 Alec Vallota-Eastman1,2
Alec Vallota-Eastman1,2 Cynthia Bui1,3
Cynthia Bui1,3 Philip M. Williams4
Philip M. Williams4 David L. Valentine2
David L. Valentine2 David Loftus5
David Loftus5 Lynn Rothschild1,3*
Lynn Rothschild1,3*- 1Space Science and Astrobiology Division, NASA Ames Research Center, Moffett Field, CA, United States
- 2Department of Marine Science, University of California, Santa Barbara, Santa Barbara, CA, United States
- 3Department of Molecular Biology, Cell Biology, and Biochemistry, Brown University, Providence, RI, United States
- 4Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, Nottingham, United Kingdom
- 5Space Biosciences Division, NASA Ames Research Center, Moffett Field, CA, United States
Biologics, such as pharmaceutical peptides, have notoriously short shelf lives, insufficient for long-duration space flight missions to the Moon or Mars. To enable the sustainable presence of humans on the Moon or Mars, we must develop methods for on-site production of pharmaceutical peptides in space, a concept we call the Astropharmacy. Here, we present a proof-of-concept for the first step needed: a low-mass system for pharmaceutical production designed to be stable in space. To demonstrate feasibility, we engineered strains of the space-hardy spore-forming bacterium, Bacillus subtilis, to secrete two pharmaceutical peptides important for astronaut health: teriparatide (an anabolic agent for combating osteoporosis) and filgrastim (an effective countermeasure for radiation-induced neutropenia). We found that the secretion peptides from the walM and yoqH genes of B. subtilis worked well for secreting teriparatide and filgrastim, respectively. In consideration of the Translational Research Institute for Space Health (TRISH) challenge to produce a dose equivalent in 24 h, dried spores of our engineered strains were used to produce 1 dose equivalent of teriparatide from a 2 mL culture and 1 dose equivalent of filgrastim from 52 mL of culture in 24 h. Further optimization of strain growth conditions, expression conditions, and promoter sequences should allow for higher production rates to be achieved. These strains provide the template for future optimization efforts and address the first step in the Astropharmacy, capable of on-site production, purification, and processing of biopharmaceutical compounds in platforms amenable for use in space.
1 Introduction
As the National Aeronautics and Space Administration (NASA) prepares for long duration missions to the Moon and beyond, it must also prepare for the reality that astronauts will get sick hundreds of thousands of miles away from the nearest pharmacy. The Artemis Program—involving NASA, the European Space Agency (ESA), the Japanese Aerospace Exploration Agency and the Canadian Space Agency—seeks to re-establish a human presence on the Moon, and seeks to establish a basecamp to facilitate the human exploration of Mars and other deep space destinations (Batcha et al., 2020; NASA, 2020). For short duration missions of up to a few months, or those where a quick return to Earth is possible, it is feasible for astronauts to take all the necessary medications they may need with them. The International Space Station (ISS) has “Med Kits” for this reason, which include small supplies of painkillers, antibiotics, antiemetics, etc. (Medical Checklist, 2006). If needed, an ISS medical emergency can be dealt with by aborting the mission and having the crew return to Earth for recovery. However, the consideration of pharmaceutical availability becomes even more important for the multi-year missions that will be required for Mars, as speedy re-supply and/or evacuation missions are not possible. Reliance on pharmaceuticals is particularly important in space since invasive procedures, such as surgeries, are exceptionally challenging during flight (Blue et al., 2019). As such, it will be necessary to build the technological capability for on-site production of critical pharmaceuticals during the duration of the Artemis program and in preparation for human Mars missions.
Relying on pharmaceuticals launched with the crew is currently not possible for a planetary mission (Blue et al., 2019) for the following reasons. First, the shelf life of many pharmaceuticals is limited (Lyon et al., 2006). Of medications flown on ISS, 87% have shelf lives of fewer than 24 months. Shelf life in space may be even shorter because of degradation caused by exposure to the space environment (Kast et al., 2017). This problem becomes even greater when considering peptide pharmaceuticals, known as biologics. Biologics play a critical role in treating many of the medical conditions that astronauts are known to, or could likely, face. Unfortunately, biologics degrade in 6 months or less, even with refrigeration, further highlighting the problem of insufficient shelf-life for a Mars mission. A second problem is that the necessary range of medication variety, dosage, and delivery forms increases as the period of time away from Earth-bound pharmacies increases. This in-turn leads to a greater up-mass requirement. An ESA report from Berry et al. (2002) calculated the probability of a medical trauma as 0.06 per person per year. Our inability to store, transport and deliver pharmacologically-active biomolecules such as vaccines, antibodies, and other medications will limit long-term human missions. This has been recognized by NASA (NASA Technology Roadmaps - TA 6: Human Health, Life Support, and Habitation Systems, 2015). Additionally, NASA’s Human Research Roadmap identifies “Risk of Ineffective or Toxic Medications Due to Long Term Storage” as a major risk. The use of medicines in space flight also varies, since the way the body processes them is altered (Williams et al., 2003), as is their efficacy (Taylor, 2015). If we are to meet mission objectives of long-duration human missions including the exploration of Mars (Denis et al., 2020), and beyond, a new paradigm of pharmaceutical provision is required. A true paradigm shift would be to use life as a means of on-site production of any pharmaceutical an astronaut may need.
To fill this vital need, we have proposed the development of an “Astropharmacy,” a compact low-mass on-demand drug production system. Our concept consists of four parts: drug synthesis, purification, testing and administration. The first step will require a means of stable production, one that is low-mass, stable, and with reduced stringency for storage conditions. The second step will be to engineer a small and user-friendly device for purification and testing of the drug. The final step will be to design a system for processing the drug to be ready for the required delivery form (e.g., intravenous, oral, dermal).
We chose Bacillus subtilis as the chassis organism for our proof of concept by engineering it to secrete filgrastim and teriparatide. While cell-free systems for peptide production were considered, a cell-based system was chosen at this point due to its intrinsic stability in space conditions (Zhang et al., 2020). Bacillus subtilis has long stood out as a promising chassis organism because B. subtilis endospores have been shown to survive the vacuum of space for nearly 6 years on NASA’s Long Duration Exposure Facility (LDEF) mission (Horneck, 1993). The endospores are also able to survive long periods of desiccation, enabling storage of nearly massless dried spores (0.7 μg per million spores (Tisa et al., 1982; Cliff et al., 2005)) at room temperature. This “microbial astronaut” was also tested for its capabilities during the PowerCell payload experiment on Eu:CROPIS (Euglena and Combined Regenerative Organic-Food Production in Space) where it was successfully transformed with a selective plasmid in space (McCutcheon et al., 2016). In addition to its innate resistance to the hostile space environment, B. subtilis stands out in industrial settings for its ability to secrete various value-added products rather than retaining them inside the cell (van Dijl and Hecker, 2013; Liu et al., 2017; Gu et al., 2018; Zhang et al., 2020, 2021). Its robust secretory system means that cell lysis is not necessary to harvest the product (Kakeshita et al., 2012; Su et al., 2020), which makes purification easier. However, the relationship of secretion tag choice to the type of protein one would like to produce is still poorly understood and a single tag cannot be used to secrete different heterologous proteins (Brockmeier et al., 2006). As a result, each peptide or protein one attempts to synthesize must be screened with all known signal peptides native to the B. subtilis genome (Yang et al., 2021). This is why we chose to evaluate a signal peptide library for secretion of our chosen therapeutic targets.
B. subtilis has long been used to express therapeutic proteins used to treat conditions ranging from diabetes to cystic fibrosis. Some examples of proteins that have been produced using B. subtilis include Human insulin, antibody fragments (Fabs), alpha-amylase and neutral protease, Human growth hormone, asparaginase, and lipase (Westers et al., 2004; Yoshida et al., 2006; Degering et al., 2010; Bento et al., 2022). Teriparatide and filgrastim are two important biologics, particularly for the perils of space flight (Figure 1). These non-glycosylated peptides, stand out as the first therapeutic targets for on-site production due to their short shelf-life and easier route to synthesis. As proteins, they can be synthesized by cells such as bacteria. Further, native secretion mechanisms that cells use to transport proteins outside of the cytoplasm can be taken advantage of to produce extracellular biologics for ease of on-site separation from cell culture and purification.
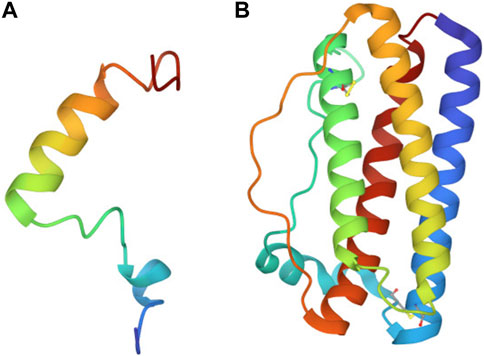
FIGURE 1. Ribbon diagram of teriparatide and filgrastim. (A) Folded structure of 34 amino acid teriparatide. (B) Folded structure of 175 amino acid filgrastim.
Teriparatide, also known as recombinant human pyrathyroid hormone (1-34) (hPTH [1-34]) is the biologically active moiety of mineral homeostasis of the native full-length hormone hPTH (1-84) (Quattrocchi and Kourlas, 2004). This single chain polypeptide is a new United States Food and Drug Administration (FDA)-approved biologic that increases bone mineralization and density. This agent could be used to counter the bone loss seen in space under reduced gravity conditions (Gabel et al., 2022). A single dose of teriparatide is 20 μg of the compound (Forteo, 2010).
