 Peter Naylor1,2,3†
Peter Naylor1,2,3† Tristan Lazard1,2,3
Tristan Lazard1,2,3 Guillaume Bataillon4†Marick Laé4,5Anne Vincent-Salomon4,6Anne-Sophie Hamy7,8,9Fabien Reyal7,8,10
Guillaume Bataillon4†Marick Laé4,5Anne Vincent-Salomon4,6Anne-Sophie Hamy7,8,9Fabien Reyal7,8,10 Thomas Walter1,2,3*
Thomas Walter1,2,3*- 1Centre for Computational Biology (CBIO), MINES ParisTech, PSL University, Paris, France
- 2Institut Curie, Paris, France
- 3INSERM U900, Paris, France
- 4Department of Diagnostic and Theranostic Medicine, Institut Curie, PSL University, Paris, France
- 5Department of Pathology, Centre Henri Becquerel, INSERM U1245, UniRouen Normandie Université, Rouen, France
- 6INSERM U934, CNRS UMR3215, Paris, France
- 7Residual Tumor and Response to Treatment Laboratory, RT2Lab, Translational Research Department, Institut Curie, Paris, France
- 8U932, Immunity and Cancer, INSERM, Institut Curie, Paris, France
- 9Department of Medical Oncology, Institut Curie, Paris, France
- 10Department of Surgery, Institut Curie, Paris, France
The automatic analysis of stained histological sections is becoming increasingly popular. Deep Learning is today the method of choice for the computational analysis of such data, and has shown spectacular results for large datasets for a large variety of cancer types and prediction tasks. On the other hand, many scientific questions relate to small, highly specific cohorts. Such cohorts pose serious challenges for Deep Learning, typically trained on large datasets. In this article, we propose a modification of the standard nested cross-validation procedure for hyperparameter tuning and model selection, dedicated to the analysis of small cohorts. We also propose a new architecture for the particularly challenging question of treatment prediction, and apply this workflow to the prediction of response to neoadjuvant chemotherapy for Triple Negative Breast Cancer.
1 Introduction
1.1 Context
Breast cancer (BC) is the most common cancer in women and the leading cause of cancer deaths with 18.2% of deaths among female cancer patients and 8% among all cancer patients (Institut National Du Cancer, 2019). Out of the four main breast cancer types, Triple Negative Breast Cancer (TNBC) represents 10% of all BC patients. This group has the worst prognostic with a five-year survival rate of around 77 percent versus 93 percent for the others. Currently no specialised treatments exists and the standard procedure consists in administrating neoadjuvant or adjuvant chemotherapy (Sakuma et al., 2011). TNBC research is still a very active field of study (Foulkes et al., 2010) and on the one hand, most works have focused on stratifying cohorts based on molecular and biological profiles (Lehmann et al., 2016). We, on the other hand, tackle the problem of predicting the response variable in a TNBC neoadjuvant chemotherapy (NACT) cohort from a histological needle-core biopsy section from the primary tumor prior to treatment. In contrast to most of the effort in cancer research, which is driven by the analysis of sequencing data, our study is based solely on the histological image data prior to treatment.
Each histological sample corresponds to tissue slides encompassing the tumor and its surrounding, stained with agents in order to highlight specific structures, such as cells, cell nuclei or collagen. The morphological properties of these elements and their spatial organisation have been linked to cancer subtypes, grades and prognosis. Even if pathologists have been trained to understand and report the evidence found in this type of data, the complexity, size and heterogeneity found in histological specimens make it highly unlikely that all relevant and exploitable patterns are known of today. Tissue images are informative about morphological and spatial patterns and are therefore inherently complementary to omics data.
Two major technological advances have triggered the emergence of the field of Computational Pathology: first, the arrival of new and powerful scanners replaced to some extent the use of conventional microscopes. Today, slides are scanned, stored and can be accessed rapidly at any moment (Huisman et al., 2010). This in turn has lead to the generation of large datasets that can also be analyzed computationally. The second element was the rise of new computer vision techniques.
Indeed, while the analysis of tissue slides has been of interest to the Computer Vision community for many years (Bartels et al., 1988), it is the advent of deep learning that has truly impacted the field. The advent of deep learning has stemmed a wide number of projects and investments. For visual systems, it is the combination of very large annotated dataset (Jia et al., 2009), hardware improvement and convolutional neural networks (CNN) that led to human-like capabilities. These outbreaks in performance have led to the creation of many annotated datasets and to the application of CNN’s to many tasks. However, for biomedical imaging in particular the application of CNN is not always straightforward:
1) The price for generating and annotating large biomedical dataset limits the progress of big data in this domain (Gurcan et al., 2009).
2) In histopathology in particular, each individual sample can be very large, one sample can be up to 60 GB uncompressed. This leads to multiple issues, again linked with the time and price needed for annotation, but also for the subsequent analysis where ad hoc methods have to be used as an entire image does not comfortably fit in RAM.
3) The nature of the data is inherently complex, each biological sample has its own individual patterns to be differentiated with relevant pathological evidence. For histopathology data, samples have a very large inter-slide, but also intra-slide variability that make the apparent signal harder to detect. In addition, the level of detail can be an additional difficulty: the relevant image features may be very fine grained, such as mitotic events, or very large such as the size of relevant image regions (necrotic, tumorous) (Janowczyk and Madabhushi, 2016).
In this paper, we set out to predict the response to NACT in TNBC from Whole Slide Images (WSI). Prediction of treatment response is one of the most difficult tasks in digital pathology (Echle et al., 2021). Unlike tasks like tumor detection and subtyping, for which high accuracies have been achieved in the past (Echle et al., 2021), there are no known morphological phenotypes that would allow for the prediction of treatment response. Moreover, datasets with treatment response are usually much smaller than datasets acquired in clinical routine for tasks such as automatic grading, metastasis detection or subtype prediction.
The contributions of this article are:
1) We benchmark several state-of-the-art architectures with respect to their performance in treatment response prediction.
2) We propose a new architecture for the prediction of treatment response and demonstrate its efficiency.
3) We propose a suitable model selection procedure that can cope with small datasets and avoids re-training, which is particularly important for deep learning. We prove its validity and efficiency on simulated data.
The paper is organised as follows: in the next Section 1.2 we describe related work. In Section 2, we describe the methodological developments. In Section 2.1, we present the limits of the current validation procedures and our alternative method used in this study. We then introduce our histopathology dataset in Section 2.3. Section 2.4 is devoted to introducing the DNN architectures which will be applied to the TNBC cohort. In Section 3, we show our results on the simulated data for our validation procedure and the application of our DNN to the TNBC cohort. Finally in Section 4 we discuss our methods and results.
