 Jasper Feng
Jasper Feng Jürgen Allgaier1
Jürgen Allgaier1 Margarita Kruteva
Margarita Kruteva Wim Pyckhout-Hintzen
Wim Pyckhout-Hintzen- 1Neutron Scattering and Biological Matter (JCNS-1/IBI-8), Forschungszentrum Jülich GmbH, Jülich, Germany
- 2Institute of Physical Chemistry, RWTH Aachen University, Aachen, Germany
Polymer networks containing transient physical and permanent chemical cross-links exhibit unique mechanical properties due to the intrinsic reassociating ability of supramolecular functional groups. Similar to supramolecular gels, these networks allow the controlled release of stored energy and can extend the life of polymer networks in practical applications. In this study, we investigated the rheology, dielectric spectroscopy, stress–strain behavior, and dynamic mechanical analysis of networks based on long polybutylene oxide (PBO) chains functionalized with randomly placed thymine (Thy) side groups. A transient network was formed by proportionally mixing this matrix with short non-entangled linear 1,3,5-diaminotriazine (DAT) head–tail modified PBO chains, exploiting the hetero-complementarity of the DAT–Thy triple hydrogen bond. This transient polymer network was further cross-linked to a dual network via a thiol-ene click reaction to form static covalent bonds. In PBO, the similar polarity of the PBO matrix and the DAT–Thy functional groups ensures that the molecular chain motion is not affected by segregation, resulting in a homogeneous polymer phase without microphase-separated functional group domains. Dielectric relaxation spectroscopy was combined with rheology to quantify the relaxation processes of the interconnected polymers and the strength of the DAT–Thy bonding interactions in the melt. The results showed two distinct plateaux in the relaxation modulus due to contributions from hydrogen and permanent bonds. In the case of the dual network, the lifetime of the hydrogen bond was prolonged and higher activation energy was observed due to the physical cross-link preventing the movement of the long chain.
1 Introduction
Rubber materials are widely utilized in numerous applications, ranging from seals and joints between different materials to tires and vibration dampers (Friedrich and Almajid, 2013; Jiang et al, 2014). Their applications play an exceedingly significant role in the area of aerospace engineering (Vahdati and Saunders, 2002), automobile engineering (Chandra and Kumar, 2017; Barrera and Tardiff, 2022), and material science (Hamed, 2000; Suslick et al, 2023). However, the limitations of conventional rubbers, such as susceptibility to fatigue, impact, abrasion, and wear, are the major causes of premature failure (Saintier et al, 2011; Awaja et al, 2016), leading to frequent replacements and substantial investments.
As a consequence, the limited service life of conventional rubbers necessitates frequent replacements, resulting in significant costs and operational downtime. Incorporating weak-bond concepts similar to those found in living matter, which prevent localized stress peaks when under high mechanical load, would be tremendously advantageous (Gold et al, 2016; Shoda et al, 2020) to extend service time and optimize ultimate properties. Achieving this goal requires an interdisciplinary approach that combines microscopic and macroscopic studies aimed at understanding fundamental principles. Non-covalent interactions, including host–guest (Lee et al, 2011; Pappalardo et al, 2012; Kawano et al, 2022), hydrogen bonds (Lange et al, 1999; van Gemert et al, 2011; Verjans et al, 2022), and metal–ligand interactions (Le Bohec et al, 2016; Mozhdehi et al, 2016; Bentz and Cohen, 2018), are now recognized to exhibit both time and inherent temperature dependencies, which are coupled with specific chemistry. The range of interactions covers a binding energy spectrum from around 1–100 kJ/mol and includes diverse types, among which hydrogen bonding interactions, as they occur in natural base-pair combinations, offer a remarkable advantage for self-healing (Murphy and Wudl, 2010; Xie et al, 2021), as well as for the development of adaptive and/or responsive materials, without the need for introducing potentially hazardous heavy ions or metals. Pre-emptive damage control, however, is a much more important mechanism than self-healing for extended use problems and a topic of recent investigations.
We had discovered in the past that the bioinspired combination of thymine (Thy) and diaminotriazine (DAT) as active H-bonding entities—as synthetic analogs to thymine–guanine nucleobase conjugation (Li et al, 2011)—in a polymer matrix of polybutylene oxide (PBO) exhibited ideal time scales for the lifetime of the closed state (Allgaier et al, 2016). An illustrative application of this from daily life is tires. Although these are already highly vulcanized and reinforced, the process of opening and re-closing bonds enables the tire to respond in a self-sufficient manner to actual road conditions. The supramolecular bonds behave as so-called sacrificial bonds and allow the modulus of the rubber to vary between soft and strong in timescales of 0.001–0.01 s in a typical temperature range between −25 and +25 degree Celsius. Taking into account typical micro- and macro-textural asperities (e.g., 0.001, 1 mm) and speeds of the order of 100 km/h, impact frequencies of the order of 100–1,000 Hz are encountered that thus fit ideally to the frequencies at which supramolecular H-bonds dissipate their stored energy and switch between higher and lower modulus. In this frequency range, the rolling resistance is optimized, fuel consumption is reduced, and premature abrasion and wear due to local overstresses is minimized. In addition, transient PBO-comb polymers, combined with monofunctional DAT-arms associating with randomly located Thy-counterparts on long chains and even transient networks of the same when bifunctional DAT-arms were used, have already been reported (Staropoli et al, 2016; Staropoli et al, 2020). However, it is important to consider the compatibility of the overall mixture, as unfavorable interactions between monomer and functional end group chemistries can lead to the formation of micelles, which often limits controlled applications. Nonetheless, if compatibility is achieved, it allows for a rather unique determination of the characteristic response times.
Although covalent networks formed through vulcanization or random linking with peroxides or radiation—with or without fillers—have good mechanical properties, especially in terms of reversibility of deformation, the addition of reversible, physical, and transient crosslinks to such permanent networks leads to the creation of (auto-) responsive dual networks. These networks exhibit novel dependencies on strain or temperature that can be essential for advanced processing methods or applications, where the transient links allow for external control (Shangguan et al, 2017; Katashima, 2021). The ability of hydrogen bonds to associate and dissociate, as demonstrated in our work, becomes a valuable tool for triggering the overall mechanical behavior of such dual networks, ranging from highly elastic to very soft. These dual networks consist of both chemical and physical crosslinks and are similar to swollen networks or hydrogels (Vereroudakis et al, 2020; Nicolella et al, 2021).
However, the practical use of gels has been limited by their relatively low elastic moduli. To overcome this limitation, the incorporation of high-density hydrogen bonding into a polymer melt can significantly enhance the significance of transient bonding. This, in turn, enables the development of new self-repairing and self-damage-avoiding materials as explained previously for tires, as well as the processing of special branched polymers where transient bonds provide an intrinsic plasticizing property.
In this study, we, therefore, aim to synthesize and characterize dual networks that take advantage of both the PBO backbone polymer and supramolecular hydrogen bonding moieties, specifically Thy and DAT, and to permanently crosslink long chains through a thiol-ene reaction. To the best of our knowledge, this work is the first in the open literature which is based on homogeneity, hetero-complementary supramolecular activity, and covalent random crosslinking in the melt state. Unlike gel systems, our transient crosslink is mobile and consists of a short polymer chain that interconnects different long polymer chains, minimizing self-looping, and thus affects the behavior of the matrix chain in a more defined and rather controlled way. We use rheology and dynamic mechanical analysis to compare the behavior of a transient network (TN) prior to, and a dual network (DN) after, bulk-crosslinking the former transient network while preserving all chain interactions like entangling and H-bonding. Dielectric relaxation spectroscopy is attracted with a special focus on the bifunctional linker. Our results show that the encaging of transient linkages within the mesh of the permanent network affects the lifetime of the H-bond positively. Finally, we successfully compare the equilibrium properties of the DN to those obtained in a non-linear stretching experiment and pave the way for future studies in this area. Our findings highlight the potential for the development of new materials with enhanced mechanical properties by exploiting transient bonding mechanisms. This research has significant implications for the design of advanced materials with unique and tunable properties.