Filgrastim, the name for human recombinant granulocyte colony stimulating factor (hG-CSF), is approved by the FDA and is now being used in the clinic to treat low-neutrophil blood count, a symptom of radiation toxicity. It is mostly used on Earth for improving radiation sickness in chemotherapy patients (Wright et al., 2017). Radiation exposure remains one of the most pressing issues in astrophysiology today (Chancellor et al., 2014). In the absence of Earth’s atmosphere, astronauts are exposed to a variety of radioactive particles, many of which cannot be effectively shielded by current shielding approaches. Astronauts on a 6 month-duration mission on ISS are exposed to approximately 75 millisieverts (mSv) of radiation–the equivalent radiation exposure of 375 chest x-rays (Hodkinson et al., 2017). Humans on Earth’s surface average 2.2 mSv of radiation per year (Hodkinson et al., 2017). By contrast, a 3-year mission to Mars, will expose astronauts to low-dose galactic cosmic rays, leading to radiation doses more than 13 times greater than astronauts experience on the ISS on average (Cucinotta and Durante, 2006; Afshinnekoo et al., 2020). This 1 Sv exposure is the equivalent of 5,000 chest x-rays. Notable known risks of excessive and/or prolonged exposure to radiation include cancer and radiation sickness. Filgrastim could be used to support the hematopoietic system by helping to restore neutrophil counts in the event of an acute radiation exposure from a solar particle event (especially if a crew member were to be un-shielded, on EVA). A single dose of G-CSF is 300 μg and it needs to be given once a day (Appelbaum, 1989).
The scope of this paper was to develop a proof of concept for the first step in this four-step process (cell-based biological production) while keeping in mind constraints imposed by the purification and processing steps that will be required in the future. In this proof of concept we screen a wide variety of B. subtilis secretion peptides for their ability to secrete filgrastim and teriparatide. We then modified the B subtilis genome to include the best-performing construct under a high constitutive expression promoter. Finally, as we were inspired by NASA’s Translational Research Institute for Space Health (TRISH) program (Mars, 2017), we set out to achieve TRISH’s goal of a production rate of at least one dose equivalent of each pharmaceutical peptide within 24 h.
2 Materials and methods
2.1 Bacterial growth
Bacterial strains used in all aspects of this project were E. coli HST08 Stellar™ Competent Cells and B. subtilis RIK1285 (A B. subtilis strain 168 derivative: trpC2, ys1, aprEdelta3, nprR2, nprE18) (TAKARA Bio Inc., San Jose, CA). Growth for both E. coli and B. subtilis was carried out at 37°C in Luria Broth (LB) containing selective antibiotics with shaking at 200 rpm unless stated otherwise. For solid media, LB plates containing 1.5% agar were used. For pBE-S-based constructs, ampicillin (Amp) [100 μg/mL] was used for plasmid propagation in E. coli and kanamycin (Km) [10 μg/mL] was used for plasmid propagation in B. subtilis. For pECE321-based plasmid propagation, ampicillin [100 μg/mL] was used for plasmid propagation in E. coli and streptomycin [100 μg/mL] was used for selecting for genomic insertions in B. subtilis. To select for double-cross over events, erythromycin [1 μg/mL] was used as a counterselection by cross patching on solid media. Difco Sporulation Medium (DSM) was prepared for spore formation, according to the recipe described by Munakata et al. (1991). DSM composition: Bacto Nutrient Broth (8g), 10% (w/v) KCl (10 mL), 1.2% (w/v) MgSO4.7H2O (10 mL), 1 M NaOH (∼1.5 mL or to pH 7.6), prepared with deionized (DI) water in a final volume of 1 L, and autoclaved at 121 C for 15 min. The media was then allowed to cool down to 50°C, in order to add the following sterile solutions of selective antibiotics: 1 M Ca(NO3)2 (1 mL), 0.01 M MnCl2 (1 mL), 1 mM FeSO4 (1 mL).
2.2 Cloning
PCR reactions were performed using PrimeSTAR® Max DNA Polymerase (TAKARA Bio Inc., San Jose, CA) according to manufacturer protocols. Homology cloning was performed using In-Fusion® Snap Assembly Master Mix for homology cloning according to manufacturer protocols (TAKARA Bio Inc., San Jose, CA). All PCR products were purified using the NucleoSpin® Gel and PCR Clean-up kit (TAKARA Bio Inc., San Jose, CA) according to manufacturer protocols. Plasmid mini-preps from E. coli strains were performed using the PureYield™ Plasmid Miniprep System (Promega Corp., Madison, WI) according to manufacturer protocols. For plasmid mini-preps from B. subtilis strains, the same kit was used, however water used to resuspend cells in the manufacturer’s protocol contained freshly dissolved lysozyme at 2.5 mg/mL final concentration and samples were incubated at 37°C for 30 min before continuing with the normal protocol. DNA was quantified using a UV-Vis NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Sanger sequencing was conducted by Elim Biopharmaceuticals, Inc. (Elim Biopharmaceuticals Inc., Hayward, CA). Sequences for primers can be found in Supplementary Table S1. Sequences for constructs can be found in Supplementary Table S2.
2.2.1 pAVE001 and pAVE003 construction
Two expression plasmids were created for expression of the drugs: pAVE001 for filgrastim and pAVE003 for teriparatide, using basic homology-based cloning. gBlocks of drug cassettes were synthesized (Integrated DNA Technologies, Coralville, IA). A linker sequence (GSSGGSSG) was incorporated between the signal peptide and the functional biologic sequence (Supplementary Table S2). pBE-S (TAKARA Bio Inc., San Jose, CA), the expression plasmid backbone, was linearized by PCR using primers F1/R1 to generate a 5,866 bp amplicon (Supplementary Table S1). For pAVE001, the filgrastim insert was amplified from its gBlock using primers F2/R2 (Supplementary Table S1) which contain ∼20 bp homologous to pBE-S. This generated a 644 bp amplicon. Similarly, for pAVE003, the teriparatide drug cassette insert was amplified from its gBlock using primers F2/R3 (Supplementary Table S1). This generated a 221 bp amplicon. Following pBE-S PCR linearization, 1 μL DpnI (New England Biolabs, Ipswich, MA) was added per 50 μL of PCR reaction and the reaction was incubated at 37°C for 2 h. Inserts and backbone were run on a Tris-borate-EDTA 1% agarose gel containing ethidium bromide to stain the DNA. Bands of expected sizes were excised from the gel and impurities were removed using a gel extraction kit (see above) and eluted in nuclease-free water. The resulting DNA was quantified and used in In-Fusion cloning reactions according to manufacturer protocol (incubation at 50°C for 15 min). In-Fusion reactions were used for transformation into E. coli.
2.2.2 Preparation of signal peptide libraries and isolation of pAVE055 and pAVE061
pAVE001 E001 and pAVE003 were mini-prepped using the PureYield™ Plasmid Miniprep System (Promega Corp., Madison, WI, USA) from single colonies of E. coli following confirmation via Sanger sequencing. pAVE001 and pAVE003 were both PCR-linearized in 50 μL reactions using primers F4/R4 (Supplementary Table S1) and the resulting PCR reactions were incubated with 1 μL DpnI at 37°C for 2 h. Following this, reactions were run on a Tris-borate-EDTA 1% agarose gel containing ethidium bromide to stain DNA. Bands of the correct size were excised from the gel and cleaned using a gel extraction kit (see above) and eluted in nuclease-free water. Expected band sizes were 6,397 bp for linearized pAVE001 and 5,974 bp for linearized pAVE003. The resulting DNA was quantified. A library of 173 unique signal peptides found in Bacillus subtilis strain 168 was inserted into this linearized backbone using In-Fusion cloning and the Signal Peptide Library DNA purchased from TAKARA Bio as a part of their B. subtilis Secretory Protein Expression System, according to the manufacturer’s protocols (TAKARA Bio Inc., San Jose, CA). Two μL of the resulting reactions were used directly to transform E. coli cells. Colony-containing plates (>2,000 colonies) were washed and scraped into 3 mL of Luria-Bertani (LB) broth three times per plate and washes were pooled into a 50 mL Falcon tube according to manufacturer’s protocol. The pooled colonies were then centrifuged, the supernatant was discarded, and the resulting cell pellets were mini-prepped using the PureYield™ Plasmid Miniprep System (Promega Corp., Madison, WI, USA) to obtain the plasmid library. The resulting library was stored at −20°C for B. subtilis transformation and subsequent screening (see HiBiT Bioassay subsection for more detail). The best performing colonies were teriparatide colony 441 and filgrastim colony 432. Plasmids pAVE055 and pAVE061 were mini-prepped from these colonies, respectively.
2.2.3 pAVE114 and pAVE118 construction
Plasmids pAVE055 and pAVE061) were mini-prepped using the PureYield™ Plasmid Miniprep System (Promega Corp., Madison, WI, USA) from colonies that exhibited the highest secretion of teriparatide and filgrastim, respectively, as evidenced from luminescent screening (see HiBiT Bioassay subsection for more detail). These plasmids were Sanger sequenced and used to clone the drug cassette and favorable signal peptide into pECE321, a suicide plasmid for ectopic integration (Guiziou et al., 2016). pECE321 was purchased from the Bacillus Genetics Stock Center (BGSC, Columbus, OH). To create pAVE114 and pAVE118, pECE321 was PCR linearized with primers F5/R5 (Supplementary Table S1) to generate an 8,199 bp amplicon. Following a DpnI digest, this backbone was gel purified. Plasmids from the best performing colonies for each drug were both amplified using primers F6/R6 (Supplementary Table S1) to generate a ∼800 bp inserts from the filgrastim library and ∼380 bp inserts from the teriparatide library. Inserts and backbone were electrophoresed on a Tris-borate-EDTA 1% agarose gel containing ethidium bromide to stain DNA. Bands of the correct size were excised from the gels and cleaned using a gel extraction kit (see above) and eluted in nuclease-free water. The resulting DNA was quantified and used in In-Fusion cloning reactions according to manufacturer’s protocols (incubation at 50°C for 15 min). A 1 μL aliquot of In-Fusion reaction was used for transformation of 50 μL of chemically competent E. coli cells.
2.2.4 pAVE147 and pAVE148 construction
The same plasmid used to create pAVE114 was mini-prepped using the PureYield™ Plasmid Miniprep System (Promega Corp., Madison, WI, USA) and used to clone the drug cassette and favorable signal peptide into pECE321. Multiple cloning efforts were carried out using E. coli as an intermediate host for replication prior to mini-prepping using the PureYield™ Plasmid Miniprep System (Promega Corp., Madison, WI, USA) for B. subtilis transformation. All genomic insertions contained deleterious mutations. One genomic insertion was confirmed to have a premature stop codon in the signal peptide region. This was used as a negative control moving forward. All other aspects of the plasmid were confirmed to be unchanged by Sanger sequencing. This control plasmid is referred to as pAVE148.