1.2 Related Work
1.2.1 Challenges in Computational Pathology
The field of research in computational pathology can be divided in three categories:
1) Preprocessing, in particular color normalisation which aims at reducing the bias introduced by staining protocols used in different centers (Ruifrok and Johnston, 2001; Bejnordi et al., 2016).
2. Detection, segmentation and classification of objects of interest, such as regions (Bejnordi et al., 2017; Chan et al., 2019; Barmpoutis et al., 2021) and nuclei (Naylor et al., 2018; Graham et al., 2019).
3) The prediction of slide variables, such as presence and detection of disease (Litjens et al., 2018; Campanella et al., 2019), grading (Niazi et al., 2016; Ström et al., 2020), survival (Zhu et al., 2017; Courtiol et al., 2019), gene expression (Binder et al., 2018; Schmauch et al., 2020), genetic mutations (Coudray et al., 2018) or genetic signatures (Kather et al., 2020; Lazard et al., 2021).
Of note, these different of Computer Vision tasks have varying degrees of difficulty. For instance, Deep Learning based tumor detection and subtyping can be achieved with high accuracies [AUC: 0.97–0.99 for detection, AUC: 0.85–0.98 for subtyping, (Echle et al., 2021)]. This is not surprising, as these tasks rely on well-known visual cues. In contrast, prediction of treatment response is deemed one of the most difficult tasks in digital pathology (Echle et al., 2021), because the related morphological patterns are unknown and could potentially be very complex. It is even not clear to which extent treatment response is actually predictable from WSI.
Pipelines for slide variable predictions are usually divided into several steps. Tiles are partitioned into smaller images, usually referred to as patches or tiles, which are then encoded by a DNN, often trained on ImageNet (Zhu et al., 2017; Courtiol et al., 2019), as depicted in Figure 2. The training on ImageNet might be surprising at first sight, as the nature of the images are very different. In addition, ImageNet samples usually have a natural orientation, where the main object of interest is usually centered and scaled to fit in the image (Raghu et al., 2019). In contrast, histopathology images have a rotationally invariant content with no prior regarding scale or positioning of the relevant structures. However, rotational invariance can be imposed (Naylor et al., 2019; Lafarge et al., 2020), and in practice ImageNet based encodings are widely used and tend to perform very well.
After encoding of all tiles, each WSI is converted into a P × ni matrix where P is the encoding size and ni the number of tiles. The last step consists in aggregating tile level encodings to perform unsupervised or supervised predictions at the slide level (Zhu et al., 2017; Courtiol et al., 2018; Campanella et al., 2019; Courtiol et al., 2019; Naylor et al., 2019).
Computational Pathology as a field has benefited from the generation of large annotated data sets, mostly with pixel-level annotations (Kumar et al., 2017; Litjens et al., 2018; Naylor et al., 2018), or cell-level annotations (Veta et al., 2015) for cell classification. The major resource for WSI with slide level annotations are the Cancer Genome Atlas (TCGA) and the Camelyon Challenge (Litjens et al., 2018). These public repositories are paralleled by many in-house datasets (thus not accessible to the public), some of which can be very large, namely in a screening context, e.g., (Campanella et al., 2019). In most cases however, the datasets tend to be very small and fall therefore in the small n large p category. This is due to the fact that often the most interesting studies focus on particular molecularly defined cancer subtypes for which only small cohorts exist. In addition, collecting the output variable might be very challenging and time-consuming, if the project is not formulated in the context of Computer Aided Diagnosis. This is particularly true for treatment response prediction.
1.2.2 Challenges in Applying DNN to Small Datasets
For all supervised learning method it is custom to use a two step procedure for estimating the performance. After dividing your dataset into three categories: train, validation and test. The first step consists in performing model selection with the training and validation set. The second step simply involves evaluating the chosen model on the test set in order to assess an unbiased estimator of the performance (Pereira et al., 2009). This is however only possible if the three categories are large enough. When the validation set is too small and the discrepancy in the data too high, one could very easily over-fit or under-fit on the dataset (Varoquaux, 2018). When the number of samples n is small, which is usually the case for biomedical data, alternative validation methods have to be found such as cross-validation (CV) and nested cross-validation (NCV). CV is mostly used for model selection or assessing performance. NCV is used when the model needs a tuning based on an external dataset, such as hyperparameter tunning for Support Vector Machines. Even if these methods have been debated (Krstajic et al., 2014; Varoquaux, 2018; Wainer and Cawley, 2018), they are widely accepted. These methods are explained in more details in Section 2.1.
1.2.3 Prediction of the Response to Neoadjuvant Chemotherapy in Triple Negative Breast Cancer
Neoadjuvant chemotherapy (NACT) responses varies among patients in TNBC and no clear biological signal has been shown. Survival in these cohorts have been correlated to the Residual Cancer Burden (RCB) variable (Symmans et al., 2007) which can be used as a proxy for response. RCB is a pathological variable based on measurements of how much the primary tumor has shrunk and of the size of metastasis in axillary lymph nodes. Finding biological evidence to NACT response would allow for adequate and specific treatment, some histological variables have been found to be correlated with survival, such as the number Ki-67 positive cells (Elnemr et al., 2016), tumor infiltrating lymphocytes (Mao et al., 2016) and the Elston and Ellis grade (Elston and Ellis, 1991). Depending on the context, some alternative treatments have been found to help overall survival, such as those based on anthracycline and taxanes (Sakuma et al., 2011), carboplatin (Pandy et al., 2019) or with olaparib and talazoparib (Won and Spruck, 2020). Some treatments have emerged with targeted immunotherapy in combination with atezolizumab (anti-PD-L1 antibody) and nanoparticle albumin-bound (nab)-paclitaxel (Won and Spruck, 2020). Most of the studies for NACT responses have been performed in clinical practices and based on pathological variables (Elnemr et al., 2016; Gass et al., 2018; Zhu et al., 2020). In addition, some studies have analysed sequencing and molecular profiles in order to better understand and stratify cohorts (Lehmann et al., 2016; García-Vazquez et al., 2017; Wang et al., 2019).
To the best of our knowledge, it remains unclear whether and to which extent NACT response can be predicted from biopsies taken prior to treatment, and only few works have addressed this question so far (Naylor et al., 2019; Ogier du Terrail et al., 2021).
2 Materials and Methods
2.1 Validation Procedure
Here, we present our procedures to replace NCV in order to train DNN in a context of small n large p. We first explain cross-validation (CV), nested cross-validation (NCV) and their limitations. We propose two different procedures, better suited and show their effectiveness on a small case study.
2.1.1 Cross-Validation
CV is a common procedure for model evaluation or model selection, specifically in situations where the data set is relatively small. CV divides the initial data set into kf folds, denoted
2.1.1.1 Cross-Validation for Model Selection
CV can be used for model selection or model tuning. The procedure that returns a tuned model
We give the pseudo code in Algorithm 1, where
Algorithm 1. Model selection, fcv.

