
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Transplant. , 17 July 2023
Sec. Transplantation Immunology
Volume 2 - 2023 | https://doi.org/10.3389/frtra.2023.1182534
 Mark Dela Cruz1
Mark Dela Cruz1 Huaiying Lin2Jiho Han1Emerald Adler2Jaye Boissiere2Maryam Khalid2Ashley Sidebottom2
Huaiying Lin2Jiho Han1Emerald Adler2Jaye Boissiere2Maryam Khalid2Ashley Sidebottom2 Anitha Sundararajan2Christopher Lehmann3Angelica Moran4Matthew Odenwald5Matthew Stutz6
Anitha Sundararajan2Christopher Lehmann3Angelica Moran4Matthew Odenwald5Matthew Stutz6 Gene Kim1Sean Pinney1Valluvan Jeevanandam7Maria-Luisa Alegre8Eric Pamer2Ann B. Nguyen1*
Gene Kim1Sean Pinney1Valluvan Jeevanandam7Maria-Luisa Alegre8Eric Pamer2Ann B. Nguyen1*
Background: Emerging evidence is revealing the impact of the gut microbiome on hematopoietic and solid organ transplantation. Prior studies postulate that this influence is mediated by bioactive metabolites produced by gut-dwelling commensal bacteria. However, gut microbial metabolite production has not previously been measured among heart transplant (HT) recipients.
Methods: In order to investigate the potential influence of the gut microbiome and its metabolites on HT, we analyzed the composition and metabolite production of the fecal microbiome among 48 HT recipients at the time of HT.
Results: Compared to 20 healthy donors, HT recipients have significantly reduced alpha, i.e. within-sample, microbiota diversity, with significantly lower abundances of key anaerobic commensal bacteria and higher abundances of potentially pathogenic taxa that have been correlated with adverse outcomes in other forms of transplantation. HT recipients have a wide range of microbiota-derived fecal metabolite concentrations, with significantly reduced levels of immune modulatory metabolites such as short chain fatty acids and secondary bile acids compared to healthy donors. These differences were likely due to disease severity and prior antibiotic exposures but were not explained by other demographic or clinical factors.
Conclusions: Key potentially immune modulatory gut microbial metabolites are quantifiable and significantly reduced among HT recipients compared to healthy donors. Further study is needed to understand whether this wide range of gut microbial dysbiosis and metabolite alterations impact clinical outcomes and if they can be used as predictive biomarkers or manipulated to improve transplant outcomes.
The human gut microbiome is composed of over a thousand microbial species that reside within the lumen of the gastrointestinal tract (1, 2). Recent metagenomic and metabolomic analyses have revealed that interactions between microbes and their human hosts may have far-reaching impacts on human health (3, 4). Medical interventions, in particular antibiotic administration, can lead to loss or suppression of beneficial commensal microbes and their associated metabolites, resulting in unstable “dysbiotic” states.
Studies in hematopoietic stem cell (HSCT), kidney, and liver transplantation have shown associations between dysbiosis and adverse outcomes, including allograft rejection and death (5–8). Fewer studies have examined the role of the gut microbiome in heart transplantation (HT) outcomes. In pre-clinical murine models, changes to the gut microbiome have an impact on cardiac allograft survival (9–11). Prior studies have shown reduced gut microbial diversity among patients with heart failure (12–14). Furthermore, gut microbiome diversity decreases with worsening heart failure and persists even after HT (15).
The mechanism for this may be through the action of gut microbial metabolites on recipient immune systems. In vitro and animal studies have utilized metabolomic analysis to demonstrate how gut microbial-derived short chain fatty acids (SCFA) and secondary bile acids may be immunomodulatory, affecting post-transplant outcomes (16–21). Such metabolomic studies are limited in humans. Instead, human studies quantify butyrate-producing bacteria or the presence of genes that encode for metabolite production pathways and associate these to post-transplant clinical outcomes (8, 22–24). In so doing, such analyses link microbial metabolites to clinical outcomes without directly quantifying stool microbial metabolite concentrations. Only in limited studies of HSCT recipients have stool concentrations of gut microbial metabolites been directly measured with quantifiable results and correlated with adverse outcomes such as graft-vs.-host disease (25, 26).
To date, no studies have examined gut microbial production of immunomodulatory metabolites at the time of HT, when the recipient immune system is first introduced to alloantigens present in the cardiac allograft. To begin to understand the potential influence of the gut microbiome on alloimmunity during this crucial period, we aimed to characterize the gut microbiome and its metabolite production in the peri-HT period. We hypothesize that, compared to healthy donors, HT recipients will demonstrate gut dysbiosis and that such dysbiosis will result in reduced production of key, potentially immunomodulatory, gut microbial metabolites.
This was a prospective cohort study of adult HT recipients at a single institution from July 2020 to February 2021. Inclusion criteria were age ≥18 years, ability to provide informed consent, and active listing for HT, including multi-organ transplantation (heart-kidney, heart-liver, and heart-liver-kidney). Subjects were excluded if they were <18 years old, unable to consent, follow for ≥2 years, or provide a stool sample within 14 days of HT. A cohort of healthy donors were also recruited through the Duchossois Family Institute at the University of Chicago. The study was approved by the Institutional Review Board at our institution. Written informed consent was obtained from all participants.
Demographic and clinical data were collected through review of the medical record. These included particular factors that could impact the gut microbiome such as length of hospitalization prior to sample collection and antibiotic exposures in the 3 months pre-HT.
Fecal samples were collected at time of study enrollment from healthy donors and HT recipients. When able to be produced by the HT subjects, samples were collected within 14 days pre- and/or post-HT.
To prevent contamination, aseptic conditions were maintained during fecal sample aliquoting and collection. Samples were immediately stored at −80°C post collection and freeze-thaw cycles were avoided to conserve microbiome diversity and prevent contamination. A unilateral workflow was maintained through designated laboratory areas for pre- and post-PCR processing. Testing with appropriate negative controls was conducted to evaluate for contamination from the reagents used for library preparation.
Fecal samples underwent next generation shot-gun DNA sequencing. To minimize biases and optimize yield of both gram-positive and gram-negative organisms (27), mechanical disruptions with a bead beater (BioSpec Product) were conducted and samples were further purified with QIAamp mini spin columns (Qiagen). Enzymatic fragmentation during library preparation ensured consistent fragment lengths and PCR-free protocols reduced biases introduced by PCR cycles. Robust libraries were generated with 200 ng DNA input using a PCR-free DNA sequence kit (QiaSeq FX DNA library kit, Qiagen).
Inputs of starting material were normalized at every step of the workflow to reduce sampling size bias. Purified DNA was quantified with a Qubit 2.0 fluorometer. DNA input for library preparation were kept consistent at 200 ng. Prior to sequencing, libraries were quantified, their sizes were determined, and pooled at equimolar concentrations to ensure even read distribution across all samples.
Samples were then sequenced on the Illumina HiSeq platform, producing around 7–8 million PE reads per sample with read length of 150 bp. Adapters were trimmed off from the raw reads, and their quality was assessed and controlled using Trimmomatic (v.0.39) (28), then human genome was removed by kneaddata (v0.7.10, https://github.com/biobakery/kneaddata). Taxonomy was profiled using metaphlan4 (29).
All aspects of the next-generation sequencing workflow were automated to increase replicability and consistency across samples. Random spot checks of previously sequenced samples were conducted to ensure taxonomic profile consistency and validate data reproducibility.
Short chain fatty acids (SCFA; butyrate, acetate, propionate, succinate) were derivatized with pentafluorobenzyl bromide and analyzed via negative ion collision induced-gas chromatography-mass spectrometry (Agilent 8,890). Eight bile acids [primary: cholic acid; conjugated primary: glycocholic acid, taurocholic acid; secondary: deoxycholic acid, lithocholic acid (LCA), isodeoxycholic acid; modified secondary: alloisolithocholic acid (alloisoLCA) and 3-oxolithocholic acid (3-oxoLCA)] were quantified (µg/ml) by negative mode liquid chromatography-electrospray ionization-quadrupole time-of-flight-MS (Agilent 6,546).
Statistical analysis was conducted using R statistical language (v4.1.1). Continuous variables were compared using Wilcoxon rank-sum test, and p-values were adjusted for multiple comparisons by following the Benjamini-Hochberg method, as the majority of variables were not normally distributed. Categorical variables were compared using Chi-Square/Fisher’s Exact test. Kendall rank correlations were performed to determine associations between measured metabolites.