To create pAVE147, pAVE148 was PCR linearized with primers F7/R7 which overlap at the mutation site to restore the proper translation frame. This generated a ∼8500 bp amplicon. Following a DpnI digest, this backbone was gel purified. This amplicon with the reversed mutation was circularized by In-Fusion® Snap Assembly according to manufacturer’s protocol. To acquire sufficient DNA for B. subtilis transformation (2 μg), the resulting reaction mix was diluted with nuclease-free water (4:1) and 1 μL of the diluted aliquot was used as the template in several 50 μL PCR reactions using primers F9/R9 (Supplementary Table S1) to amplify the portion of the plasmid necessary for homologous recombination into the B. subtilis genome (3,654 bp amplicon). The PCR reactions were then gel electrophoresed and DNA bands of expected sizes were removed from the gel using a gel extraction kit (see above) and eluted in nuclease-free water. The resulting DNA was quantified and 2 μg was used for transformation of 600 μL of naturally competent B. subtilis cells.
2.2.5 B. subtilis colony PCR to confirm AE147 and AE148
B. subtilis cells were transformed using products from Section 2.2.4 to allow for integration of the constructs present in pAVE147 and pAVE148 into the B. subtilis genome at the amyE locus. This was done to enable stable transgenic expression of teriparatide.
To confirm correct ectopic integration of final cassettes into the B. subtilis genome, colony PCR of strain genomic DNA was carried out. In brief, individual colonies were picked and scraped into PCR tubes containing 10 μL of nuclease-free water. Colonies were also used to inoculate selective LB media for making glycerol stocks. PCR tubes containing the cell-water suspensions were then incubated on ice for 5 min, then microwaved for 1 min, then chilled on ice for 30 s. The 1 min microwaving and 30 s ice incubation process was then repeated for a total three times in the microwave oven. Tubes were then chilled on ice for 5 min. A 1 μL aliquot of microwaved cell-water suspension was then used in a 50 µL PCR reaction. PCR reactions were done according to manufacturer’s protocol for PrimeStar DNA Polymerase (TAKARA Bio Inc., San Jose, CA) with the following changes: a 10-minute incubation at 95°C was added to the beginning (before cycling). Immediately after the PCR reaction was complete, EDTA was added to a final concentration of 50 mM to prevent heat resistant DNAse degradation released from B. subtilis cytoplasm. Colonies that demonstrated expected sequences from colony PCR and Sanger sequencing were named strains AE147 and AE148.
2.3 Bacterial transformation
2.3.1 E. coli transformation
A 100 µL aliquot of E. coli Stellar™ Competent Cells (TAKARA Bio Inc., San Jose, CA) was added to pre-chilled centrifuge tubes and chilled on ice for 10 min. In-Fusion Reaction Master Mix (2 µL) was then added into each tube and chilled on ice for 30 min. Competent cells were heat shocked on a thermal block halfway filled with water to 42°C for 45 s, then chilled on ice for 5 min. A 900 µL volume of SOC medium (TAKARA Bio Inc., San Jose, CA), pre-warmed to 37°C, was added into each tube and placed in a shaking incubator at 37°C and 250 rpm for 60 min. For signal peptide library creation, several 200 µL aliquots were plated on LB + Amp [100 μg/mL] selective plates. The plates were incubated at 37°C for 16 h.
2.3.2 B. subtilis transformation
SPI medium, SPII medium and 100 mM EGTA (pH 7.0) were prepared according to the manufacturer’s instructions for the B. subtilis Secretory Protein Expression System (TAKARA Bio Inc., San Jose, CA). For signal peptide library transformation into B. subtilis, all steps in manufacturer protocol were followed. For our inserted target genes, 600 µL plating volume yielded a sufficient number of wells-spaced colonies for screening. Cells were plated on LB + Km [10 μg/mL] selective plates.
For ectopic integration, all steps outlined in the aforementioned protocol were followed, however, after inoculating SPII medium and incubating at 37°C for 90 min, the cell suspension was divided in 600 µL aliquots in microcentrifuge tubes. Glycerol was added to a final concentration of 10% (v/v) and aliquots were flash frozen in liquid nitrogen before being stored at −80°C for later use. When ready to use in transformations, tubes were incubated at 37°C until completely thawed and 6 µL of 100 mM EGTA (pH 7.0) was added. Tubes were then shaken at 37°C, 90–100 rpm for 5–10 min before adding 2 µg of plasmid to be recombined into the host genome. After DNA addition, the cell suspensions were incubated at 37°C, 90–100 rpm for 90 min. A 200 µL aliquot was then used for plating on spectinomycin (Sp) [100 μg/mL] selective LB plates overnight. Individual colonies were also streaked on erythromycin (Em) [1 μg/mL] selective LB plates for counterselection. Colonies which grew on Sp selective plates but did not grow on Em selective plates were picked for colony PCR. Colony PCR was done to amplify the genomic DNA using primers F10/R10 (Supplementary Table S1) and resulting amplicons were sent for Sanger sequencing using sequencing primers F11 and R11 (Supplementary Table S1).
2.4 High-throughput bioassay for detecting protein secretion
A 150 μL volume of LB + Km [10 μg/mL] was inoculated with single B. subtilis colonies in 96-well plates. Border wells were not inoculated as they experienced significant evaporation during growth. Three control colonies were added to each individual plate to use as a reference and to monitor “plate-to-plate” variability in data. These controls were: B. subtilis strains harboring pAVE001 and pAVE003 (pBE-S containing HiBiT-tagged filgrastim and teriparatide cassettes with a known low-level secretion signal peptide from the aprE gene) and a B. subtilis strain harboring an empty pBE-S backbone (no drug or HiBiT tag inserted). Each plate also contained a blank LB control. Cultures were grown at 37°C for 24–48 h.
Following growth, HiBiT reagent was prepared according to manufacturer directions (Promega Corp., Madison, WI). Using a multichannel pipette, 25 μL of freshly made HiBiT reagent (Promega Corp., Madison, WI) was added to new 96-well white opaque plates (Fisher Scientific, Waltham, MA) used to detect bioluminescence. Corresponding wells in the growth plates were mixed well by pipetting up and down before transferring the same volume (25 μL) of culture. Following culture additions, dispensed cultures were mixed well with the HiBiT reagent by pipetting. The culture-reagent mixture was allowed to incubate at room temperature for no longer than 10–12 min before being measured on a plate luminometer using default instrument parameters.
Following the luminescent reading, luminescence values read from blank LB controls were averaged and the average value was subtracted from all other well readings to correct background luminescence. Luminescence data was then analyzed, and the brightest colonies were saved from the remainder of culture plates to make 15% (w/v) glycerol stocks stored at −80°C. All stored colonies were streaked on LB + Km [10 μg/mL] plates to isolate single colonies for miniprepping and further screening.
2.5 Secretion vs. temperature
Two separate pre-cultures (5 mL LB + Sp [100 μg/mL]) were inoculated from glycerol stocks of B. subtilis strains AE147 and AE148 and grown for 16 h at 28°C. Following the 16-h incubation, six 5 mL expression cultures were started for each of the two strains by inoculating 5 mL of Sp containing LB with 50 μL of pre-culture. These were incubated at temperatures ranging from 15°C to 40°C in 5°C increments for 24 h with shaking at 200 rpm. After growth, triplicate 50 μL aliquots were taken from each culture and optical density readings at 600 nm (OD600) were taken. Following OD600 readings, triplicate 25 μL aliquots of each culture were added to white, opaque 96-well plates and reagents were added according to the manufacturer’s protocols for the Nano-Glo® HiBiT Extracellular Detection System (Promega Corp., Madison, WI). After a 10-minute incubation step, luminescence was read from plate wells. Triplicate blank LB readings were also conducted and the average of these readings were subtracted from all other luminescence values prior to analyses.
2.6 Spore suspension preparation and quantification
Spores were prepared by adapting a previously published protocol (Munakata et al., 1991) as follows. A colony was inoculated into 25 mL of freshly prepared DSM and grown at 37°C and 150 rpm for 2 h. This pre-culture was diluted 1:10 into 250 mL of prewarmed (37°C) DSM in a 2 L flask. This larger culture was incubated for 48 h at 37°C and 150 rpm. The entire culture was then centrifuged for 10 min at 9,400 xg at 4°C and the supernatant was carefully discarded. The resulting pellet was washed with 200 mL of cold (4°C) sterile DI water via resuspension. The culture was then centrifuged again, and the supernatant was carefully discarded. The pellet was once again resuspended in 200 mL of fresh cold sterile DI water and the resuspended pellet was incubated overnight 4°C. Following overnight incubation, water/spore suspension was again centrifuged at 9,400xg, supernatant was carefully discarded, and the pellet was resuspended in 200 mL cold sterile DI water. The overnight incubation at 4°C was repeated once more. Spores were then examined microscopically with a Zeiss Axio Imager Z1 under phase contrast optics and the vast majority of suspension was observed to be free-living spores. Finally, the suspension was centrifuged once more, the supernatant was discarded, and the pellet was resuspended in 20 mL of 70% ethanol and the lids of storage tubes were wrapped with parafilm to prevent ethanol evaporation. Spore suspensions were allowed to incubate at 4°C for 1 h prior to plating to ensure vegetative cell death. Aliquots of each spore suspension were diluted 1-million-fold in 70% ethanol and 20 μL of this 1-million-fold dilution was plated on selective solid media, then incubated at 37°C overnight. Resulting colonies were counted to determine the number of colony forming units (CFU)/mL of each spore suspension.
For paper blots containing B. subtilis endospores, ∼2 cm2 pieces of normal printer paper were cut into small squares, folded into aluminum foil, and autoclaved. Aliquots of 10 μL of spore suspensions were blotted onto each piece of paper and allowed to dry in a sterile culture flow hood. These pieces of paper were then re-wrapped in sterile aluminum foil, labeled, and stored in 50 mL Falcon tubes at room temperature for future use.
2.7 Pharmaceutical peptide production
For analysis of optimal expression temperature, a single bacterial colony was picked from each plate and suspended separately in 5 mL of streptomycin-containing LB broth and incubated overnight at 37°C. A 10 mL volume of starter culture was added to 50 mL antibiotic-containing LB in a 250 mL conical flask and incubated at 37°C for 24–48 h. The mixture was transferred to a 50 mL Falcon tube and centrifuged at max speed for 5 min; supernatant was collected in a clean 50 mL Flacon tube for purification.
