 Lisa De Lorenzi
Lisa De Lorenzi Pietro Parma
Pietro Parma
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Water , 14 September 2022
Sec. Environmental Water Quality
Volume 4 - 2022 | https://doi.org/10.3389/frwa.2022.1008838
This article is part of the Research Topic Wastewater and Microbial Contamination of Water Courses View all 9 articles
Characterization of the microbial community of a river can provide various indications, such as its general state of health or the presence of contamination. Furthermore, the study of Bacteroidetes, which have a high degree of host specificity, can provide information on the species involved in any fecal contamination. The analysis of the 16S rRNA was used to characterize the bacterial community of the Lambro river (Italy) through. The results, which were obtained by analyzing water from 15 sampling points, show a reduction in the complexity of the bacterial community as the river enters a densely populated region. The cause could be a source of chemical or physical contamination that carries out a positive selection toward some bacterial species and negative toward others. In addition, a notable increase in the percentage of Bacteroidetes was reported, especially when the river enters regions characterized by high agricultural and livestock activity, such as cattle and pig farming. However, in the samples taken from this area, no Bacteroidetes ascribable to these two species or to the other species considered (i.e., human, dog, and cat) were found. Surprisingly, suspected bacterial contamination of swine origin was identified in a sparsely populated region characterized by small family farms. Finally, the efficient treatment of urban wastewater was confirmed as no markers of fecal pollution of human origin were identified.
Water is a crucial element for the life of plants and animals, and the availability of water sources free of microorganisms is a fundamental condition for community development as it reduces the likelihood of bacterial infections. The analysis of the microbial communities present along a river can therefore represent a useful tool to determine its state of health, identify any sources of contamination, learn more about the origin of the contamination, and prepare any necessary remediation plans (Hwang et al., 2012; Kwon et al., 2018). Water can be polluted both from natural causes, for example volcanic eruptions, as well as from those related to human activities, including fecal contamination caused by non-compliant treatment of urban and livestock waste. However, it is important to define the origin of fecal contamination (human vs. animal). Escherichia coli has always been used as a marker of fecal contamination, but its high genetic diversity, unrelated to a specific host and associated with the ability to replicate outside of a host, limits the possibility to define the source of contamination (Simpson et al., 2002). Bacteria belonging to the Bacteroidetes phylum represent a better alternative to E. coli, as they make up a large part of the fecal bacterial population (Madigan et al., 2003). Specifically, they have a poor ability to replicate in the environment (Fiksdal et al., 1985), and they exhibit a very high degree of specificity for the host (Dionisi et al., 2003). For this reason, Bacteroidetes have often been used in the past to identify the species involved in fecal contamination (Bernhard and Field, 2000; Layton et al., 2006; Villemur et al., 2015; Staley et al., 2018; Cruz et al., 2021; Yasar et al., 2021).
The Lambro is a river with a total length of about 130 km. The source of the river is located in Alpe del Piano Rancio (942 m a.s.l., 45°55′05.26″N, 9°14′24.06″E), and it ends near Orio Litta (51 m a.s.l., 45°08′07.86″N, 9°32′45.55″E). During its relatively short path, it crosses six Lombardy provinces, which include Como, Lecco, Monza/Brianza, Milano, Pavia, and Lodi (Supplementary Figure 1). It also spans different geographical regions, including mountainous and densely populated areas as well as those ones where zootechnical activity is intensively represented. Therefore, it represents an interesting model for investigating the relationships between the environment and microbial communities. In this work the bacterial communities of the Lambro along its path was analyzed and this fact represents a novelty: until now no river had been characterized in all its length, from source to mouth. The supervisory authorities have declared the complete isolation of urban and livestock waste so fecal contamination, whether of animal or human origin, was expected to be excluded from any Lambro sample. Moreover, a detailed analysis of Bacteroidetes was conducted to identify the eventual origin of any fecal contamination.