Alpha-diversity (a reflection of the number of unique bacterial taxa and their relative abundances) of fecal samples was estimated using Inverse Simpson Index.
Beta-diversity (compositional similarity between cohorts) analysis was performed with Uniform Manifold Approximation and Projection (UMAP), and difference in Bray-Curtis distances between groups were tested by Permutational Multivariate Analysis of Variance (PERMANOVA).
The linear discriminant analysis effect size (LEfSe) method was utilized to identify bacterial taxa more abundant within one cohort compared to another (30). The LEfSe algorithm uses the non-parametric Kruskal Wallis statistical test to compare all taxa at different taxonomic levels between groups, and paired Wilcoxon Rank Sum to test among subgroups. It then builds a linear discriminant analysis (LDA) model which utilizes continuous independent variables (e.g., bacterial abundance) to predict one dependent variable (e.g., healthy donors vs. HT recipients) and provides an effect size for the significantly different taxa (30). Higher LEfSe indicates that the dependent variable (e.g., healthy donors) has increased abundance of that specific microbial species compared to the other dependent variables (e.g., HT recipients).
To investigate the impact of clinical factors to the gut microbiome, additional analyses were conducted on HT recipients stratified based on transplant type (single vs. multi-organ), tertiles of alpha-diversity, and butyrate and bile acid production. Further analysis was conducted based on median pre-sample hospital length of stay and antibiotic exposure to high impact antibiotics within 3 months pre-HT. Adjusted p-values ≤0.05 were considered significant.
58 HT recipients were enrolled in the study, of whom 48 (33 HT, 15 multi-organ transplants) were able to produce stool samples for analysis. Demographic and clinical characteristics were compared to those of 20 healthy donors (Table 1). HT recipients were significantly older [57 (20–71) vs. 31.5 (18–63) years, p < 0.001], and were more likely to be male or black compared to healthy donors (Table 1).
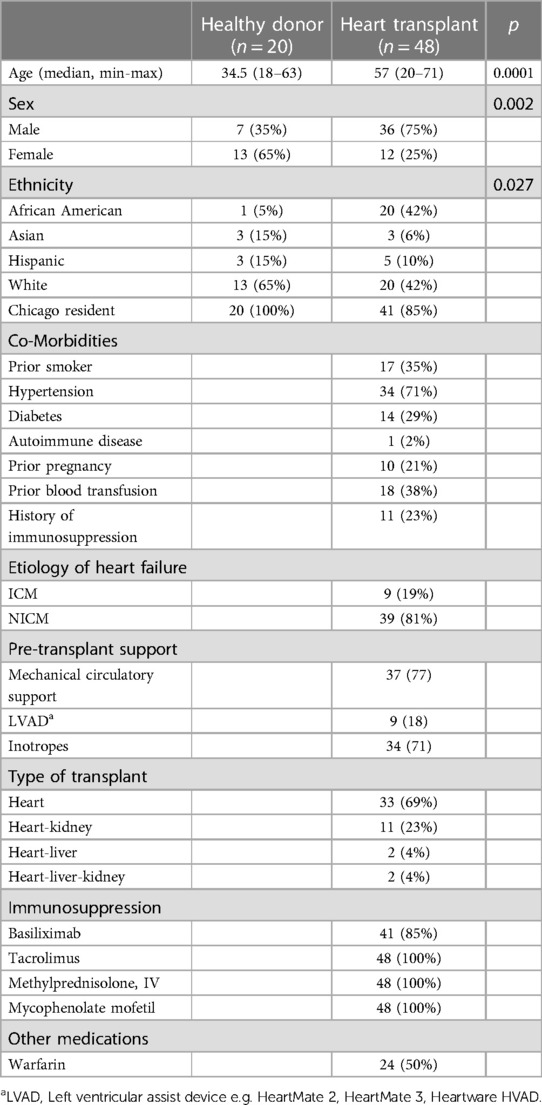
Table 1. Cohort demographic and clinical characteristics.
Clinical co-morbidities were only available for the HT cohort (Table 1), with the most common etiology of heart failure being non-ischemic cardiomyopathy (81%). The median pre-HT hospital length-of-stay was 16 days (range = 0–62 days). Thirty-seven patients (77%) were supported with mechanical circulatory support devices and 34 (71%) were on intravenous continuous inotropes prior to HT (Supplementary Table S1).
Twenty-five (52%) of HT recipients received antibiotic therapy within 3 months pre-HT (Supplementary Table S2). All HT recipients received prophylactic intravenous antibiotics per protocol, including cefazolin (unless already receiving another therapeutic dose of cephalosporin) and vancomycin (Figure 1).
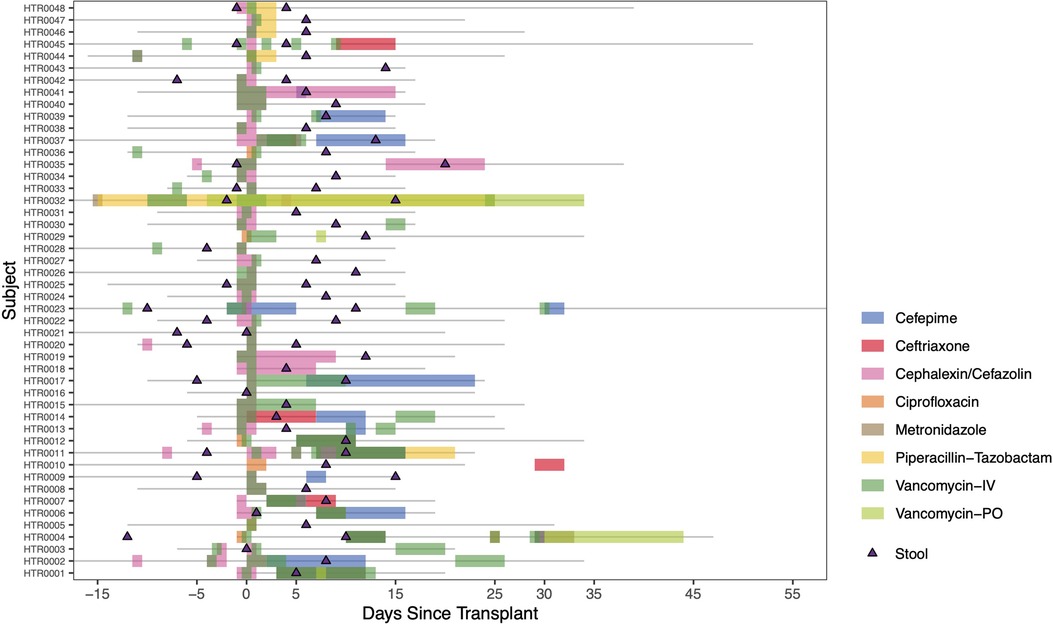
Figure 1. Heart transplant timelines. Individual timelines of admission duration (grey lines) and peri- and post-transplant antibiotic administration in reference to day of transplantation (Day 0) are shown here. Triangles indicate date of stool sample collections. Separate colors indicate notable antibiotic courses received during this timeframe.
Peri-operatively, 85% of HT recipients received induction with basiliximab, and 100% received high dose steroid therapy with methylprednisolone and anti-metabolite therapy with mycophenolate mofetil (Table 1). Calcineurin inhibitor therapy with tacrolimus was initiated within 24–72 h following transplantation at the discretion of the clinical team.
Each participant’s gut microbiome composition is depicted in Figure 2. Similar to previously published data on the normal human gut microbiome (3), the gut microbiome of healthy donors is diverse with a high relative abundance of commensal anaerobic bacterial taxa belonging to the phyla Bacteroidetes and Firmicutes, including the families Ruminococcaceae and Lachnospiraceae of the class Clostridia.
