 Hai Wang
Hai Wang Yongfeng Men
Yongfeng Men Kohji Tashiro
Kohji Tashiro- 1Deparment of Polymer Materials and Engineering, School of Chemical Engineering, Dalian University of Technology, Dalian, China
- 2State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Changchun, China
- 3Aichi Synchrotron Radiation Center, Knowledge Hub Aichi, Minami-Yamaguchi, Seto, Japan
- 4Department of Future Industry-Oriented Basic Science and Materials, Toyota Technological Institute, Nagoya, Japan
The studies of time-dependent structural changes in the solvent-induced crystallization and phase transition phenomena have been reviewed by focusing mainly on syndiotactic polystyrene (SPS) and its relatives having the functional groups on the phenyl rings. The time-resolved measurement of X-ray diffraction and vibrational spectra has revealed the structural evolution process in the solvent-induced crystallization of SPS, which depends on the type (polarity, bulkiness, etc.) of the solvent molecules. The heating of the SPS-solvent complexes causes the complicated phase transitions from the δ form (complex) to the γ form and to the α (or β) form. The introduction of such a polar functional group as OCH3 or halogen units on the phenyl rings enhances the interactions between the SPS and the solvent, the strength of which depends on the substitution position of the OCH3 units on the phenyl ring. For example, the ortho- or para-substitution dissolves the sample quite easily at room temperature, while the meta-substitution makes it possible to create the solvent complexes. The discussion has been made for the structural relation before and after the formation of the solvent-complexes.
1 Introduction
Solvent-induced crystallization and phase transition behavior of polymers have been attracting much attention in both the academic research and the industrial production. Many polymers have been found to exhibit the solvent-induced crystallization phenomenon, including polyethylene terephthalate (PET) (Desai and Wilkes, 1974; Durning et al., 1986; Ouyang et al., 2002; Ouyang et al., 2004), polybutylene terephthalate (PBT) (Waywood and Durning, 1987), polycarbonate (Sheldon and Blakey, 1962; Park and Hong, 1997), nylon (Liu et al., 1995), poly(ether ether ketone) (PEEK) (Cornélis et al., 1996) and so on. In some cases, the spatial orientations of the crystallites are controlled through the interactions between the polymer chains and the solvent molecules. The well-known example is seen for an electronically-conductive polymer, poly(3-hexylthiophene) (P3HT) (Tremel and Ludwigs, 2014); the folded polymer chains stand vertically (end-on) or lie horizontally on the substrate (edge-on or face-on), which are dependent on the solvent evaporation process. The chain orientations affect the electronic transport performance of this polymer.
In many cases, the solvent molecules play a role as a kind of accelerator of the cold-crystallization and they do not remain in the thus-created crystalline lattices. They may be assumed to be a kind of plasticizer. In another type of solvent-induced crystallization, the whole system is stabilized through the polymer-solvent interactions and these two species are coexistent in the common crystal lattices, that is, the formation of the so-called polymer-solvent complex. This type of complex is not only unique and important from the scientific point of view but also useful for the industrial application. For example, the solvent molecules trapped in the crystal lattices can be displaced easily with the other kinds of solvent. This behavior can be utilized as a molecular filter (Mensitieri et al., 2003). In addition, the heating of these complexes may purge the solvents with or without keeping the spaces originally occupied by the solvent molecules and cause the drastic change in the higher-order structures, which cannot be realized by the usual melt-crystallization process. As a result, the physicochemical properties become quite different between these two cases. The typical examples are seen for polylactic acid (PLA) (Wang and Tashiro, 2022) and syndiotactic polystyrene (SPS) and its relatives (Gowd et al., 2009). PLA, as one of the very popular renewable source-based polymers, produces the complex (ε form) with CO2 (Marubayashi et al., 2012a; Marubayashi et al., 2012b) or organic solvent like cyclopentanone (CPO) (Rizzo et al., 2015; Shaiju et al., 2016) and N,N-dimethylformamide (DMF) (Praveena et al., 2022a; Praveena et al., 2022b) from the amorphous or meso phase. By controlling the type of the solvent and/or the pressure of the solvent atmosphere, the phase transition is induced among the different PLA/solvent complexes (Marubayashi et al., 2013).