Water samples (400 ml) were taken from 15 different sampling points (Supplementary Figures 1, 2). Sampling was carried out as close as possible to the central part of the river in order to avoid collecting material from the banks. All samples were obtained on the same day. The samples were collected in sterile plastic bottles, transported to the laboratory at 4°C and subsequently stored at −80°C. Finally, animal and Human fecal samples were also used.
First, the samples were filtered through a paper filter to eliminate soil and/or plant contaminants, and subsequently, the bacteria were collected on a 0.22 μm membrane. The bacteria were then resuspended by stirring the filters in 3 ml of sterile H2O for 1 h, and a subsequent centrifugation at 12,000 rpm for 30 min collected a bacterial pellet. Bacterial DNA was extracted using the NucleoSpin Tissue kit (Qiagen) following the supplier's directions. The DNA was initially resuspended in a volume of 40 μl of sterile H2O and subsequently diluted to obtain a final concentration of 5 ng/μl. To create a reference of the microbial communities of fecal samples, samples of human, dog, cat, pig, and bovine feces were analyzed. In this case, the bacterial DNA was obtained using the NucleoSpin Soil kit (Macherey-Nagel) starting from 25 mg of material.
A portion of the 16S rRNA was amplified using 5 μl of the extracted DNA in a final reaction volume of 25 μl using Platinum Taq DNA Polymerase High Fidelity (Thermofisher) in accordance with the manufacturer's instructions. The amplifications were performed for 26 cycles of the following amplification profile: 95°C-60 s (denaturation), 55°C-60 s (annealing) and 72°C-60 s (extension). The following primers were used: Pro341F: 5′-CCTACGGGNBGCASCAG-3′ and Pro805R: Rev 5′-GACTACNVGGGTATCTAATCC-3′ (Takahashi et al., 2014). These primers produce an amplicon of ~445 bp (depending on the amplified bacterial species). The overhang adapter (forward overhang: 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-locus specific sequence and reverse overhang: 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-locus specific sequence; from BMR Genomics srl-Italy) was added and followed by library construction. The libraries were purified with Beads Amplure XP 0.8X, amplified with Indexes Nextera XT Illumina, and then normalized, mixed, and loaded on Miseq with 2 × 300 bp (paired-end).
The raw sequences were verified and filtered by quality, trimmed by the primers, and fused with Qiime2 v. 8 software (Bolyen et al., 2019). DADA2 (Qiime2) software isolated the OTUs (Operational Taxonomic Unit), whose sequences were compared against Greengenes v. 13-8 database to obtain the taxonomic assignment. A similarity analysis between the species detected in the feces and those ones in the water was performed using the BLAST program, available at NCBI (Altschul et al., 1990).
The interpretation of the results regarding the percentage of the different bacterial species was carried out using Excel 365. PCA (Principal Components Analysis) analysis was performed using StataMp v. 17 software. The statistical evaluation of the different Shannon–Weaver index obtained (SWi) was calculated with the two sample Mann–Whitney test.
The complexity of the bacterial populations was evaluated using the Shannon–Weaver index (SWi; Figure 1A). A higher value was identified at sampling D (8.06), while a lower value was identified at sampling R (4.37). These values are consistent with what has been observed in other watercourses (Cruz et al., 2021; Tee et al., 2021). The trend of the SWi at the different sampling points indicates that the maximum values were near the source of the river, while the minimum values were near the end of the path. The greatest complexity change occurred between points H and I. This fact could be explained by considering that between those two sampling points, the Lambro river passes from a sparsely inhabited area to a highly inhabited area (municipality of Milan) and then crosses an area densely characterized by farms. The median of the SWi values obtained from the A–H samplings, 7.48, was significantly higher (p < 0.001) than the median of the values obtained at the I–R sampling points, which was 4.94 (Figure 1B). A great variety in the bacterial populations of a watercourse is an indication of good health and represents also a main factor that allows the ecosystem to react with adverse phenomena (Loreau et al., 2001; Cardinale, 2011; Zinger et al., 2012). What has been observed supports the hypothesis that the quality of the water of Lambro worsens significantly when it crosses areas with high anthropic activities.