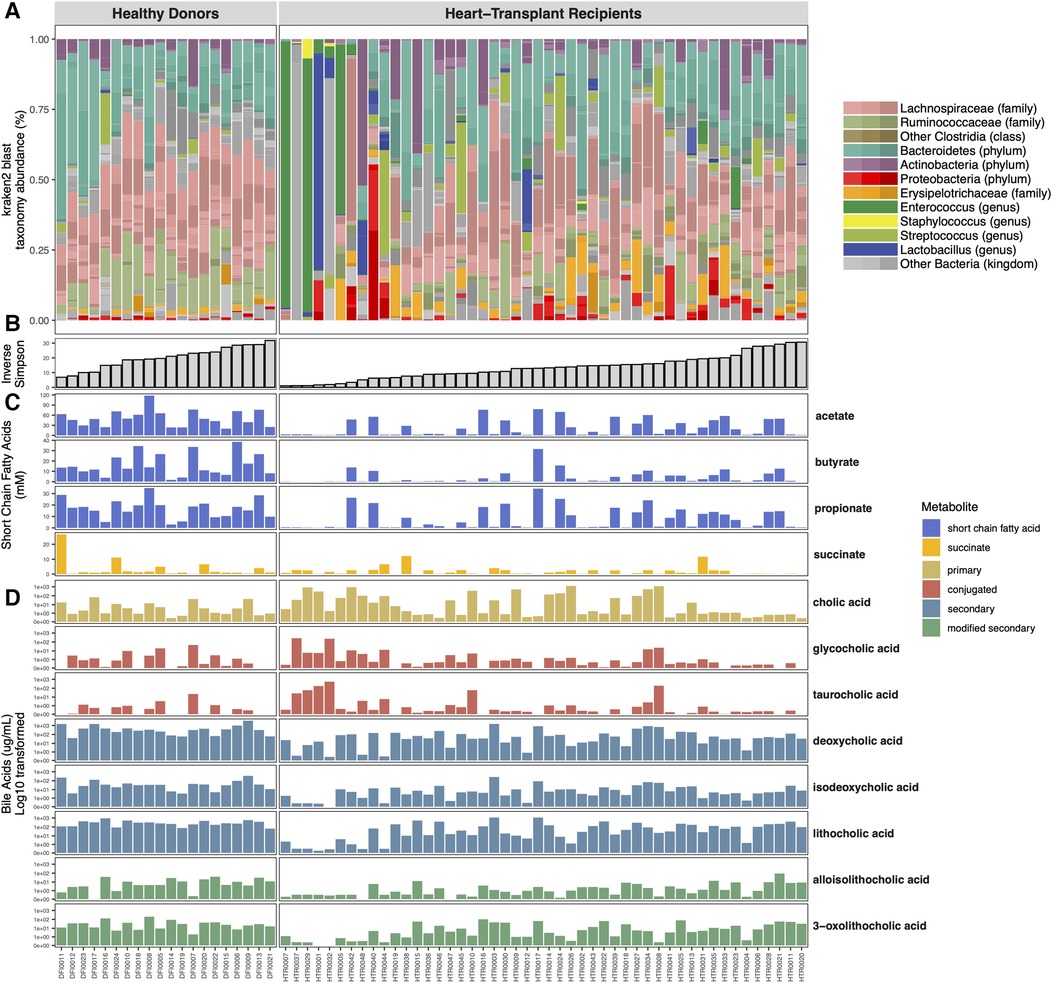
Figure 2. Shotgun metagenomic and metabolomic analysis of the microbiome. Vertical columns represent individual subjects and metagenomic sequencing data is color-coded with the relative abundance of specific bacterial taxa comprising their microbiome (A). Subjects are organized in ascending order of within-sample (alpha) diversity (B). Mass-spectrometry quantification of gut microbial metabolite levels demonstrates variable concentrations of bile acids (C) and short chain fatty acids within the fecal samples (D).
HT recipients also exhibit a wide range of gut microbial compositions. Many express a similar abundance of commensal anaerobic bacteria found among healthy donors. However, the microbiomes of some HT recipients were marked by loss of commensal anaerobic bacteria and consequent expansion of 1 or 2 bacterial genera, including Enterococcus, Enterocloster, Ligilactobacillus, Escherichia, and Klebsiella (Figure 2A). The gut dysbiosis among HT recipients is reflected in the significantly reduced alpha diversity in this cohort compared to healthy donors (Inverse Simpson 14.42 vs. 18.86, p = 0.05) (Figure 3A).
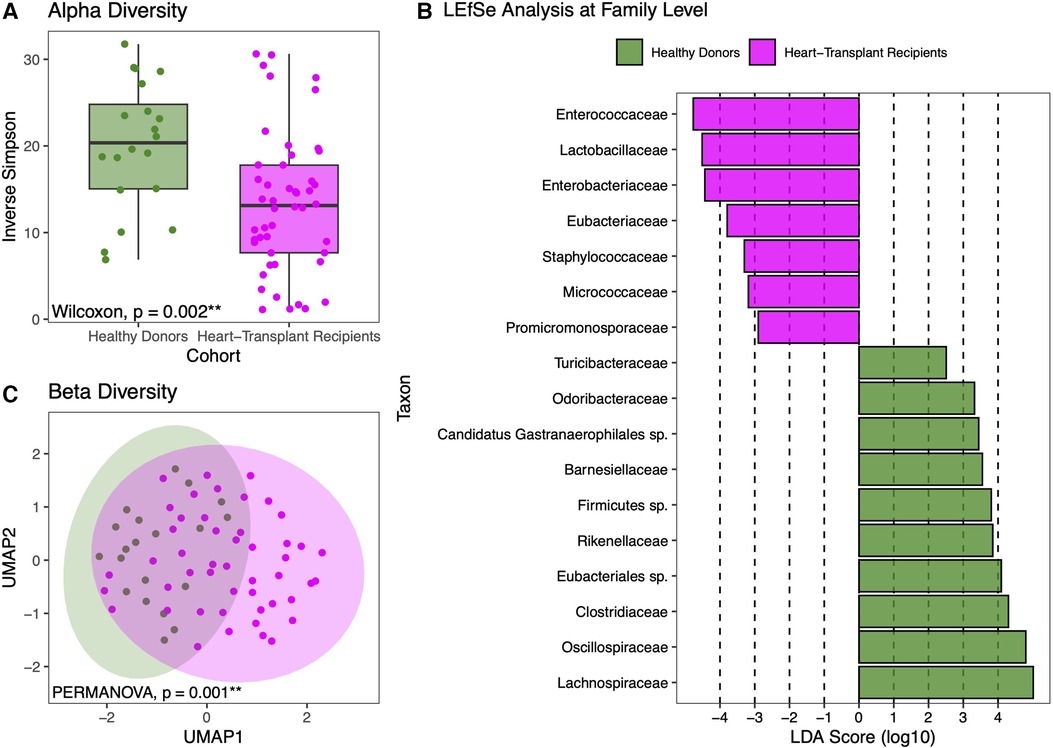
Figure 3. Diversity and compositional analysis. Healthy donors have more diversity in their individual samples than heart transplant recipients (A). LEfSe analysis showing statistically significant differences between the two cohorts at the family level (B). UMAP-based beta-diversity analysis further demonstrating that heart transplant recipients have gut microbiomes that are more compositionally similar to each other than to that of healthy donors, even after adjustment for age, sex, and race (C).
LEfSe analysis revealed that at the phylogenetic family level, HT recipients had higher abundances of Enterococcaceae, Lactobacillaceae, Enterobacteriaceae, Staphylococcaceae, Micrococcaceae, and Promicromonosporaceae (Figure 3B). To examine compositional differences between HT recipients and healthy donors, beta-diversity analysis was performed and revealed distinct clustering, even after adjusting for age, sex, and race, (PERMANOVA p = 0.001) (Figure 3C) indicating that the gut microbiome compositions of healthy donors are more similar to each other than to that of HT recipients.
Healthy donors exhibit marked differences in metabolite concentrations compared to HT recipients. Whereas healthy donors produce similar levels of SCFA between individuals, we observed a wide range of fecal SCFA concentrations among HT recipients (Figure 4A). HT recipients produce significantly lower median levels of fecal butyrate (0.695 vs. 13.04 mM, p < 0.001), acetate (4.98 vs. 48.98 mM, p < 0.001), and propionate (1.26 vs. 17.95 mM, p < 0.001) compared to healthy donors (Figure 4A). Consistent with previously published studies, individuals who express greater loss of normal commensal bacteria and domination with 1–2 bacterial taxa produce lower concentrations of fecal SCFA (31–34).
