 Joohyun Kim1*
Joohyun Kim1* Seung-Keun Hong1
Seung-Keun Hong1 Yongqiang Yang1
Yongqiang Yang1 Alice Lee1
Alice Lee1 Karin M. Hoffmeister2Benjamin N. Gantner3
Karin M. Hoffmeister2Benjamin N. Gantner3 Jong-In Park4
Jong-In Park4
- 1Department of Surgery, Division of Transplant Surgery, Medical College of Wisconsin, Milwaukee, WI, United States
- 2Versiti Translational Glycomics Center, Blood Research Institute and Medical College of Wisconsin, Milwaukee, WI, United States
- 3Department of Medicine, Division of Endocrinology, Medical College of Wisconsin, Milwaukee, WI, United States
- 4Department of Biochemistry, Medical College of Wisconsin, Milwaukee, WI, United States
Introduction: Machine perfusion is increasingly being utilized in liver transplantation in lieu of traditional cold static organ preservation. Nevertheless, better understanding of the molecular mechanisms underlying the ischemia-reperfusion injury (IRI) during ex vivo perfusion is necessary to improve the viability of liver grafts after transplantation using machine perfusion technology. Since key cellular signaling pathways involved in hepatic IRI may allow a chance for designing a promising approach to improve the clinical outcomes from this technology, we determined how warm ischemia time (WIT) during procurement affects the activity of mitogen-activated protein kinase (MAPK) and perfusate concentration of cytokines in an ex vivo rat liver machine perfusion model.
Methods: Male Sprague-Dawley rats underwent in situ hepatic ischemia with varying WIT (0, 10, 20, 30 min, n = 5 each), and subsequently 3 h of cold ischemia time and 2 h of machine perfusion prior to determining the degree of MAPK activation-phosphorylation and cytokine concentration in liver tissue and perfusates, respectively.
Results: Our data revealed a strong correlation between incremental WIT and a series of liver injury markers, and that prolonged WIT increases ERK1/2 and p54 JNK phosphorylation during machine perfusion. Notably, specific cytokine levels (MCP-1, MIP-2, GRO/KC, IL-10, and IL-5) were inversely correlated with the phosphorylation levels of ERK1/2, p38 MAPK, and p46/p54 JNK.
Discussion: These results suggest that MAPK activation, specifically ERK1/2 and p54 JNK phosphorylation, have potential as a biomarker for hepatic IRI pathophysiology during machine perfusion. Elucidation of their functional significance may lead to designing a novel strategy to increase the clinical benefit of machine perfusion.
Introduction
Currently, the only definitive treatment for end-stage liver disease is liver transplantation (LT). However, the shortage of available organs remains a major challenge in this field (1). A potential solution to this problem may stem from the use of marginal grafts which are typically discarded due to their poor tolerance to hepatic ischemia-reperfusion injury (IRI) (2). Machine perfusion systems have been introduced to mitigate IRI in marginal livers, especially those from donation after circulatory death, which shows an increased degree of injury under the increased warm ischemia time (WIT) during the organ donation process (3). Nevertheless, there is still limited understanding of the underlying pathophysiology of IRI during machine perfusion for LT, which hinders the development of diagnostic or therapeutic methods required for improving the viability of livers for LT (4). Therefore, it is critical to identify a molecular mechanism underlying the hepatic IRI. Identifying the cellular signaling pathways involved in the injury may lead to a promising strategy to improve patient outcomes after LT.
Mitogen-activated protein kinases (MAPKs) play crucial roles in responding to various stress signals, including hypoxia, and subsequent regulation of cellular responses critical to survival, proliferation, growth, migration, differentiation, and metabolism (5–7). Among the 14 mammalian MAPKs, extracellular signal-regulated kinase (ERK) 1/2, p38 MAPK, and c-Jun N-terminal kinase (JNK) have been hypothesized as key regulators of hepatic IRI given the strong correlation between their activity and the injury (5). In support of this notion, a recent study of human livers during normothermic machine perfusion has demonstrated that ERK1/2 activity increases in livers with prolonged cold ischemia time (CIT) (8), suggesting involvement of the kinases in the pathophysiology of IRI. Because different MAPKs have distinct roles (5), it is possible that certain MAPKs support liver cell survival from IRI while others contribute to the liver damage (9).
The dual nature of cell signals during hepatic IRI, which can be both injurious and reparative, has been attributed to the function of innate immunity (10). In this context, MAPKs are involved in regulating cytokine production during innate immune responses (11, 12). MAPKs regulate diverse cytokine responses, including both pro-inflammatory (13–17) and anti-inflammatory cytokines (18). In hepatic IRI, Kupffer cells initiate the production and release of cytokines (10). Kupffer cells release tissue necrosis factor alpha (TNF-α), which induces the expression of adhesion molecules, facilitating the adhesion of circulating neutrophils to the endothelial cells, resulting in cell extravasation (19, 20). TNF-α and interleukin-1 (IL-1) released by activated Kupffer cells also upregulate the expression of adhesion proteins on the surface of neutrophils and promote chemotaxis (21). TNF-α derived from Kupffer cells can induce chemokines of hepatocytes, mobilizing neutrophils towards injured areas (22).
Prior research has primarily concentrated on the role of cytokines on in situ IRI, rather than ex vivo machine perfusion (23–25). Exposure of the liver to ischemia under ex vivo conditions may develop different cellular responses. During ex vivo machine perfusion, decreased numbers of Kupffer cells (26–28) and neutrophils (29) may alter the immune response associated with cytokine release in the liver. A recent study on discarded human livers highlighted the increase in cytokine levels during normothermic machine perfusion, although their significance under the condition remains unclear (29). Therefore, investigating MAPK activation and cytokine production in response to incremental liver damage during ex vivo perfusion may provide insights into the involvement of the MAPK pathways in machine perfusion. This study determined the correlation between WIT, MAPK phosphorylation, and cytokine levels in an ex vivo rat liver machine perfusion model.
Materials and methods
Animals
Male Sprague-Dawley rats were housed in an animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. They had ad libitum access to Purina Lab Diet 5,001 and reverse osmosis water. Surgical procedures were conducted under isoflurane inhalation anesthesia (1%–3%; oxygen flow 1 L/min). The rats were placed on a far infrared warming pad (Kent Scientific, Torrington, CT) set to maintain a temperature of approximately 39°C. All animals received humane care in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee at the Medical College of Wisconsin approved all experiments (animal use application number 00004857).
Total hepatectomy for ex vivo perfusion model
Male rats (median age of 8.5 weeks and median body weight of 330 g, interquartile range of 8.0–9.2 weeks and 310–363 g, respectively, Charles River Laboratories) were used for this study. There were four experimental groups (n = 5 each) based on WIT: 0 min, 10 min, 20 min, and 30 min. The procedure involved opening the abdomen with a cruciate incision, mobilizing the whole liver by incising ligaments, and ligating and dividing the phrenic vein and the right adrenal vein using 7-0 silk ties. Heparin (100 units, Sagent Pharmaceuticals) was injected into the intrahepatic inferior vena cava (IVC). Subsequently, the hepatic artery and the portal vein were dissected and ligated. In situ WIT was started, and the abdomen was temporarily closed using 2-0 nylon continuous stitches. After the designated WIT, the abdomen was reopened, and the portal vein was cannulated with a 16-gauge catheter while the infrahepatic IVC was ligated. The chest wall was opened, and the suprahepatic IVC was partially incised to exsanguinate the rat. The liver was flushed with 50 ml of cold lactated Ringer's solution with heparin (50 units) through the portal vein, and the portal vein cannula was secured in place. The suprahepatic IVC was cannulated using a 5 mm segment of polyethylene tubing (internal diameter 0.2794 mm and outer diameter 0.6096 mm, PE10, Braintree Scientific, Inc.). The whole rat liver was dissected and removed from the body. The liver was weighed and submerged in cold lactated Ringer's solution in ice for 3 h until it was connected to the normothermic machine perfusion system.