Expression cultures used to produce Table 1 were made by inoculating fresh media with dried spore blots and allowing the cultures to grow for 24 h before purifying the secreted protein. Dried B. subtilis spores prepared from strains AE068 (pBE-S_YoqHSP-HiBiT-Filgrastim) and AE147 (
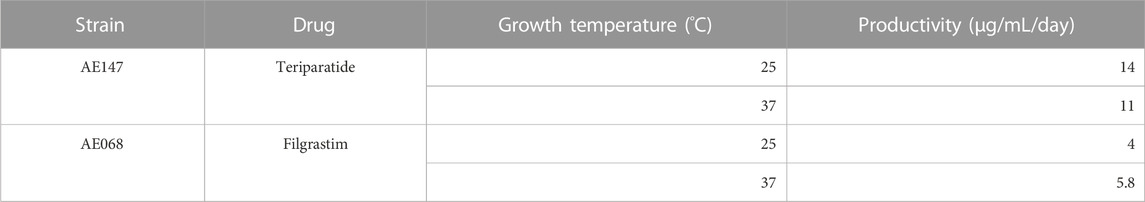
TABLE 1. Maximum productivity rates observed from dried B. subtilis spores.
2.8 Protein purification and quantification
An 8 mL aliquot of each supernatant was added to the same volume of standard equilibration buffer (20 mM Tris-HCl, 300 mM NaCl, antiproteases, pH 7.5). This mixture was then loaded onto a HisPurTM Ni-NTA Spin Column and the protein was purified according to the manufacturer’s instruction with the following buffer changes: Wash buffer used was 20 mM Tris-HCl, 300 mM NaCl, antiproteases, pH 7.5. Elution buffer used was 20 mM Tris-HCl, 300 mM NaCl, 250 mM Imidazole, antiproteases, pH 7.5. The flowthrough, column wash, and elution fractions were collected and analyzed by SDS-PAGE to check for sufficient purification of filgrastim and teriparatide (Figure 2). Marker used was Novex Sharp Pre-Stained Protein Standard (Thermo Fisher Scientific, Waltham, MA) SDS-PAGE gel analysis showed isolated protein bands at the expected molecular weights in the elution fractions. Following His-tag purification, the eluted fractions were combined and concentrated to 2–4 mL final volume using a 5–20 mL, 3K MWCO Pierce Protein Concentrator according to manufacturer’s instruction. Imidazole was removed using a 10 mL 7K MWCO ZebaSpin Desalting Column according to manufacturer’s instruction. During desalting, a buffer exchange was conducted to transfer to protein to a 2x storage buffer (40 mM Tris-HCl, 600 mM NaCl, antiproteases, pH 7.5). All of the resulting buffer exchanged protein was then concentrated to ∼75 μL final volume using 0.5 mL, 3kDa Amicon Ultra-Centrifugal Filters according to manufacturer’s instructions. Protein quantification was conducted via the Thermo Scientific Pierce™ Rapid Gold BCA Protein Assay Kit in 96-well plates using 10 μL of each purified protein solution. Following quantification, protein solutions were diluted 1:1 with glycerol for a final concentration of 50% glycerol (w/v) and 1 x storage buffer. Proteins were then stored at −20°C.
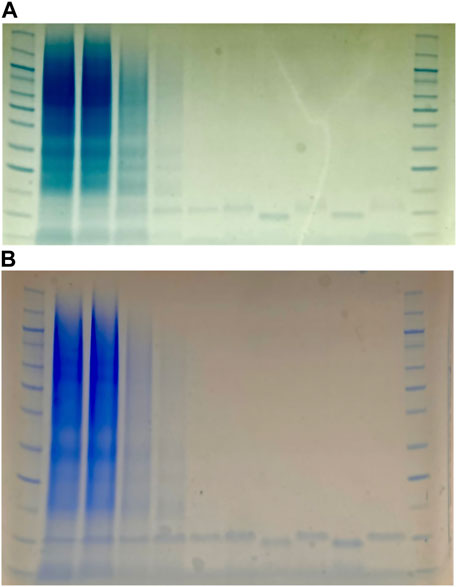
FIGURE 2. SDS-PAGE gels of filgrastim purification (A) and teriparatide purification (B). Purifications were done via His-tag spin column. First and last columns are protein markers (Novex Sharp Pre-Stained Protein Standard from Invitrogen). Columns 2–3 are flow-through. Columns 4–6 are washes. Columns 7, 9, and 11 are elution columns. Columns 8 and 10 are denatured elutions containing dithiothreitol (DTT).
3 Results
3.1 Secretion peptide screening
Both filgrastim and teriparatide were fused to a library of secretion peptides with a HiBiT reporter tag to detect secretion via luminescence. In the first round of colony screening, 277 colonies expressing filgrastim constructs and 368 colonies expressing teriparatide constructs were individually picked, grown, and screened from the resulting library. This corresponded to three 96-well plates for filgrastim colonies and four 96-well plates for teriparatide colonies (Figures 3, 4). Each plate contained its own set of controls including: 1) a positive control colony harboring either filgrastim or teriparatide fused to a HiBiT reporter tag and a secretion peptide from the aprE gene inserted in pBE-S (i.e., aprESP_Fil/Ter). 2) A negative control colony harboring empty pBE-S with no insert (i.e., Empty_BB). 3) Uninoculated media for subtracting background luminescence from readings. Following a 24-hour growth period, plates were screened for luminescence, indicative of secretion. In this initial screen, colonies which had stronger luminescent signal compared to the aprESP_Fil/Ter controls were considered for further screening and sequencing. It is important to note that the first pass in screening cold not, and therefore did not contain replicates. As such no error bars are shown (Figures 3, 4).
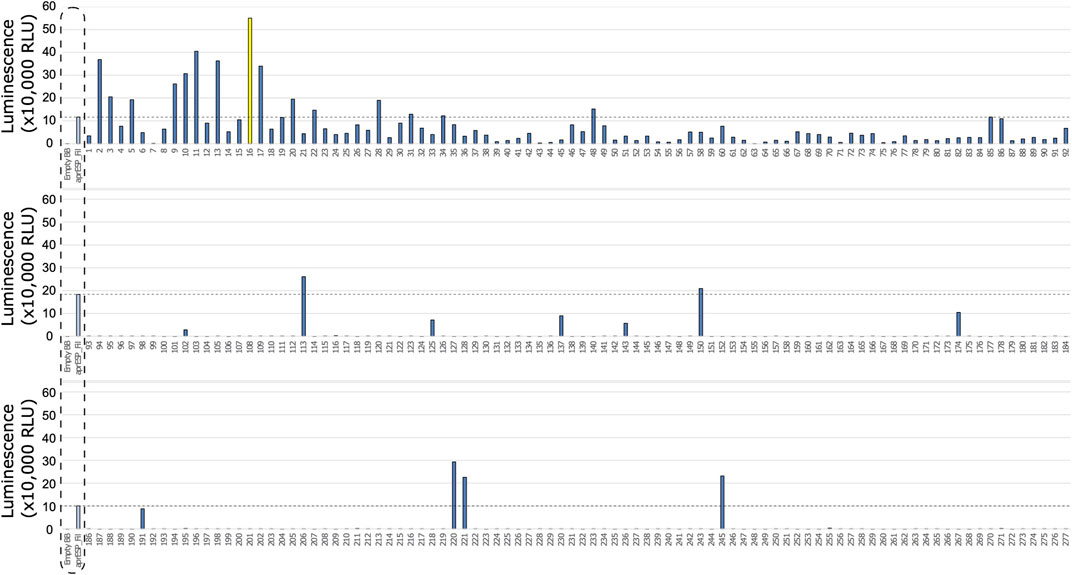
FIGURE 3. First round of colony screening of B. subtilis colonies secreting filgrastim fused to a HiBiT reporter and SP library. Each row shows 1 plate screened. Colonies are numbered below x-axis. Dotted box shows negative and positive controls (from l left to right). Controls were included separately in every plate. Negative control is an empty backbone control without filgrastim fusion peptide insert. Positive control are cells expressing filgrastim fusion peptide tagged with the HiBiT reporter and a secretion peptide from the aprE gene. Dotted line shows luminescent signal from positive control. Colonies with luminescence above this contained secretion tags with better performance compared to the positive control. Colony 16 is shown in yellow and was considered the best performing candidate to take forward for further screening and sanger sequencing.
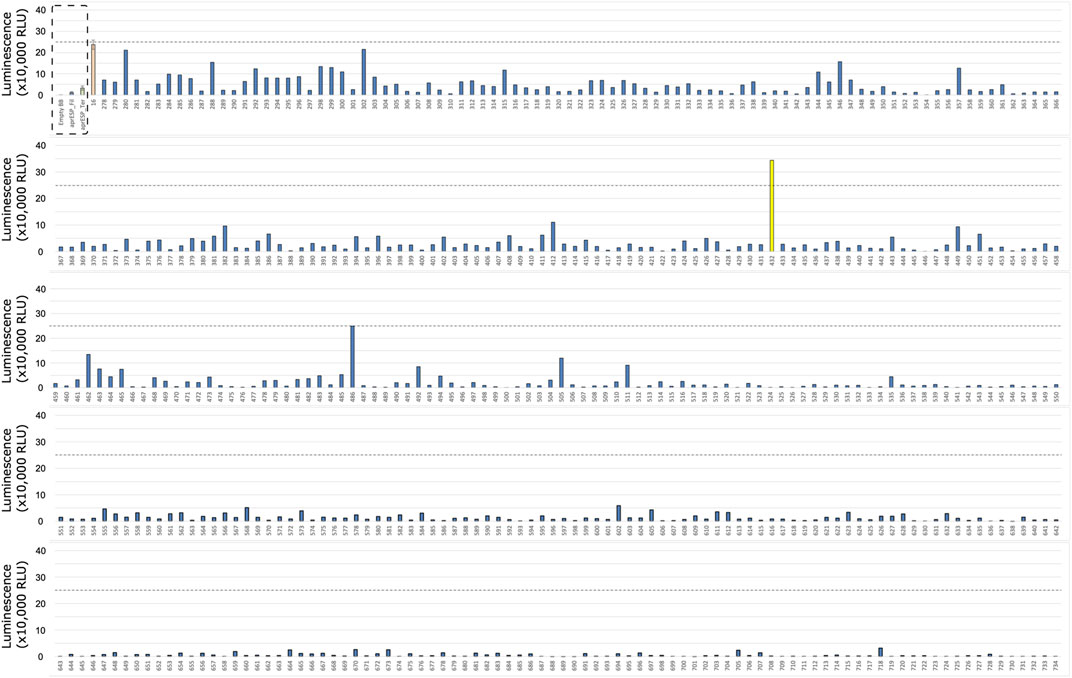
FIGURE 4. First Round of Colony screening of B. subtilis colonies secreting teriparatide fused to a HiBiT tag and one of 167 secretion peptides. Each row shows 1 plate screened. Colonies are numbered below x-axis. Dotted box shows negative and positive controls (from left to right). Controls were included separately in every plate. Negative control is an empty backbone construct without teriparatide fusion peptide insert. Positive control are cells expressing teriparatide fusion peptide tagged with the HiBiT reporter and a secretion peptide from the aprE gene. Dotted line shows luminescent signal from positive control. Colonies with luminescence above this level contained secretion tags with better performance compared to the positive control. Colonies 109, 123, 266, and 292 are shown in yellow and were considered the best performing candidate to take forward for further screening and sanger sequencing.