The most widely studied solvent complexes are those of SPS. Many different types of crystal modifications have been revealed so far, which are formed variously depending on the crystallization conditions: in the crystallization from the melt (α’, α”, β’, β” phases etc.) and in the atmosphere of the solvent vapors (γ, δ, δe, and ε phases) (Gowd et al., 2009). Figures 1–4 show, respectively, the chain conformations and crystal structures of the representative crystal forms. When an (oriented) amorphous SPS sample is exposed to a solvent atmosphere, it forms the complex with solvent. The chain conformation is contracted from the fully-extended planar-zigzag form (Figure 1). By washing the complex using acetone, for example, the so-called empty δ (δe) is obtained (Figure 2), (De Rosa et al., 1997) which keeps the empty spaces (cavities) originally occupied by the solvent molecules. The heating of the δe form induces the transition to the γ form without any voids in the unit cell. The heating at higher temperature results in the formation of the α and β forms consisting of the closely-packed planar-zigzag chains. The latter two phases are obtained by the melt-crystallization. However, as mentioned, the heat treatment of the solvent-related crystalline phases causes the phase transition to the crystalline phases of the zigzag chains.
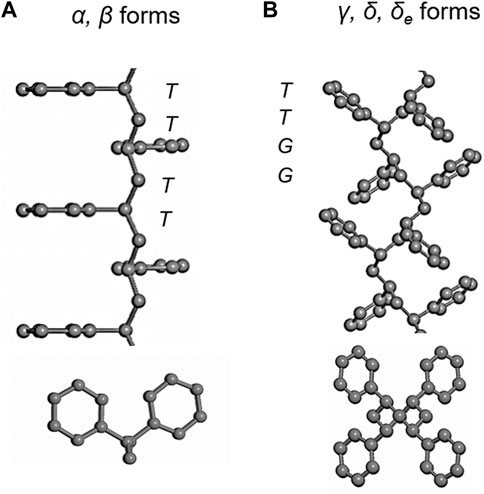
FIGURE 1. Chain conformations of SPS in the various crystalline forms. (A) Planar-zigzag conformation of the α and β forms. (B) TTGG helical conformation in the δ, δe and γ forms (Gowd et al., 2009).
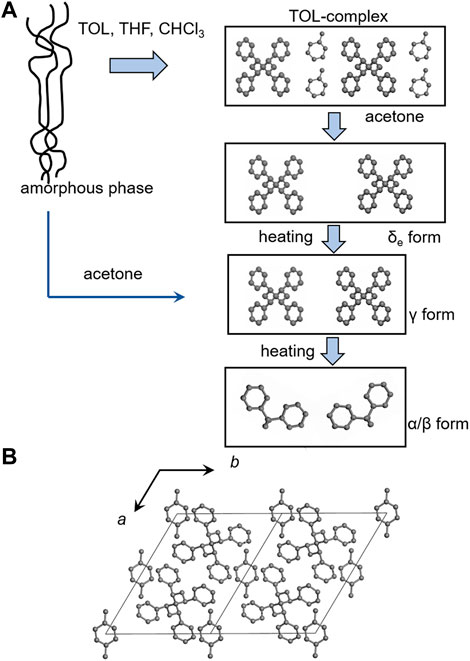
FIGURE 2. (A) The schematic illustrations of the crystal structures of SPS in the solvent-induced and thermally-induced crystallization phenomena. Starting from the (oriented) amorphous state, the crystallization to the δ form occurs when the sample is exposed in the atmosphere of good solvent like toluene, chloroform, etc. The δ form changes to the empty δ (δe) by washing with acetone. By heating, the δe transforms to the γ form and then to the α and β forms. The supply of poor solvent like acetone to the amorphous SPS sample causes the direct crystallization into the γ form (Gowd et al., 2009). (B) The crystal structure of the δ form analyzed by the X-ray diffraction data analysis (Chatani et al., 1993a).