2.1.1.2 Cross-Validation for Model Evaluation
We can use CV to evaluate a given model and a HP set h. The procedure is similar to the pseudo code given in Algorithm 1, however we give in input a model and only one hyperparameter set and return
2.1.1.3 Nested Cross-Validation
NCV is a procedure that allows one to tune a model and effectively report an unbiased estimation of the performance of the tuned model.
Given sets of HP and a data set
Algorithm 2. Nested cross-validation.
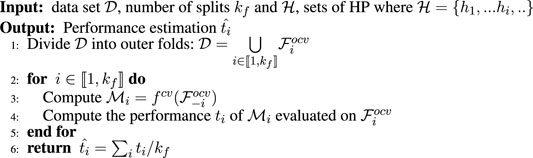
Another possible view is to see NCV as a simple CV for a model selection algorithm. For NCV, the model selection algorithm would be fcv.It is important to note that as we are training DNN, we do not use a fixed hyperparameter set,
2.1.2 Limitations
DNN training suffers from inherent randomness, as the loss function is highly non-convex and possess many symmetries (Bishop, 2006). In addition, there are some stochastic differences between training runs, such as the random initialization of the weight parameters and the data shuffling, naturally leading to different solutions. Especially for small datasets, these stochastic variations lead to notable differences in performance when we repeat training with the same hyperparameters.
In the classical setting, CV provides us with a set of hyperparameters that lead to a model with optimal performance, as estimated in the inner loop. For DNN trained on small datasets, there is no guarantee that the same set of hyperparameters will lead to similar performance, and for this reason retraining is not guaranteed to lead to a very good solution.
Another problem with the retraining in line 10 in Algorithm 1 is the use of early stopping. Early stopping is a very powerful regularization procedures that choses experimentally the point between the under- and over-fitting regime, but for this it requires a validation set. Early stopping would therefore not be applicable in the traditional CV-scheme with retraining.
2.1.3 Nested Ensemble Cross-Validation
Due to the incompatibility between NCV and early stopping we propose to modify the model selection procedure, i.e., function fcv shown in Algorithm 1. In particular we do not perform retraining and return an ensemble of the models used during CV. Similarly to NCV, we perform a CV where we propose to modify fcv into a better suited procedure, named fecv (ensemble cross-validation), shown in Algorithm 3.
Algorithm 3. Model selection, fecv.

The main difference between fcv and fecv is that we remove the final model retraining, i.e., line 10 of Algorithm 1 and give back the full set of kf models trained for all folds for the maximizing hyperparameters; the prediction is obtained by ensembling these models.The advantage of this procedure is that we omit the retraining step which allows us to use early stopping for all individual models. In addition, we add another level of regularization by the ensembling. Of note, fecv can be used in an inner loop, too. This then leads to Nested Ensemble cross-validation (NECV).
2.1.4 Nested Best Fold Ensemble Cross-Validation
Similarly to the procedure NECV, we can define another procedure that we name NBFCV. NBFCV is based on fbfcv defined in Algorithm 4. Compared to the preceding procedure, we inverted the two for-loops and retain the best model per fold. It then returns the ensemble of these selected models.
Algorithm 4. Model selection, fbfcv.

2.2 Simulations
In order to compare and validate our procedure presented in Section 2.1 we propose to conduct a series of simulation studies where the results will be given in Section 3.1. In particular, we wish to demonstrate that DNN training given a set of HP can lead to inconsistent models and that NCV therefore might provide under-performing models compared to NECV and NBFCV, the proposed validation procedures.
2.2.1 Data Set Simulation
We simulated a simple balanced binary data, of size N = 350 in a medium to high dimensional setting with p = 256. We have ∀i ∈ [(1, N)], Yi ∈ (−1, 1) and
FIGURE 1. Data set simulation with p = 256 and model, (A) σ = 2, (B) σ = 6, (C) σ = 10, (D) σ = 14. (E) Simple two layer DNN model for predicting the class label.
2.2.2 Model
We apply a simple DNN model, composed of two layers, a first hidden layer with 256 hidden nodes and a classification layer with two nodes, the model is depicted in Figure 1E.
In particular, we minimise a cross-entropy error with an Adam optimiser and a batch size of 16. The tunable parameters are the weight-decay, drop out and learning rate. As an extra regularisation we use batch normalisation.
2.3 Application to Histopathology Data
2.3.1 Data Generation and Annotation
The data set used was generated at the Curie Institute and consists of annotated H&E stained histology needle core biopsy sections at 40× magnification sampled from a patient suffering from TNBC. In this paper, we evaluate the prediction of the response to treatment based solely on a biopsy sectioned prior to the treatment. As discussed in the introduction, not all patients respond to NACT, and we are therefore aiming at predicting the response to NACT based on the biopsy. In particular, each section was quality checked by expert histopathologists.
For each patient, we also collect WSI after surgery, allowing an expert pathologist to establish the residual cancer burden, as a proxy for treatment success. Out of the 336 samples that populate our data set, 167 were annotated as RCB-0, 27 as RCB-I, 113 as RCB-II and 29 as RCB-III. This data set is twice as large as the data set used in our previous study (Naylor et al., 2019). Similarly to this study, we refine the number of classes in order to avoid the problem of under-represented class. We investigate two prediction settings: 1) pCR (no residuum) vs. RCB (some residuum) and 2) pCR-RCB-I vs. RCB-II-III, which is clinically more relevant, as it is informative of a patient’s prognosis.
2.3.2 Data Encoding
As each biopsy section is relatively big, we wish to reduce the computational burden of feeding the entire biopsy to our algorithms. Instead, given a magnification factor, we divide each biopsy into tiles of equal sizes, 224 × 224 and project this tile into a lower dimensional space. We use a pre-trained DNN on ImageNet (Jia et al., 2009) such as ResNet (He et al., 2016) which produces a encoding of size 2048. This process is illustrated in Figure 2 where each biopsy section is converted into an encoded matrix of size ni × P where P is the size of the resulting encoding and ni the number of tiles tissue extracted from tissue i,
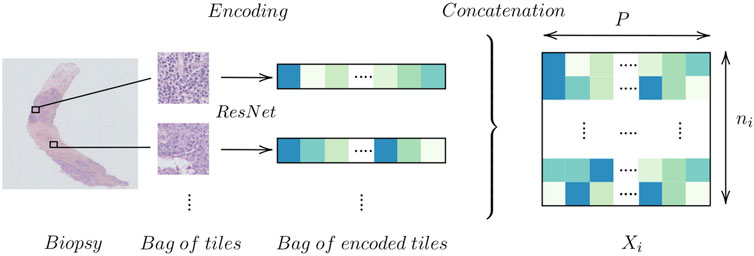
FIGURE 2. Encoding a biopsy.