2 Experiment methods
2.1 Synthesis
Supplementary Schemes S1, S2 in the Supplementary file show the reaction synthesis scheme for the long- and short-chain polymers, respectively, while the previous literature (Allgaier et al, 2016) and Supplementary Section 1.3 in the Supplementary file provide further details on their synthesis. The structure of the polymer precursors and networks are indicated in Figure 1. The average number of vinyl groups per P(BO/EOc)120k(Thy20) chain is 28.2. The functionalization of the end-standing double bonds of the comonomer to thymine leads to an estimated number of thymine units of 19.7 in P(BO/EOc)120k(Thy20) as determined by the integral of the corresponding peaks in the NMR spectrum of the long chain. The remaining unreacted vinyl groups are used for further thiol-ene crosslinking reactions. The crosslinking method for the network is presented in the Supplementary file, Section 2.
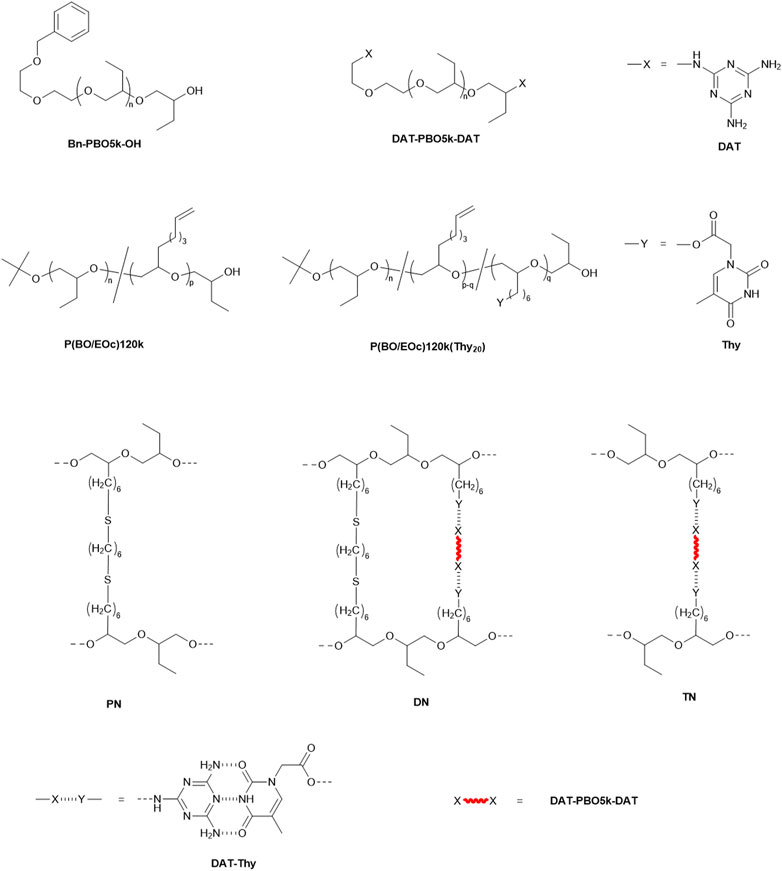
FIGURE 1. Structures of PBO precursors, functionalized samples, and network structures. Unfunctionalized short-chain (Bn-PBO5k-OH) and long-chain (P(BO/EOc)120k) components and functionalized short-chain (DAT-PBO5k-DAT) and long-chain (P(BO/EOc)120k(Thy20)) were investigated as reference polymers.
2.2 Characterization
2.2.1 Rheology
Oscillatory rheology has been used to probe the flow properties of the polymer melt. The measurements were performed on both ARES (Rheometric Sci) and ARES G2 (TA Instruments) systems in dynamic mode using standard 8-mm parallel plates. Frequency–temperature sweep tests were conducted in the frequency region with ω = 0.1…100 rad/s and a temperature ramp between −55°C and 25°C in steps of 5° for TN samples and 10° for all other polymer melt samples. The temperature was controlled by a liquid nitrogen blanket. The soaking time to achieve thermal equilibrium was 300 s for each temperature. The strain amplitude was 1% with the sample loading thickness of roughly 1 mm. Master curves with a reference temperature T0 = −25°C were generated by the TRIOS software, and a 2D shifting algorithm was applied.
2.2.2 Broadband dielectric spectroscopy
Dielectric properties were measured using a dielectric spectrometer (Novocontrol Technologies) equipped with an alpha impedance analyzer (Novocontrol Technologies) covering a frequency range from 10¬7 to 10–2 Hz. The temperature was controlled by a Quattro system (Novocontrol Technologies) from −55°C to 25°C, with temperature steps of 10°C for each sample. Melt samples and two 0.5-mm diameter glass fiber spacers were placed between the 20-mm diameter Au-coated electrodes. Network samples were directly placed in between the electrodes.
2.2.3 Size-exclusion chromatography
To determine the molecular weight of polymers, size-exclusion chromatography (SEC) was conducted on an Agilent 1260 Infinity SEC instrument with Wyatt DAWN Heleos II light scattering (LS) detector and an Optilab T-rex differential refractive index detector. The experiment was carried out using three PolyPore columns at 50°C and a solvent mixture consisting of tetrahydrofuran (THF), N,N-dimethylacetamide (DMA), and acetic acid (84:15:1 by volume) at a flow rate of 1 mL/min. The differential refractive index of polymer samples was calculated by the ASTRA software with the value of 0.0049.
2.2.4 Differential scanning calorimetry
Differential scanning calorimetry (DSC) analysis was carried out using a Q2000 DSC analyzer (TA Instruments). A temperature range from −120°C to 20°C with a heating/cooling rate of 10°C/min was analyzed.
2.2.5 Dynamical mechanical analysis
Dynamic mechanical analysis (DMA) was carried out on a Q800 DMA analyzer (TA Instruments) in tension mode. Networks were cut into a rectangular shape with dimensions of 25 mm × 4 mm × 1 mm. The storage modulus and loss modulus were obtained in dynamic oscillatory tests. Data were collected as a function of temperature in the range −55 to +25°C, covering a frequency range from 0.1 to 100 Hz with a 5 min soaking time and 1% strain amplitude. All the measurements were performed under a nitrogen atmosphere.
2.2.6 Mechanical characterization
Tensile tests were conducted on an ARES (Rheometric Sci) system, cooled with liquid nitrogen, exploiting the normal force measured by the 2KFRTN1 transducer and using home-made clamps. The samples were cut to a shape of 10 mm × 1 mm × 1 mm. Tensile deformation was performed within a range of strain rates varying from 0.106 s-1 to 0.0067 s-1 with careful temperature control. The deformed samples were reused to perform the same strain rate-dependent experiments.
The degree of cross-linking of a polymer network can be characterized from equilibrium swelling experiments using the Flory–Rehner equation, which is expressed in gravimetric terms as follows:
where
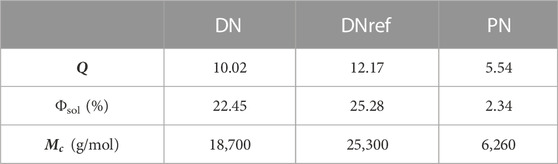
TABLE 1. Swelling degree and calculated mesh size. The correlation between soluble material and the fraction of short chains is virtually 100%. In the PN network, the measured sol fraction is mostly related to an unused HDT crosslinker and no chain scission was detected.