Normothermic machine perfusion
The normothermic machine perfusion system was primed with 100 ml of perfusate (Table 1) in a glass reservoir, which was placed on a magnetic stirrer (Corning Pyroceram Top Digital Stirrer, Fischer Scientific) maintaining a temperature of approximately 38°C using a heated recirculator (Model 210, PolyScience). The system was connected to a peristaltic pump (Masterflex® Ismatec® Reglo, Avantor) and the oxygenator (fiber/membrane oxygenator, Harvard Apparatus) with a flow rate of 0.5 L/min using a mixture of 95% oxygen and 5% carbon dioxide. Previous studies have shown that red blood cells from different species can be used as oxygen carriers in rat liver machine perfusion experiments (30). To avoid the use of up to 100 rats for whole blood collection in this study, porcine red blood cells were utilized as oxygen carriers in the perfusate. Fresh pig blood was obtained from a local farm (Wilson's Prairie View Farm, Inc., Elkhorn, WI) and heparin (10,000 U/ml, Sagent Pharmaceuticals) and antibiotics (Penicillin-Streptomycin 10,000 IU/ml, ThermoFisher Scientific) were added before keeping the blood on ice. The whole blood was then filtered using sterilized gauze and centrifuged at 1,500 g for 5 min. After removing the supernatant, the pellet was filtered using a leukocyte filter (LeukoGuard® BC2 cardioplegia filter, Pall Corporation), and stored in a blood container (Transfer-pack, Fenwal Inc.) at 4°C for up to 2 weeks. The complete list of perfusate contents is summarized in Table 1.
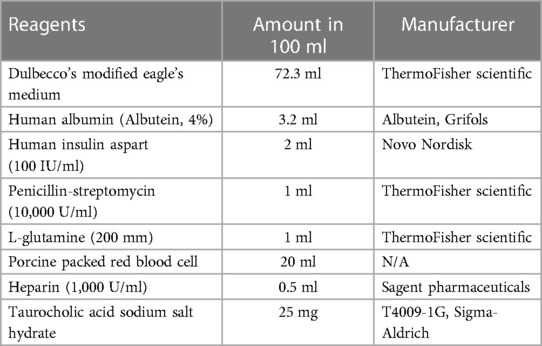
Table 1. Perfusate component for normothermic machine perfusion.
After the designated WIT and CIT, the rat liver was placed in a heated organ moist chamber (Harvard Apparatus) in the normothermic machine perfusion system. The portal vein was connected to the inflow metal tube connector, and the suprahepatic IVC was connected to the outflow metal tube connector. Polyethylene tubing placed during total hepatectomy was used to connect the blood vessels and metal connectors. The initial flow rate was set at 5 ml/min and was increased by 5 ml/min every 5 min up to a maximum of 30 ml/min or 12 mmHg of portal venous pressure, as measured by a monitor (PM-4 perfusion pressure monitor, Living Systems Instrumentation). After 2 h of machine perfusion, both perfusate and liver tissue samples were harvested. Approximately 0.5 ml of perfusate was retrieved from the glass reservoir situated above the magnetic stirrer, which was then centrifuged at 3,000 rpm for 10 min at room temperature to isolate the supernatant. These collected serum samples were promptly frozen at −20°C and subsequently stored at −80°C until further analysis.
Immunoblot analysis
Proteins were extracted from snap frozen liver samples and quantified using the BCA reagent (Pierce, 23,225) (31). For SDS-PAGE analysis, 50 μg of protein was resolved and transferred to a polyvinylidene difluoride membrane filter (Bio-Rad, 162-0177). Membrane filters were then blocked in 0.1 M Tris (pH 7.5)-0.9% NaCl-0.05% Tween 20 with 5% nonfat dry milk and incubated with the following primary antibodies: ERK1/2 (Cell Signaling, 4,695, 1:2,000), phospho-ERK1/2 (Thr202/Tyr204, Cell Signaling, 4,370, 1:2,000), SAPK/JNK (Cell Signaling, 9,252, 1:2,000), phospho-SAPK/JNK (Thr183/Tyr185, Cell Signaling, 46,682, 1:2,000), p38 MAPK (Cell Signaling, 8,690, 1:2,000), phospho-p38 MAPK (Thr180/Tyr182, Cell Signaling, 4,511, 1:2,000), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Cell Signaling, 2,118, 1:5,000). Detection of the signal was performed using the SuperSignal West Femto chemiluminescence kit (Pierce) and the ChemiDoc MP imaging system (Bio-Rad). Following the detection of phosphorylated protein forms, the same blot was stripped and reprobed for GAPDH, serving as a loading control, as well as the total MAPK proteins. We quantified protein phosphorylation as a ratio of phosphorylated to total protein expression, using densitometry with Image Lab 6.1 software (Bio-Rad, Hercules, CA). Expression levels within each WIT group were normalized against those in the 0 min WIT group, which served as the control for the standardization of densitometry data (Supplementary Figure S1). Specifically, we normalized the expression levels of phosphorylated MAPKs against the optical density (O.D.) of the 0 min WIT group. This involved dividing the O.D. of the phosphorylated protein by the O.D. of the total protein, and subsequently normalizing this ratio using the mean value of the control group. As a result, the expressions at WIT 10, 20, and 30 min are presented as relative expressions in comparison to that at WIT 0 min.
Multiplex cytokine array
We used the Rat Cytokine 27-Plex Discovery Assay (Eve Technologies, Calgary, AB, Canada) to perform cytokine and chemokine profiling on sera from perfusate samples. The assay detected 27 different markers, including Eotaxin (CCL11), Epidermal Growth Factor (EGF), Fractalkine (CX3CL1), Interferon-γ (IFN-γ), Interleukin-1α) (IL-1α), Interleukin-1beta (IL-1β), Interleukin-2 (IL-2), Interleukin-4 (IL-4), Interleukin-5 (IL-5), Interleukin-6 (IL-6), Interleukin-10 (IL-10), Interleukin-12 (IL-12), Interleukin-13 (IL-13), Interleukin-17A (IL-17A), Interleukin-18 (IL-18), Interferon-gamma-inducible protein 10 (IP-10/CXCL10), Growth-Related Oncogene/Keratinocyte Chemoattractant (GRO/KC/CXCL1), TNF-α, Granulocyte Colony-Stimulating Factor (G-CSF), Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF), Monocyte Chemoattractant Protein-1 (MCP-1/CCL2), Leptin, Lipopolysaccharide-Induced CXC Chemokine (LIX/CXCL5), Macrophage Inflammatory Protein-1α (MIP-1α/CCL3), Macrophage Inflammatory Protein-2 (MIP-2/CXCL2), Regulated on Activation, Normal T-cell Expressed and Secreted (RANTES/CCL5), and Vascular Endothelial Growth Factor (VEGF).
Liver injury profiling
To evaluate the extent of liver injury, we measured the levels of perfusate aspartate aminotransferase (AST), α-glutathione S-transferase (α-GST), and arginase1 (ARG1) at the end of the experiment using the Rat Liver Injury 5-Plex Featured Assay (Eve Technologies, Calgary, AB, Canada). AST is a marker of hepatocyte injury, commonly used in conjunction with other markers. α-GST is a specific marker for hepatocellular damage, particularly effective in early detection (32). ARG1 is a marker of alternative activated (anti-inflammatory) macrophage (33, 34).