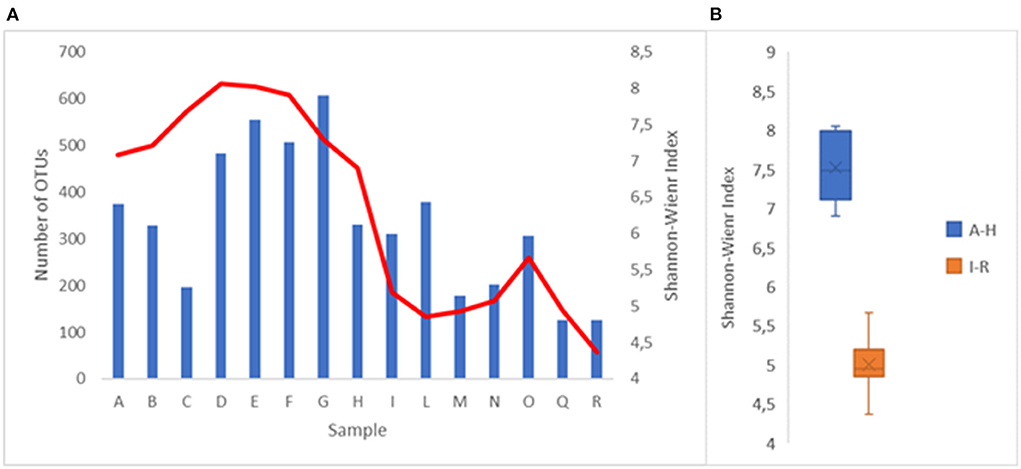
Figure 1. (A) Number of OTU's (blue bar) and Shannon-Wiener index (red line) at the different withdrawal points. (B) Representation of the Shannon-Wiener index values in the sampling points A–H and I–R.
The deterioration of the water quality was also confirmed by observing the number of OTUs identified (Figure 1A). The number of OTUs, standardized on 15,000 sequences, varied from 126 (Q and R sample points) to 607 (G sample point). The average number of OTUs in the first seven withdrawals (from A to G) was approximately double of the average of the last eight withdrawals (from I to R) at 423 compared to 220, respectively.
Considering all 15 sampling points, bacteria belonging to 37 different phyla were identified, even if only 16 of them were present at a percentage >0.5% in at least one sample (Figure 2A). The most represented phyla were Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria. They all represent from a minimum of 79.05% (point C) to a maximum of 99.99% (point R) of the entire bacterial community; furthermore, after sampling point D, their percentage was always higher than 90% (Figure 2B). Proteobacteria represented the majority phylum at all sampling points, and their percentage never dropped below 50%. The bacteria belonging to the Bacteroidetes, in contrast, showed an almost constant trend upward to sampling point L and then an increase to the last sampling point. Sampling point L marks the entrance of the Lambro in the region largely characterized by the presence of farms. The Firmicutes exhibited an opposite trend, in fact their presence was appreciable up to point I and then disappeared in the subsequent sampling points, while the Actinobacteria showed a constant presence in all sampling points, except points I and L, where they reached higher percentage. Finally, regarding the other phyla observed (always among those present in at least 0.5% of at least one sampling point), the data are shown in Supplementary Figure 3.