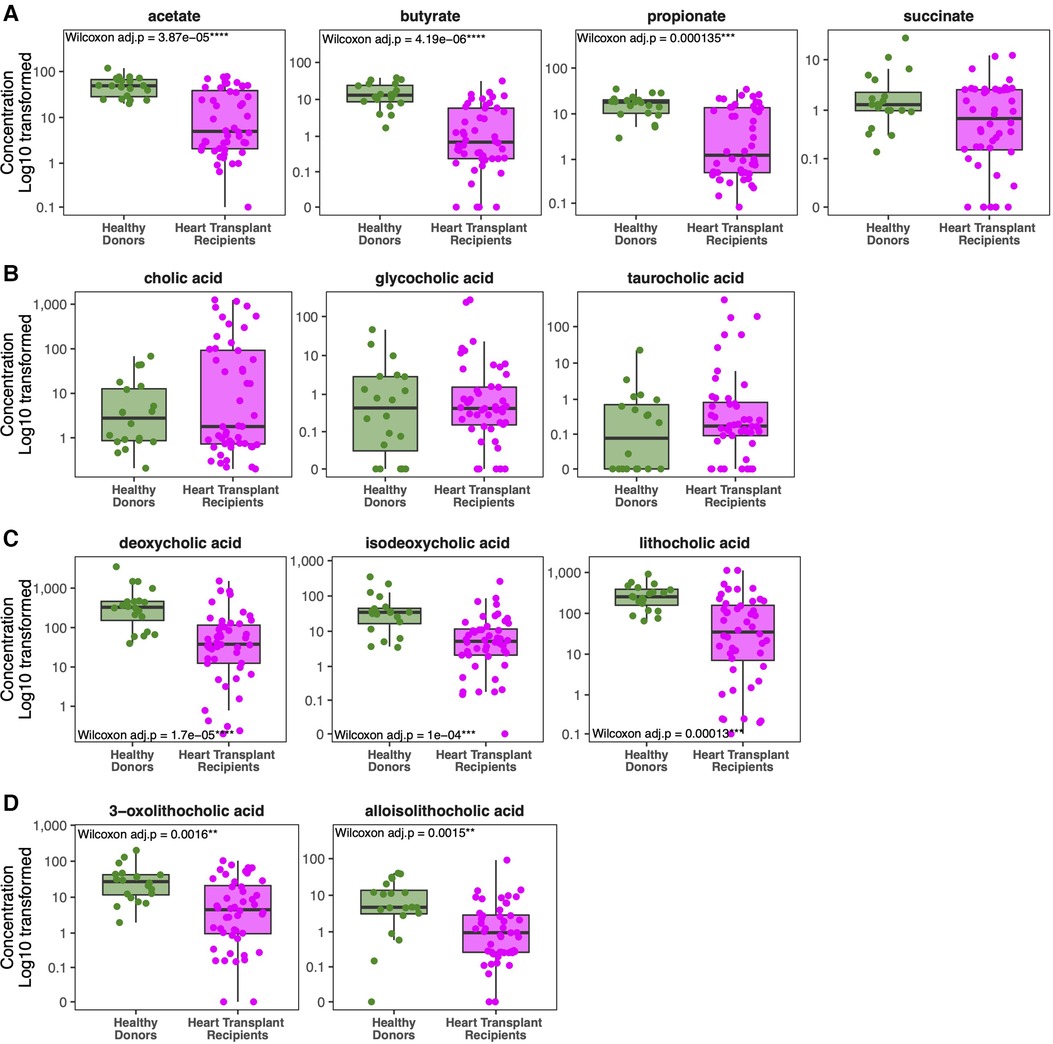
Figure 4. Comparison of metabolite concentrations between cohorts. Heart transplant recipients exhibit lower concentrations of short chain fatty acids (A) compared to healthy donors. Although they exhibit similar levels of primary and conjugated primary bile acids (B), heart transplant recipients exhibit lower levels of secondary bile acids (C) and modified secondary bile acids (D) compared to healthy donors.
While healthy donors generally have similar concentrations of fecal bile acids across individuals, there is significant inter-individual variability in secondary bile acid concentrations among HT recipients, as seen with SCFAs (Figure 4C–D). Patients with microbiota domination by non-obligate anaerobes have lower secondary bile acid concentrations. While concentrations of host-derived primary and conjugated bile acids are similar between HT recipients and healthy donors (Figure 4B), HT recipients have significantly lower median levels of the secondary bile acid LCA (34.99 vs. 254.01 μg/ml, p = 0.003), deoxycholic acid (37.80 vs. 327.50 μg/ml, p < 0.001), and isodeoxycholic acid (5.21 vs. 34.52 μg/ml, p = 0.002) compared to healthy donors (Figure 4C). Modified from LCA, alloisoLCA (0.95 vs. 4.71 μg/ml, p = 0.036) and 3-oxoLCA (4.51 vs. 27.20 μg/ml, p = 0.032) were also significantly lower among HT recipients compared to healthy donors (Figure 4D).
To determine whether there is concordance between SCFA and bile acid producers, Spearman correlations were performed between SCFA and bile acid levels (Figure 5). Although there are strong correlations between the concentrations of individual SCFA to other SCFA as well as secondary bile acids to other bile acids, SCFA are only moderately correlated to levels of either secondary or modified secondary bile acids. This suggests that factors resulting in elevated SCFA and bile acid levels are likely independent of each other.
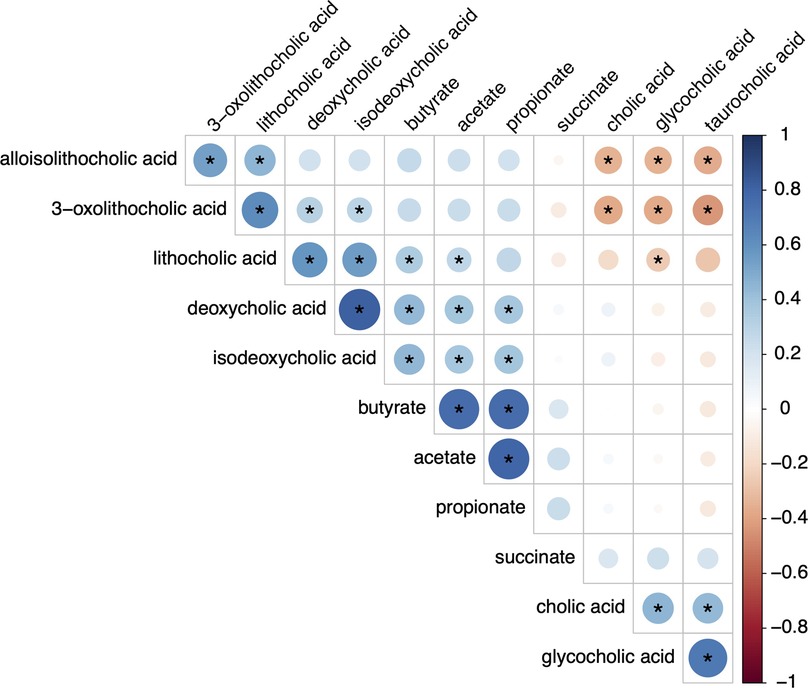
Figure 5. Spearman correlations of metabolites. This plot demonstrates correlations between the short chain fatty acids and bile acids. Blue indicates positive correlations and red indicates negative correlations. Deeper hues indicate stronger correlations with the (*) indicating significance.
To examine the potential clinical drivers of these compositional and metabolite differences, the HT cohort was divided into tertiles based on (1) inverse Simpson index, (2) butyrate production, (3) secondary bile acid production, and (4) modified secondary bile acid production. When divided into tertiles of low, intermediate, or high inverse Simpson index, there were no differences between groups in terms of age, sex, race, comorbidities, heart failure etiology, or other clinical factors (Supplementary Figure S1). Similarly, no differences were found between tertiles of secondary and modified secondary bile acid production. Analysis of butyrate revealed no association to demographic and comorbid factors.
To analyze the potential impacts of prolonged hospitalization prior to sample collection, a separate subgroup analysis based on median pre-sample length-of-stay was conducted. Subjects who experienced short hospitalization [median = 10, IQR (6, 14) days] were compared to long hospitalization [median = 22, IQR (18, 32) days]. There were no significant differences between the groups in terms of alpha or beta diversity (Supplementary Figure S2). Subjects with a long pre-HT hospitalization had significantly lower levels of LCA (67.6 vs. 251.0, p = 0.02) but exhibited no differences in SCFA and other secondary bile acid levels (Supplementary Figure S3).
To understand the impact of antibiotics on the gut microbiome, antibiotic exposures 3 months pre-sample were also analyzed (Supplementary Table S2). 52 of the 58 HT subjects were exposed to antibiotics within 7 days pre-sample and, thus, we focused our analysis on this timeframe. There were no significant differences in gut microbial diversity or metabolite production between HT recipients when exposures to all antibiotics were considered, 7 days pre-sample collection (Supplementary Figures S4, 5).