Statistical analysis
The data were presented as the median with interquartile ranges. P < 0.05 was considered statistically significant. The relationship between two variables was assessed using simple linear regression, and the P and R2 values were calculated using Prism 9 for Windows (GraphPad Software, Inc., San Diego, CA).
Results
Increasing WIT corresponds to higher perfusate injury markers in the ex vivo rat liver perfusion model
To induce various degrees of hepatic IRI, we exposed the rat liver to incremental periods of in situ WIT ranging from 0 to 30 min (10 min intervals between each group; n = 5 per group) before CIT and machine perfusion (Figure 1A). We evaluated liver injury by measuring perfusate markers of liver damage, such as AST, α-GST, and ARG1. We found that the duration of WIT is strongly correlated with the level of the liver injury markers AST (R2 = 0.5585, Figure 1B), α-GST (R2 = 0.5797, Figure 1C), and ARG1 (R2 = 0.8180, Figure 1D). These data suggest that WIT is an important factor in determining liver injury ensued from 3 h of CIT and 2 h of ex vivo machine perfusion.
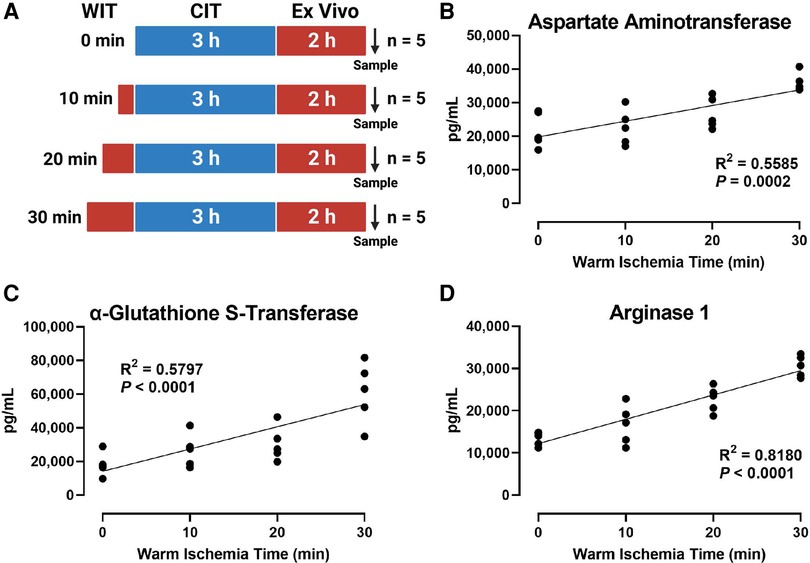
Figure 1. Effect of WIT on the levels of liver injury markers in the perfusate during machine perfusion. (A) Scheme of four experimental groups (n = 5 each). The rat livers underwent designated WIT of 0, 10, 20, and 30 min, followed by 3 h of cold ischemia time and 2 h of normothermic machine perfusion. Perfusate levels of (B) aspartate Aminotransaminase, (C) α-glutathione S-transferase, and (D) arginase1 demonstrated a significant correlation with WIT. The correlation was determined by the Spearman's rank correlation coefficient (R). WIT, warm ischemia time; CIT, cold ischemia time.
WIT differentially induces MAPK activity in the ex vivo rat liver machine perfusion model
To determine how WIT affects MAPK activity in the ex vivo rat liver machine perfusion model, we analyzed the activation-loop phosphorylation of different MAPKs, a bona-fide surrogate maker for MAPK activity (35), by Western blotting of rat liver extracts collected at the end of machine perfusion. Our analysis revealed that the duration of in situ WIT prior to CIT and machine perfusion is strongly correlated with certain MAPK phosphorylation. Most notably, the activation-loop phosphorylation of ERK1 (Thr 202 and Tyr 204) and ERK2 (Thr 185 and Tyr 187) was substantially increased in a strong correlation with WIT (Figure 2, R2 = 0.4236 and 0.4385 for ERK1 and ERK2, respectively). Similarly, the activation-loop phosphorylation of p54 JNK (Thr 183 and Tyr 185) was also increased in a strong correlation with WIT (R2 = 0.4784). In contrast, the activation-loop phosphorylation of p46 JNK (Thr 183 and Tyr 185) and p38 MAPK (Thr 180 and Tyr 182) was not increased and did not show any correlation with WIT. These data suggest that the activity of certain MAPKs, such as ERK1/2 and p54 JNK, increase upon prolonging WIT.
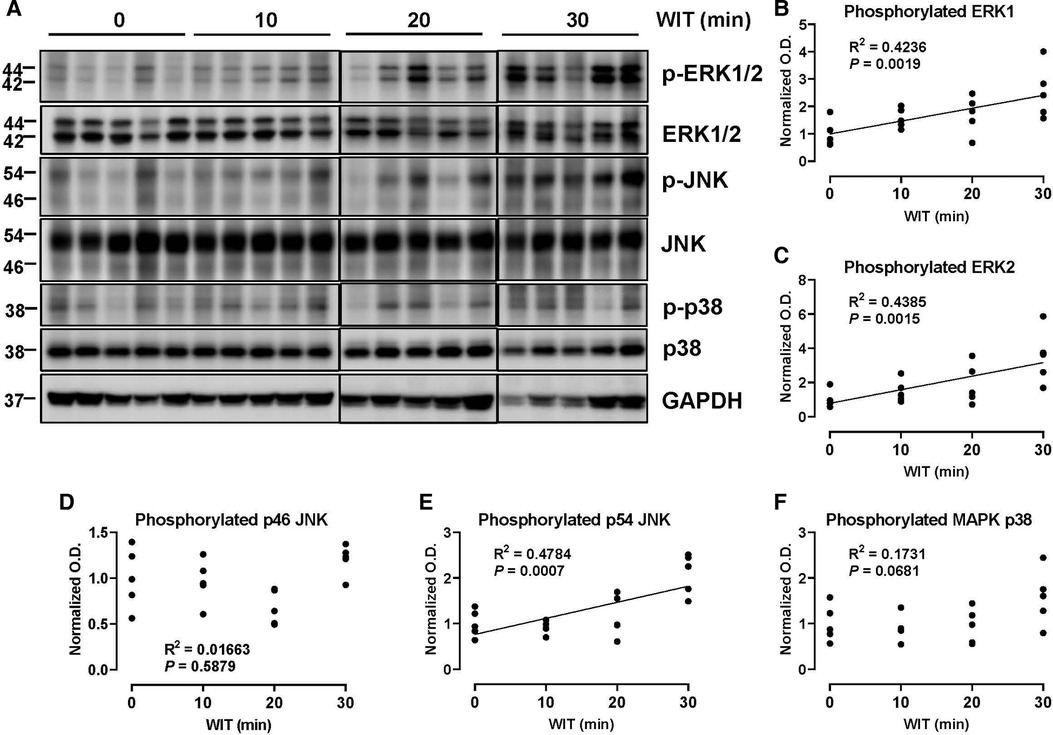
Figure 2. Effect of WIT on MAPK phosphorylation during machine perfusion. (A) Protein expression of phosphorylate ERK1/2, ERK1/2, phosphorylated p46/p54 JNK, p46/p54 JNK, phosphorylated p38 MAPK, p38 MAPK, and GAPDH. (B–F) The level of phosphorylated MAPKs relative to that of total MAPKs was presented by densitometry normalized to the mean value in the control group. O.D., optical density. Spearman's rank correlation coefficient (R) was used to determine the correlation (n = 20). WIT, warm ischemia time.