3 Results and discussion
From the perspective of molecular design, mixtures of P(BO/EOc)120k(Thy20) and DAT-PBO5k-DAT form hydrogen bonds through a variety of complementary interactions, including homo-complementary interactions between thymine and thymine groups (Thy–Thy) and between diaminotriazine and diaminotriazine groups (DAT–DAT), as well as hetero-complementary interactions between thymine and diaminotriazine groups (Thy–DAT) (Herbst et al, 2010; Cortese et al, 2011; Cortese et al, 2012; Brás et al, 2013; Krutyeva et al, 2015). The transient network (TN) obtained from these active components already showed higher viscosity and longer relaxation times at room temperature than the non-functionalized polymer melt. The photo-induced thiol-ene reaction, carried out in the polymer melt state at room temperature, generated a stable dual network (DN) with both transient and permanent bonds and quenches the different topologies. Although the system contains multiple components and relaxation mechanisms, preliminary SAXS (Supplementary Figures S17, S18) at low temperatures confirm a typical RPA or block-copolymer peak, which is the consequence of a correlation gap between correlated blocks. The Thy–DAT complex is treated as a single block and two such blocks are separated by the flexible 5 k PBO chain. The correlation with further blocks in the chain or network is negligible due to the separation distance to the neighboring Thy–DAT unit. It is shown that the scattering vector at high-Q wing follows a Q−2 dependence, which is typical of a random walk of the polymer chain, in clear contrast to an expected ∼ Q−4 decay for microphase-separated Thy–DAT domains. Therefore, the rheological properties of TN are first investigated to gain a better understanding of the behavior of DN.
3.1 Rheology master curve of TN
The material under investigation, TN, is a blend based on P(BO/EOc)120k(Thy20) and DAT-PBO5k-DAT with a stoichiometry of [Thy]/[DAT] = 1. The tentative rheology master curve is shown in Figure 2A at the reference temperature T = −25
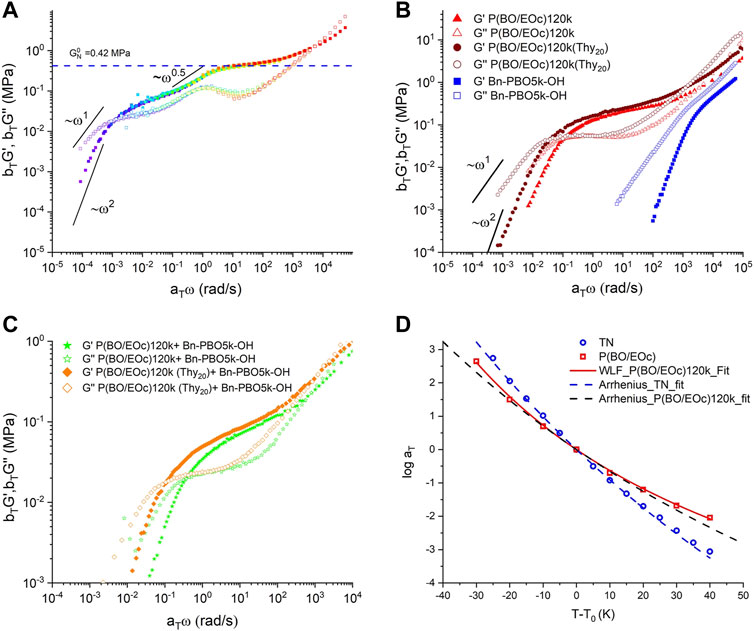
FIGURE 2. (A) Best rheology master curve of storage (solid squares) and loss modulus (hollow squares) of the TN sample at T = 248 K. (B) Storage and loss modulus master curve of unfunctionalized (P(BO/EOc)120 k, Bn-PBO5k-OH, functionalized precursor polymers (P(BO/EOc)120 k(Thy20)), and (C) master curve of their binary mixture.(D) Shear modulus time–temperature superposition shift factor of the TN (blue circle) and P(BO/EOc) 120k (red square). The reference temperature
The effective storage modulus of TN shows two plateaux, the first occurring in the region around
The equation involves several variables, where
Considering the TN case, the association number of supramolecular pairs affects the dilution effect of the entanglement modulus
The volume fraction is determined by the stoichiometry of the initial mixture
where
To discuss the deceleration of the TN terminal time, the sticky reptation model (Leibler et al, 1991) could be used to describe the complex system. To begin with the simplest system, P(BO/EOc)120k(Thy20), the Thy–Thy association pairs can also form a TN. The whole curve would shift horizontally to lower frequencies, i.e., longer times. The plateau modulus is apparently not increased by the Thy–Thy association, indicating that the Thy–Thy bonds must have already dissociated in the intermediate-frequency range. Considering the PBO structure with a lower number of thymine groups (Allgaier et al, 2016), a smaller shift factor was observed consequently. In both cases, the Thy–Thy lifetime is shorter than the Rouse relaxation time of a strand between the sticky points. (Chen et al, 2016). The shift toward longer times could be the result of increased ‘smeared’ friction and a change in
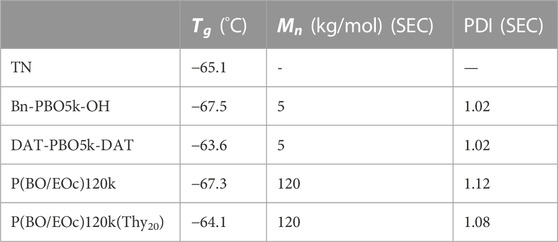
TABLE 2. Glass transition temperature and molecular weight for different polymers.
In accordance with the sticky reptation model on the TN sample (Figure 2A), the storage modulus displays a power law of
In Figure 2A, the TTS master curve is not strictly applicable in the region
In addition to the WLF equation, an Arrhenius correlation can also be used to determine the activation energy for a particular temperature range:
Here, the gas constant is denoted by
Assuming that a single activation process takes place in the interval of hydrogen bonding from the temperature region
3.2 Dielectric spectroscopy for dipole relaxation
Dielectric analysis could provide further microscopic insight into the molecular mechanism. The dipole moments of the PBO monomers are aligned parallel to the chain backbone and summed up, which classifies PBO as a type A polymer according to the Stockmayer classification (Stockmayer, 1967). The sum of these dipole moments gives an end-to-end net polarization vector, and the loss rate curve (
The peak position is obtained from
In our case, the instrumental dielectric frequency is reported in rad/s. In some of the samples, the low-frequency relaxation response is smeared out due to the contribution of DC conductivity. Hence, the derivative of storage permittivity response which is not affected by conductivity (See Figure 3B) is used to identify the corresponding peak position as follows (Wübbenhorst and van Turnhout, 2002):
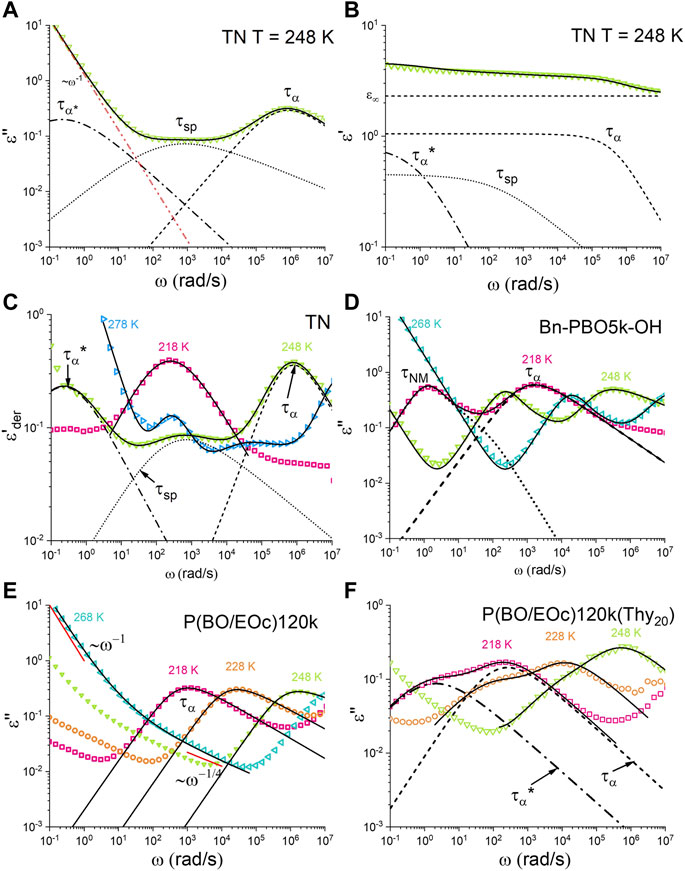
FIGURE 3. Dielectric (A) loss, (B) storage, and (C) derivative of the storage spectrum of the TN sample at T = 248 K. Dashed lines present
This approach improved the resolution of peaks in the spectrum, allowing for the detection of relaxation processes with low intensities or those occurring at low frequencies. The fitting function used in this method is based on the literature (Wübbenhorst and van Turnhout, 2002). In summary, the dielectric response of the samples can be described as a combination of a power-law fit for the low-frequency DC conductivity and two HN functions for the high-frequency
The dielectric spectrum of the TN sample is presented in Figure 3A. P(BO/EOc)120k and P(BO/EOc)120k(Thy20) are presented in Figures 3E, F.