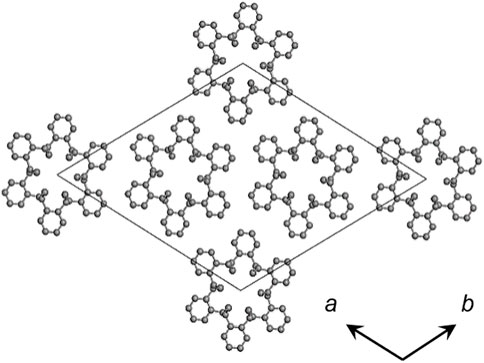
FIGURE 3. Crystal structure of SPS α form (De Rosa et al., 1991).
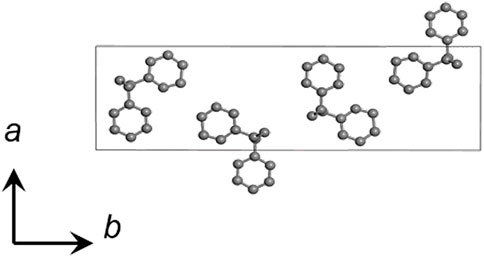
FIGURE 4. Crystal structure of SPS β form (Chatani et al., 1993b).
These behaviors are sensitively changed depending on the shape, polarity and/or thermal activity of the solvent molecules (Tarallo et al., 2010). Not only the solvent, but also the modification of the parent polymer itself is important for the control of the solvent complexation. The introduction of functional groups into the phenyl rings of SPS modifies the physicochemical property of the original SPS remarkably. For example, methoxy group (-OMe) has been found to remarkably affect the chain aggregation state and the solvent solubility of SPS (Wang et al., 2022). Besides, the substitution positions of the OMe groups on the phenyl ring, i.e., the ortho, meta and para-substitutions were found to change the chain conformation and the chain packing structure drastically. In this way, the phenomenon of the SPS-solvent complexation is quite complicated and is affected sensitively by the change of the solvent species and also by the introduction of the functional groups to the polymer chain.
In the long history of the scientific and industrial development of synthetic polymers, the role of solvent is indispensably important. In order to understand this intimate relation between polymer and solvent, we need to reveal their behaviors from the molecular level. The study of the liquid state of polymers in the solution gives us the averaged information of the spatial relation between the polymer and solvent molecules from the phenomenological point. The concrete information of the polymer chain conformation, the spatial distribution of the solvent molecules, and their interactions with polymers may be obtained in more detail by focusing on the solid-state system, i.e., the polymer-solvent complexes. The study of the solvent complexes of SPS and its relatives may be a good chance for us to know these information.
In the present article, we will review the studies of SPS and phenyl-substituted SPS by focusing on the solvent-induced crystallization behaviors and the associated phase transition processes from the viewpoints of microscopic structure level.
Before the concrete description of the detailed stories, it is needed to note about the naming of the various crystalline forms of SPS. In general, the naming of the various crystalline forms has not yet been established even in the IUPAC (International Union of Pure and Applied Chemistry). In many literatures of the solvent-SPS system, too, we encounter the variously different names: “complex,” “clathrate,” “clathrate complex,” “adduct,” “inclusion compound,” “co-crystals,” etc. The “clathrate” meant originally the cage structure with small molecules (like water) trapped inside. The concept of “clathrate” is now becoming wider and may cover even the general inclusion complex composed of the several different kinds of molecules. The several researchers are now using “clathrate” for the SPS-solvent system. The term “complex” is more general and is used in a variety of inorganic and organic compounds. Anyway, rather more important point is that we have to avoid the confusions in the discussions by using the variously different kinds of terminologies. Then, we will unify the naming by using the term “complex” through the manuscript.
