNA gene
Citation: Rathore R, Dowling DN, Forristal PD, Spink J, Cotter PD and Germaine KJ (2021) Conservation Strip Tillage Leads to Persistent Alterations in the Rhizosphere Microbiota of Brassica napus Crops. Front. Soil Sci. 1:659454. doi: 10.3389/fsoil.2021.659454
Received: 27 January 2021; Accepted: 19 July 2021;
Published: 18 August 2021.
Edited by:
Adnane Bargaz, Mohammed VI Polytechnic University, MoroccoReviewed by:
Michelle M. Wander, University of Illinois at Urbana-Champaign, United StatesCopyright © 2021 Rathore, Dowling, Forristal, Spink, Cotter and Germaine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kieran J. Germaine, a2llcmFuLmdlcm1haW5lQGl0Y2FybG93Lmll
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.