Figures 3D–F also show the dielectric loss spectrum, i.e.,
where
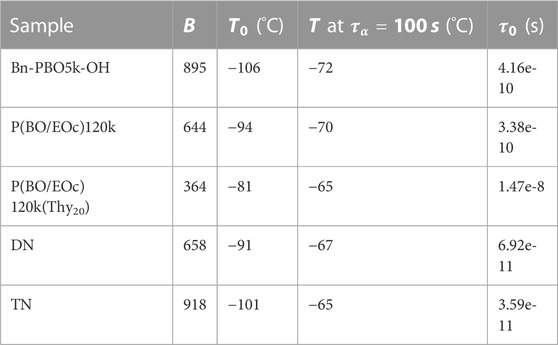
TABLE 3. Vogel–Fulcher–Tammann fit parameters of the segmental relaxation.
Comparing the dielectric relaxation of P(BO/EOc)120k (Figure 3E) with that of P(BO/EOc)120k(Thy20) (Figure 3F) reveals the presence of an additional relaxation peak present at frequencies about 2 orders of magnitude lower than those of the alpha relaxation process. This additional peak, termed as
On the other hand, when the short-chain DAT-PBO5k-DAT is added to form the TN, the
The TN sample in Figure 3A or the derivative of the real part of the permittivity in Figure 3C, e.g., at T = 248 K, revealed three peaks. In Figure 3C, but best visible at higher temperatures, a peak appears, which correlates with the normal mode relaxation of the DAT-PBO5k-DAT polymer. As the temperature increases, the dissociation of the supramolecular groups induces a normal mode relaxation of the PBO5k chain. This Thy–DAT
Figure 4A represents the relaxation time from different temperatures. The relaxation process
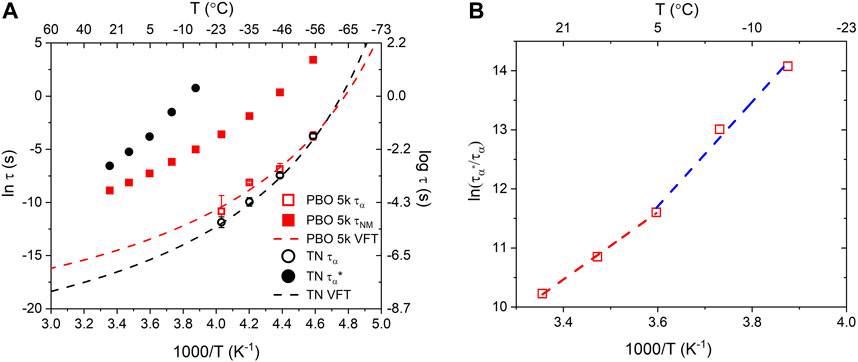
FIGURE 4. (A) Dielectric peak relaxation time of the Bn-PBO5k-OH
The fitted slope in Figure 4B allows the estimation of the association activation energy for TN. The activation energy shows two slopes in the high- and low-temperature regions, corresponding to activation energies of 47.3 and 73.5 kJ/mol, respectively. This indicates that the hydrogen bonds in the TN remain active up to +5°C, whereas at higher temperatures, all bonds are open and the equilibrium state for the supramolecular group would be dissociated. The difference between the energies is 26.2 kJ/mol, which corresponds to the energy scale for triple hydrogen bonds. The higher activation energy at low temperatures suggests that chain dynamics is also involved in the transient binding system (Monkenbusch et al, 2016), and additional energy is required to separate the functional group to a sufficient distance.
The same analysis could now be ideally performed on the DN sample. Crosslinking involves the formation of chemical bonds between polymer chains and effectively freezes in or quenches the orientation of the dipole moments. Likewise, it limits the mobility of charge carriers and results in a reduction in the overall conductivity due to the permanent crosslinks. These facts considerably complicate the analysis of such networks and less sensitivity is expected. Nevertheless, Figure 5 summarizes the difference between the TN and DN with the derivative of the storage permittivity.
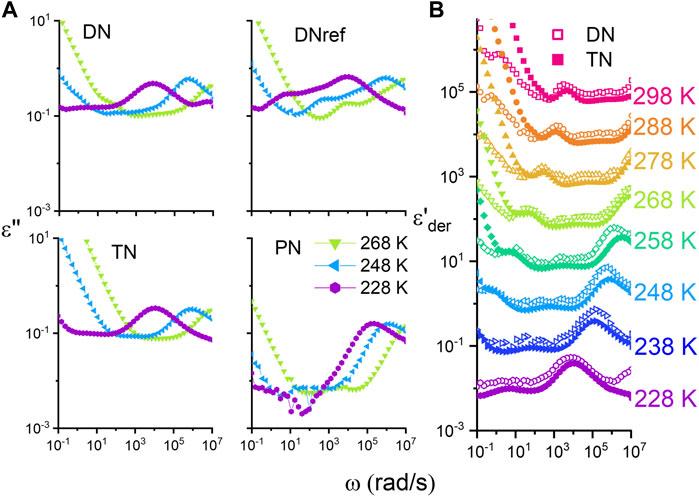
FIGURE 5. (A) Dielectric loss permittivity of network samples at different temperatures. (B) Derivative of storage modulus of the DN and TN. The curves for the different temperatures are shifted by 1 decade upwards for clarity reasons.
Using the derivative of the storage permittivity, at least the DC conductivity can be adequately corrected for.
In this way, only dipolar relaxation processes (
At the chosen rheological reference temperature of 248 K, the DN sample broadens and smears toward lower frequencies in comparison to the TN. This is physical and becomes more pronounced at even lower temperatures. This indicates a different slowing down or another temperature dependence compared to the higher T. For T = 258 K and above, the shape of the function of the TN and DN peaks is very congruent and reveals no discrepancies, except for the stronger parasitic polarization effects in the TN. Apparently, the reason for the broadening has vanished, and therefore it could be concluded that the broadening toward longer characteristic times is correlated to the presence of permanent crosslinks.