2 Solvent-induced crystallization and phase transition of syndiotactic polystyrene
Immediately after the SPS was synthesized using a metallocene catalyst, the solvent-induced crystallization phenomenon of SPS has been discovered (Ishihara et al., 1986; Ishihara et al., 1988). The solvents used are categorized into non-guest and guest solvents (Immirzi et al., 1988; Vittoria et al., 1988).
2.1 Role of non-guest solvents
There are several types of non-guest solvents that accelerate the crystallization and phase transition of SPS (Rizzo et al., 2005), among which acetone (Naddeo et al., 1999) and super-critical-fluid (SCF) CO2 (Handa et al., 1997) were usually used. Non-guest solvent works as a plasticizer and accelerates the crystallization of SPS, which induces the formation of the γ form, not the δ form. As mentioned above, the γ form consists of the closest packing of the helical chains of TTGG conformation and it does not contain any solvent molecules. Generally, the γ form is produced by heating the solvent-complex [δ form (Chatani et al., 1993a)] so that the solvent is purged out (Figure 2). The γ form is formed more easily by immersing the SPS sample in the SCF-CO2 atmosphere compared with in the SCF-N2 atmosphere. This is because the free volume in the amorphous region is expanded more effectively in the SCF-CO2 environment (Ma et al., 2004; Ma et al., 2008).
In this way the crystallization of the γ form is induced by supplying the non-guest solvent. The growth of the crystallite size of the γ form is enhanced by annealing the thus-created nuclei of the γ form. Figure 5 shows the experimental result about the relation between the lamellar thickness (dc) evaluated by the SAXS data analysis and the heat treatment temperature. The data may be classified into the three temperature regions: the stable lamellae region (25–90°C), melt-recrystallization region (90–185°C), and the region of pre-phase transition to the α and β forms (185–195°C) (Wang et al., 2018). These data are fitted well to the Gibbs-Thomson equation (Wunderlich, 1980).
where
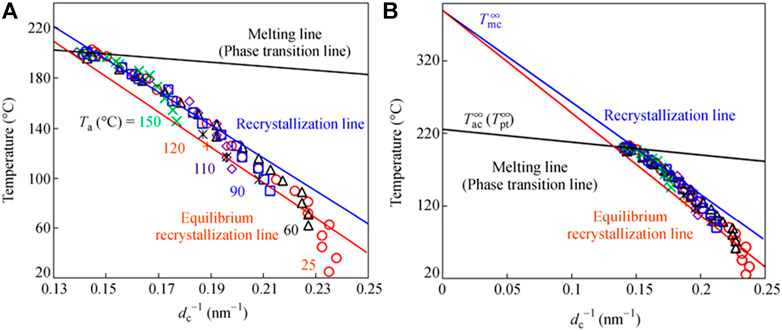
FIGURE 5. (A) Plot of temperature against dc−1 of sPS γ form annealed at various temperatures in the range of 20–190°C. (B) The extrapolation of the curves to the high temperature region to estimate the equilibrated melting point. The phase transition to the α and β forms did not occur in these temperature regions. (Reprinted with permission from Wang et al. (2018) Chin. J. Polym. Sci. 2018, 36, 749–755. Copyright 2018, Springer Nature).
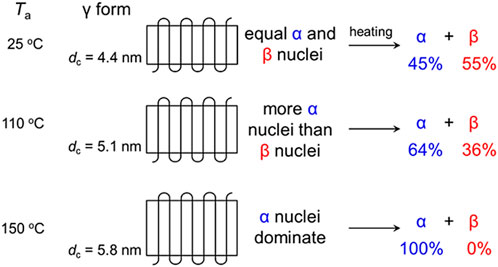
FIGURE 6. The relationship between the initial lamellar thickness (dc) and the crystal modifications obtained by heat treatment of the γ form. (Reprinted with permission from Wang et al. (2018) Macromolecules 2018, 51, 497–503. Copyright 2018, American Chemical Society).