One colony stood out from the filgrastim library: colony 16 (Figure 3). This colony gave a reading of >550,000 relative luminescence units (RLUs) (Supplementary File S1). This corresponded to an over 5-fold increase in RLU signal as compared to the positive control. Due to the higher positive control variability seen in the teriparatide library plates, 4 colonies were selected for further screening (Figure 4). For the teriparatide library, colonies 109, 123, 266, and 292 all exhibited RLU values well above the aprESP_Ter positive control (∼3.5, 11, 3, and 1 million RLUs, respectively). These readings corresponded to an over 3, 10, 4, and 18-fold increase in RLU compared to positive controls, respectively. Sanger sequencing revealed that filgrastim colony 16 contained the secretion peptide from the mpr gene (UniProt: P39790). Secretion peptides from teriparatide colonies 109, 123, 266, and 292 were from genes phrG, sacC, yncM, ypuA (UniProt: O32295, P05656, O31803, P31847), respectively.
Following the first round of colony screening, an additional 457 filgrastim colonies and 448 teriparatide colonies (corresponding to five 96-well plates per drug) were screened in the second round (Figures 5, 6). Colonies selected from the previous round of screening were grown up alongside unknown colonies in the next screening round in triplicate for comparison, along with 10 replicates of previously included positive and negative controls (one per plate). Previously identified colonies were confirmed to exhibit higher luminescence compared to the aprESP_Fil/Ter positive controls and teriparatide colony 123 showed the highest average luminescence of the previous set.
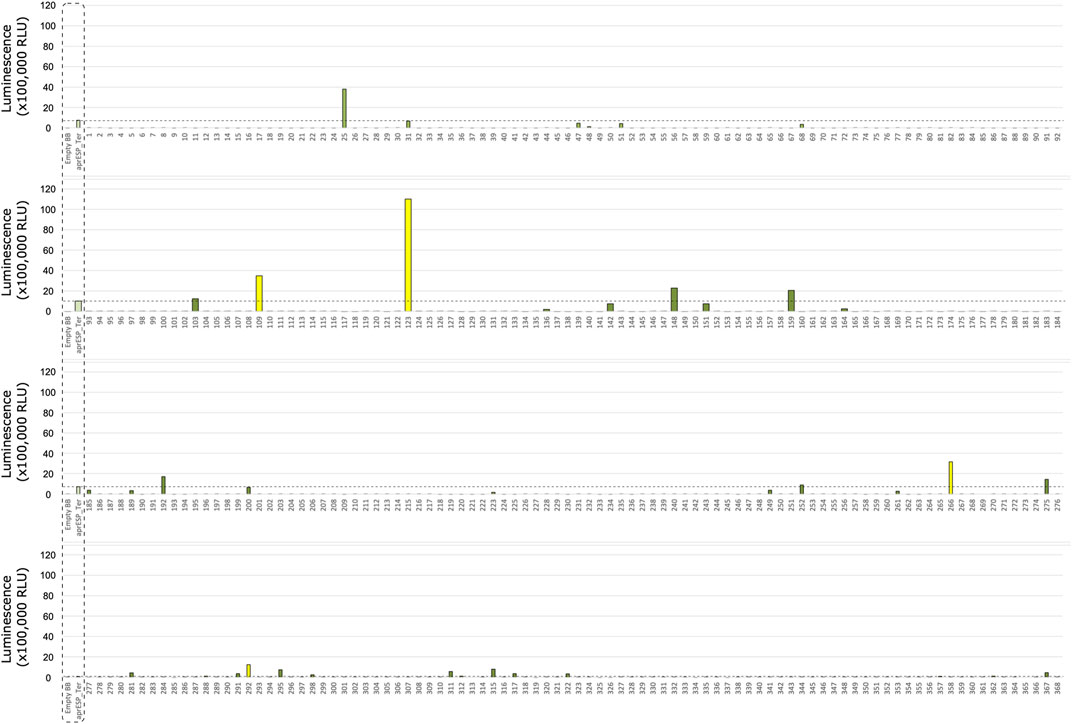
FIGURE 5. Second Round of colony screening of B. subtilis colonies secreting filgrastim fused to a HiBiT tag and one of 167 secretion peptides. Each row shows 1 plate screened. Colonies are numbered below x-axis. Dotted box shows negative and positive controls for filgrastim and teriparatide (from left to right). Negative control is an empty backbone construct without filgrastim fusion peptide insert. Positive controls are cells expressing either filgrastim or teriparatide fusion peptides tagged with the HiBiT reporter and a secretion peptide from the aprE gene. Controls were included separately in every plate and averaged. Orange bar is average luminescence reading of best-performing colony from first screen. Dotted line shows luminescent signal from most luminescent colony from previous screen (i.e., Colony 16) (see Figure 3). Colonies with luminescence above this level contained secretion tags with better performance compared to Colony 16. Colony 432 is shown in yellow and was considered the best performing candidate to take forward for further screening and sanger sequencing. Error bars show standard error. For error bars of positive and negative controls, n = 10. For error bars of Colony 16 (orange), n = 3.
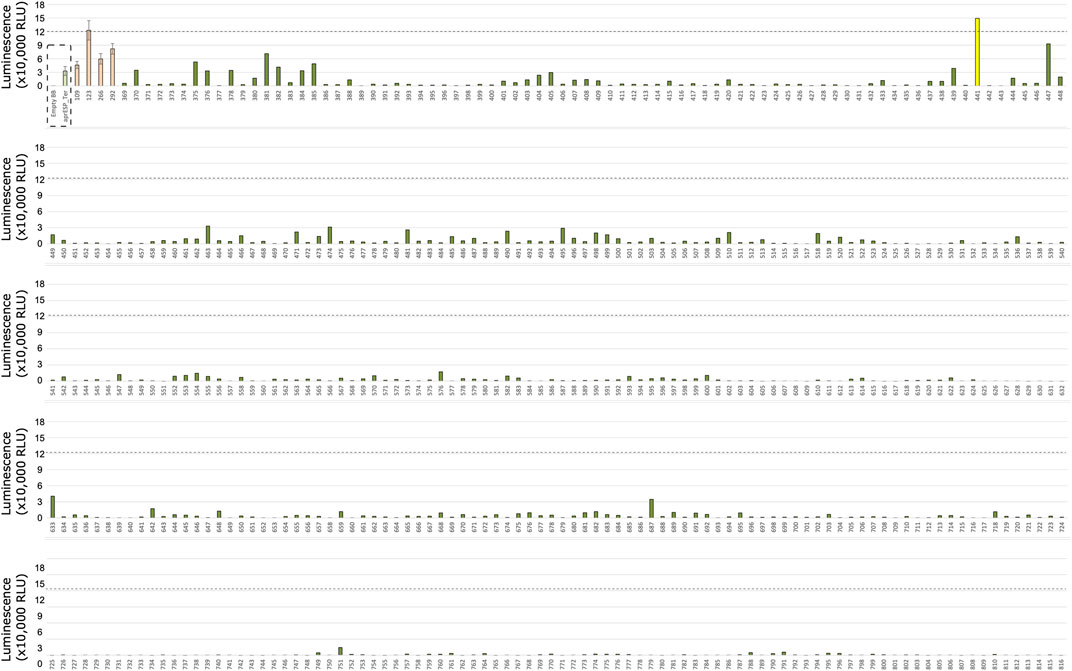
FIGURE 6. Second Round of Colony screening of B. subtilis colonies secreting teriparatide fused to a HiBiT tag and one of 167 secretion peptides. Each row shows 1 plate screened. Colonies are numbered below x-axis. Dotted box shows negative and positive controls for teriparatide (from left to right). Negative control is an empty backbone control without teriparatide fusion peptide insert. Positive controls are cells expressing teriparatide fusion peptides tagged with the HiBiT reporter and a secretion peptide from the aprE gene. Controls were included separately in every plate and averaged. Orange bars are average luminescence readings of best-performing colonies from first screen. Dotted line shows luminescent signal from most luminescent colony from previous screen (i.e., Colony 123) (see Figure 4). Colonies with luminescence above this level contained secretion tags with better performance compared to Colony 123. Colony 441 is shown in yellow and was considered the best performing candidate to take forward for further screening and Sanger sequencing. Error bars show standard error. For error bars of positive and negative controls, n = 10. For error bars of colonies from first screen (orange), n = 3.
Teriparatide colony screening identified one colony which exhibited significantly higher RLU readings compared to teriparatide colony 123: colony 441. This colony gave an RLU reading of ∼150,000 which corresponded to a ∼1.2-fold increase in RLU compared to colony 123. Following sanger sequencing, colony 441 was found to contain the secretion peptide gene walM (UniProt: A0A6M3Z808) and plasmid pAVE055 was isolated from it (Figure 7A).