A full comparison of the investigated networks and mixtures is also summarized in Figure 5 (A). For TN, DN, DNref, and PN, the
3.3 DMA analysis on DN and TN
Figure 6 presents a comparison between the DMA and rheology curves of the DN and TN at varying temperatures. In order to match the dynamic Young’s modulus
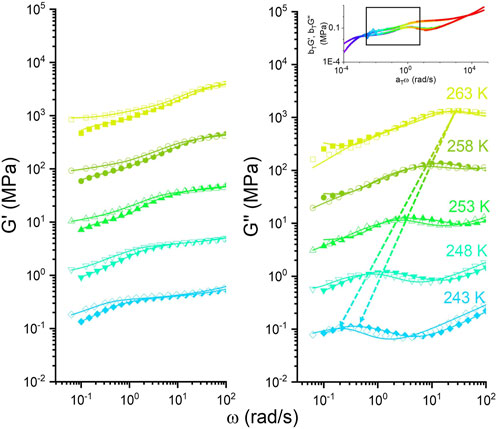
FIGURE 6. Storage modulus of the TN (hollow) and DN (solid) samples derived from dynamic mechanical analysis and small amplitude oscillatory shear rheology. The relation
This may be attributed to the static crosslink hindering the Rouse motion of the elastic chains between them for the DN compared to the TN. At lower temperatures, these permanent static bonds form a cage-like prison or confinement, which impedes the relative motion of the long backbone and extends the lifetime of transient bonds. As a result of the permanent bonds suppressing the basic polymer relaxation, the probability of Thy–DAT sticker pairs opening and attaching to new stickers decreases on small length scales. This is equivalent to the increase in the activation energy barrier for bond cleavage and favors the reassociation ability due to the short PBO chains living in a tube-like cage. To investigate the increase of activation energy between the DN and TN, a phenomenological multi-Maxwell model is used to fit the relaxation processes at different temperatures. For the TN, a contribution of two types of Maxwell models can be considered at low (reptation) and high frequency (hydrogen bonds).
The characteristic time for the sticker is fitted at each temperature separately without the interference of a possible slightly erroneous TTS shifting. The parameter
To extrapolate the activation energy from the hydrogen bond relaxation time, the Arrhenius equation is applied as follows:
where
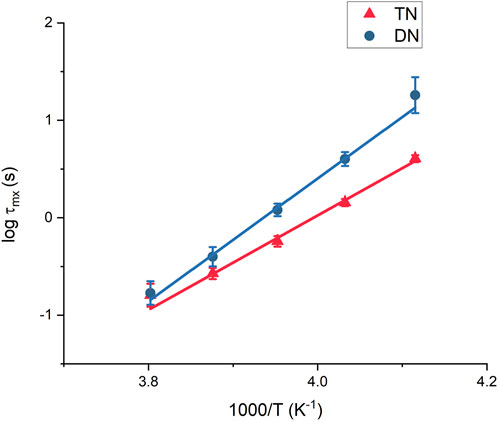
FIGURE 7. Extracted relaxation time (
3.4 Small deformation stretch
The relationship between the Thy–DAT bond lifetime, activation energy, and modulus in a quiescent state has been discussed at length in the previous sections. To discuss the characteristics of transient bonds in energy dissipation (Hu et al, 2017) and the relationship of bond lifetime in anisotropic stretching, the recent Dobrynin–Konkolewicz model (Carrillo et al, 2013; Bennett et al, 2022) is applied. The microscale bond lifetime and the macroscale rubber mechanical properties of the DN in the small deformation range of a stress–strain experiment with varying strain rates lead to
The model assumes that transient bonds within a covalent network will cause the total crosslink density to diminish during uniaxial stretching. The lifetime or relaxation time of the TN, denoted as
where
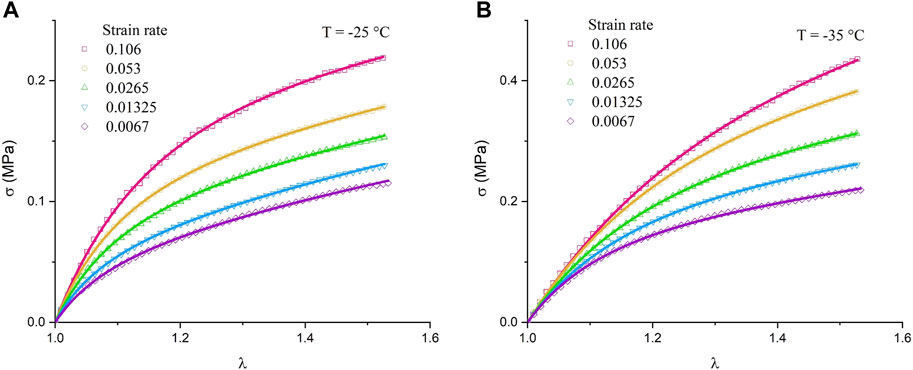
FIGURE 8. Stress–strain curve of the DN sample at different strain rates (A) T = −25
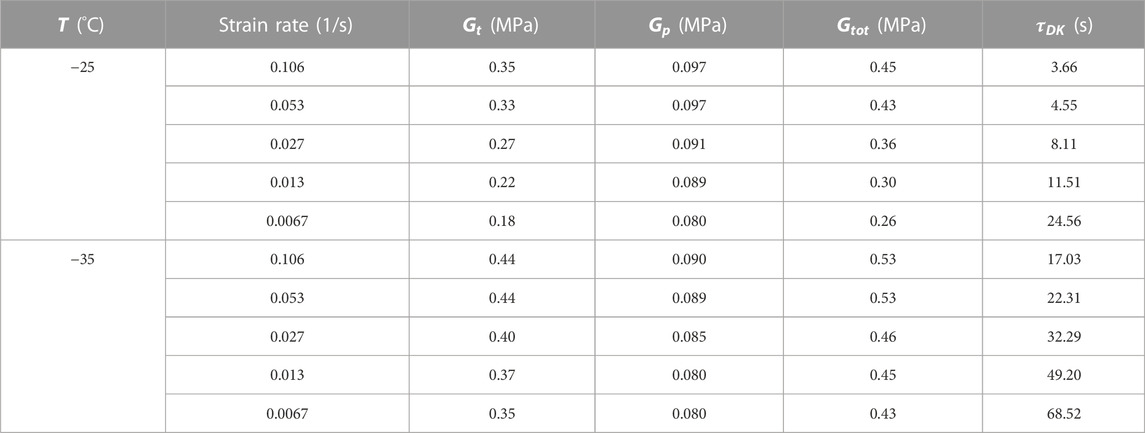
TABLE 4. Dobrynin–Konkolewicz fitting parameters for Figures 8A, B.
The fitting results show a good agreement with the permanent
Figures 9A, B present the strain rate relationship with
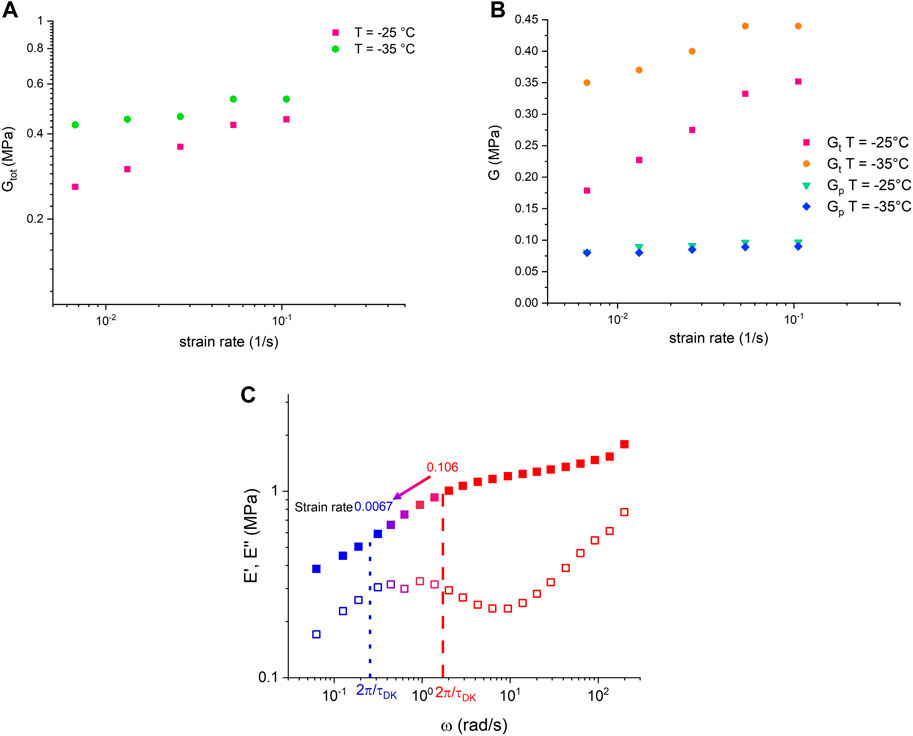
FIGURE 9. (A)
4 Conclusion and outlook
In the former, we have explored the evolution of dynamic properties in the transition from inactive polymer blends to supramolecular-active TNs, and finally to dual networks that combine all aspects simultaneously. The value of such a system over conventional elastomers lies in a novel responsiveness to both strain and time. The introduction of permanent crosslinks to an H-bonded chain mixture, where the different topologies that are present in the bulk are preserved and quenched, induces a clear shift toward considerably longer bonding times of the Thy–DAT complex. We refer to a summary of parameters in Tables 5, 6. The activation energy of dissociation of the Thy–DAT stickers and the lifetime of the closed state are consistently higher as the stickers have a higher probability to remain at the same partner. All independent methods in this study are in agreement with this unexpected result. Apparently, the increased confinement or at least the stronger restriction of the configuration space within the elastic meshes is responsible for the longer stability of the triple H-bond that is formed in the Thy–DAT association. Despite the individually different sensitivities of the methods to the original aim of the investigation, i.e., to produce an idealized model-dual dynamic network, conclusions can be safely drawn: supramolecular hydrogen bonds can be introduced as sacrificial but repeatable units to control the modulus of the rubber between soft and strong. For frequencies of the order of 1 Hz, or strain rates of the order of 1.0/s, the critical strain-at-rupture of the triple H-bonds is identified to be about 10% in our example. Although the hetero-association of both nucleobase-like groups certainly depends on the immediate environment (matching polarity, competing interactions, solvents…), this strain should be a parameter that is grossly independent of the environment and could be verified from ab initio methods or molecular dynamics simulations.