Specific cytokines decrease proportionately to the duration of WIT after machine perfusion
In the ex vivo machine perfusion model, we examined 27 cytokine markers to find that seven of those decreased in a significant correlation with the duration of WIT, including TNF-α (R2 = 0.2328), IL-1α (R2 = 0.2399), MCP-1 (R2 = 0.4064), MIP-1α (R2 = 0.2464), MIP-2 (R2 = 0.4728), GRO/KC (R2 = 0.3071), and IL-5 (R2 = 0.3229, Figure 3). The inverse correlation between the cytokine levels and WIT suggests that prolonged WIT in the livers in ex vivo conditions decreases the production of these cytokines, especially MCP-1 and MIP-2. However, there was no strong correlation between WIT and IL-6, IL-4, IL-10, IP10 and LIX, suggesting that the production of these cytokines is not sensitive to WIT in the liver perfusion model.
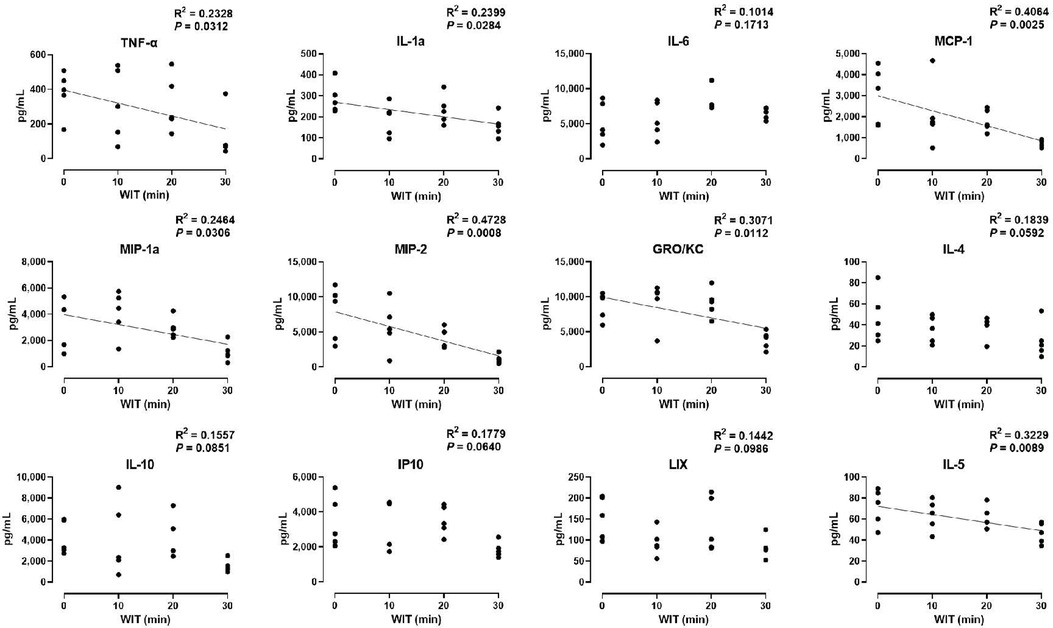
Figure 3. Effect of WIT on perfusate levels of cytokines in machine perfusion. Spearman's rank correlation coefficient (R) was used to determine the correlation (n = 20). A trend line is displayed only when the relationship is statistically significant. WIT, warm ischemia time.
ERK1/2 phosphorylation correlates positively with liver injury markers but inversely with specific cytokines
We examined the relationship between MAPK phosphorylation and perfusate levels of liver injury markers during ex vivo machine perfusion. Our data revealed a strong positive correlation between ERK1 phosphorylation and the liver injury markers, AST (R2 = 0.4705), α-GST (R2 = 0.02009), and ARG1 (R2 = 0.0.3530, Figure 4B). Similar as ERK1, ERK2 phosphorylation was also strongly correlated with AST (R2 = 0.5003), α-GST (R2 = 0.4436), and ARG1 (R2 = 0.0.5190, Figure 4D). These data show that ERK1/2 activity during machine perfusion may be a marker for the severity of hepatic IRI.
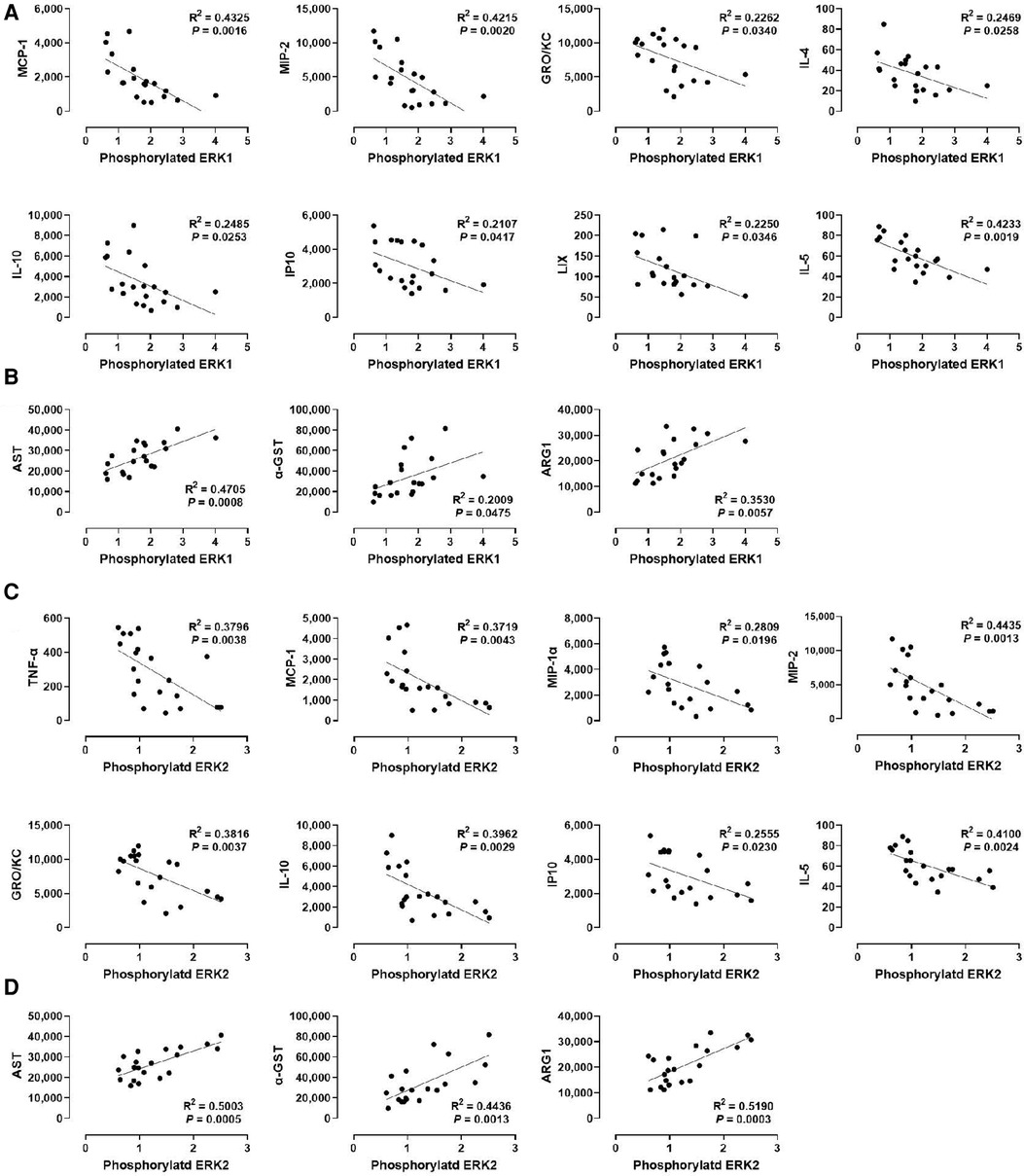
Figure 4. Correlation between ERK1/2 phosphorylation and perfusate levels of cytokines and liver injury markers. (A) ERK1 and cytokines, (B) ERK1 and liver injury markers, (C) ERK2 and cytokines, and (D) ERK2 and liver injury markers. Spearman's rank correlation coefficient (R) was used to determine the correlation (n = 20). The level of phosphorylated ERK1/2 relative to that of total ERK1/2 was presented by densitometry normalized to the mean value in the control group.