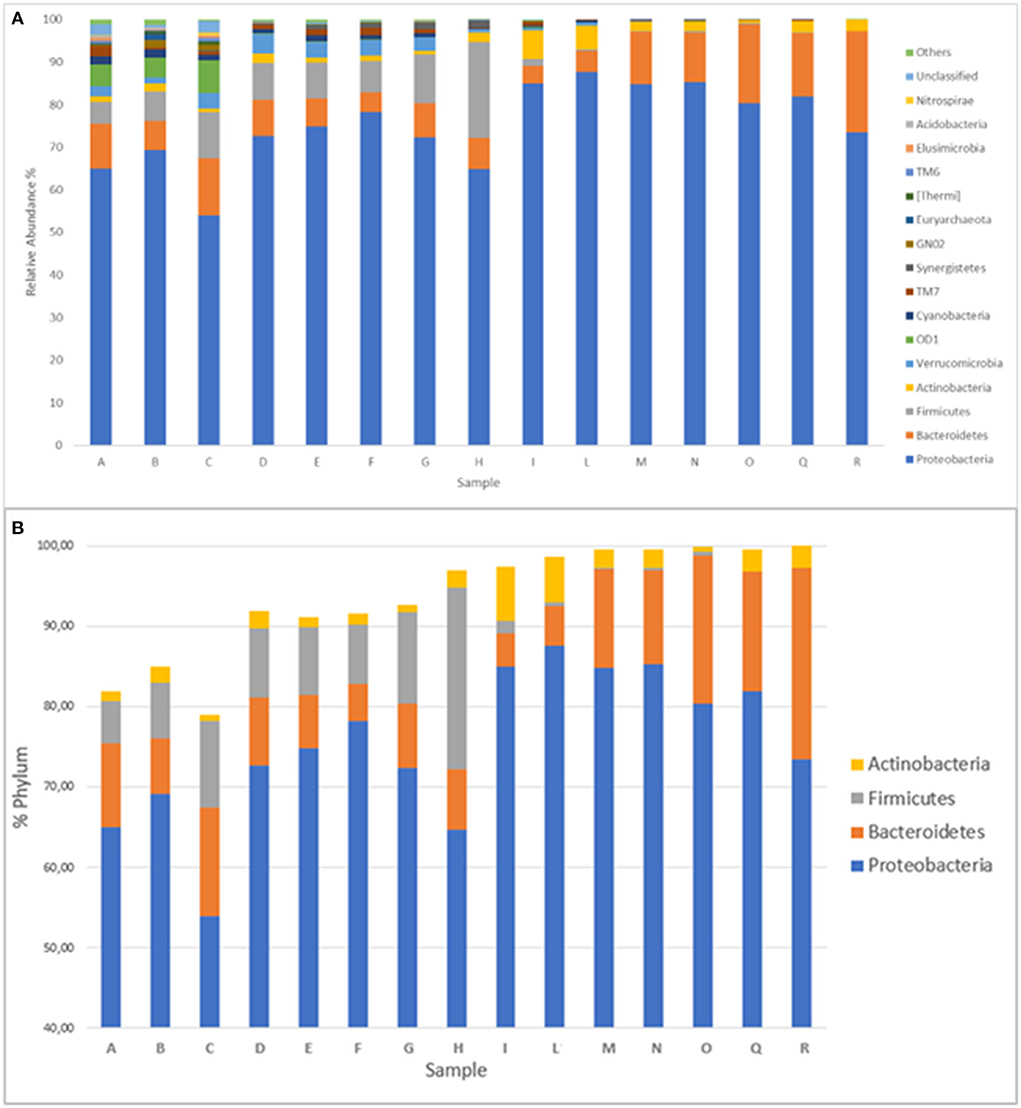
Figure 2. (A) Distribution of the 16 phyla identified in at least a percentage higher than 0.5% in at least one sampling point. Phyla with a lower percentage are grouped under the category “others.” (B) Distribution of the presence of the four main phyla (Proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria) in the 15 sampling points.
The analysis of the bacterial species identified in the 15 samples revealed that 87 of them showed a presence higher than 1% in at least one single sample (the list of these species is reported in Supplementary Figure 4). Considering the total percentage of these species at sampling points, it is possible to note an increasing trend from the source (60%) to the end of the path (92%; Supplementary Figure 5). An opposite trend could be observed by considering the number of species that possessed a presence higher than 1% at the individual sampling points, which included 15/20 near the source to 8/10 near the last sampling points. In both cases, the variations were not progressive but occurred abruptly between the withdrawal points H and I. The observed result confirms what has already been seen observed previously regarding the sudden change in the quality of the Lambro when crossing it crosses the most populated area (municipality of Milan).