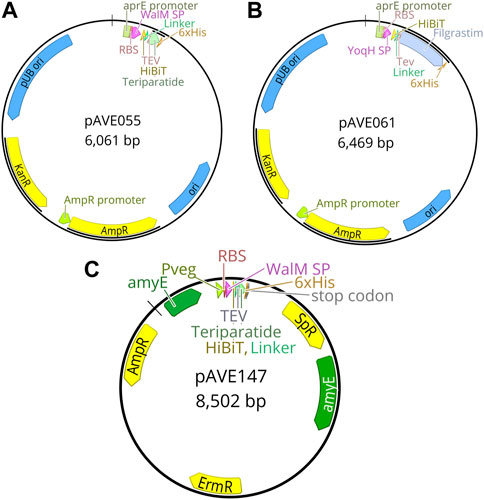
FIGURE 7. Plasmid maps of final constructs used in this study. (A) pAVE055 was created by fusing the secretion peptide from the walM gene with a downstream HiBiT tag followed by a linker sequence and a cleavage site recognizable by TEV protease, to teriparatide tagged with a C-terminal 6x-His tag. This teriparatide fusion construct was inserted into plasmid pBE-S. (B) pAVE061 was created by fusing the secretion peptide from the yoqH gene with a downstream HiBiT tag followed by a linker sequence and a cleavage site recognizable by TEV protease, to filgrastim tagged with a C-terminal 6x-His tag. This filgrastim fusion construct was inserted into plasmid pBE-S. (C) pAVE147 was created by cloning the insert from pAVE055 into the suicide plasmid, pECE321. This plasmid has left and right-hand homology sites to the amyE locus of the Bacillus subtilis genome and functions to integrate the insert along with a spectinomycin (Sp) resistance cassette for selection into this locus.
Filgrastim colony screening identified two colonies which exhibited higher RLU readings compared to filgrastim colony 16 from the first round of screening: colonies 432 and 486 gave RLU readings of ∼340,000 and ∼250,000 respectively, corresponding to a ∼1.5 and ∼1.1-fold increase compared to fillgrastim colony 16 (Supplementary File S2). Following sanger sequencing, both colonies were found to contain the same secretion peptide from the yoqH gene (UniProt: O34999) and plasmid pAVE061 was isolated from it (Figure 7B).
3.2 Genomic integration of constructs into Bacillus subtilis
Following identification of the best-performing secretion peptides for the filgrastim and teriparatide constructs, the constructs were inserted into the genome of B. subtilis. Several rounds of genomic insertion were conducted and over 40 colonies per construct were sequenced. All colonies sequenced contained frameshift mutations and premature stop codons in the secretion peptide region of the construct (data not shown). To overcome this issue, the intermediate cloning step was eliminated and replaced with PCR amplification and linearization of the construct, the product of which could directly be used for B. subtilis transformation (see Methods 2.2.4). This yielded a non-mutated colony for the WalMSP-Teriparatide construct (pAVE147) (Figure 7C) which was able to be integrated into the B. subtilis genome successfully to produce strain AE147 (Figure 8A). A mutant of this strain harboring a frameshift mutation in the secretion peptide region of the gene, and thus a premature stop codon, was also isolated to function as a negative control (AE148) (Figure 8B). YoqHSP-Filgrastim construct integration showed no success using this method as dozens of colonies still possessed premature stop codons in the secretion peptide region.
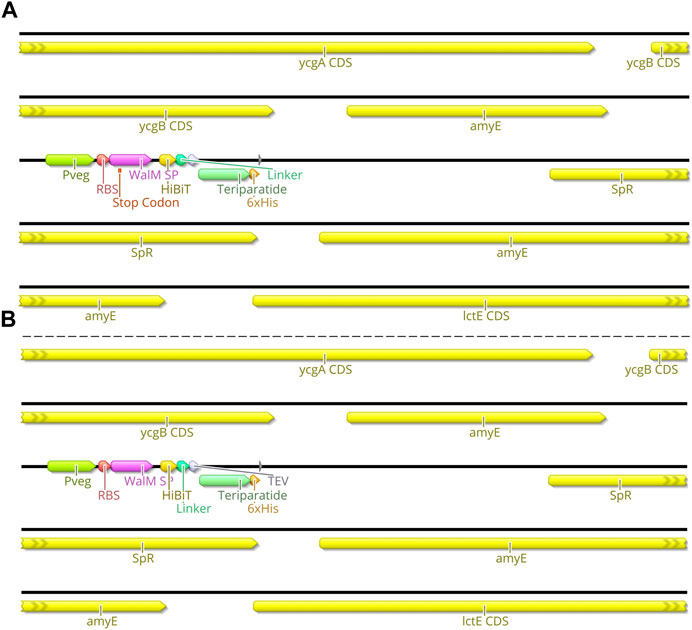
FIGURE 8. Genome Map of AE147 and AE148. This schematic shows the insertion site of the transgenic component of Bacillus subtilis strains AE147 and AE148. Integration was accomplished at the amyE locus. (A) Strain AE148 consists of a truncated teriparatide secretion cassette with a single base mutation within WalM SP, resulting in a premature stop codon in the same motif. (B) Strain AE147 consists of a fully functional teriparatide secretion cassette.
3.3 Temperature-dependent teriparatide secretion
To assess the optimal constitutive expression temperature for secretion of teriparatide, AE147 and AE148 were grown in triplicate at various temperatures and the luminescent signal was read after a 24 h incubation at these temperatures. Optical Density at the 600 nm wavelength (OD600) was also obtained after this 24 h incubation period (Figure 9). Results showed the highest luminescent signal in cultures grown at room temperature (25°C). Conversely, cell density was highest at 40°C. The negative control strain showed minimal luminescence across the temperature range tested and the highest cell density was obtained when grown at 35°C.
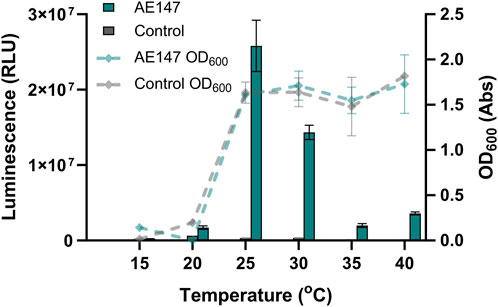
FIGURE 9. Effect of growth temperature on AE147 teriparatide secretion. Measurements are for B. subtilis strain AE147. This strain has a teriparatide fusion peptide inserted into the amyE locus. This teriparatide peptide is fused to a secretion peptide from the walM gene and a HiBiT reporter tag. A control strain harboring the same inserted fusion peptide but with a premature stop codon in the walM secretion peptide was also measured (B. subtilis strain AE148). Triplicate cultures were grown for 24 h at temperatures ranging from 15°C to 40°C. Left y-axis shows luminescence values from aliquots of cultures after 24 h incubation (bar chart). Right y-axis relates to absorbance values at OD600 taken at same time and from same cultures as luminescence values (lines chart). Error bars show standard error (n = 3).
3.4 Teriparatide and filgrastim purification
To test the expression stability of our strains with an eye towards implementation in our Astropharmacy, we produced spore stocks of strains AE068 and AE147. After overnight storage in 70% ethanol to rid the stocks of all vegetative cells, we blotted the spore suspension on pieces of paper and allowed the spores to air dry in the hood for 20 min before storing in 50 mL Falcon tubes at room temperature. Paper blots were stored for at least 7 days before being used to inoculate expression cultures. Expression cultures in Table 1 were made by inoculating fresh media with dried spore blots and allowing the cultures to grow for 24 h before purifying the secreted protein. The flowthrough, column wash, and elution fractions were collected and analyzed by SDS-PAGE to check for sufficient purification of filgrastim and teriparatide (Figure 2). SDS-PAGE analysis showed isolated protein bands at the expected molecular weights in the elution fractions. Sporulation and storage of our strains in desiccated conditions did not eliminate the ability of our strains to secrete filgrastim and teriparatide. The eluted fractions were then combined, and protein concentration was quantified to obtain production rates shown in Table 1.
4 Discussion
By thinking of life as a technology–truly programmable, capable of self-replication and self-repair, and in some cases, dormancy (Rothschild, 2016) – we seek to develop solutions to the challenges of pharmaceutical expiration to enable more ambitious space exploration. We developed a proof of concept for a cell-based “Astropharmacy.” This platform for protein expression and secretion, where protein is manufactured according to instructions written in strands of an organism’s DNA, could produce a therapeutic dose of any protein-based biopharmaceutical within hours. Want a different drug? Use a different DNA sequence. This is the first step in developing a fully functioning Astropharmacy: the rapid on-site, on-demand production of protein-based therapeutics, and in principle, all biopharmaceuticals.
In our work, the production rate of filgrastim was less than that of teriparatide (by about 2-fold). This points to difficulties regarding filgrastim production in general which is compounded by the fact that the dose requirement for the compound is 10-fold greater than that of teriparatide. In addition to low production, we were unsuccessful in being able to move the successful filgrastim fusion-peptide into the genome of B. subtilis. After screening dozens of colonies for successful insertion, sequencing results showed single nucleobase frameshift mutations in the secretion peptide region of the construct. All frameshift mutations lead to premature stop codons before the CDS for filgrastim itself. This problem was thought to be a result of gene toxicity to the intermediate cloning strain being used (E. coli) prior to plasmid transfer to B. subtilis. A non-replicative cloning strategy was attempted to get around this issue. While this non-replicative cloning strategy worked for insertion of the teriparatide construct, the filgrastim construct showed the same issue in B. subtilis cells as it had in E. coli cells and, again, frameshift mutations were found in all of the dozens of B. subtilis colonies sequenced. This signifies that filgrastim itself is cytotoxic to B. subtilis cells as there seems to be strong selective pressure toward functional deletions of the recombinant filgrastim cassette when controlled with a strong genomically-localized promoter (Pveg). This cytotoxicity was alleviated when filgrastim was expressed in Bacillus subtilis cells via a transformed plasmid. This could be because the promoter we used for plasmid expression (PaprE) results in lower expression rates, allowing for cell proliferation despite filgrastim expression.