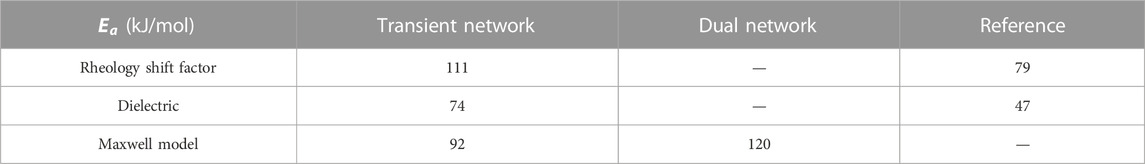
TABLE 5. Activation energy according to different extrapolation methods.
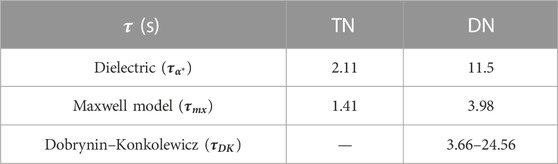
TABLE 6. Hydrogen bond relaxation time at
Furthermore, whereas the timely response of the DN is already of interest, e.g., shock damping materials or common tire specifications, this level of strain—under static or dynamic excitation—is also important. We have already discussed in the introduction how most common daily life elastomers can benefit from our study. The tire tread rubber is highly filled, and many reinforcing mechanisms are involved to arrive at the final properties. These typical properties are, however, counter-acting and if some are optimized, the others are influenced negatively. The well-known Payne effect in filled rubbers, which is due to the breaking-and-reforming of filler aggregates, is connected with a reduction of the storage modulus and loss of elastic behavior which becomes clear through a pronounced dissipation peak. The engine will consume more fuel as the resistance with the road is increased. The strain range, at which the loss modulus peaks in the thread, coincides with the estimated breaking strain from the static stress–strain model. In addition, the microscopic times applied to a tire running at an average speed of 100 km/h and typical loading degrees of the filler of about 100 phr are well-caught. The confinement that these fillers induce seems to be of the same order of magnitude as the spacing in our model system, i.e., elastic chain lengths of the order of 1,000–4,000 g/mol corresponding to ∼2–8 nm. Thus, considerable improvements could be obtained in the future. The aforementioned example does not involve any further time-dependent mechanisms. Modifying the thread component with supramolecular functions comparable to the Thy–DAT combination would be able to compensate for the (low frequency) Payne effect by adding the transient modulus to the chemical crosslinking modulus without destroying the high-frequency properties like wet skid that come into play at large deformations where the H-bonds have sacrificed already. Since the modulus, however, decreases with temperature, an increase of the elastic modulus due to the H-bonding interactions could have a decisive positive effect on the overall performance of such a dual-rubber-based tire. In the future, H-bonding groups like ours can be grafted onto a typical silica filler surface and dispersed into a non- or supramolecular-modified elastomer. The direct consequence of all these is the reduction of hysteresis at small strains, restoring elasticity while minimizing cracks and its development at large deformations. Key processes may be developed to adjust the desired mechanical properties for useful materials in day-to-day life. A forthcoming study will focus on the structure and its change upon uniaxial deformation using in situ small angle X-ray and neutron scattering.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
Conceptualization, WP-H; data acquisition, JF, JA, and MK; discussion, JF, JA, MK, SF, and WP-H; formal analysis, JF; investigation, JF and WP-H; visualization, JF; writing the original draft, JF; writing—review and editing, JF, JA, and WP-H; supervision, SF and WP-H; funding acquisition, SF and WP-H; project administration, SF and WP-H. All authors contributed to the article and approved the submitted version.
Acknowledgments
JF expresses gratitude to Dr. Aakash Sharma and Dr. Tulika Sharma for their valuable contributions in the form of insightful discussions and meticulous proofreading. Dr. Andreas Raba is thanked for the synthesis of the DAT-F component.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frsfm.2023.1221803/full#supplementary-material
References
Ahmadi, M., Jangizehi, A., Saalwächter, K., and Seiffert, S. (2023). Effect of Junction aggregation on the dynamics of supramolecular polymers and networks. Macromol. Chem. Phys. 224, 2200389. doi:10.1002/macp.202200389
Ahmadi, M., Jangizehi, A., and Seiffert, S. (2022). Backbone polarity tunes sticker clustering in hydrogen-bonded supramolecular polymer networks. Macromolecules 55, 5514–5526. doi:10.1021/acs.macromol.2c00645
Allgaier, J., Hövelmann, C. H., Wei, Z., Staropoli, M., Pyckhout-Hintzen, W., Lühmann, N., et al. (2016). Synthesis and rheological behavior of poly(1,2-butylene oxide) based supramolecular architectures. R. Soc. Chem. 6, 6093–6106. doi:10.1039/c5ra24547h
Awaja, F., Zhang, S., Tripathi, M., Nikiforov, A., and Pugno, N. (2016). Cracks, microcracks and fracture in polymer structures: Formation, detection, autonomic repair. Prog. Mater. Sci. 83, 536–573. doi:10.1016/j.pmatsci.2016.07.007
Barrera, C. S., and Tardiff, J. L. (2022). Static and dynamic properties of eggshell filled natural rubber composites for potential application in automotive vibration isolation and damping. J. Clean. Prod. 353, 131656. doi:10.1016/j.jclepro.2022.131656
Bennett, C., Hayes, P. J., Thrasher, C. J., Chakma, P., Wanasinghe, S. V., Zhang, B., et al. (2022). Modeling approach to capture hyperelasticity and temporary bonds in soft polymer networks. Macromolecules 55, 3573–3587. doi:10.1021/acs.macromol.1c02319
Bentz, K. C., and Cohen, S. M. (2018). Supramolecular metallopolymers: From linear materials to infinite networks. Angew. Chem. Int. Ed. 57, 14992–15001. doi:10.1002/anie.201806912
Brás, A. R., Hövelmann, C. H., Antonius, W., Teixeira, J., Radulescu, A., Allgaier, J., et al. (2013). Molecular approach to supramolecular polymer assembly by small angle neutron scattering. Macromolecules 46, 9446–9454. doi:10.1021/ma401714r
Carrillo, J. M. Y., Mackintosh, F. C., and Dobrynin, A. V. (2013). Nonlinear elasticity: From single chain to networks and gels. Macromolecules 46, 3679–3692. doi:10.1021/ma400478f
Chandra, A. K., and Kumar, N. R. (2017). “Polymer nanocomposites for automobile engineering applications,” in Properties and applications of polymer nanocomposites: Clay and carbon based polymer nanocomposites. Editors D. K. Tripathy, and B. P. Sahoo (Berlin, Heidelberg: Springer Berlin Heidelberg), 139–172.