Among all the species observed, only one, Acinetobacter johnsonii, was present in quantities higher than 1% in all 15 samples. This bacterium made up 4.5% of the bacterial community in sample A and 60.7% in sample R; however, in this case, the growth was not gradual and constant but there was a sudden growth between withdrawal H (7.7%) and withdrawal I (54.8). The same trend, albeit with lower percentages, could be observed for an unidentified bacterium belonging to the genus Acinetobacter. Finally, in addition to the two bacteria previously mentioned, only four other bacteria had a presence higher than 10% in at least one of the sampling points: a bacterium of the genus Enhydrobacter (32.2% in sample B), a bacterium of the genus Delftia (24.1% in sample A), a bacterium belonging to the genus Flavobacterium (20.9% in sample R), and, finally, a bacterium of the genus Arcobacter (12.9% in sample H). Acinetobacter johnsonii is a very common bacterium in the environment, so it is not surprising that it was the most common microorganism found (Guardabassi et al., 1999). Moreover, it rarely poses a health hazard. Bacteria of the genus Deftia, and in particular Deftia acidovorans, prefers an environment with low salt concentrations that are typical of high-altitude springs and known for their ability to aggregate gold and produce small nuggets (Johnston et al., 2013).
A further interesting fact emerged from the principal coordinates analysis (PCA; Figure 3). The results clearly revealed two clusters of withdrawal points. The first group was associated with the samples taken before the entrance of the Lambro into highly populated areas (red circle in Figure 3), while the second group included sample points belonging to the region with the highest agricultural density (green area in Figure 3). This result is very interesting as there is a close relationship between the geographic position of the sampling point and the composition of the microbial community. Observing the phylums identified in these two groups and the variations (Supplementary Figure 6) it can be observed that only three phylum undergo a percentage increase (Proteobacteria, Bacteroidetes and Actinobacteria), while all the others undergo a strong decrease. This observation is consistent with what was previously observed regarding the decrease in bacterial variability after sampling point H.
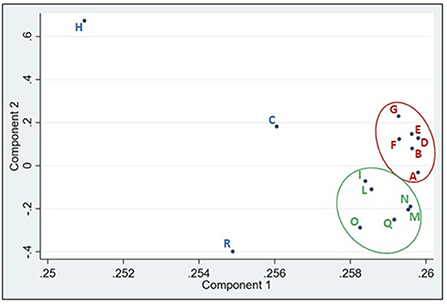
Figure 3. Result of the PCA analyzes. The red circle clearly groups the sampling points that are before the Milan region and those ones included in the green circle those that are located in the agricultural-zootechnical region.
To obtain information about the Bacteroidetes present in feces, samples belonging to five different species were analyzed: human, cattle, pig, dog, and cat. The phyla identified in the five different types of feces are shown in Supplementary Figure 7A. In the samples, most of the microbial components were made up of Firmicutes and Bacterioidetes, which together never comprised less than 90% of the total microbial flora. With regard to the feces samples, the number of OTUs (standardized to 15,000 sequences) and the Shannon–Wiener index are indicated in Supplementary Figure 7B. The composition observed was consistent with what was previously published (Bermingham et al., 2018; Mehta et al., 2018; Lourenco et al., 2020; Pilla and Suchodolski, 2020; Wylensek et al., 2020). By analyzing the species of Bacteroidetes present at a percentage higher than 2.5% in the stool samples, it was possible to confirm the great specificity of this phylum with the origin of the feces (Table 1). Of the 38 species considered, only five were present in the feces of at least two species, and four involved man, dog, and cat. In fact, the species present in pig and bovine feces were strictly specie-specific. The analysis of the similarities between these specie of Bacteroidetes and those identified in the water withdrawals indicated that there were only six similarities higher than 99% (Table 2). The results show that a porcine-specific Bacteroidetes (SS4, 3.8%) was identified at a percentage higher than 0.1% at sampling point C. Furthermore, at the same sampling point, a specie was found that had a high similarity with another specific specie of the pig (SS1, 8.4%). Finally, it was noted that at the sampling points M, N and Q, a bacterial specie very similar to Bacteroidetes specific to man (HS3, 6.2) and cat (FC10, 3.6) was identified. Sampling point C corresponds to a municipality where it is possible to hypothesize the presence of family-type pig farms.