The problem of pharmaceutical toxicity to cells is a phenomenon that is seen sometimes in cell-based production systems. Not all protein-based pharmaceuticals will be amenable to recombinant expression in every strain. As a result, a large cloning effort, trying various host cells, may have to be undertaken for each new pharmaceutical needed. Conversely, cell-free expression systems do not have this problem. A great advantage of cell-free systems is that there is no need to maintain the complex physiological homeostasis of a living organism. Therefore, these systems allow for more flexibility in the type of recombinant protein it produces. However, cell-free systems have their own disadvantages. The stability of all the components in cell-free systems for recombinant protein expression is usually insufficient, particularly for the mission critical role that a life support technology, such as an Astropharmacy, would require. Whilst the PT7 expression system, which comprises just the ribosomes and 35 bacterial proteins, has shown a year-long room temperature shelf-life in lyophilized form (Pardee et al., 2014), cell extract expression systems boast shelf-life stability on the order of weeks (Gregorio et al., 2020). Even with new advances in the discovery of low-cost excipients for enhanced stability (Warfel et al., 2023), cell-free expression systems have only been shown to be stable at room temperature for up to 4-weeks, currently. In contrast, our cell-based expression system has stability at least several orders of magnitude higher given rates of B. subtilis survivability in their spore state (Horneck, 1993). Future work should seek to address issues of toxicity of certain compounds while making stability of the expression system a priority.
One solution to get around the cytotoxicity of recombinant proteins would be stringent regulatory control via promoter optimization (Gu et al., 2018; Souza et al., 2021). While an inducible expression system would likely lead to insufficient production rates on the scale of 24 h, weaker constitutive promoters may allow for successful genomic integration, thereby increasing stability of the expression system, and still allow for sufficient production rates through increased culture volumes. Another solution may be optimization of the expression conditions via growth at lower temperatures. Our data indicates a significant difference in luminescent signal, indicative of recombinant protein secretion, when strains were grown at lower temperatures (25°C) (Figure 9). When the pharmaceutical peptides were purified, yields were comparable at 25°C as compared to the same strains grown at 37°C (Table 1). One explanation for this may be proper peptide folding in the lower temperature condition which leads to increased solubility, secretion, and HiBiT function. However, the growth rate of B. subtilis at this lower temperature suffers, leading to a decrease in overall cell number and therefore a decrease in the mass of protein secreted. More work will have to be done to address this hypothesis but if supported, it signifies a large benefit to optimizing B. subtilis for optimal growth at lower temperatures using techniques such as adaptive laboratory evolution (ALE). ALE could not only decrease the problem of gene toxicity of recombinant proteins, but it could, at the same time, increase production rates, soluble yields, and the bioactivity of protein produced.
Further optimization of strain growth conditions, expression conditions, and promoters used could achieve higher production rates (Kang et al., 2014; Souza et al., 2021), however, these strains provide the template for further optimizations in the future. We can now use the methodology outlined to produce other drug candidates by B. subtilis secretion. All will have to undergo individual screening for the best performing secretion tag of a given peptide. Due to the cytotoxicity problems observed, it is likely that some pharmaceuticals will need to be localized on a plasmid, while others may be chromosomally integrated.
Bacillus subtilis is a promising platform for drug production for several reasons. In addition to the inherent space tolerance of its spores (Horneck, 1993), marine strains of this same host have also been shown to have an increased capacity for secondary metabolite secretion (Ivanova et al., 1999; Mondol et al., 2013; Pandey et al., 2014; Velupillaimani and Muthaiyan, 2019). This is thought to be a result of selective evolution in the highly competitive marine environment where secretion of toxic secondary metabolites as a defense mechanism is beneficial. Marine strains of this species are also salt tolerant. Previous studies have shown that high concentrations of some salts predicted to be present in Martian brines provide UV radiation protection to B. subtilis cells in simulated Mars conditions (Godin et al., 2021). Aside from UV protection, brine present on Mars could be used with minimal desalination to grow B. subtilis cells on-site if marine strains are selected that can tolerate these higher salt concentrations. Thus, tools developed in the lab strain (strain 168) could be further transferred to marine strains with more favorable phenotypes for resistance and secretion to develop additional benefits to our engineered strains and a potential solution to potable water consumption on-site.
5 Conclusion
We have completed a proof of concept for the first step in our Astropharmacy: the production and secretion of peptide biologics using B. subtilis as a production platform. The pharmaceutical peptides chosen, teriparatide and filgrastim, are important for astronaut health. While B. subtilis has been used to produce pharmaceutical peptides for use on Earth, this effort represents the first time that these peptide pharmaceuticals, with uses in aerospace medicine, have been recombinantly produced in a space-tolerant microbe. Using high-throughput screening we identified the two secretion peptides that were most useful for teriparatide (walM gene) and filgrastim (yoqH gene) secretion. The resulting teriparatide-producing strain (AE147) produced 1 dose-equivalent (20 μg) of teriparatide in 24 h from as little as 1.5 mL of cell culture. The filgrastim-producing strain (AE068) was able to produce 1-dose equivalent (300 μg) of filgrastim in 24 h from 52 mL of cell culture. We showed that these production rates were unaffected by sporulation of the host cells, and subsequent storage in a desiccated state, indicating high stability and reliability in B. subtilis. Finally, we identified several considerations, previously unknown for recombinant expression of these pharmaceutical peptides in Bacillus subtilis that should be explored in future development of a cell-based Astropharmacy. Further optimization of strain growth conditions, expression conditions, and promoters could enable higher production rates to be achieved. These strains provide templates that can be further optimized in follow-on work. Successful demonstration of B. subtilis as a peptide pharmaceutical production platform paves the way for the development of novel purification technology and drug delivery devices so that the vision of an Astropharmacy can be fully realized.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
Conceptualization, AV-E and LR; methodology, AV-E and LR; formal analysis, AV-E and LR; lab work, AV-E and CB; writing—original draft preparation, AV-E; writing, review and editing, AV-E, LR, PW, DL, and DLV; visualization, AV-E; project administration, AV-E and LR. All authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the NASA Innovative Advanced Concepts (NIAC) Phase I, the National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) under Grant No. 1650114, the NSF California Louis Stokes Alliances for Minority Participation (LSAMP) Bridge to the Doctorate (BD) Fellowship under Grant No. HRD-1701365, and the NSF Non-Academic Research Internships for Graduate Students (INTERN) Supplemental Funding Opportunity under Grant No. INTERN NSF DCL 21-013.
Acknowledgments
This research originally appeared as a pre-print submitted to bioRxiv (Vallota-Eastman et al., 2023).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor JH declared a shared affiliation with the author AV-E at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frspt.2023.1181843/full#supplementary-material
References
Afshinnekoo, E., Scott, R. T., MacKay, M. J., Pariset, E., Cekanaviciute, E., Barker, R., et al. (2020). Fundamental biological features of spaceflight: advancing the field to enable deep-space exploration. Cell 183, 1162–1184. doi:10.1016/j.cell.2020.10.050
Appelbaum, F. R. (1989). The clinical use of hematopoietic growth factors. Semin. Hematol. 26, 7–14.
Batcha, A. L., Williams, J., Dawn, T. F., Gutkowski, J. P., Widner, M. V., Smallwood, S. L., et al. (2020). Artemis I trajectory design and optimization.
Bento, H. B. S., Paiva, G. B., Almeida, M. R., Silva, C. G., Carvalho, P. J., Tavares, A. P. M., et al. (2022). Aliivibrio fischeri L-Asparaginase production by engineered Bacillus subtilis: a potential new biopharmaceutical. Bioprocess Biosyst. Eng. 45, 1635–1644. doi:10.1007/s00449-022-02769-x
Berry, I., Berthier, A., Marescaux, J., Mutter, D., Bouabene, A., Magee, P., et al. (2002). MARSTECHCARE NECESSARY BIOMEDICAL TECHNOLOGIES FOR CREW HEALTH CONTROL DURING LONG-DURATION INTERPLANETARY MANNED MISSIONS. Available at: https://www.documentcloud.org/documents/3900622-MARSTECHCARE-FINAL-REPORT-2 (Accessed November 22, 2022).
Blue, R. S., Bayuse, T. M., Daniels, V. R., Wotring, V. E., Suresh, R., Mulcahy, R. A., et al. (2019). Supplying a pharmacy for NASA exploration spaceflight: challenges and current understanding. Npj Microgravity 5, 14–12. doi:10.1038/s41526-019-0075-2
Brockmeier, U., Caspers, M., Freudl, R., Jockwer, A., Noll, T., and Eggert, T. (2006). Systematic screening of all signal peptides from Bacillus subtilis: a powerful strategy in optimizing heterologous protein secretion in gram-positive bacteria. J. Mol. Biol. 362, 393–402. doi:10.1016/j.jmb.2006.07.034
Chancellor, J. C., Scott, G. B. I., and Sutton, J. P. (2014). Space radiation: the number one risk to astronaut health beyond low Earth orbit. Life 4, 491–510. doi:10.3390/life4030491
Cliff, J. B., Jarman, K. H., Valentine, N. B., Golledge, S. L., Gaspar, D. J., Wunschel, D. S., et al. (2005). Differentiation of spores of Bacillus subtilis grown in different media by elemental characterization using time-of-flight secondary ion mass spectrometry. Appl. Environ. Microbiol. 71, 6524–6530. doi:10.1128/AEM.71.11.6524-6530.2005
Cucinotta, F. A., and Durante, M. (2006). Cancer risk from exposure to galactic cosmic rays: implications for space exploration by human beings. Lancet Oncol. 7, 431–435. doi:10.1016/S1470-2045(06)70695-7
Degering, C., Eggert, T., Puls, M., Bongaerts, J., Evers, S., Maurer, K.-H., et al. (2010). Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides. Appl. Environ. Microbiol. 76, 6370–6376. doi:10.1128/AEM.01146-10
Denis, G., Alary, D., Pasco, X., Pisot, N., Texier, D., and Toulza, S. (2020). From new space to big space: how commercial space dream is becoming a reality. Acta Astronaut. 166, 431–443. doi:10.1016/j.actaastro.2019.08.031
Forteo (2010). (teriparatide) package insert. Available at: http://pi.lilly.com/us/forteo-pi.pdf (Accessed November 29, 2022).