In Table 1 we show the average number of tiles,
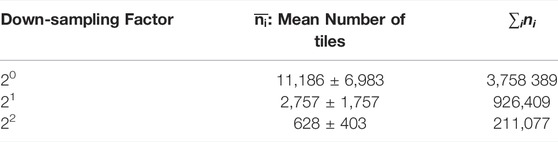
TABLE 1. Mean number of tiles.
The size of the data remains relatively large even after this reduction. We further reduce the size of the tile encoding with a PCA (Jolliffe, 2011), and project each tile encoding into a space approximately 10× smaller. By keeping 256 components, we keep 93.2% at 20, 94.0% at 21 and 94.3% at a magnification factor of 22 of the explained variance.
2.3.3 Mathematical Framework
The data set will be denoted by
2.4 Neural Network Architectures
Today, DNN models for WSI classification usually consist in three steps: starting from encodings that are usually provided by pre-trained networks, a reduction layer might be applied, followed by an aggregation step that computes a slide level representation from the tile level representations and a final module that maps the slide level representation to the output variable.
In Figure 3, we show a basic example for such an architecture along these lines, with the three algorithmic blocks highlighted in gray. At the tile-level computation, we use 1D convolutions to transform the input encodings Xi into a more compact representation
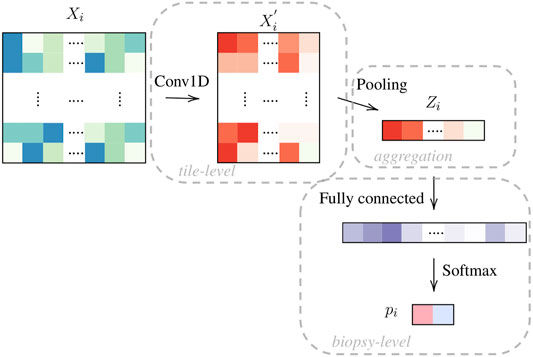
FIGURE 3. Model OneOne: tile encodings Xi from a pretrained network are projected to a more compact representations
In this article, we test several encoding and agglomeration strategies which are explained in the next sections.
2.4.1 Encoding Projection
In Figure 3, the baseline architecture is illustrated (OneOne), with a 1D convolution for the tile level encoding and one fully connected layer at the slide level. In Figure 4, we test a deeper architecture for the encoding projections, consisting in three consecutive 1D convolutions, including bottleneck layers (depicted in orange), according to best practice in deep learning (Huang et al., 2017).
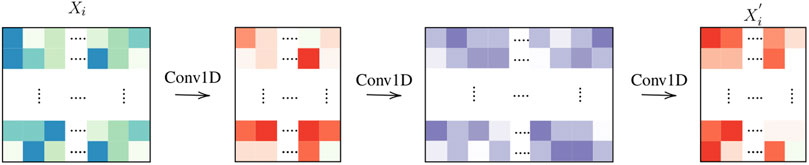
FIGURE 4. Tile computation: Three layers.
Furthermore, we also experiment with skip connections by concatenating the first tile representations to the final representation

FIGURE 5. ThreeTwoSkip.
2.4.2 Pooling Layers
In terms of pooling layers, we experiment with: average pooling shown in Figure 6A, WELDON (Durand et al., 2016) shown in Figure 6B, a modified version of WELDON shown in Figure 6C and the concatenation of the first and the third is named WELDON-C (for context). The DNN that uses WELDON-C will be named CONAN2.

FIGURE 6. Aggregation layers: (A) Average pooling; (B) WELDON pooling; (C) WELDON-C pooling which is WELDON concatenated with previous tile encoding.
The WELDON pooling is an attention-based layer which filters tiles based on a 1D convolution score. In particular, it retains the top and lowest
We recap all the tested models in Table 2.
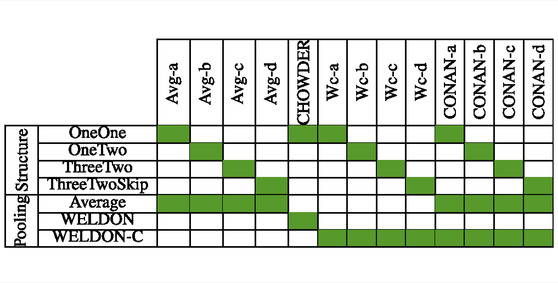
TABLE 2. Model architecture and pooling strategy, as described in the text. CHOWDER has been published previously (Courtiol et al., 2018).
2.4.3 Baseline Approach
In addition to comparing our proposed architectures to CHOWDER, we also compare them to a much simpler approach where we propagate the slide label to the tile level. If a slide is positive, then we assume all extracted tiles from this slide are positive, if a slide is negative, we assume that all tiles are negative. This is the simplest form of MIL. The training set is therefore huge, and we also known that there will be many errors, as many tiles do not contain any useful information regarding treatment response. Nevertheless, such an approach can work if there is a large fraction of informative tiles.
2.4.4 Model Tuning
We perform a random grid search for most parameters and only in suitable ranges. For the learning rate and weight decay we perform a random log sampling for a random scale associated to a random digit. We range from a scale of 10−6 to 10−3 for the learning rate and from 10−4 to 10−1 for the weight decay. We randomly sample a drop out from a uniform
3 Results
3.1 Simulation Results
3.1.1 High Performance Variability in DNN Training
In Figure 7A, we show the average variance of our model with increasing standard deviation σ. In particular, for each σ, we generate 100 simulated dataset with a standard deviation of σ and train 1000 DNN on the same data and with the same HP. As this is simulated data, we evaluate the performance of each training on a large independently simulated test set instead of using the outer CV loop (Varoquaux, 2018). We found that setting the learning rate to 1.10−4, the weight decay to 5.10−3 and drop out to 0.4 tend to always return reasonable results for our simulation setting.