2.2 Role of guest solvents in the complex formation
In order to understand the intimate relation between SPS chains and solvent molecules, we need to clarify the behavior of the solvent molecule themselves in detail. We may imagine that the solvent molecules are absorbed (or penetrate) into the SPS sample, migrate (diffuse) in the amorphous matrix of SPS chains, interact with them and are trapped by forming the stable (or unstable) SPS-solvent complexes. Let us see the concrete behaviors of the solvents.
The penetration rate of the solvent into the SPS film was estimated on the basis of the time-dependent 2D IR spectral data measured in the solvent-induced crystallization process by using a rapid-scan-type transmission 2D FTIR microscope equipped with a 2-dimensionally-arrayed focal plane detectors (Varian Fast Image FTIR microscope®) (Tashiro et al., 2006). A drop of TOL liquid was supplied on the edge of a thin amorphous SPS film. The solvent started to diffuse into the film, during which the IR images were collected at a time span of about 10 s. The crystallization-sensitive IR bands were observed to increase their intensities. Figure 7 shows the time dependence of the integrated absorbance estimated for the various IR bands. When the inner position distanced about 80 μm from the film edge was focused, the crystalline band increased in intensity in parallel to the TOL band, and the amorphous band decreased the intensity instead as the solvent penetration was proceeded. The infrared band absorbances (A) are expressed as a function of the position (x) and the time (t), which were analyzed on the basis of the so-called Fick’s diffusion equation to obtain the diffusion constant D of the solvent passing through the film.
where erf refers to the error function. The D constant was about 4.4 × 10−7 cm2/s at room temperature. This value is comparable to that of the TOL molecule penetrating into the amorphous atactic-PS (APS) sample, 3 × 10−7 cm2/s measured at 110°C and 1 × 10−6 cm2/s measured at 160°C higher than the glass transition temperature. (Duda et al., 1979) The diffusion rate of TOL molecules is almost common to both of SPS and amorphous APS. However, in the former case, the crystallization occurs immediately once when the solvent molecules contact with the amorphous chains of SPS even at a temperature below the glass transition point.
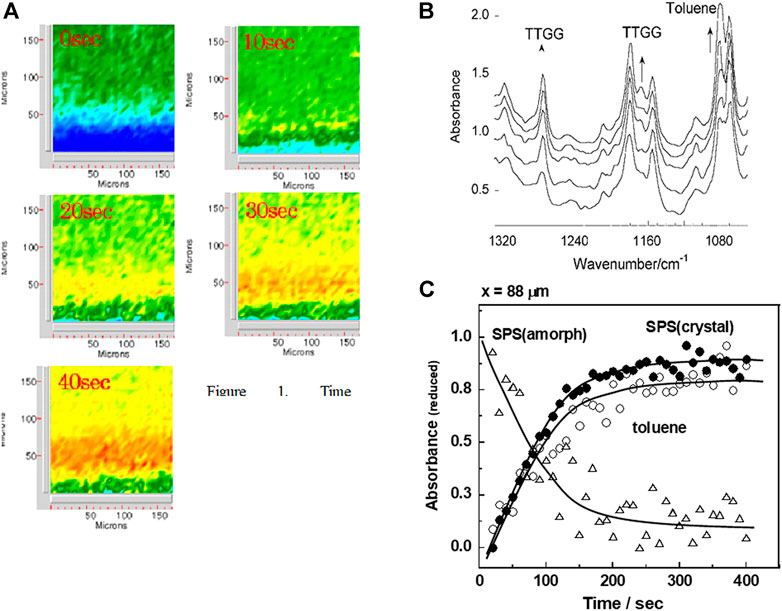
FIGURE 7. (A) The time-dependent 2D-IR images measured in the solvent-induced crystallization process of SPS film. (B) The IR spectral change at a fixed position of the film in (A). (C) Time dependence of the integrated absorbance estimated for the various IR bands of SPS in the presence of TOL. The diffusion constant D of toluene molecule in the amorphous SPS matrix is estimated from the curve. (From Tashiro et al. (2006) the Polymer Reprints, the Society of Polymer Science, Japan, 2006).