We next examined whether a correlation exists between ERK1/2 phosphorylation and various cytokines (Figures 4A,C). We found that the levels of MCP-1, MIP-2, GRO/KC, IL-1, IP10, and IL-5 were inversely correlated with the levels of ERK1 and ERK2 phosphorylation, at similar degrees. In contrast, the levels of IL-4 and LIX were inversely correlated only with ERK1 phosphorylation (R2 = 0.2469 and 0.2250, respectively), whereas the levels of TNF-α and MIP-1α were inversely correlated only with ERK2 phosphorylation (R2 = 0.3796 and 0.2809, respectively). These data suggest that cytokine production decreases as ERK1/2 activity increases in the liver during machine perfusion.
P38 MAPK phosphorylation also correlates positively with liver injury markers but inversely with specific cytokines
We also analyzed the correlation between p38 MAPK phosphorylation and liver injury markers in the perfusate (Figure 5B). Although p38 MAPK did not show any correlation with WIT (Figure 2F), there was a significant positive correlation between p38 MAPK phosphorylation and the perfusate levels of α-GST (R2 = 0.3135) and ARG1 (R2 = 0.2953, Figure 5B). However, there was no significant correlation between p38 MAPK phosphorylation and AST.
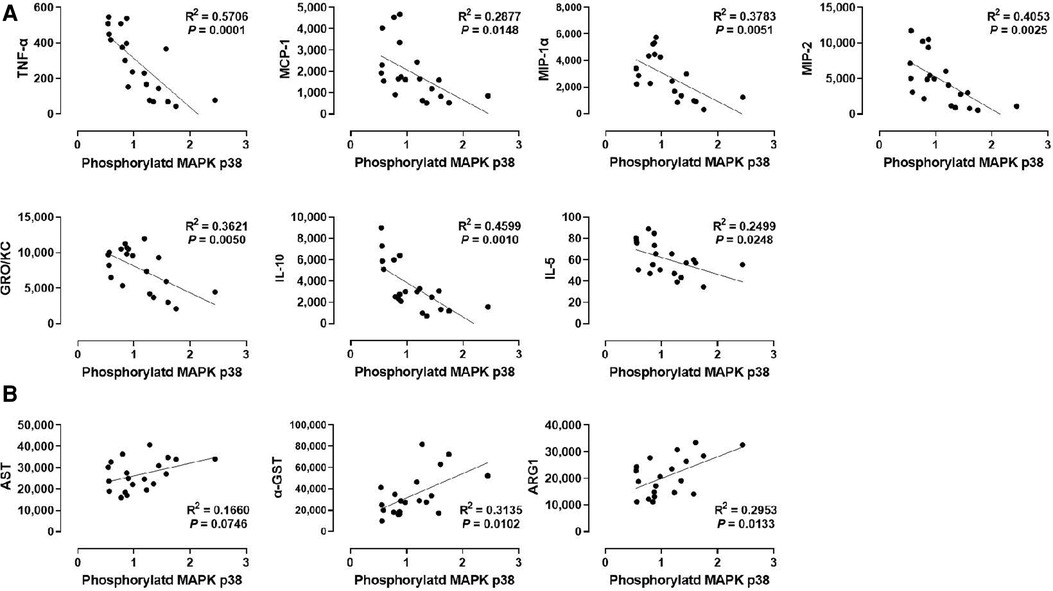
Figure 5. Correlation between p38 MAPK phosphorylation and perfusate levels of cytokines and liver injury markers. (A) p38 MAPK and cytokines and (B) ERK1 and liver injury markers. Spearman's rank correlation coefficient (R) was used to determine the correlation (n = 20). The level of phosphorylated p38 MAPK relative to that of total p38 MAPK was presented by densitometry normalized to the mean value in the control group.
Next, we examined the correlation between p38 MAPK phosphorylation levels in the liver tissues and the cytokine levels in the perfusate (Figure 5A). Like ERK1/2, p38 MAPK phosphorylation was inversely correlated with TNF-α, MCP-1, MIP-1α, MIP-2, GRO/KC, IL-1β, and IL-5. Interestingly, the correlation of these cytokines, especially TNF-α (R2 = 0.5706) and MIP-1α (R2 = 0.3783), with p38 MAPK were more like their correlation with ERK2 phosphorylation (Figure 4C) than with ERK1 phosphorylation (Figure 4A). Moreover, similar to ERK2 phosphorylation, p38 MAPK phosphorylation was not correlated with IL-4 and LIX. These data suggest that cytokine production during machine perfusion also decreases as p38 MAPK activity increases in the liver. Unlike its association with the liver injury markers and cytokine levels, p38 MAPK phosphorylation was not correlated with WIT (Figure 2F). Therefore, one may speculate that hepatic IRI, MAPK activation, and cytokine production are not in a unidirectional causal relationship.
Phosphorylation of p54 JNK, but not p46 JNK, correlates with injury markers while various cytokines show inverse correlations with the JNK isoforms’ phosphorylation
Lastly, we determined whether JNK phosphorylation is correlated with injury markers and cytokine levels by examining p46 JNK and p54 JNK phosphorylation. We found that phosphorylation of p54 JNK, but not p46 JNK, was strongly correlated with all injury markers (R2 = 0.5003 for AST, R2 = 0.4436 for α-GST, and R2 = 0.5190 for ARG1; Figures 6B,D). These data suggest that p54 JNK activity, but not p46 JNK activity, correlates with the severity of hepatic IRI during machine perfusion. Interestingly, despite this differential correlation, phosphorylation of both JNK isoforms was similarly inverse-correlated with TNF-α, MCP-1, MIP-2, GRO/KC, IL-10, and IL-5 (Figures 6A,C). The only difference was that p46 JNK phosphorylation was inversely correlated with IL-6 whereas p54 JNK phosphorylation was inversely correlated with MIP-1α and IP10. These data suggest that cytokine production also decreases as JNK activity increases in the liver during machine perfusion.