Gabel, L., Liphardt, A.-M., Hulme, P. A., Heer, M., Zwart, S. R., Sibonga, J. D., et al. (2022). Pre-flight exercise and bone metabolism predict unloading-induced bone loss due to spaceflight. Br. J. Sports Med. 56, 196–203. doi:10.1136/bjsports-2020-103602
Godin, P. J., Schuerger, A. C., and Moores, J. E. (2021). Salt tolerance and UV protection of Bacillus subtilis and Enterococcus faecalis under simulated martian conditions. Astrobiology 21, 394–404. doi:10.1089/ast.2020.2285
Gregorio, N. E., Kao, W. Y., Williams, L. C., Hight, C. M., Patel, P., Watts, K. R., et al. (2020). Unlocking applications of cell-free biotechnology through enhanced shelf life and productivity of E. coli extracts. ACS Synth. Biol. 9, 766–778. doi:10.1021/acssynbio.9b00433
Gu, Y., Xu, X., Wu, Y., Niu, T., Liu, Y., Li, J., et al. (2018). Advances and prospects of Bacillus subtilis cellular factories: from rational design to industrial applications. Metab. Eng. 50, 109–121. doi:10.1016/j.ymben.2018.05.006
Guiziou, S., Sauveplane, V., Chang, H.-J., Clerté, C., Declerck, N., Jules, M., et al. (2016). A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 44, 7495–7508. doi:10.1093/nar/gkw624
Hodkinson, P. D., Anderton, R. A., Posselt, B. N., and Fong, K. J. (2017). An overview of space medicine. Br. J. Anaesth. 119, i143–i153. doi:10.1093/bja/aex336
Horneck, G. (1993). Responses of Bacillus subtilis spores to space environment: results from experiments in space. Orig. Life Evol. Biosphere J. Int. Soc. Study Orig. Life 23, 37–52. doi:10.1007/BF01581989
Ivanova, E. P., Vysotskii, M. V., Svetashev, V. I., Nedashkovskaya, O. I., Gorshkova, N. M., Mikhailov, V. V., et al. (1999). Characterization of Bacillus strains of marine origin. Int. Microbiol. 2 (4), 267–271.
Kakeshita, H., Kageyama, Y., Ozaki, K., Nakamura, K., Ara, K., Kakeshita, H., et al. (2012). Improvement of heterologous protein secretion by Bacillus subtilis. IntechOpen. doi:10.5772/29256
Kang, Z., Yang, S., Du, G., and Chen, J. (2014). Molecular engineering of secretory machinery components for high-level secretion of proteins in Bacillus species. J. Ind. Microbiol. Biotechnol. 41, 1599–1607. doi:10.1007/s10295-014-1506-4
Kast, J., Yu, Y., Seubert, C. N., Wotring, V. E., and Derendorf, H. (2017). Drugs in space: pharmacokinetics and pharmacodynamics in astronauts. Eur. J. Pharm. Sci. 109, S2–S8. doi:10.1016/j.ejps.2017.05.025
Liu, Y., Li, J., Du, G., Chen, J., and Liu, L. (2017). Metabolic engineering of Bacillus subtilis fueled by systems biology: recent advances and future directions. Biotechnol. Adv. 35, 20–30. doi:10.1016/j.biotechadv.2016.11.003
Lyon, R. C., Taylor, J. S., Porter, D. A., Prasanna, H. R., and Hussain, A. S. (2006). Stability profiles of drug products extended beyond labeled expiration dates. J. Pharm. Sci. 95, 1549–1560. doi:10.1002/jps.20636
Mars, K. (2017). The translational research Institute for space health (TRISH). NASA. Available at: http://www.nasa.gov/hrp/tri (Accessed November 20, 2022).
McCutcheon, G., Kent, R., Paulino-Lima, I., Pless, E., Ricco, A., Mazmanian, E., et al. (2016). “PowerCell payload on Eu:CROPIS - measuring synthetic biology in space,” in Small Satell. Conf. Available at: https://digitalcommons.usu.edu/smallsat/2016/TS11SciPayload1/4.
Medical Checklist (2006). Medical checklist. Available at: https://www.nasa.gov/centers/johnson/pdf/359892main_125_MED_G_K_4_E1.pdf.
Mondol, M. A. M., Shin, H. J., and Islam, M. T. (2013). Diversity of secondary metabolites from marine Bacillus species: chemistry and biological activity. Mar. Drugs 11, 2846–2872. doi:10.3390/md11082846
Munakata, N., Saito, M., and Hieda, K. (1991). Inactivation action spectra of BACILLUS subtilis spores in extended ultraviolet wavelengths (50–300nm) obtained with synchrotron radiation. Photochem. Photobiol. 54, 761–768. doi:10.1111/j.1751-1097.1991.tb02087.x
NASA (2020). ARTEMIS plan: NASA’s lunar exploration program overview. Available at: https://www.nasa.gov/sites/default/files/atoms/files/artemis_plan-20200921.pdf (Accessed December 16, 2022).
NASA Technology Roadmaps - TA 6: Human Health, Life Support, and Habitation Systems (2015). NASA technology Roadmaps. Hum. Health 217.
Pandey, S., Sree, A., Sethi, D. P., Kumar, C. G., Kakollu, S., Chowdhury, L., et al. (2014). A marine sponge associated strain of Bacillus subtilis and other marine bacteria can produce anticholinesterase compounds. Microb. Cell Factories 13, 24. doi:10.1186/1475-2859-13-24
Pardee, K., Green, A. A., Ferrante, T., Cameron, D. E., DaleyKeyser, A., Yin, P., et al. (2014). Paper-based synthetic gene networks. Cell 159, 940–954. doi:10.1016/j.cell.2014.10.004
Quattrocchi, E., and Kourlas, H. (2004). Teriparatide: a review. Clin. Ther. 26, 841–854. doi:10.1016/S0149-2918(04)90128-2
Rothschild, L. J. (2016). Synthetic biology meets bioprinting: enabling technologies for humans on Mars (and Earth). Biochem. Soc. Trans. 44, 1158–1164. doi:10.1042/BST20160067
Souza, C. C. de, Guimarães, J. M., Pereira, S., dos, S., and Mariúba, L. A. M. (2021). The multifunctionality of expression systems in Bacillus subtilis: emerging devices for the production of recombinant proteins. Exp. Biol. Med. 246, 2443–2453. doi:10.1177/15353702211030189
Su, Y., Liu, C., Fang, H., and Zhang, D. (2020). Bacillus subtilis: a universal cell factory for industry, agriculture, biomaterials and medicine. Microb. Cell Factories 19, 173. doi:10.1186/s12934-020-01436-8
Taylor, P. W. (2015). Impact of space flight on bacterial virulence and antibiotic susceptibility. Infect. Drug Resist. 8, 249–262. doi:10.2147/IDR.S67275
Tisa, L. S., Koshikawa, T., and Gerhardt, P. (1982). Wet and dry bacterial spore densities determined by buoyant sedimentation. Appl. Environ. Microbiol. 43, 1307–1310. doi:10.1128/aem.43.6.1307-1310.1982
Vallota-Eastman, A., Bui, C., Williams, P. M., Valentine, D. L., Loftus, D., and Rothschild, L. (2023). Bacillus subtilis engineered for aerospace medicine: a platform for off-planet production of pharmaceutical peptides. bioRxiv. 2023.02.22.529550. doi:10.1101/2023.02.22.529550
van Dijl, J., and Hecker, M. (2013). Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb. Cell Factories 12, 3. doi:10.1186/1475-2859-12-3
Velupillaimani, D., and Muthaiyan, A. (2019). Potential of Bacillus subtilis from marine environment to degrade aromatic hydrocarbons. Environ. Sustain. 2, 381–389. doi:10.1007/s42398-019-00080-2
Warfel, K. F., Williams, A., Wong, D. A., Sobol, S. E., Desai, P., Li, J., et al. (2023). A low-cost, thermostable, cell-free protein synthesis platform for on-demand production of conjugate vaccines. ACS Synth. Biol. 12, 95–107. doi:10.1021/acssynbio.2c00392
Westers, L., Westers, H., and Quax, W. J. (2004). Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1694, 299–310. doi:10.1016/j.bbamcr.2004.02.011
Williams, P. M., Fowler, S. B., Best, R. B., Toca-Herrera, J. L., Scott, K. A., Steward, A., et al. (2003). Hidden complexity in the mechanical properties of titin. Nature 422, 446–449. doi:10.1038/nature01517
Wright, C. R., Ward, A. C., and Russell, A. P. (2017). Granulocyte colony-stimulating factor and its potential application for skeletal muscle repair and regeneration. Mediat. Inflamm. 2017, 1–9. doi:10.1155/2017/7517350
Yang, H., Qu, J., Zou, W., Shen, W., and Chen, X. (2021). An overview and future prospects of recombinant protein production in Bacillus subtilis. Appl. Microbiol. Biotechnol. 105, 6607–6626. doi:10.1007/s00253-021-11533-2
Yoshida, K., Yamaguchi, M., Morinaga, T., Ikeuchi, M., Kinehara, M., and Ashida, H. (2006). Genetic modification of Bacillus subtilis for production of d-chiro-Inositol, an investigational drug candidate for treatment of type 2 diabetes and polycystic ovary syndrome. Appl. Environ. Microbiol. 72, 1310–1315. doi:10.1128/AEM.72.2.1310-1315.2006
Zhang, Q., Wu, Y., Gong, M., Zhang, H., Liu, Y., Lv, X., et al. (2021). Production of proteins and commodity chemicals using engineered Bacillus subtilis platform strain. Essays Biochem. 65, 173–185. doi:10.1042/EBC20210011
Keywords: Astropharmacy, biologics, space, medicine, NASA, pharmaceuticals, teriparatide, filgrastim
Citation: Vallota-Eastman A, Bui C, Williams PM, Valentine DL, Loftus D and Rothschild L (2023) Bacillus subtilis engineered for aerospace medicine: a platform for on-demand production of pharmaceutical peptides. Front. Space Technol. 4:1181843. doi: 10.3389/frspt.2023.1181843
Received: 08 March 2023; Accepted: 16 November 2023;
Published: 30 November 2023.
Edited by:
Mark Sephton, Imperial College London, United KingdomReviewed by:
Sarfaraz Niazi, University of Illinois, United StatesApurv Mhatre, Oak Ridge National Laboratory (DOE), United States
Copyright © 2023 Vallota-Eastman, Bui, Williams, Valentine, Loftus and Rothschild. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lynn Rothschild, bHlubi5qLnJvdGhzY2hpbGRAbmFzYS5nb3Y=