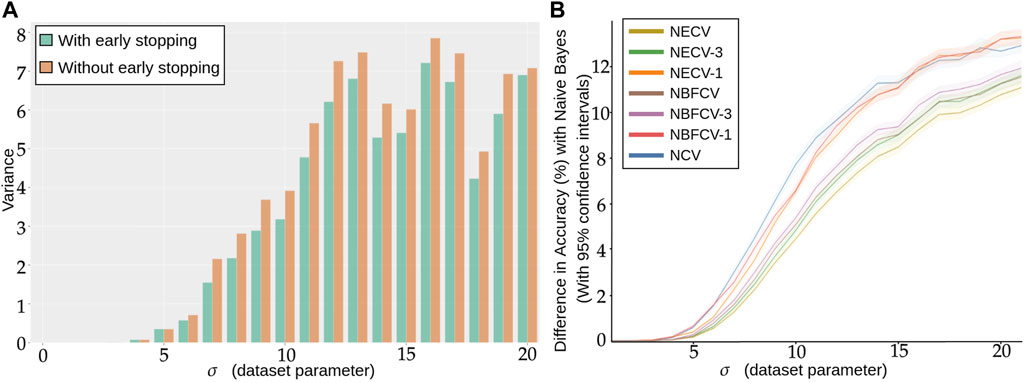
FIGURE 7. (A) Repeated training, with and without early stopping: (A) Performance Variance, as measured on an independently simulated test set, (B) Comparison of models trained with NCV, NECV and NBFCV. On the y-axis we have the difference in accuracy between the Naive Bayes model and a given validation approach. As the data distribution is known, the Naive Bayes is a theoretical upper limit of the achievable performance. Curves are shown with 95% confidence intervals. A lower curve implies a better score.
As the standard deviation σ of the simulated data increases, we expect more overlapping between our two classes and naturally, the classification accuracy decreases. For lower σ, regardless of using early stopping or not, the models reaches perfect scores.
In Figure 7, we observe that the more difficult the problem (larger σ), the lower the accuracy, but also the larger the variance: not only do we predict less well, but also does the performance variation increase, such that by retraining a model with the same hyperparameters is not guaranteed at all to provide a model with similar performance. We also see from Figure 7A that early stopping alleviates this problem and consistently reduces the variance in performance, in particular for higher σ.
3.1.2 Nested Cross-Validation Leads to Under-performing Models
We compare the performance of the proposed validation procedures to NCV with the number splits kf = 5 in Figure 7B with 95% confidence intervals around the estimator. On the x-axis we have the standard deviation σ of the simulated data and on the y-axis we have the difference between the Naive Bayes estimator with the corresponding performance, therefore the lower the curve the better the procedure. In addition to Figure 7B, we give the corresponding accuracy score for each σ and validation procedure explicitly in Supplementary Table S1. As the dataset distribution is known, the Naive Bayes is the best classification rule that can be implemented and can be viewwed as an upper bound of the performance. For each σ, we collect 100 estimators with Algorithm 1, Algorithm 3 and Algorithm 4. Next, we compare several strategies for NECV and NBFCV: NECV-1/NBFCV-1, where we keep the best scoring model of the ensemble, NECV-3/NBFCV-3, where we keep the top 3 models and NECV/NBFCV, where we keep all five models.
We first notice that the NCV curve is lower or equal to any of the NECV/NBFCV curves. The best performing model is NECV—i.e., the average of all selected models from the inner CV. In particular NECV has a higher Accuracy than NCV by at least 2%. NBFCV under-performs by a slight margin compared to NECV and seems to be equivalent to NECV-3 in terms of performance.
We conclude that retraining the model without an outer validation score leads to lower overall performance and early stopping is a very useful regularization technique for small sample size problems.
3.2 Prediction of Response to Neoadjuvant Chemotherapy
We next applied the different architectures detailed in Section 2.4 (also summarized in Table 2) to the problem of the prediction of response to neoadjuvant chemotherapy in TNBC. We tested 3 different image resolutions (0, 1, 2), 0 being the highest resolution. In order to get realistic estimations of the performance, while using early stopping, we perform the validation proposed in Section 2.1. In Figures 8A,B we show the average AUC ROC performance on the residual and prognostic prediction tasks, for all methods shown in Table 2 and for all resolution levels.
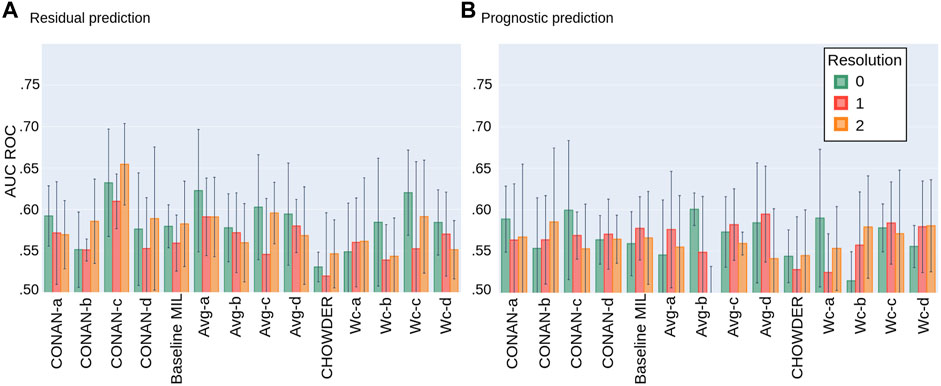
FIGURE 8. Average AUC ROC performance with standard deviation on five fold NECV on the task of predicting: (A) residual cancer and (B) patient prognostic in TNBC.
For the task of predicting the residual cancer, the best performing model would be the CONAN-c model at resolution 2 with an AUC of 0.654 ± 0.049. Others model’s performance range in between 0.55 and 0.60 of AUC with higher standard deviations. Models at resolution 0 seem to generally achieve higher scores then those at lower resolutions. Model architecture c seem to be better suited for this task than the others. The Average concatenated to WELDON-C pooling seems to perform slightly better then the rest. The method CHOWDER which gave excellent results on CAMELYON for cancer detection (Courtiol et al., 2018) and which has been a state-of-the-art solution in the field under-performs on our dataset for response prediction.
For the task of predicting the patient prognostic, the best performing model would be the Avg-b model at resolution 0 with an AUC of 0.601 ± 0.019. CONAN-c at resolution 2 performs similarly but with a much higher standard deviation. Neither resolution, nor model architecture and pooling layer seem to unanimously be better then the others. However, CHOWDER under-performs compared to the other proposed methods.
4 Discussion
In this study, we set out to predict the response to neoadjuvant chemotherapy in TNBC from biopsies taken before treatment. A system that would allow to predict this response with high accuracy could help identifying patients with no or little benefit of the treatment and therefore spare them the heavy burden of the therapy.
From a methodological point of view, this is particularly challenging for three reasons: first, we do not know to which extent the relevant information is actually present in the image data. In addition, even if the relevant information is contained in the slide, the complexity of the related patterns is unclear. Second, biopsies only capture a part of the relevant information, as they are only a localized sample of the tumor. Third, as this is a project regarding a specific subtype, the cohort is relatively small, unlike many pan-cancer cohorts used in large Computational Pathology projects (Campanella et al., 2019).
In order to solve this problem, we have developed the model CONAN, that combines the power of selecting K tiles (top and bottom), but keeps both the ranking scores and the full tile descriptions to build the slide representation. We have compared this model with a number of different architectures, and achieved an AUC of 0.65.