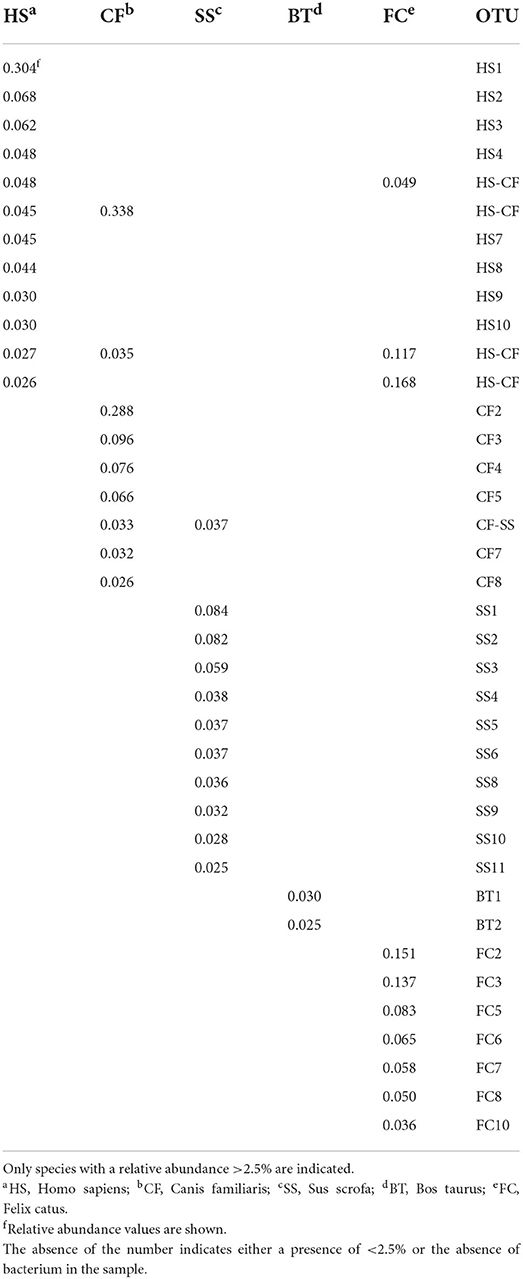
Table 1. Species of Bacteroidetes identified in the feces.
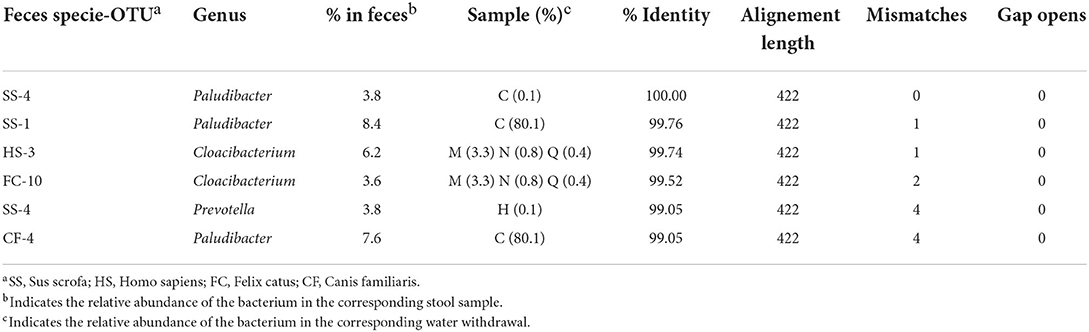
Table 2. OTUs identified in stool samples (with a relative abundance >2.5%) that showed >99% identity are reported.