2.2.1 Effect on the thermal motion of chains
As mentioned above, the solvent molecules and SPS chains interact to form the crystalline complex finally. Since the structural reconstruction must occur in the crystallization process, the micro-Brownian motion of the amorphous chains may be enhanced by the solvents. As a result, the short helical segments are formed and grow longer with time. A series of these processes must occur at room temperature, far below the glass transition temperature (Tg ∼ 90°C) of the original SPS sample. We may speculate that the solvent molecules reduce the glass transition temperature through the strong interactions between the SPS chains and the solvent (Tashiro et al., 2003). In fact, as confirmed by the temperature-dependent crystallization experiment (Yoshioka and Tashiro, 2003a) and the molecular dynamics calculation (Yoshioka and Tashiro, 2004), the magnitude of the depression of Tg was found to be remarkably large, as shown in Figure 8. In that experiment, the crystallization rate (k) of the solvent-induced crystallization was estimated from the time-dependence of the IR spectral change at the various temperature (see Figure 8A). The extrapolation of the k values to almost negligibly small value gives the glass transition temperature (Tg) below which no crystallization occurs any more (Figure 8B). In the thermally-induced crystallization of the amorphous SPS sample, the Tg is about 90°C. The Tg was found to decrease to about -40°C in the toluene (TOL) atmosphere (Figure 8B). The comparison is made in Figure 8C) among the various kinds of solvent. The shift of Tg from the thermally-induced crystallization is about 130°C for TOL, 180°C for benzene, and 200°C for chloroform! The size and polarity of the solvents are important factors for the effective acceleration of the micro-Brownian motion of the chains.
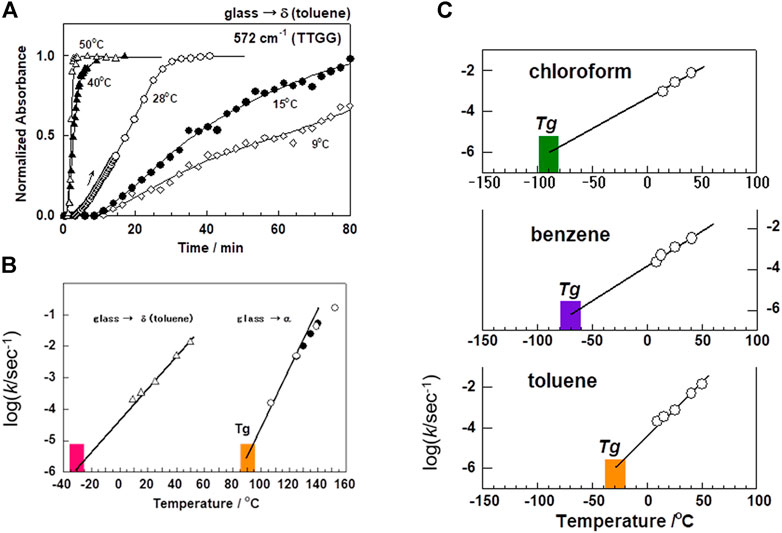
FIGURE 8. Comparison of the crystallization rate among the various kinds of solvents. (A) Time dependence of the growth of the crystallization-sensitive IR band at 572 cm−1, which is assigned to the vibrational mode characteristic of the TTGG form, measured at the various temperatures in the atmosphere of toluene vapor. (B) The rate constants (k) of the solvent-induced crystallization were evaluated from the steepest slope of the individual curves, and plotted against the temperature. The extrapolation of log(k) to the zero value gives the apparent glass transition temperature below which no crystallization occurs. (C) The similar plot is compared among the three different kinds of solvent. The Tg is found to be different remarkably depending on the kind of solvent. (Reprinted with permission from Yoshioka and Tashiro, Polymer 2003, 44, 6681–6688. Copyright 2003, American Chemical Society; Reprinted with permission from Yoshioka and Tashiro, Macromolecules 2004, 37, 467–472. Copyright 2004, American Chemical Society).