Chen, Q., Zhang, Z., and Colby, R. H. (2016). Viscoelasticity of entangled random polystyrene ionomers. J. Rheology 60, 1031–1040. doi:10.1122/1.4955432
Cortese, J., Soulie-Ziakovic, C., Cloitre, M., Tencé-Girault, S., and Leibler, L. (2011). Order-disorder transition in supramolecular polymers. J. Am. Chem. Soc. 133, 19672–19675. doi:10.1021/ja209126a
Cortese, J., Soulie-Ziakovic, C., Tencé-Girault, S., and Leibler, L. (2012). Suppression of mesoscopic order by complementary interactions in supramolecular polymers. J. Am. Chem. Soc. 134, 3671–3674. doi:10.1021/ja2119496
Doi, M., and Edwards, S. (1978). Dynamics of concentrated polymer systems. Part 1.—brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 74, 1789–1801. doi:10.1039/f29787401789
Friedrich, K., and Almajid, A. A. (2013). Manufacturing aspects of advanced polymer composites for automotive applications. Appl. Compos. Mater. 20, 107–128. doi:10.1007/s10443-012-9258-7
Gałązka, M., and Osiecka-Drewniak, N. (2022). Electric conductivity and electrode polarization as markers of phase transitions. Crystals 12, 1797. doi:10.3390/cryst12121797
Ge, S., Tress, M., Xing, K., Cao, P.-F., Saito, T., and Sokolov, A. P. (2020). Viscoelasticity in associating oligomers and polymers: Experimental test of the bond lifetime renormalization model. Soft Matter 16, 390–401. doi:10.1039/c9sm01930h
Gerstl, C., Schneider, G. J., Pyckhout-Hintzen, W., Allgaier, J., Richter, D., Alegría, A., et al. (2010). Segmental and normal mode relaxation of poly(alkylene oxide)s studied by dielectric spectroscopy and rheology. Macromolecules 43, 4968–4977. doi:10.1021/ma100384j
Glomann, T., Schneider, G. J., Brás, A. R., Pyckhout-Hintzen, W., Wischnewski, A., Zorn, R., et al. (2011). Unified description of the viscoelastic and dielectric global chain motion in terms of the tube theory. Macromolecules 44, 7430–7437. doi:10.1021/ma200674z
Gold, B. J., Hövelmann, C. H., Weiss, C., Radulescu, A., Allgaier, J., Pyckhout-Hintzen, W., et al. (2016). Sacrificial bonds enhance toughness of dual polybutadiene networks. Polymer 87, 123–128. doi:10.1016/j.polymer.2016.01.077
Goldansaz, H., Fustin, C.-A., Wübbenhorst, M., and Van Ruymbeke, E. (2016). How supramolecular assemblies control dynamics of associative polymers: Toward a general picture. Macromolecules 49, 1890–1902. doi:10.1021/acs.macromol.5b01535
Hamed, G. R. (2000). Reinforcement of rubber. Rubber Chem. Technol. 73, 524–533. doi:10.5254/1.3547603
Havriliak, S., and Negami, S. (1967). A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 8, 161–210. doi:10.1016/0032-3861(67)90021-3
Herbst, F., Schröter, K., Gunkel, I., Gröger, S., Thurn-Albrecht, T., Balbach, J., et al. (2010). Aggregation and chain dynamics in supramolecular polymers by dynamic rheology: Cluster formation and self-aggregation. Macromolecules 43, 10006–10016. doi:10.1021/ma101962y
Hu, X., Zhou, J., Daniel, W. F. M., Vatankhah-Varnoosfaderani, M., Dobrynin, A. V., and Sheiko, S. S. (2017). Dynamics of dual networks: Strain rate and temperature effects in hydrogels with reversible H-bonds. Macromolecules 50, 652–659. doi:10.1021/acs.macromol.6b02422
Jacobs, M., Liang, H., and Dobrynin, A. V. (2021). Theory and simulations of hybrid networks. Macromolecules 54, 7337–7346. doi:10.1021/acs.macromol.1c00774
Jiang, Y., Wang, Y. X., Wang, S. Z., and Su, X. W. (2014). “Frequency dependence stiffness and damping of "O" rubber ring,” in Advanced materials research, 368–372.
Katashima, T. (2021). Rheological studies on polymer networks with static and dynamic crosslinks. Polym. J. 53, 1073–1082. doi:10.1038/s41428-021-00505-y
Kawano, S., Nakano, K., Sato, H., Muraoka, M., and Shizuma, M. (2022). Photo- and thermo-responsive supramolecular polymer networks via in situ polymerization using homoternary macrocyclic host with coumarin monomers in water. Polym. Chem. 13, 5820–5828. doi:10.1039/d2py00952h
Krutyeva, M., Brás, A. R., Antonius, W., Hövelmann, C. H., Poulos, A. S., Allgaier, J., et al. (2015). Association behavior, diffusion, and viscosity of end-functionalized supramolecular poly(ethylene glycol) in the melt state. Macromolecules 48, 8933–8946. doi:10.1021/acs.macromol.5b02060
Lange, R. F. M., Van Gurp, M., and Meijer, E. W. (1999). Hydrogen-bonded supramolecular polymer networks. J. Polym. Sci. Part A Polym. Chem. 37, 3657–3670. doi:10.1002/(sici)1099-0518(19991001)37:19<3657::aid-pola1>3.0.co;2-6
Le Bohec, M., Banère, M., Piogé, S., Pascual, S., Benyahia, L., and Fontaine, L. (2016). Sol–gel reversible metallo-supramolecular hydrogels based on a thermoresponsive double hydrophilic block copolymer. Polym. Chem. 7, 6834–6842. doi:10.1039/c6py01639a
Lee, M., Moore, R. B., and Gibson, H. W. (2011). Supramolecular pseudorotaxane graft copolymer from a crown ether polyester and a complementary paraquat-terminated polystyrene guest. Macromolecules 44, 5987–5993. doi:10.1021/ma201241t
Leibler, L., Rubinstein, J. M., and Colby, R. H. (1991). Dynamics of reversible networks. Macromolecules 24, 4701–4707. doi:10.1021/ma00016a034
Li, X., Kuang, Y., Lin, H.-C., Gao, Y., Shi, J., and Xu, B. (2011). Supramolecular nanofibers and hydrogels of nucleopeptides. Angew. Chem. Int. Ed. 50, 9365–9369. doi:10.1002/anie.201103641
Mcleish, T. C. B. (2002). Tube theory of entangled polymer dynamics. Adv. Phys. 51, 1379–1527. doi:10.1080/00018730210153216
Monkenbusch, M., Krutyeva, M., Pyckhout-Hintzen, W., Antonius, W., Hövelmann, C. H., Allgaier, J., et al. (2016). Molecular view on supramolecular chain and association dynamics. Phys. Rev. Lett. 117, 147802. doi:10.1103/physrevlett.117.147802
Mozhdehi, D., Neal, J. A., Grindy, S. C., Cordeau, Y., Ayala, S., Holten-Andersen, N., et al. (2016). Tuning dynamic mechanical response in metallopolymer networks through simultaneous control of structural and temporal properties of the networks. Macromolecules 49, 6310–6321. doi:10.1021/acs.macromol.6b01626
Murphy, E. B., and Wudl, F. (2010). The world of smart healable materials. Prog. Polym. Sci. 35, 223–251. doi:10.1016/j.progpolymsci.2009.10.006