We also tackled an important problem of model selection with cross-validation, a crucial step in particular for small datasets. We found that the retraining step in classical Nested cross-validation can lead to lower performances for small N, because the training is highly variable, and a network retrained with the optimal set of hyperparameters is not guaranteed at all to be optimal itself. We therefore have proposed a new cross-validation procedure relying on ensembling rather than retraining, and thus allowing to use early stopping as a regularization method.
Nevertheless, we must conclude that the prediction of treatment response is probably one of the hardest problems in Computational Pathology, and that even though we see that there is some degree of predictability, the results still seem far from clinical applicability. Also, another problem that is not addressed in this study is the applicability of trained networks across different centers. Clearly, we need more data to tackle these challenging questions. But it is also likely that even with much more data, AUCs will not reach very high levels by looking at biopsies alone. A promising avenue would therefore be to use other kinds of data in addition to histopathology data.
Data Availability Statement
The datasets presented in this article are not readily available because they are the property of Institut Curie. Requests to access the datasets should be directed to dGhvbWFzLndhbHRlckBtaW5lcy1wYXJpc3RlY2guZnI=.
Author Contributions
PN was main contributor, developed the methods and code and generated the results. TL helped with the code. FR and TW designed the project. GB, ML, AVC, A-SH, and FR generated the cohort. GB and ML provided interpretation of the biological results. TW supervised the project. PN and TW wrote the paper.
Funding
PN was funded by the Ligue Contre le Cancer. TL was supported by a Q-Life PhD fellowship (Q-life ANR-17-CONV-0005). Furthermore, this work was supported by the French government under man-agement of Agence Nationale de la Recherche as part of the “Investissements d’avenir” program, reference ANR-19-P3IA-0001 (PRAIRIE 3IA Institute).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank André Nicolas from the PathEx platform, Service the Pathologie, Institut Curie, Paris, where the images have been acquired.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frsip.2022.851809/full#supplementary-material
Footnotes
1For Magnification Factor.
2Context cOncatenated tile selection NeurAl Network
References
Barmpoutis, P., Di Capite, M., Kayhanian, H., Waddingham, W., Alexander, D. C., Jansen, M., et al. (2021). Tertiary Lymphoid Structures (TLS) Identification and Density Assessment on H&E-stained Digital Slides of Lung Cancer. Plos one 16, e0256907. doi:10.1371/journal.pone.0256907
Bartels, P. H., Weber, J. E., and Duckstein, L. (1988). Machine Learning in Quantitative Histopathology. Anal. Quant Cytol. Histol. 10, 299–306.
Bejnordi, B. E., Lin, J., Glass, B., Mullooly, M., Gierach, G. L., Sherman, M. E., et al. (2017). Deep Learning-Based Assessment of Tumor-Associated Stroma for Diagnosing Breast Cancer in Histopathology Images.” in 2017 IEEE 14th international symposium on biomedical imaging (ISBI 2017), 929. doi:10.1109/isbi.2017.7950668
Bergstra, J., and Bengio, Y. (2012). Random Search for Hyper-Parameter Optimization. J. machine Learn. Res. 13, 281–305.
Binder, A., Bockmayr, M., Hägele, M., Wienert, S., Heim, D., Hellweg, K., et al. (2018). Towards Computational Fluorescence Microscopy: Machine Learning-Based Integrated Prediction of Morphological and Molecular Tumor Profiles. arXiv preprint arXiv:1902.07208.
Campanella, G., Hanna, M. G., Geneslaw, L., Miraflor, A., Werneck Krauss Silva, V., Busam, K. J., et al. (2019). Clinical-grade Computational Pathology Using Weakly Supervised Deep Learning on Whole Slide Images. Nat. Med. 25, 1301–1309. doi:10.1038/s41591-019-0508-1
Chan, L., Hosseini, M. S., Rowsell, C., Plataniotis, K. N., and Damaskinos, S. (2019). Histosegnet: Semantic Segmentation of Histological Tissue Type in Whole Slide Images.” in Proceedings of the IEEE/CVF International Conference on Computer Vision. 10662doi:10.1109/iccv.2019.01076
Chollet, F. (2015). Keras. GitHub. Available at: https://github.com/fchollet/keras.
Coudray, N., Ocampo, P. S., Sakellaropoulos, T., Narula, N., Snuderl, M., Fenyö, D., et al. (2018). Classification and Mutation Prediction from Non-small Cell Lung Cancer Histopathology Images Using Deep Learning. Nat. Med. 24, 1559–1567. doi:10.1038/s41591-018-0177-5
Courtiol, P., Maussion, C., Moarii, M., Pronier, E., Pilcer, S., Sefta, M., et al. (2019). Deep Learning-Based Classification of Mesothelioma Improves Prediction of Patient Outcome. Nat. Med. 25, 1519–1525. doi:10.1038/s41591-019-0583-3
Courtiol, P., Tramel, E. W., Sanselme, M., and Wainrib, G. (2018). Classification and Disease Localization in Histopathology Using Only Global Labels: A Weakly-Supervised Approach. arXiv preprint arXiv:1902.07208.