However, previous studies have demonstrated that certain “high impact” antibiotics drastically reduce the concentrations of healthy commensal anaerobic gut microbes (35–38). Of these, oral vancomycin, cefepime, piperacillin/tazobactam, metronidazole, and ciprofloxacin were observed in our cohort. 16 HT subjects were treated with one or more of these high impact antibiotics in the 7 days prior to sample production. These subjects had significantly lower within sample diversity (Figure 6A) compared to healthy donors and those who had not been exposed to these specific antibiotics (p = 0.0014). These subjects also had gut microbial compositions that were more similar to each other than to those who were not exposed to these antibiotics (Figure 6B, p = 0.001). The “high impact” antibiotic cohort consistently demonstrated significantly reduced metabolite levels of SCFA and all secondary and modified secondary bile acids, especially when compared to healthy donors (Figure 7).
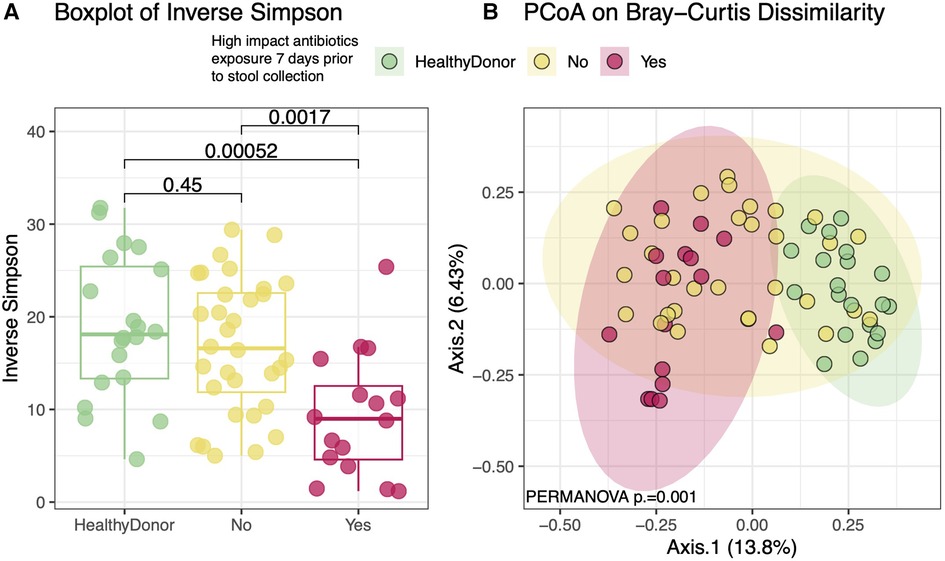
Figure 6. Diversity and compositional analysis between “high impact” vs No significant antibiotic exposure 7 days Pre-sample collection. Heart transplant recipients who were exposed to high impact antibiotics had significantly lower within-sample diversity when compared to either healthy donors or transplant recipients who were not exposed to such antibiotics (A). Heart transplant recipients who received any of the high impact antibiotics appear to have compositionally similar gut microbiomes that distinguish them from either healthy donors or those transplant recipients who did not receive these antibiotics (B). Of note, heart transplant recipients who were not exposed to these antibiotics were also more compositionally similar to each other than healthy donors (p = 0.001). High impact antibiotics = oral vancomycin, cefepime, piperacillin/tazobactam, metronidazole, and ciprofloxacin.
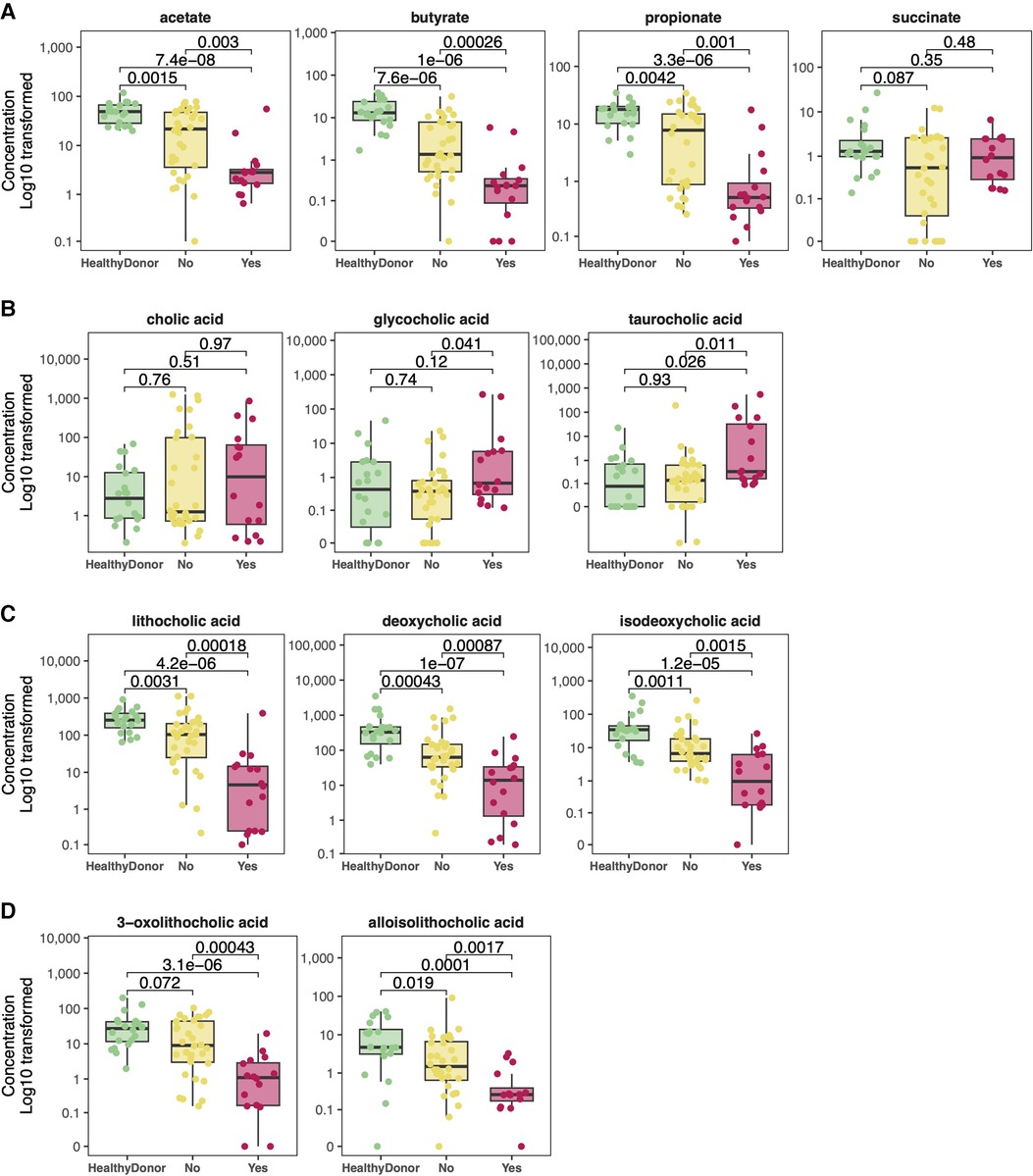
Figure 7. Comparison of metabolite concentrations by high impact antibiotic exposure. Significantly lower levels of short chain fatty acids (A), as well as secondary (C), and modified secondary bile acids (D) were noted amongst the recipients who had been exposed to high impact antibiotics as compared to either healthy donors or to those recipients who had not been exposed.
To analyze the impact of the peri-HT period on the gut microbiome, sequential pre- and post-HT samples from the same HT recipients were compared. Successful pre- and post-HT stool collection was achieved for 17 HT recipients, a median of 5 days before and 10 days after HT (Figure 1). There was no difference in alpha or beta diversity between pre- and post-HT samples (Supplementary Figure S6). There were also no significant differences in metabolite production between these two timepoints (Supplementary Figure S7).
Given the possible differences in comorbidities, illness severity, and antibiotic exposure, the entire cohort was stratified based on single vs. multi-organ transplant (11 heart-kidney, 2 heart-liver, and 2 heart-liver-kidney). Compared to the 33 heart-alone recipients, multi-organ transplant recipients had a similarly wide variability in gut microbial composition with a wide spectrum of alpha-diversity. Within-sample diversity was significantly different between all groups (Figure 6A). Multi-organ transplant recipients had the lowest alpha-diversity compared to healthy donors (10.9 vs. 20.0, p = 0.0012; Figure 8A). The three subgroups each had significantly increased abundance of specific bacterial taxa (Figure 8B). Metabolite analysis showed that SCFA and secondary bile acid production was overall similar between the single and multi-organ transplant recipients but remained lower than that of healthy donors (Supplementary Figure S8). Beta-diversity analysis showed that there was a significant difference in gut microbial composition between healthy donors and HT recipients but no significant difference between single and multi-organ transplant recipients (Figure 8C).