Nicolella, P., Lauxen, D., Ahmadi, M., and Seiffert, S. (2021). Reversible hydrogels with switchable diffusive permeability. Macromol. Chem. Phys. 222, 2100076. doi:10.1002/macp.202100076
Pappalardo, A., Ballistreri, F. P., Destri, G. L., Mineo, P. G., Tomaselli, G. A., Toscano, R. M., et al. (2012). Supramolecular polymer networks based on calix[5]arene tethered poly(p-phenyleneethynylene). Macromolecules 45, 7549–7556. doi:10.1021/ma3015239
Riedel, C., Alegría, A., Tordjeman, P., and Colmenero, J. (2010). High and low molecular weight crossovers in the longest relaxation time dependence of linear cis-1,4 polyisoprene by dielectric relaxations. Rheol. Acta 49, 507–512. doi:10.1007/s00397-010-0433-1
Saintier, N., Cailletaud, G., and Piques, R. (2011). Cyclic loadings and crystallization of natural rubber: An explanation of fatigue crack propagation reinforcement under a positive loading ratio. Mater. Sci. Eng. A 528, 1078–1086. doi:10.1016/j.msea.2010.09.079
Schaefer, C., Laity, P. R., Holland, C., and Mcleish, T. C. B. (2020). Silk protein solution: A natural example of sticky reptation. Macromolecules 53, 2669–2676. doi:10.1021/acs.macromol.9b02630
Shahid, T., Huang, Q., Oosterlinck, F., Clasen, C., and Van Ruymbeke, E. (2017). Dynamic dilution exponent in monodisperse entangled polymer solutions. Soft Matter 13, 269–282. doi:10.1039/c6sm01083k
Shangguan, Y., Yang, J., and Zheng, Q. (2017). Rheology of nitrile rubber with hybrid crosslinked network composed of covalent bonding and hydrogen bonding. RSC Adv. 7, 15978–15985. doi:10.1039/c7ra01106g
Shoda, Y., Aoki, D., Tsunoda, K., and Otsuka, H. (2020). Polybutadiene rubbers with urethane linkages prepared by a dynamic covalent approach for tire applications. Polymer 202, 122700. doi:10.1016/j.polymer.2020.122700
Staropoli, M., Kruteva, M., Allgaier, J., Wischnewski, A., and Pyckhout-Hintzen, W. (2020). Supramolecular dimerization in a polymer melt from small-angle X-ray scattering and rheology: A miscible model system. Polymers 12, 880. doi:10.3390/polym12040880
Staropoli, M., Raba, A., Hövelmann, C. H., Krutyeva, M., Allgaier, J., Appavou, M.-S., et al. (2016). Hydrogen bonding in a reversible comb polymer architecture: A microscopic and macroscopic investigation. Macromolecules 49, 5692–5703. doi:10.1021/acs.macromol.6b00978
Stockmayer, W. H. (1967). Dielectric dispersion in solutions of flexible polymers. Pure Appl. Chem. 15, 539–554. doi:10.1351/pac196715030539
Suslick, B. A., Hemmer, J., Groce, B. R., Stawiasz, K. J., Geubelle, P. H., Malucelli, G., et al. (2023). Frontal polymerizations: From chemical perspectives to macroscopic properties and applications. Chem. Rev. 123, 3237–3298. doi:10.1021/acs.chemrev.2c00686
Urakawa, O., Yamane, M., Tomie, S., Inoue, T., Shikata, T., and Adachi, K. (2018). Relationship between global and segmental dynamics of poly(butylene oxide) studied by broadband dielectric spectroscopy. J. Chem. Phys. 148, 034904. doi:10.1063/1.5006364
Vahdati, N., and Saunders, L. K. L. (2002). High frequency testing of rubber mounts. ISA Trans. 41, 145–154. doi:10.1016/s0019-0578(07)60074-3
Van Gemert, G. M. L., Peeters, J. W., Söntjens, S. H. M., Janssen, H. M., and Bosman, A. W. (2011). Self-healing supramolecular polymers in action. Macromol. Chem. Phys. 213, 234–242. doi:10.1002/macp.201100559
Van Krevelen, D. W., and Te Nijenhuis, K. (2009). “Chapter 7 - cohesive properties and solubility,” in Properties of polymers. Editors D. W. Van Krevelen, and K. Te Nijenhuis 4 (Amsterdam: Elsevier), 189–227.
Van Ruymbeke, E., Shchetnikava, V., Matsumiya, Y., and Watanabe, H. (2014). Dynamic dilution effect in binary blends of linear polymers with well-separated molecular weights. Macromolecules 47, 7653–7665. doi:10.1021/ma501566w
Vereroudakis, E., Bantawa, M., Lafleur, R. P. M., Parisi, D., Matsumoto, N. M., Peeters, J. W., et al. (2020). Competitive supramolecular associations mediate the viscoelasticity of binary hydrogels. ACS Central Sci. 6, 1401–1411. doi:10.1021/acscentsci.0c00279
Verjans, J., André, A., Van Ruymbeke, E., and Hoogenboom, R. (2022). Physically cross-linked polybutadiene by quadruple hydrogen bonding through side-chain incorporation of ureidopyrimidinone with branched alkyl side chains. Macromolecules 55, 928–941. doi:10.1021/acs.macromol.1c01908
Vogel, D. H. (1921). Das temperaturabhaengigkeitsgesetz der Viskositaet von Fluessigkeiten. Phys. Z. 22, 645. doi:10.4236/ojn.2014.43019
Watanabe, H. (2001). Dielectric relaxation of type-A polymers in melts and solutions. Macromol. Rapid Commun. 22, 127–175. doi:10.1002/1521-3927(200102)22:3<127::aid-marc127>3.0.co;2-s
Wu, S., and Chen, Q. (2022). Advances and new opportunities in the rheology of physically and chemically reversible polymers. Macromolecules 55, 697–714. doi:10.1021/acs.macromol.1c01605
Wübbenhorst, M., and Van Turnhout, J. (2002). Analysis of complex dielectric spectra. I. One-dimensional derivative techniques and three-dimensional modelling. J. Non-Crystalline Solids 305, 40–49. doi:10.1016/s0022-3093(02)01086-4
Xie, Z., Hu, B.-L., Li, R.-W., and Zhang, Q. (2021). Hydrogen bonding in self-healing elastomers. ACS Omega 6, 9319–9333. doi:10.1021/acsomega.1c00462
Yamane, M., Hirose, Y., and Adachi, K. (2005). Dielectric normal and segmental modes in undiluted poly(butylene oxide). Macromolecules 38, 9210–9215. doi:10.1021/ma0516384
Keywords: rheology, dielectric spectroscopy, dual network, DMA, supramolecular polymer, Thy-DAT, hydrogen bonding
Citation: Feng J, Allgaier J, Kruteva M, Förster S and Pyckhout-Hintzen W (2023) Constraining effects on polymer chain relaxation in crosslinked supramolecular dual networks. Front. Soft. Matter 3:1221803. doi: 10.3389/frsfm.2023.1221803
Received: 12 May 2023; Accepted: 28 June 2023;
Published: 18 July 2023.
Edited by:
Kay Saalwächter, Martin-Luther-University Halle-Wittenberg, GermanyReviewed by:
Damien Montarnal, Laboratoire de chimie polymérisation procédés et matériaux, FranceMostafa Ahmadi, Johannes Gutenberg University Mainz, Germany
Copyright © 2023 Feng, Allgaier, Kruteva, Förster and Pyckhout-Hintzen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jasper Feng, ai5mZW5nQGZ6LWp1ZWxpY2guZGU=; Wim Pyckhout-Hintzen, dy5weWNraG91dEBmei1qdWVsaWNoLmRl