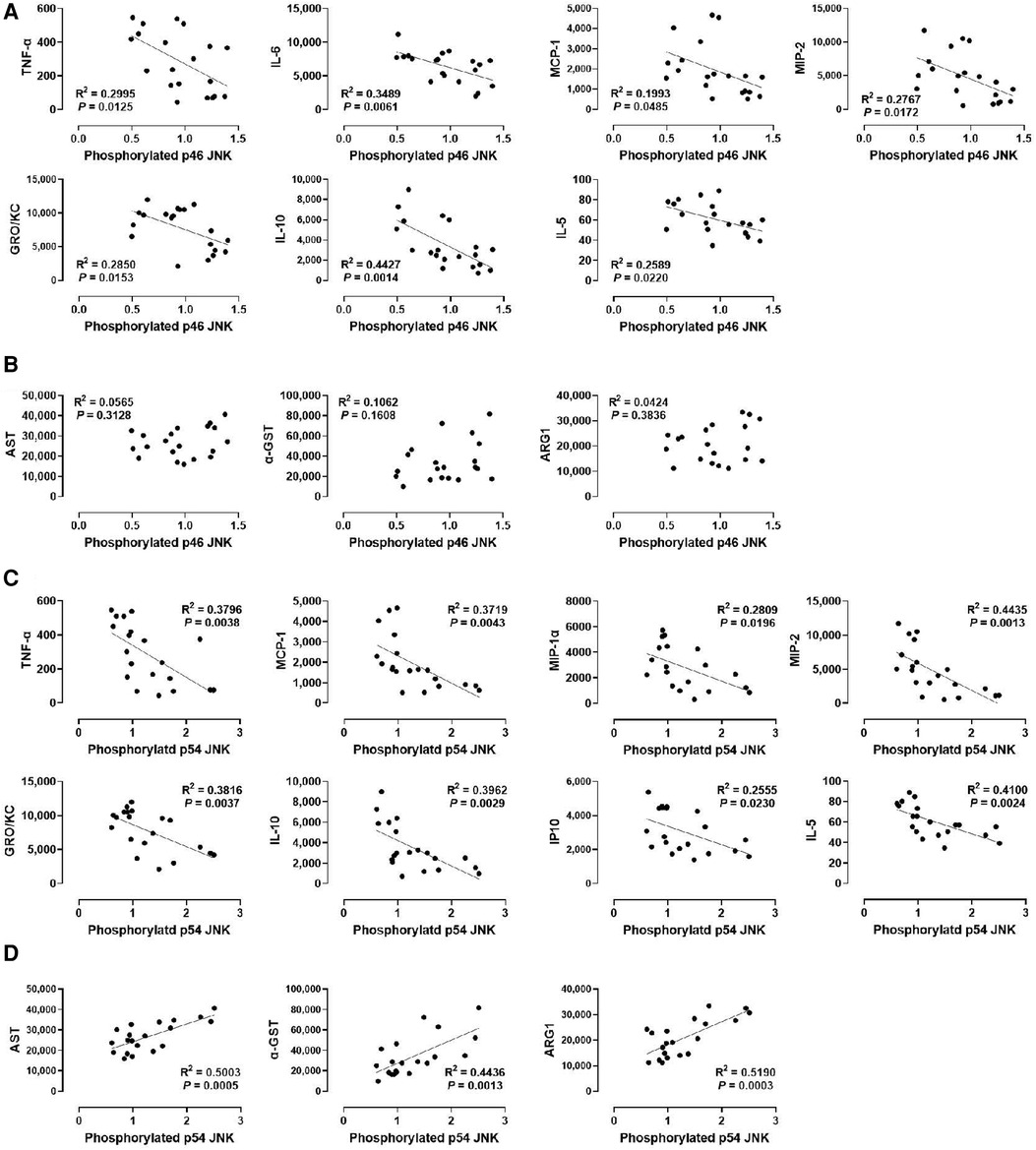
Figure 6. Correlation between JNK phosphorylation and perfusate levels of cytokines and liver injury markers. (A) p46 JNK and cytokines, (B) p46 JNK and liver injury markers, (C) p54 JNK and cytokines, and (D) p54 JNK and liver injury markers. Spearman's rank correlation coefficient (R) was used to determine the correlation (n = 20). A trend line is displayed only when the relationship is statistically significant. The level of phosphorylated p46/p54 JNK relative to that of total p46/p54 JNK was presented by densitometry normalized to the mean value in the control group.
Discussion
Given the brief period of machine perfusion application for donor livers, typically only a few hours (4), it is essential to understand rapid cellular signal changes in response to injury during liver machine perfusion. MAPKs, which show an immediate stress response through phosphorylation (9, 36), serve as promising biomarkers. In an ex vivo rat liver machine perfusion model, this study examined the relationship between WIT, MAPK activation, and cytokine production. Our data reveal novel positive correlations between WIT and injury markers in the initial 2 h of perfusion along with relationships between WIT with ERK1/2 and p54 JNK phosphorylation levels corresponding to injury severity.
Understanding the association between MAPKs and cytokines during liver injury is important, as they can significantly influence each other's activities. In the early phase of acute liver injury, Kupffer cells release cytokines and chemokines, which are sensed by circulating immune cells (37, 38). However, it is unclear whether this pathophysiological context can be applied to ex vivo machine perfusion conditions. Cytokine production will be compromised during ex vivo machine perfusion due to the absence of bone marrow-derived monocytes (27), extrahepatic macrophages (10), or neutrophils (39) in the ex vivo circuit, unless the native Kupffer cell function is preserved. The decrease in perfusate cytokine levels with increasing WIT in our findings appears counterintuitive, considering that in a clinical context, prolonged WIT typically results in more pronounced injury and presumed elevated inflammation. This unexpected observation can be explained by the lack of key inflammatory contributors during machine perfusion. The impacts of different WIT durations on IRI become evident following reperfusion with host blood, which contains a complete array of blood cells, including neutrophils and platelets. These elements are crucial for triggering a comprehensive inflammatory cascade and its eventual resolution (10). The combined impact of cytokine-mediated cellular interactions on IRI, following macrophage-driven cytokine production, can be dualistic, potentially leading to either damage or restoration. This is due to the multi-faceted role of macrophages that extends beyond merely triggering inflammation; they also play a significant part in its regulation and resolution (40). A recent study using discarded human livers found that high levels of cytokine in the perfusate may indicate recovery of local immune cell function during normothermic machine perfusion (29). In our study, perfusate cytokine levels showed an inverse correlation with WIT, whereas liver injury markers showed a positive correlation with WIT. This suggests that preserving Kupffer cells can also be protective against IRI after LT. For example, Kupffer cell depletion resulted in increased susceptibility to hepatic IRI, whereas ablation of circulating monocytes did not (41). Furthermore, Kupffer cells can be categorized into immunoregulatory and pro-inflammatory subsets based on gene signature (38, 42). Kupffer cell-originated HO-1 mediates the protected phenotype (43), and switches macrophages from classical (pro-inflammatory) to alternate (anti-inflammatory) activation via MAPK activation (44).
Our study found a significant inverse correlation between the activation-phosphorylation of MAPKs and levels of cytokines modulating immune cell behavior (i.e., MCP-1, MIP-2, GRO/KC, IL-10, and IL-5; Figures 4–6) in the perfusate. The relationship between cytokines and MAPKs during hepatic IRI is complex and involves various interacting factors. For example, cytokines can activate JNK and p38, which can subsequently induce pro-inflammatory cytokine production (45). In contrast, ERK1/2 activation is associated with anti-inflammatory cytokines such as IL-10 production and decreased IL-12 production (46–49). Additionally, MAPKs can impact each other, such as phosphorylated p38 inhibiting ERK1/2 during hepatic IRI (50). MAPK activation can also directly affect intracellular signals of hepatocytes during hepatic IRI, such as JNK's association with the endoplasmic reticulum (ER) stress response, the mitochondrial pathway of apoptosis, and autophagy (51). Moreover, MAPK isoforms may have distinct roles during hepatic IRI. For instance, TNF-α preferentially activates isoforms within the p46 JNK subfamily during macrophage regulation (52), while specific isoforms of ERK1 and ERK2 are responsible for determining the survival and proliferation of hepatocytes, respectively (52). Our data also suggests that roles of ERK1 and ERK2 may not be entirely redundant during hepatic IRI. For example, given the similarity between ERK2 and p38 MAPK in our results, p38 MAPK function may be more closely associated with ERK2 than with ERK1 during machine perfusion. Therefore, the relationship between MAPK activation and cytokines can be direct and indirectly mediated via other MAPK, different isoforms of MAPK, and extra-cellular interactions. Decoding cell signals associated with cytokine interaction in hepatic IRI will require further mechanistic studies based on the specific kinase inhibitors (53–56). Such knowledge will aid in the development of advanced organ resuscitation methods such as a combination targeted therapy in the MAPK pathway during machine perfusion.