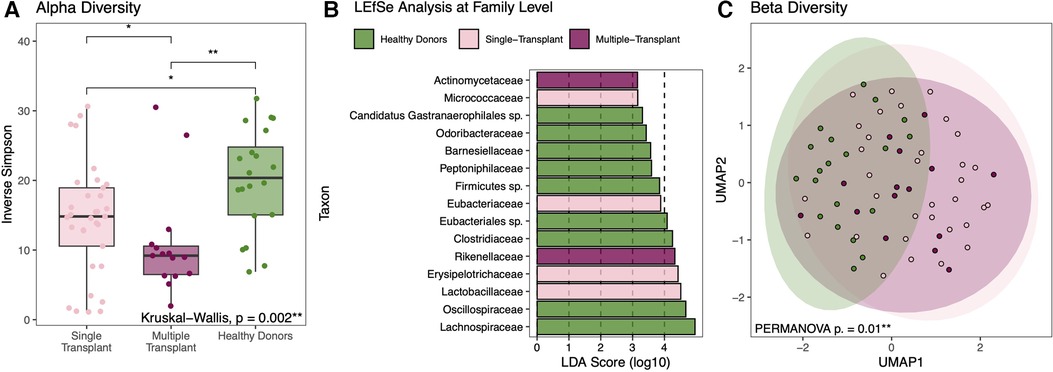
Figure 8. Diversity and compositional analysis between heart alone vs multi-organ transplant vs healthy donors. Although not significantly different when compared to heart-alone recipients, within-sample diversity is significantly lowest among multi-organ transplant recipients compared to healthy donors (A). Each group is marked by increased abundance of specific bacterial taxa with the phylogenetic family level selectively shown here (B). When comparing composition across groups, there is a significant degree of compositional differences between healthy donors and either transplant group but not between the single or multi-organ transplant recipients (C).
This study is the first to characterize the gut microbiome along with its bioactive byproducts among heart and multi-organ transplant recipients at the time of transplantation. To our knowledge, this is the first report directly measuring stool metabolites amongst solid organ transplant recipients. Our analysis adds to the growing body of evidence that, compared to healthy donors, the gut microbiome of HT recipients is marked by reduced within-sample diversity and increased dysbiosis. Such dysbiosis results in lower stool concentrations of key immunomodulatory gut microbial metabolites such as SCFA and secondary bile acids, when compared to levels in healthy donors. There is also a notable degree of inter-individual variability in gut microbial diversity that appears driven by heart failure illness severity and exposure to certain high impact antibiotics.
Heart failure has been associated with reduced within-sample or alpha diversity in a number of cohort studies (15, 39). The degree of dysbiosis appears to worsen with worsening heart failure (15, 40). Having the most severe forms of heart failure illness, our HT recipients unsurprisingly demonstrate significantly dysbiotic gut microbiomes. Overall disease severity appears to further these patterns as gut microbial dysbiosis appears particularly pronounced among multi-organ transplant recipients. Demographic factors and other comorbidities do not appear to impact this variability, contrary to what has been previously described (41). Nor do they appear to be significantly associated with pre-HT hospitalization length or the interventions administered in the peri-HT period.
It is likely that beyond illness severity, a primary driver of gut dysbiosis among HT recipients is exposure to high impact antibiotics. We demonstrate that even in the absence of such exposures, HT recipients have significantly reduced gut microbial diversity and metabolite production. Exposure to specific high impact antibiotics exacerbate these trends. As most of the exposures to antibiotics occurred within 7 days of sample production in our cohort, the duration with which these alterations persist warrants further study. Prior studies among healthy individuals have demonstrated high inter-individual variability in gut microbial recovery rates after antibiotic discontinuation, with some individuals experiencing changes that persist for months (38, 42). Whether HT recipients who have baseline gut dysbiosis are able to achieve similar levels of gut microbial recovery after exposure to high impact antibiotics remains unclear. Most importantly, whether these observations ultimately impact clinical HT outcomes through a gut microbial-dependent mechanism warrants further investigation.
Compared to healthy donors, our HT recipients have reduced alpha-diversity, partially due to significantly lower abundance of the phylum Firmicutes, including the species F. prausnitzii. F. prausnitzii serves as a marker of gut microbial health (43) and exerts anti-inflammatory effects, the loss of which may lead to a variety of inflammatory disease states (44–47). In post-liver transplant patients, low F. prausnitzii levels have been observed (48).
The reduced abundance of commensal anaerobic bacteria that we observe amongst HT recipients compared to healthy donors often coincides with the expansion of certain bacterial taxa, including known pathogens. In other forms of transplantation, similar patterns result in significant clinical outcomes. In allo-HSCT recipients, higher levels of Enterobacteriaceae were correlated to increased mortality (49). In one kidney transplant cohort, acute rejection within 90 days of transplantation was associated with higher relative abundance of Lactobacillales and Enterococcus (7). Similarly, liver transplant recipients with acute cellular rejection had increased abundance of Enterobacteriaceae, Streptococcaceae, and Bifidobacteriaeceae (6). Further study is needed to understand whether the strikingly similar patterns of dysbiosis among our HT recipients could result in similar clinical correlations.
The link between gut dysbiosis and transplantation outcomes may stem from the increasingly evident role of the gut microbiome on regulatory T cell (Treg) differentiation. Tregs may promote allograft tolerance by suppressing immune responses leading to rejection (50). Specific commensal species e.g., Clostridia (51, 52) and F. prausnitzii (44) or the presence of a stereotypical commensal anaerobic bacterial community (53, 54) appear to induce Treg maturation, likely through the impact of the metabolites they produce (55).
An important class of gut-derived immunomodulatory metabolites, SCFA are produced as gut microbes ferment dietary fiber. Of these, butyrate has been associated with reduced inflammation and oxidative stress (56) and increased Treg maturation (17). In mouse models of HSCT, increased butyrate has been associated with increased Treg populations (21). Exogenous butyrate administration in mice has been shown to mitigate graft-vs.-host disease in HSCT (21) and increase renal allograft survival, likely through a Treg-dependent mechanism (18). In our study, HT recipients have reduced abundance of Roseburia, Ruminococcus, and F. prausnitzii, some of the most potent butyrate-producing bacteria in the human colon (57). Consequently, we observe a wide range of butyrate production amongst our HT recipients, with many producing significantly less than healthy donors.
Bile acids have also emerged as an important mediator of gut microbial influence on adaptive immunity. Synthesized from cholesterol in the liver and then secreted into the gut to aid in digestion (58), primary bile acids that evade enterohepatic recirculation are metabolized solely by gut microbes into secondary bile acids and their derivatives (59, 60). Secondary bile acids such as LCA, and its derivatives 3-oxoLCA and isoalloLCA, have been shown to promote differentiation of Tregs (19). Hang et al. demonstrated that exogenous administration of 3-oxoLCA and isoalloLCA to mice increased Treg differentiation (19). Compared to healthy donors, our HT recipients had lower abundance of Clostridium spp, one of the main genera responsible for the modification of primary into secondary bile acids (61), and had lower levels of secondary bile acids.
Our study adds to the limited data demonstrating reduced gut microbial SCFA among patients with heart failure (62). We are also the first to describe reduced secondary bile acid concentrations in stool samples of patients with chronic heart failure. In the only other study directly measuring bile acids in chronic heart failure, Mayerhofer et al. demonstrated the opposite, citing an elevation of secondary bile acids in this population (63). The reasons for and implications of this discrepancy are unclear.
To our knowledge, direct measurements of stool gut microbial metabolite concentrations have not previously been performed among human solid organ transplant recipients. Without metabolomic analysis, prior studies in kidney and liver transplantation have inferred the impact of gut-derived metabolites based on surrogates such as quantification of known butyrate-producing bacteria or presence of genomic sequences with the potential to encode known metabolic pathways. In contrast, limited direct analyses of stool metabolite concentrations have been performed among HSCT recipients. Among HSCT, lower butyrate stool concentrations have been correlated to an increased risk for graft-vs.-host disease (26) and higher stool butyrate concentrations to an 80% reduction in lower respiratory tract infections (25). These studies highlight how metabolomic analyses may further our mechanistic understanding of the influence of the gut microbiome on outcomes of HT recipients.