Couture, H. D., Marron, J. S., Perou, C. M., Troester, M. A., and Niethammer, M. (2018). Multiple Instance Learning for Heterogeneous Images: Training a CNN for Histopathology. Lecture Notes Comp. Sci., 11071, 254–262. doi:10.1007/978-3-030-00934-2_29
di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., and Notredame, C. (2017). Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 35, 316–319. doi:10.1038/nbt.3820
Durand, T., Thome, N., and Cord, M. (2016). “WELDON: Weakly Supervised Learning of Deep Convolutional Neural Networks,” in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, Las Vegas, Nevada, June 27–30, 4743–4752. doi:10.1109/CVPR.2016.513
Echle, A., Rindtorff, N. T., Brinker, T. J., Luedde, T., Pearson, A. T., and Kather, J. N. (2021). Deep Learning in Cancer Pathology: a New Generation of Clinical Biomarkers. Br. J. Cancer 124, 686–696. doi:10.1038/s41416-020-01122-x
Ehteshami Bejnordi, B., Litjens, G., Timofeeva, N., Otte-Höller, I., Homeyer, A., Karssemeijer, N., et al. (2016). Stain Specific Standardization of Whole-Slide Histopathological Images. IEEE Trans. Med. Imaging 35, 404–415. doi:10.1109/TMI.2015.2476509
Elnemr, G. M., El-Rashidy, A. H., Osman, A. H., Issa, L. F., Abbas, O. A., Al-Zahrani, A. S., et al. (2016). Response of Triple Negative Breast Cancer to Neoadjuvant Chemotherapy: Correlation between Ki-67 Expression and Pathological Response. Asian Pac. J. Cancer Prev. 17, 807–813. doi:10.7314/apjcp.2016.17.2.807
Elston, C. W., and Ellis, I. O. (1991). Pathological Prognostic Factors in Breast Cancer. I. The Value of Histological Grade in Breast Cancer: Experience from a Large Study with Long-Term Follow-Up. Histopathology 19, 403–410. doi:10.1111/j.1365-2559.1991.tb00229.x
Foulkes, W. D., Smith, I. E., and Reis-Filho, J. S. (2010). Triple-negative Breast Cancer. N. Engl. J. Med. 363, 1938–1948. doi:10.1056/NEJMra1001389
García-Vazquez, R., Ruiz-García, E., Meneses García, A., Astudillo-De La Vega, H., Lara-Medina, F., Alvarado-Miranda, A., et al. (2017). A Microrna Signature Associated with Pathological Complete Response to Novel Neoadjuvant Therapy Regimen in Triple-Negative Breast Cancer. Tumour Biol. 39, 1010428317702899. doi:10.1177/1010428317702899
Gass, P., Lux, M. P., Rauh, C., Hein, A., Bani, M. R., Fiessler, C., et al. (2018). Prediction of Pathological Complete Response and Prognosis in Patients with Neoadjuvant Treatment for Triple-Negative Breast Cancer. BMC cancer 18, 1051–1058. doi:10.1186/s12885-018-4925-1
Graham, S., Vu, Q. D., Raza, S. E. A., Azam, A., Tsang, Y. W., Kwak, J. T., et al. (2019). Hover-Net: Simultaneous Segmentation and Classification of Nuclei in Multi-Tissue Histology Images 58. doi:10.1016/j.media.2019.101563
Gurcan, M. N., Boucheron, L. E., Can, A., Madabhushi, A., Rajpoot, N. M., and Yener, B. (2009). Histopathological Image Analysis: A Review. IEEE Rev. Biomed. Eng. 2, 147–171. doi:10.1109/rbme.2009.2034865
He, K., Zhang, X., Ren, S., and Sun, J. (2016). “Deep Residual Learning for Image Recognition,” in Proceedings of the IEEE conference on computer vision and pattern recognition, Las Vegas, Nevada, June 27–30, 770–778. doi:10.1109/CVPR.2016.90
Huang, G., Liu, Z., Van Der Maaten, L., and Weinberger, K. Q. (2017). “Densely Connected Convolutional Networks,” in Proceedings - 30th IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, Hawaii, July 22–25. (Los Alamitos, California: IEEE Computer Society, Conference Publishing Services), 2261–2269. Available at: https://www.worldcat.org/title/proceedings-30th-ieee-conference-on-computer-vision-and-pattern-recognition-21-26-july-2016-honolulu-hawaii/oclc/1016407672. doi:10.1109/CVPR.2017.243
Huisman, A., Looijen, A., van den Brink, S. M., and van Diest, P. J. (2010). Creation of a Fully Digital Pathology Slide Archive by High-Volume Tissue Slide Scanning. Hum. Pathol. 41, 751–757. doi:10.1016/j.humpath.2009.08.026
Janowczyk, A., and Madabhushi, A. (2016). Deep Learning for Digital Pathology Image Analysis: A Comprehensive Tutorial with Selected Use Cases. J. Pathol. Inform. 7, 29. doi:10.4103/2153-3539.186902
Jia, D., Dong, W., Socher, R., Li, L., Li, K., and Fei-Fei, Li. (2009). ImageNet: A Large-Scale Hierarchical Image Database. CVPR, 248–255. doi:10.1109/cvprw.2009.5206848
Kather, J. N., Heij, L. R., Grabsch, H. I., Loeffler, C., Echle, A., Muti, H. S., et al. (2020). Pan-cancer Image-Based Detection of Clinically Actionable Genetic Alterations. Nat. Cancer 1, 789–799. doi:10.1038/s43018-020-0087-6
Krstajic, D., Buturovic, L. J., Leahy, D. E., and Thomas, S. (2014). Cross-validation Pitfalls when Selecting and Assessing Regression and Classification Models. J. Cheminform 6, 10–15. doi:10.1186/1758-2946-6-10
Kumar, N., Verma, R., Sharma, S., Bhargava, S., Vahadane, A., and Sethi, A. (2017). A Dataset and a Technique for Generalized Nuclear Segmentation for Computational Pathology. IEEE Trans. Med. Imaging 36, 1550–1560. doi:10.1109/TMI.2017.2677499
Lafarge, M. W., Bekkers, E. J., Pluim, J. P. W., Duits, R., and Veta, M. (2020). Roto-Translation Equivariant Convolutional Networks: Application to Histopathology Image Analysis. Med. Image Anal. 68, 101849. doi:10.1016/j.media.2020.101849
Lazard, T., Bataillon, G., Naylor, P., Popova, T., Bidard, F.-C., Stoppa-Lyonnet, D., et al. (2021). Deep Learning Identifies New Morphological Patterns of Homologous Recombination Deficiency in Luminal Breast Cancers from Whole Slide Images. Preprint, Cancer Biol. doi:10.1101/2021.09.10.459734
Lehmann, B. D., Jovanović, B., Chen, X., Estrada, M. V., Johnson, K. N., Shyr, Y., et al. (2016). Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PloS one 11, e0157368. doi:10.1371/journal.pone.0157368
Litjens, G., Bandi, P., Ehteshami Bejnordi, B., Geessink, O., Balkenhol, M., Bult, P., et al. (2018). 1399 H&E-stained sentinel Lymph Node Sections of Breast Cancer Patients: the CAMELYON Dataset. GigaScience 7, 65. doi:10.1093/gigascience/giy065
Mao, Y., Qu, Q., Chen, X., Huang, O., Wu, J., and Shen, K. (2016). The Prognostic Value of Tumor-Infiltrating Lymphocytes in Breast Cancer: a Systematic Review and Meta-Analysis. PloS one 11, e0152500. doi:10.1371/journal.pone.0152500
McInnes, L., Healy, J., Saul, N., and Großberger, L. (2018). UMAP: Uniform Manifold Approximation and Projection. Joss 3, 861. doi:10.21105/joss.00861
Naylor, P., Boyd, J., Laé, M., Reyal, F., and Walter, T. (2019). Predicting Residual Cancer burden in a Triple Negative Breast Cancer Cohort.” in Proceedings - International Symposium on Biomedical Imaging, doi:10.1109/ISBI.2019.8759205
Naylor, P., Laé, M., Reyal, F., and Walter, T. (2019). Segmentation of Nuclei in Histopathology Images by Deep Regression of the Distance Map. IEEE Trans. Med. Imaging 38, 448–459. doi:10.1109/TMI.2018.2865709
Niazi, M. K. K., Keluo Yao, K., Zynger, D. L., Clinton, S. K., Chen, J., Koyutürk, M., et al. (2016). Visually Meaningful Histopathological Features for Automatic Grading of Prostate Cancer. IEEE J. Biomed. Health Inform. 21, 1027–1038. doi:10.1109/JBHI.2016.2565515
Ogier du Terrail, J., Leopold, A., Joly, C., Beguier, C., Andreux, M., Maussion, C., et al. (2021). Collaborative Federated Learning behind Hospitals’ Firewalls for Predicting Histological Response to Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. medRxiv. Available at: https://www.medrxiv.org/content/10.1101/2021.10.27.21264834v1.