Interpretation of the results warrants certain considerations. Factors such as the choice of preservation and machine perfusion conditions, including the initial cold liver flush [using lactated Ringer's solution (57, 58)] and the components of the perfusate (as detailed in Table 1), could potentially impact the study outcomes. In this study, only male rats were utilized to obtain sufficiently large size of livers for our machine perfusion system from the age-matched rats. Consequently, further investigations under varying conditions may be necessary to substantiate our findings. Additionally, the intrinsic complexities of ex vivo liver machine perfusion complicate the establishment of controls because obtaining perfusate samples from a liver without ischemic time is impracticable. In response to these challenges, we derived a linear correlation from outcomes across four incrementally longer WIT durations in addition to the duration of CIT and machine perfusion, providing an alternative assessment of specific factors’ influences.
In summary, our results revealed a positive relationship between WIT and perfusate injury markers, linked to ERK1/2 and p54 JNK phosphorylation in an ex vivo rat liver machine perfusion model. Moreover, an inverse correlation was observed between MAPK phosphorylation and perfusate levels of cytokines modulating immune cell behavior. These findings indicate that MAPK activation is an early response during machine perfusion and strongly correlates to the pre-existing injury level of the liver. To further determine the role of MAPK activation on cytokine production during hepatic IRI, a study design that specifically regulates each MAPK would be necessary, and further comprehensive research is needed to translate the utility of MAPK regulation in hepatic IRI from the laboratory to clinical application, which may provide an opportunity to development of a novel pharmacological strategy during machine perfusion.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The Institutional Animal Care and Use Committee at the Medical College of Wisconsin approved all experiments (animal use application number 00004857).
Author contributions
The authors’ contributions are: JK conceived and designed the study; JK, SKH, and YY conducted data acquisition, analysis, and interpretation; and JK, AL, KH, BG, and JIP were involved in drafting and critically revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The project described was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, Award Number UL1 TR001436. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors JK, KH and JIP declared that they were editorial board members of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frtra.2023.1215182/full#supplementary-material
Supplementary Figure 1
Immunoblotting analysis of liver tissue lysates. Protein expression of phosphorylated ERK1/2, ERK1/2, phosphorylated p46/p54 JNK, p46/p54 JNK, phosphorylated p38 MAPK, p38 MAPK, and GAPDH were examined in each group. Samples from each WIT group and the control group (0 min) were run together to compare the differences. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control.
References
1. Wertheim JA, Petrowsky H, Saab S, Kupiec-Weglinski JW, Busuttil RW. Major challenges limiting liver transplantation in the United States. Am J Transplant. (2011) 11(9):1773–84. doi: 10.1111/j.1600-6143.2011.03587.x
2. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation–from bench to bedside. Nat Rev Gastroenterol Hepatol. (2013) 10(2):79–89. doi: 10.1038/nrgastro.2012.225
3. Gaurav R, Butler AJ, Kosmoliaptsis V, Mumford L, Fear C, Swift L, et al. Liver transplantation outcomes from controlled circulatory death donors: SCS vs in situ NRP vs ex situ NMP. Ann Surg. (2022) 275(6):1156–64. doi: 10.1097/SLA.0000000000005428
4. Kim J, Zimmerman MA, Hong JC. Emerging innovations in liver preservation and resuscitation. Transplant Proc. (2018) 50(8):2308–16. doi: 10.1016/j.transproceed.2018.03.080
5. Jimenez-Castro MB, Cornide-Petronio ME, Gracia-Sancho J, Casillas-Ramirez A, Peralta C. Mitogen activated protein kinases in steatotic and non-steatotic livers submitted to ischemia-reperfusion. Int J Mol Sci. (2019) 20(7):1785. doi: 10.3390/ijms20071785
6. Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, et al. A lactate-induced response to hypoxia. Cell. (2015) 161(3):595–609. doi: 10.1016/j.cell.2015.03.011
7. Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. (2020) 21(10):607–32. doi: 10.1038/s41580-020-0255-7
8. Xu M, Zhou F, Ahmed O, Randle LV, Shin JK, Zhu Y, et al. Dual lactate clearance in the viability assessment of livers donated after circulatory death with ex situ normothermic machine perfusion. Transplant Direct. (2021) 7(12):e789. doi: 10.1097/TXD.0000000000001243
9. Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. (1996) 8(2):205–15. doi: 10.1016/s0955-0674(96)80067-6
10. Hirao H, Nakamura K, Kupiec-Weglinski JW. Liver ischaemia-reperfusion injury: a new understanding of the role of innate immunity. Nat Rev Gastroenterol Hepatol. (2021) 19(4):239–56. doi: 10.1038/s41575-021-00549-8
11. Lucas RM, Luo L, Stow JL. ERK1/2 in immune signalling. Biochem Soc Trans. (2022) 50(5):1341–52. doi: 10.1042/BST20220271
12. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
13. Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, et al. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. (1999) 19(1):21–30. doi: 10.1128/MCB.19.1.21
14. Yang SH, Galanis A, Sharrocks AD. Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol Cell Biol. (1999) 19(6):4028–38. doi: 10.1128/MCB.19.6.4028
15. Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. (1997) 386(6622):296–9. doi: 10.1038/386296a0
16. Gilley R, March HN, Cook SJ. ERK1/2, but not ERK5, is necessary and sufficient for phosphorylation and activation of c-fos. Cell Signal. (2009) 21(6):969–77. doi: 10.1016/j.cellsig.2009.02.006
17. Chalmers CJ, Gilley R, March HN, Balmanno K, Cook SJ. The duration of ERK1/2 activity determines the activation of c-fos and fra-1 and the composition and quantitative transcriptional output of AP-1. Cell Signal. (2007) 19(4):695–704. doi: 10.1016/j.cellsig.2006.09.001
18. Pattison MJ, Naik RJ, Reyskens K, Arthur JSC. Loss of Mef2D function enhances TLR induced IL-10 production in macrophages. Biosci Rep. (2020) 40(8):BSR20201859. doi: 10.1042/BSR20201859
19. Yazdani HO, Kaltenmeier C, Morder K, Moon J, Traczek M, Loughran P, et al. Exercise training decreases hepatic injury and metastases through changes in immune response to liver ischemia/reperfusion in mice. Hepatology. (2021) 73(6):2494–509. doi: 10.1002/hep.31552
20. Yang H, Li N, Du Y, Tong C, Lu S, Hu J, et al. Neutrophil adhesion and crawling dynamics on liver sinusoidal endothelial cells under shear flow. Exp Cell Res. (2017) 351(1):91–9. doi: 10.1016/j.yexcr.2017.01.002
21. Sumagin R, Prizant H, Lomakina E, Waugh RE, Sarelius IH. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J Immunol. (2010) 185(11):7057–66. doi: 10.4049/jimmunol.1001638
22. Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-alpha promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis. (2018) 9(3):323. doi: 10.1038/s41419-018-0377-4
23. Zhang JK, Ding MJ, Liu H, Shi JH, Wang ZH, Wen PH, et al. Regulator of G-protein signaling 14 protects the liver from ischemia-reperfusion injury by suppressing TGF-beta-activated kinase 1 activation. Hepatology. (2022) 75(2):338–52. doi: 10.1002/hep.32133
24. Neri AA, Dontas IA, Iliopoulos DC, Karatzas T. Pathophysiological changes during ischemia-reperfusion injury in rodent hepatic steatosis. In Vivo. (2020) 34(3):953–64. doi: 10.21873/invivo.11863
25. Loi P, Yuan Q, Torres D, Delbauve S, Laute MA, Lalmand MC, et al. Interferon regulatory factor 3 deficiency leads to interleukin-17-mediated liver ischemia-reperfusion injury. Hepatology. (2013) 57(1):351–61. doi: 10.1002/hep.26022
26. McDonald B, Kubes P. Innate immune cell trafficking and function during sterile inflammation of the liver. Gastroenterology. (2016) 151(6):1087–95. doi: 10.1053/j.gastro.2016.09.048
27. Yue S, Zhou H, Wang X, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Prolonged ischemia triggers necrotic depletion of tissue-resident macrophages to facilitate inflammatory immune activation in liver ischemia reperfusion injury. J Immunol. (2017) 198(9):3588–95. doi: 10.4049/jimmunol.1601428
28. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
29. Lee ACH, Edobor A, Lysandrou M, Mirle V, Sadek A, Johnston L, et al. The effect of normothermic machine perfusion on the immune profile of donor liver. Front Immunol. (2022) 13:788935. doi: 10.3389/fimmu.2022.788935
30. Dondossola D, Santini A, Lonati C, Zanella A, Merighi R, Vivona L, et al. Human red blood cells as oxygen carriers to improve ex-situ liver perfusion in a rat model. J Clin Med. (2019) 8(11):1918. doi: 10.3390/jcm8111918
31. Hong SK, Yoon S, Moelling C, Arthan D, Park JI. Noncatalytic function of ERK1/2 can promote raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem. (2009) 284(48):33006–18. doi: 10.1074/jbc.M109.012591
32. Maina I, Rule JA, Wians FH, Poirier M, Grant L. Lee WM. Alpha-glutathione S-transferase: a new biomarker for liver injury? J Appl Lab Med. (2016) 1(2):119–28. doi: 10.1373/jalm.2016.020412
33. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
34. Amin SN, Sakr HI, El Gazzar WB, Shaltout SA, Ghaith HS, Elberry DA. Combined saline and vildagliptin induced M2 macrophage polarization in hepatic injury induced by acute kidney injury. PeerJ. (2023) 11:e14724. doi: 10.7717/peerj.14724
35. Prowse CN, Deal MS, Lew J. The complete pathway for catalytic activation of the mitogen-activated protein kinase, ERK2. J Biol Chem. (2001) 276(44):40817–23. doi: 10.1074/jbc.M105860200
36. Kocieniewski P, Faeder JR, Lipniacki T. The interplay of double phosphorylation and scaffolding in MAPK pathways. J Theor Biol. (2012) 295:116–24. doi: 10.1016/j.jtbi.2011.11.014
37. Li P, He K, Li J, Liu Z, Gong J. The role of Kupffer cells in hepatic diseases. Mol Immunol. (2017) 85:222–9. doi: 10.1016/j.molimm.2017.02.018
38. Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. (2019) 3(6):730–43. doi: 10.1002/hep4.1356
39. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. (2014) 5:508. doi: 10.3389/fimmu.2014.00508
40. Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. (2010) 10(6):427–39. doi: 10.1038/nri2779
41. Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, et al. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol Ther. (2009) 17(1):65–72. doi: 10.1038/mt.2008.237
42. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. (2018) 9(1):4383. doi: 10.1038/s41467-018-06318-7
43. Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, et al. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. (2005) 5(8):1793–800. doi: 10.1111/j.1600-6143.2005.00932.x
44. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. (2000) 6(4):422–8. doi: 10.1038/74680
45. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. (2014) 1843(11):2563–82. doi: 10.1016/j.bbamcr.2014.05.014
46. Richardson ET, Shukla S, Sweet DR, Wearsch PA, Tsichlis PN, Boom WH, et al. Toll-like receptor 2-dependent extracellular signal-regulated kinase signaling in Mycobacterium tuberculosis-infected macrophages drives anti-inflammatory responses and inhibits Th1 polarization of responding T cells. Infect Immun. (2015) 83(6):2242–54. doi: 10.1128/IAI.00135-15
47. Bouhamdan M, Bauerfeld C, Talreja J, Beuret L, Charron J, Samavati L. MEK1 dependent and independent ERK activation regulates IL-10 and IL-12 production in bone marrow derived macrophages. Cell Signal. (2015) 27(10):2068–76. doi: 10.1016/j.cellsig.2015.07.015
48. Ma X, Yan W, Zheng H, Du Q, Zhang L, Ban Y, et al. Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000Res. (2015) 4:1465. doi: 10.12688/f1000research.7010.1
49. Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. (2010) 10(3):170–81. doi: 10.1038/nri2711
50. Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. (2006) 58(11):621–31. doi: 10.1080/15216540600957438
51. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. (2013) 59(3):583–94. doi: 10.1016/j.jhep.2013.03.033
52. Chan ED, Winston BW, Jarpe MB, Wynes MW, Riches DW. Preferential activation of the p46 isoform of JNK/SAPK in mouse macrophages by TNF alpha. Proc Natl Acad Sci U S A. (1997) 94(24):13169–74. doi: 10.1073/pnas.94.24.13169
53. Yong HY, Koh MS, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs. (2009) 18(12):1893–905. doi: 10.1517/13543780903321490
54. Wu PK, Park JI. MEK1/2 inhibitors: molecular activity and resistance mechanisms. Semin Oncol. (2015) 42(6):849–62. doi: 10.1053/j.seminoncol.2015.09.023
55. Wu QN, Liao YF, Lu YX, Wang Y, Lu JH, Zeng ZL, et al. Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. (2018) 412:243–55. doi: 10.1016/j.canlet.2017.10.007
56. Wu Q, Wu W, Jacevic V, Franca TCC, Wang X, Kuca K. Selective inhibitors for JNK signalling: a potential targeted therapy in cancer. J Enzyme Inhib Med Chem. (2020) 35(1):574–83. doi: 10.1080/14756366.2020.1720013
57. Oldani G, Lacotte S, Morel P, Mentha G, Toso C. Orthotopic liver transplantation in rats. J Visualized Exp. (2012) (65):4143. doi: 10.3791/4143
Keywords: liver viability, warm ischemia time, cytokine, chemokine, machine perfusion
Citation: Kim J, Hong SK, Yang Y, Lee A, Hoffmeister KM, Gantner BN and Park JI (2023) Prolonged warm ischemia time increases mitogen-activated protein kinase activity and decreases perfusate cytokine levels in ex vivo rat liver machine perfusion. Front. Transplant. 2:1215182. doi: 10.3389/frtra.2023.1215182
Received: 1 May 2023; Accepted: 10 August 2023;
Published: 25 August 2023.
Edited by:
Sam Kesseli, Duke University Health System, United StatesReviewed by:
Laura Giuseppina Di Pasqua, University of Pavia, ItalyKeri E. Lunsford, The State University of New Jersey, United States
© 2023 Kim, Hong, Yang, Lee, Hoffmeister, Gantner and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joohyun Kim am9raW1AbWN3LmVkdQ==