2.2.2 Effect of solvent type on the crystallization rate
Figure 9 shows the experimental results to clarify the effect of solvent on the crystallization phenomenon of amorphous SPS sample, where chloroform, benzene, toluene and acetone were used as the solvents (Tashiro et al., 2003). The induction time, that is, the waiting time for the start of crystallization is longer in the following order.
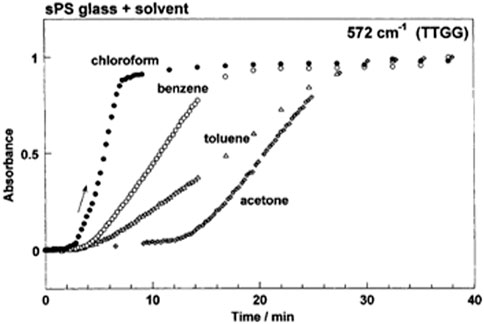
FIGURE 9. Time dependence of the IR absorbance of the crystallization-sensitive band (572 cm−1) in the solvent-induced crystallization phenomenon at room temperature. (Reprinted with permission from Tashiro, Yoshioka and Hashida, Macromolecular Symposium, 2003, 203, 13-25. Copyright 2003, John and Wiley).
The degree of crystallinity increased with time. The slope of the curve, or the crystallization rate depends on the solvent type in the following order.
As pointed out above, the solvent molecules diffuse in the amorphous matrix and interact with the polymer chains. Roughly speaking, the relative size of the solvent molecules must be smaller for the higher diffusion motion, which is.
However, this geometrical order is not very well associated with both of the observed induction time and crystallization rate. Rather the interaction may be more important. The solubility of SPS chain, as a measure of interaction, is in the following order.
The quantitative estimation of the relation between the solubility and crystallization rate should be a future theme.
The actual solvent-induced crystallization phenomenon is quite complicated and cannot be easily understood simply from the above-listed several factors (size, thermal activity and interaction of solvent with polymer chain). The whole amount of the penetrated solvent molecules might be another factor which will increase the starting points of the crystallization phenomenon in the amorphous matrix and may decrease the Tg of the polymer chains more effectively (Yoshioka and Tashiro, 2004).
The capacity to accept the solvents of various sizes is much larger than our expectation. In fact, even the relatively long n-alkanes molecules (n = 6–10) (Uda et al., 2004; Kaneko et al., 2006; Gowd and Tashiro, 2011; Tarallo et al., 2011), the polyethylene glycol (Kaneko and Sasaki, 2011), and the crown ether molecules (Kaneko et al., 2011) can form the complexes with SPS. These alkane molecules trapped in the complex were found to take an extended zigzag form (Gowd and Tashiro, 2011; Tarallo et al., 2011). The consideration of the higher-order structure composed of the crystalline and amorphous phases is also important as a future theme.
2.2.3 Solvent exchange
Another useful hint for the above discussion is seen in the solvent-exchange phenomenon. Because of such relatively high flexibility of the host polymer chains, the exchange of the different solvent molecules can be induced relatively easily. Figure 10 shows one experimental report, in which the exchanging phenomena between the several solvents are demonstrated (Yoshioka and Tashiro, 2003b). For example, as shown in Figure 10A, when chloroform is supplied to the sample of SPS-TOL complex, the SPS-chloroform complex is immediately formed by purging out the original TOL molecules. The exchange rate was not very much different among these three kinds of solvents. However, this phenomenon becomes a principle for the industrial application of the molecular filter. In general, the order of solvent-exchange rate is almost parallel to that of the crystallization rate (Yoshioka and Tashiro, 2003b; Tashiro, 2005). The unique columnar structure and not-very-strong interactions between SPS chains and solvent molecules are speculated to be the main reason for this solvent exchange phenomenon (Gowd et al., 2002; Yoshioka and Tashiro, 2003b).