Additionally, elevated SCFA and secondary bile acid levels are likely independent of each other. We found only moderate correlations between SCFA and secondary or their derived bile acid levels, consistent with previously published research indicating that the bacterial taxa responsible for producing SCFA or secondary bile acids are distinct (31, 33, 59). This may also be due to inter-individual differences in fiber intake or cholesterol metabolism. Understanding the gut microbial characteristics that can generate higher concentrations of these potentially beneficial metabolites may lead to novel treatments for transplant recipients.
Our results should be interpreted with significant limitations in mind. First, the small cohort size at a single center may limit its generalizability to other patient populations. Second, the limited follow-up period did not allow for exploration of the impact of the findings on clinical outcomes such as rejection or mortality. Further, we were not able to capture granular health-related comorbidities amongst our healthy donors. Potentially and most importantly, we were unable to account for the impact of health-related behaviors such as diet and other lifestyle factors on the gut microbiome. We aim to address these and many other issues with further recruitment for future analyses from this study.
In conclusion, we demonstrate that HT recipients have patterns of gut dysbiosis and metabolite production that distinguish them from healthy donors. They also have a significant degree of inter-individual variability in gut microbial composition which result in marked differences in measurable concentrations of metabolites. Many of these patterns have been associated with immune consequences that have been linked to adverse outcomes in other transplant populations. Larger and more longitudinal studies are necessary to understand the true nature of the interactions between the gut microbiome, its metabolites, and outcomes in HT. Gaining this knowledge may present a unique modality to improve the care of HT recipients.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject Number: PRJNA845905.
The studies involving human participants were reviewed and approved by Institutional Review Board of the University of Chicago. The patients/participants provided their written informed consent to participate in this study.
MD participated in the research design, performance of the research, data analysis, and the writing of the paper. HL participated in the research design, data analysis, and writing of the paper. JH participated in the writing of the paper. EA participated in the performance of the research and the writing of the paper. JB participated in the performance of the research and the writing of the paper. MK participated in the performance of the research and the writing of the paper. ASi participated in the research design, performance of the research, and the writing of the paper. ASu participated in the performance of the research and the writing of the paper. CL participated in the research design and the writing of the paper. AM participated in the research design and the writing of the paper. MO participated in the research design and the writing of the paper. MS participated in the research design and the writing of the paper. GK participated in the research design and the writing of the paper. SP participated in the research design and the writing of the paper. VJ participated in the research design and the writing of the paper. M-LA participated in the research design and the writing of the paper. EP participated in the research design, data analysis, and the writing of the paper. AN participated in the research design, data analysis, the performance of the research, and the writing of the paper. All authors contributed to the article and approved the submitted version.
Funding for this work was provided by the Duchossois Family Institute. MD was supported by NIH Grant 5T32HL007381-42. MO was supported by NIH Grant 5T32DK007074-48. MS was supported by NIH Grant 5T32HL007605.
We dedicate this to Eric Littmann whose contributions were invaluable to this project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frtra.2023.1182534/full#supplementary-material
HSCT, hematopoietic stem cell transplant; HT, heart transplant; LCA, lithocholic acid; LDA, linear discriminant analysis; LVAD, left ventricular assist device; PERMANOVA, permutational multivariate analysis of variance; SCFA, short chain fatty acid; Treg, regulatory T cells; UMAP, uniform manifold approximation and projection; VA-ECMO, veno-arterial extracorporeal mechanical oxygenation.
1. Marchesi JR, Adams DH, Fava F, Hermes GD, Hirschfield GM, Hold G, et al. The gut microbiota and host health: a new clinical frontier. Gut. (2016) 65(2):330–9. doi: 10.1136/gutjnl-2015-309990
2. Rajilić-Stojanović M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. (2014) 38(5):996–1047. doi: 10.1111/1574-6976.12075
3. The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486(7402):207–14. doi: 10.1038/nature11234
4. Tang WH, Kitai T, Hazen SL. Gut Microbiota in cardiovascular health and disease. Circ Res. (2017) 120(7):1183–96. doi: 10.1161/CIRCRESAHA.117.309715
5. Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N Engl J Med. (2020) 382(9):822–34. doi: 10.1056/NEJMoa1900623
6. Kato K, Nagao M, Miyamoto K, Oka K, Takahashi M, Yamamoto M, et al. Longitudinal analysis of the intestinal Microbiota in liver transplantation. Transplant Direct. (2017) 3(4):e144. doi: 10.1097/TXD.0000000000000661
7. Lee JR, Muthukumar T, Dadhania D, Toussaint NC, Ling L, Pamer E, et al. Gut microbial community structure and complications after kidney transplantation: a pilot study. Transplantation. (2014) 98(7):697–705. doi: 10.1097/TP.0000000000000370
8. Swarte JC, Li Y, Hu S, Björk JR, Gacesa R, Vich Vila A, et al. Gut microbiome dysbiosis is associated with increased mortality after solid organ transplantation. Sci Transl Med. (2022) 14(660):eabn7566. doi: 10.1126/scitranslmed.abn7566
9. Lei YM, Chen L, Wang Y, Stefka AT, Molinero LL, Theriault B, et al. The composition of the microbiota modulates allograft rejection. J Clin Invest. (2016) 126(7):2736–44. doi: 10.1172/JCI85295
10. Rey K, Manku S, Enns W, Van Rossum T, Bushell K, Morin RD, et al. Disruption of the Gut Microbiota with antibiotics exacerbates acute vascular rejection. Transplantation. (2018) 102(7):1085–95. doi: 10.1097/TP.0000000000002169
11. Bromberg JS, Hittle L, Xiong Y, Saxena V, Smyth EM, Li L, et al. Gut microbiota-dependent modulation of innate immunity and lymph node remodeling affects cardiac allograft outcomes. JCI Insight. (2018) 3(19):e121045. doi: 10.1172/jci.insight.121045
12. Luedde M, Winkler T, Heinsen FA, Rühlemann MC, Spehlmann ME, Bajrovic A, et al. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. (2017) 4(3):282–90. doi: 10.1002/ehf2.12155
13. Jennings DL, Bohn B, Zuver A, Onat D, Gaine M, Royzman E, et al. Gut microbial diversity, inflammation, and oxidative stress are associated with tacrolimus dosing requirements early after heart transplantation. PLoS One. (2020) 15(5):e0233646. doi: 10.1371/journal.pone.0233646
14. Yuzefpolskaya M, Bohn B, Javaid A, Mondellini GM, Braghieri L, Pinsino A, et al. Levels of trimethylamine N-oxide remain elevated long term after left ventricular assist device and heart transplantation and are independent from measures of inflammation and gut dysbiosis. Circ Heart Fail. (2021) 14(6):e007909. doi: 10.1161/CIRCHEARTFAILURE.120.007909
15. Yuzefpolskaya M, Bohn B, Nasiri M, Zuver AM, Onat DD, Royzman EA, et al. Gut microbiota, endotoxemia, inflammation, and oxidative stress in patients with heart failure, left ventricular assist device, and transplant. J Heart Lung Transplant. (2020) 39(9):880–90. doi: 10.1016/j.healun.2020.02.004
16. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science. (2013) 341(6145):569–73. doi: 10.1126/science.1241165
17. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504(7480):446–50. doi: 10.1038/nature12721
18. Wu H, Singer J, Kwan TK, Loh YW, Wang C, Tan J, et al. Gut microbial metabolites induce donor-specific tolerance of kidney allografts through induction of T regulatory cells by short-chain fatty acids. J Am Soc Nephrol. (2020) 31(7):1445–61. doi: 10.1681/ASN.2019080852
19. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T. Nature. (2019) 576(7785):143–8. doi: 10.1038/s41586-019-1785-z
20. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature. (2020) 581(7809):475–9. doi: 10.1038/s41586-020-2193-0
21. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. (2016) 17(5):505–13. doi: 10.1038/ni.3400
22. Lee JR, Huang J, Magruder M, Zhang LT, Gong C, Sholi AN, et al. Butyrate-producing gut bacteria and viral infections in kidney transplant recipients: a pilot study. Transpl Infect Dis. (2019) 21(6):e13180. doi: 10.1111/tid.13180
23. Michonneau D, Latis E, Curis E, Dubouchet L, Ramamoorthy S, Ingram B, et al. Metabolomics analysis of human acute graft-versus-host disease reveals changes in host and microbiota-derived metabolites. Nat Commun. (2019) 10(1):5695. doi: 10.1038/s41467-019-13498-3
24. Meedt E, Hiergeist A, Gessner A, Dettmer K, Liebisch G, Ghimire S, et al. Prolonged suppression of butyrate-producing Bacteria is associated with acute gastrointestinal graft-vs-host disease and transplantation-related mortality after allogeneic stem cell transplantation. Clin Infect Dis. (2022) 74(4):614–21. doi: 10.1093/cid/ciab500
25. Haak BW, Littmann ER, Chaubard JL, Pickard AJ, Fontana E, Adhi F, et al. Impact of gut colonization with butyrate-producing microbiota on respiratory viral infection following allo-HCT. Blood. (2018) 131(26):2978–86. doi: 10.1182/blood-2018-01-828996
26. Romick-Rosendale LE, Haslam DB, Lane A, Denson L, Lake K, Wilkey A, et al. Antibiotic exposure and reduced short chain fatty acid production after hematopoietic stem cell transplant. Biol Blood Marrow Transplant. (2018) 24(12):2418–24. doi: 10.1016/j.bbmt.2018.07.030
27. Guo F, Zhang T. Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl Microbiol Biotechnol. (2013) 97(10):4607–16. doi: 10.1007/s00253-012-4244-4
28. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
29. Blanco-Miguez A, Beghini F, Cumbo F, McIver LJ, Thompson KN, Zolfo M, et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species with MetaPhlAn 4. bioRxiv (2022):2022.08.22.504593. doi: 10.1101/2022.08.22.504593
30. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12(6):2011–12. doi: 10.1186/gb-2011-12-6-r60
31. Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol. (2010) 12(2):304–14. doi: 10.1111/j.1462-2920.2009.02066.x
32. Fu X, Liu Z, Zhu C, Mou H, Kong Q. Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria. Crit Rev Food Sci Nutr. (2019) 59(sup1):S130–S52. doi: 10.1080/10408398.2018.1542587
33. Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. (2009) 294(1):1–8. doi: 10.1111/j.1574-6968.2009.01514.x
34. Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, et al. Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem. (2010) 285(29):22082–90. doi: 10.1074/jbc.M110.117713
35. Stein-Thoeringer CK, Saini NY, Zamir E, Blumenberg V, Schubert ML, Mor U, et al. A non-antibiotic-disrupted gut microbiome is associated with clinical responses to CD19-CAR-T cell cancer immunotherapy. Nat Med. (2023) 29(4):906–16. doi: 10.1038/s41591-023-02234-6
36. Bhalodi AA, van Engelen TSR, Virk HS, Wiersinga WJ. Impact of antimicrobial therapy on the gut microbiome. J Antimicrob Chemother. (2019) 74(Suppl 1):i6–i15. doi: 10.1093/jac/dky530
37. Kumari R, Yadav Y, Misra R, Das U, Das Adhikari U, Malakar P, et al. Emerging frontiers of antibiotics use and their impacts on the human gut microbiome. Microbiol Res. (2022) 263: 127127. doi: 10.1016/j.micres.2022.127127
38. Isaac S, Scher JU, Djukovic A, Jiménez N, Littman DR, Abramson SB, et al. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother. (2017) 72(1):128–36. doi: 10.1093/jac/dkw383
39. Kummen M, Mayerhofer CCK, Vestad B, Broch K, Awoyemi A, Storm-Larsen C, et al. Gut Microbiota signature in heart failure defined from profiling of 2 independent cohorts. J Am Coll Cardiol. (2018) 71(10):1184–6. doi: 10.1016/j.jacc.2017.12.057
40. Mayerhofer CCK, Kummen M, Holm K, Broch K, Awoyemi A, Vestad B, et al. Low fibre intake is associated with gut microbiota alterations in chronic heart failure. ESC Heart Fail. (2020) 7(2):456–66. doi: 10.1002/ehf2.12596
41. Redondo-Useros N, Nova E, González-Zancada N, Díaz LE, Gómez-Martínez S, Marcos A. Microbiota and lifestyle: a special focus on diet. Nutrients. (2020) 12(6):1776. doi: 10.3390/nu12061776
42. Rashid MU, Zaura E, Buijs MJ, Keijser BJ, Crielaard W, Nord CE, et al. Determining the long-term effect of antibiotic administration on the human normal intestinal Microbiota using culture and pyrosequencing methods. Clin Infect Dis. (2015) 60(Suppl 2)):S77–84. doi: 10.1093/cid/civ137
43. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. (2011) 473(7346):174–80. doi: 10.1038/nature09944
44. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. (2008) 105(43):16731–6. doi: 10.1073/pnas.0804812105
45. Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. (2013) 498(7452):99–103. doi: 10.1038/nature12198
46. Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. (2011) 6(1):0016393. doi: 10.1371/journal.pone.0016393
47. Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H, et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. (2013) 16(3):255–61. doi: 10.1016/j.mib.2013.06.003
48. Wu ZW, Ling ZX, Lu HF, Zuo J, Sheng JF, Zheng SS, et al. Changes of gut bacteria and immune parameters in liver transplant recipients. Hepatobiliary Pancreat Dis Int. (2012) 11(1):40–50. doi: 10.1016/S1499-3872(11)60124-0
49. Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. (2014) 124(7):1174–82. doi: 10.1182/blood-2014-02-554725
50. McMurchy AN, Bushell A, Levings MK, Wood KJ. Moving to tolerance: clinical application of T regulatory cells. Semin Immunol. (2011) 23(4):304–13. doi: 10.1016/j.smim.2011.04.001
51. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. (2013) 500(7461):232–6. doi: 10.1038/nature12331
52. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. (2011) 331(6015):337–41. doi: 10.1126/science.1198469
53. Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med. (2014) 6(220):3008051. doi: 10.1126/scitranslmed.3008051
54. Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. (2011) 34(5):794–806. doi: 10.1016/j.immuni.2011.03.021
55. Bartman C, Chong AS, Alegre ML. The influence of the microbiota on the immune response to transplantation. Curr Opin Organ Transplant. (2015) 20(1):1–7. doi: 10.1097/MOT.0000000000000150
56. Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. (2011) 17(12):1519–28. doi: 10.3748/wjg.v17.i12.1519
57. Flint HJ, Duncan SH, Scott KP, Louis P. Links between diet, gut microbiota composition and gut metabolism. Proc Nutr Soc. (2015) 74(1):13–22. doi: 10.1017/S0029665114001463
58. Fiorucci S, Distrutti E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol Med. (2015) 21(11):702–14. doi: 10.1016/j.molmed.2015.09.001
59. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. (2014) 30(3):332–8. doi: 10.1097/MOG.0000000000000057
60. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. (2006) 47(2):241–59. doi: 10.1194/jlr.R500013-JLR200
61. Kriaa A, Bourgin M, Potiron A, Mkaouar H, Jablaoui A, Gérard P, et al. Microbial impact on cholesterol and bile acid metabolism: current status and future prospects. J Lipid Res. (2019) 60(2):323–32. doi: 10.1194/jlr.R088989
62. Cui X, Ye L, Li J, Jin L, Wang W, Li S, et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep. (2018) 8(1):635. doi: 10.1038/s41598-017-18756-2
Keywords: gut microbiome, heart transplant, gut microbial metabolites, short chain fatty acids, secondary bile acid, gut microbial diversity
Citation: Dela Cruz M, Lin H, Han J, Adler E, Boissiere J, Khalid M, Sidebottom A, Sundararajan A, Lehmann C, Moran A, Odenwald M, Stutz M, Kim G, Pinney S, Jeevanandam V, Alegre M-L, Pamer E and Nguyen AB (2023) Reduced immunomodulatory metabolite concentrations in peri-transplant fecal samples from heart allograft recipients. Front. Transplant. 2:1182534. doi: 10.3389/frtra.2023.1182534
Received: 8 March 2023; Accepted: 30 June 2023;
Published: 17 July 2023.
Edited by:
Marilia Cascalho, University of Michigan, United StatesReviewed by:
Naoka Murakami, Harvard Medical School, United States© 2023 Dela Cruz, Lin, Han, Adler, Boissiere, Khalid, Sidebottom, Sundararajan, Lehmann, Moran, Odenwald, Stutz, Kim, Pinney, Jeevanandam, Alegre, Pamer and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ann B. Nguyen YW5ndXllbjRAdWNoaWNhZ28uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.