The analyses carried out show a sudden change in the complexity of the bacterial population between the withdrawal points H (Brugherio) and I (Parco Lambro-Milano), which corresponds to the entrance of the Lambro into the area of competence of the municipality of Milan. This is an important observation, as a decrease in the complexity of a bacterial community has been associated with a deterioration in water quality. We can hypothesize that between these two sampling points the Lambro River undergoes chemical and/or physical contamination which favors the development of some bacterial species to the detriment of others. A further result emerged from the PCA analysis: in this case there is a correspondence between the microbial characteristics and the geographical location of the samples. Finally, the analyses have shown that the contamination of Bactroidetes in almost all cases cannot be attributed to the species considered, except at sampling point C, where contamination of pig origin could be detected. In the future, it is recommended that further analyses be performed near sampling points C, H, and I to better understand the possible source of these changes in water quality or contamination.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://doi.org/10.5061/dryad.95x69p8nv.
LD and PP made the withdrawals, managed the results of the analyzes, and wrote the paper. BC extracted the bacterial DNA from the samples, performed the PCRs, and prepared the samples for sequencing. The sequencing was carried out at an external service (BMR Genomics SRL, Padova, Italy). All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frwa.2022.1008838/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bermingham, E. N., Young, W., Butowski, C. F., Moon, C. D., Maclean, P. H., Rosendale, D., et al. (2018). The fecal microbiota in the domestic cat (Felis catus) is influenced by interactions between age and diet; a five year longitudinal study. Front. Microbiol. 9, 1231. doi: 10.3389/fmicb.2018.01231
Bernhard, A. E., and Field, K. G. (2000). A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Appl. Environ. Microbiol. 66, 4571–4574. doi: 10.1128/AEM.66.10.4571-4574.2000
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Cardinale, B. J. (2011). Biodiversity improves water quality through niche partitioning. Nature 472, 86–89. doi: 10.1038/nature09904
Cruz, A. F., Wijesekara, R. G. S., Jinadasa, K. B. S. N., Gonzales, B. J., Ohura, T., Guruge, K. S., et al. (2021). Preliminary investigation of microbial community in wastewater and surface waters in Sri Lanka and the Philippines. Front. Water 3, 730124. doi: 10.3389/frwa.2021.730124
Dionisi, H. M., Harms, G., Layton, A. C., Gregory, I. R., Parker, J., Hawkins, S. A., et al. (2003). Power analysis for real-time PCR quantification of genes in activated sludge and analysis of the variability introduced by DNA extraction. Appl. Environ. Microbiol. 69, 6597–6604. doi: 10.1128/AEM.69.11.6597-6604.2003
Fiksdal, L., Maki, J. S., LaCroix, S. J., and Staley, J. T. (1985). Survival and detection of Bacteroides spp., prospective indicator bacteria. Appl. Environ. Microbiol. 49, 148–150. doi: 10.1128/aem.49.1.148-150.1985
Guardabassi, L., Dalsgaard, A., and Olsen, J. E. (1999). Phenotypic characterization and antibiotic resistance of Acinetobacter spp. isolated from aquatic sources. J. Appl. Microbiol. 87, 659–667. doi: 10.1046/j.1365-2672.1999.00905.x
Hwang, C., Ling, F., Andersen, G. L., LeChevallier, M. W., and Liu, W.-T. (2012). Microbial community dynamics of an urban drinking water distribution system subjected to phases of chloramination and chlorination treatments. Appl. Environ. Microbiol. 78, 7856–7865. doi: 10.1128/AEM.01892-12
Johnston, C. W., Wyatt, M. A., Li, X., Ibrahim, A., Shuster, J., Southam, G., et al. (2013). Gold biomineralization by a metallophore from a gold-associated microbe. Nat. Chem. Biol. 9, 241–243. doi: 10.1038/nchembio.1179
Kwon, M. J., Ham, B., and Kwon, J.-S. (2018). “Geochemical characteristics and microbial community compositions in groundwater from underground waste storage sites,” in: EGU General Assembly Conference Abstracts, Vol. 20, 7057.