Pandy, J. G. P., Balolong-Garcia, J. C., Cruz-Ordinario, M. V. B., and Que, F. V. F. (2019). Triple Negative Breast Cancer and Platinum-Based Systemic Treatment: a Meta-Analysis and Systematic Review. BMC cancer 19, 1065–1069. doi:10.1186/s12885-019-6253-5
Pereira, F., Mitchell, T., and Botvinick, M. (2009). Machine Learning Classifiers and Fmri: a Tutorial Overview. Neuroimage 45, S199–S209. doi:10.1016/j.neuroimage.2008.11.007
Raghu, M., Zhang, C., Kleinberg, J., and Bengio, S. (2019). “NeurIPS 2019,” in Proceedings of the 33rd International Conference on Neural Information Processing Systems, December 2019, 3347–3357.
Ruifrok, A. C., and Johnston, D. A. (2001). Quantification of Histochemical Staining by Color Deconvolution. Anal. Quant Cytol. Histol. 23, 291.
Sakuma, K., Kurosumi, M., Oba, H., Kobayashi, Y., Takei, H., Inoue, K., et al. (2011). Pathological Tumor Response to Neoadjuvant Chemotherapy Using Anthracycline and Taxanes in Patients with Triple-Negative Breast Cancer. Exp. Ther. Med. 2, 257–264. doi:10.3892/etm.2011.212
Schmauch, B., Romagnoni, A., Pronier, E., Saillard, C., Maillé, P., Calderaro, J., et al. (2020). A Deep Learning Model to Predict RNA-Seq Expression of Tumours from Whole Slide Images. Nat. Commun. 11, 4. doi:10.1038/s41467-020-17678-4
Ström, P., Kartasalo, K., Olsson, H., Solorzano, L., Delahunt, B., Berney, D. M., et al. (2020). Artificial Intelligence for Diagnosis and Grading of Prostate Cancer in Biopsies: a Population-Based, Diagnostic Study. Lancet Oncol. 21, 222–232. doi:10.1016/s1470-2045(19)30738-7
Symmans, W. F., Peintinger, F., Hatzis, C., Rajan, R., Kuerer, H., Valero, V., et al. (2007). Measurement of Residual Breast Cancer burden to Predict Survival after Neoadjuvant Chemotherapy. Jco 25, 4414–4422. doi:10.1200/JCO.2007.10.6823
Varoquaux, G. (2018). Cross-validation Failure: Small Sample Sizes lead to Large Error Bars. Neuroimage 180, 68–77. doi:10.1016/j.neuroimage.2017.06.061
Veta, M., van Diest, P. J., Willems, S. M., Wang, H., Madabhushi, A., Cruz-Roa, A., et al. (2015). Assessment of Algorithms for Mitosis Detection in Breast Cancer Histopathology Images. Med. Image Anal. 20, 237–248. doi:10.1016/j.media.2014.11.010
Wainer, J., and Cawley, G. (2018). “Nested Cross-Validation when Selecting Classifiers Is Overzealous for Most Practical Applications,” in Expert Systems with Applications 182, 115222. doi:10.1016/j.eswa.2021.115222
Wang, D. Y., Jiang, Z., Ben-David, Y., Woodgett, J. R., and Zacksenhaus, E. (2019). Molecular Stratification within Triple-Negative Breast Cancer Subtypes. Sci. Rep. 9, 19107–19110. doi:10.1038/s41598-019-55710-w
Won, K. A., and Spruck, C. (2020). Triple-negative Breast Cancer Therapy: Current and Future Perspectives. Int. J. Oncol. 57 (6), 1245–1261. doi:10.3892/ijo.2020.5135
Xu, Y., Li, Y., Shen, Z., Wu, Z., Gao, T., Fan, Y., et al. (2017). Parallel Multiple Instance Learning for Extremely Large Histopathology Image Analysis. BMC Bioinformatics 18, 360. doi:10.1186/s12859-017-1768-8
Xu, Y., Zhang, J., Chang, E. I.-C., Lai, M., and Tu, Z. (2012). Context-constrained Multiple Instance Learning for Histopathology Image Segmentation. Lecture Notes Comp. Sci. 751, 623–630. doi:10.1007/978-3-642-33454-2_77
Zhu, M., Yu, Y., Shao, X., Zhu, L., and Wang, L. (2020). Predictors of Response and Survival Outcomes of Triple Negative Breast Cancer Receiving Neoadjuvant Chemotherapy. Chemotherapy 65, 1–9. doi:10.1159/000509638
Zhu, X., Yao, J., Zhu, F., and Huang, J. (2017). “WSISA: Making Survival Prediction from Whole Slide Histopathological Images,” in Proceedings - 30th IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, Hawaii, July 22–25 (Los Alamitos, California: IEEE Computer Society, Conference Publishing Services), 6855–6863. doi:10.1109/CVPR.2017.725
Keywords: breast cancer, digital pathology, whole slide images, treatment response, cross-validation, deep learning, multiple instance learning, small sample size
Citation: Naylor P, Lazard T, Bataillon G, Laé M, Vincent-Salomon A, Hamy A-S, Reyal F and Walter T (2022) Prediction of Treatment Response in Triple Negative Breast Cancer From Whole Slide Images. Front. Sig. Proc. 2:851809. doi: 10.3389/frsip.2022.851809
Received: 10 January 2022; Accepted: 21 March 2022;
Published: 22 June 2022.
Edited by:
Elaine Wan Ling Chan, International Medical University, MalaysiaReviewed by:
Jónathan Heras, University of La Rioja, SpainPanagiotis Barmpoutis, University College London, United Kingdom
Copyright © 2022 Naylor, Lazard, Bataillon, Laé, Vincent-Salomon, Hamy, Reyal and Walter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Walter, dGhvbWFzLndhbHRlckBtaW5lcy1wYXJpc3RlY2guZnI=
†Present Address: Peter Naylor, RIKEN AIP, Chuo-ku, Japan Guillaume Bataillon, Department of Pathology, IUCT, Toulouse, France