Layton, A., McKay, L., Williams, D., Garrett, V., Gentry, R., and Sayler, G (2006). Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Appl. Environ. Microbiol. 72, 4214–4224. doi: 10.1128/AEM.01036-05
Loreau, M., Naeem, S., Inchausti, P., Bengtsson, J., Grime, J. P., Hector, A., et al. (2001). Biodiversity and ecosystem functioning: current knowledge and future challenges. Science 294, 804–808. doi: 10.1126/science.1064088
Lourenco, J. M., Kieran, T. J., Seidel, D. S., Glenn, T. C., Silveira, M. F. D., Callaway, T. R. Jr., et al. (2020). Comparison of the ruminal and fecal microbiotas in beef calves supplemented or not with concentrate. PLoS ONE 15, e0231533. doi: 10.1371/journal.pone.0231533
Madigan, M. M., Martinko, J. M., and Parker, J. (2003). Brock Biology of Microorganisms, 10th ed. Upper Saddle River, NJ: Prentice Hall.
Mehta, R. S., Abu-Ali, G. S., Drew, D. A., Lloyd-Price, J., Subramanian, A., Lochhead, P., et al. (2018). Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 3, 347–355. doi: 10.1038/s41564-017-0096-0
Pilla, R., and Suchodolski, J. S. (2020). The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front. Vet. Sci. 6, 498. doi: 10.3389/fvets.2019.00498
Simpson, J. M., Santo Domingo, J. W., and Reasoner, D. J. (2002). Microbial source tracking: state of the science. Environ. Sci. Technol. 36, 5279–5288. doi: 10.1021/es026000b
Staley, Z. R., Chuong, J. D., Hill, S. J., Grabuski, J., Shokralla, S., Hajibabaei, M., et al. (2018). Fecal source tracking and eDNA profiling in an urban creek following an extreme rain event. Sci. Rep. 8, 14390. doi: 10.1038/s41598-018-32680-z
Takahashi, S., Tomita, J., Nishioka, K., Hisada, T., and Nishijima, M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PLoS ONE 9, e105592. doi: 10.1371/journal.pone.0105592
Tee, H. S., Waite, D., Lear, G., and Handley, K. M. (2021). Microbial river-to-sea continuum: gradients in benthic and planktonic diversity, osmoregulation and nutrient cycling. Microbiome 9, 190. doi: 10.1186/s40168-021-01145-3
Villemur, R., Imbeau, M., Vuong, M. N., Masson, L., and Payment, P. (2015). An environmental survey of surface waters using mitochondrial DNA from human, bovine and porcine origin as fecal source tracking markers. Water Res. 69, 143–153. doi: 10.1016/j.watres.2014.10.063
Wylensek, D., Hitch, T. C. A., Riedel, T., Afrizal, A., Kumar, N., Wortmann, E., et al. (2020). A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 11, 6389. doi: 10.1038/s41467-020-19929-w
Yasar, S. A., Mills, T. J. T., Uluturk, Z. I., Ruszczyk, J. M. S., LeBard, R. J., and Neilan, B. A. (2021). Quantitative detection of human- and canine-associated Bacteroides genetic markers from an urban coastal lagoon. Water. Sci. Technol. 84, 1732–1744. doi: 10.2166/wst.2021.341
Keywords: PCR, microbial communities, fecal contamination, Lambro river, 16S rRNA
Citation: De Lorenzi L, Carimati B and Parma P (2022) Analysis of the 16S rRNA gene for the characterization of the bacterial community of the Lambro river (Italy). Front. Water 4:1008838. doi: 10.3389/frwa.2022.1008838
Received: 01 August 2022; Accepted: 16 August 2022;
Published: 14 September 2022.
Edited by:
Amin Mojiri, Hiroshima University, JapanReviewed by:
Abid Ali Khan, Jamia Millia Islamia, IndiaCopyright © 2022 De Lorenzi, Carimati and Parma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pietro Parma, cGlldHJvLnBhcm1hQHVuaW1pLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.