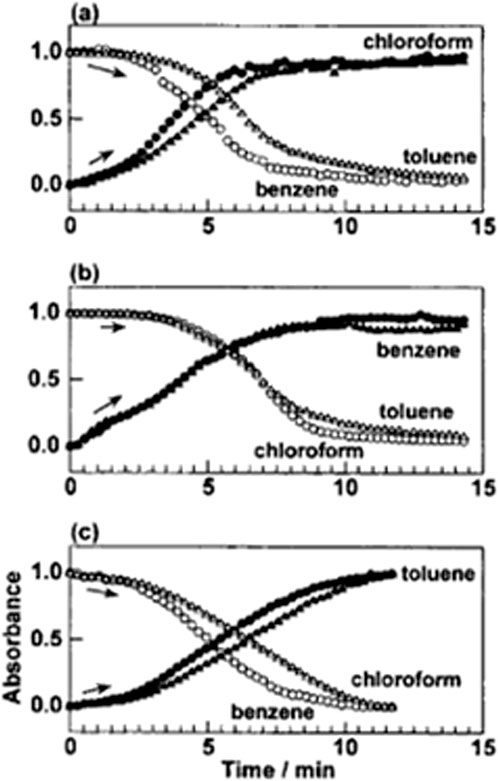
FIGURE 10. Time dependence of the absorbances of the IR bands characteristic of the solvent molecules in the solvent exchange phenomena of SPS-solvent complexes (Reprinted with permission from Yoshioka and Tashiro Macromolecules 2003, 36, 3593-3600. Copyright 2003, American Chemical Society).
2.2.4 Transition of the δ form
The solvent trapped in the complex is not permanently stable. By washing the SPS δ form using acetone (De Rosa et al., 1997), a solvent-free δe form (the empty δ form) is obtained. The spaces of the original solvent molecules are kept empty, being different from the γ form in which the helical chains are packed closely. This phenomenon may be assumed also as a kind of the above-mentioned solvent exchange phenomena; the acetone molecules push out the original solvents but, since the SPS-acetone complex is not very stable, the acetone molecules evaporate immediately to remain the empty spaces.
Both of the δ and δe forms transform to the γ form by heating at around 100°C. The further heating above 190°C causes the transition of the γ form to the α and β forms, as already mentioned in the preceding section. The effects of temperature, type of solvent, and higher-order structure on the δ/δe—γ –α/β phase transitions are reported in several references (Gowd et al., 2003; Gowd et al., 2006; Gowd and Tashiro, 2007; Gowd et al., 2008a; Gowd et al., 2008b; Gowd and Tashiro, 2011).
3 Solvent-induced crystallization and phase transition of syndiotactic polystyrene relatives
In the previous sections, the complex formation of the various kinds of solvents has been described for a unique polymer, SPS. Inversely, we need to know the effect of the parent polymer species on the polymer-solvent complex formation phenomenon. Unfortunately, however, this type of research was limited only in a few cases (Dell’Isola et al., 1997; Petraccone et al., 1998; Loffredo et al., 2001; La Camera et al., 2001; Loffredo et al., 2003; Girolamo et al., 2003; Petraccone and Tarallo, 2004). In this section we will see at first the case of SPS derivatives with the polar OMe groups introduced on the phenyl rings. The SPS derivatives with methyl or halogen groups will be also reviewed as the other cases.
3.1 Methoxy-substituted syndiotactic polystyrene
The substitution of a H atom with an -OCH3 (OMe) group on the phenyl ring is possible in the three ways; the ortho-, meta-, and para-substitutions as shown in Figure 11. They are denoted as the ortho-OMe-SPS, meta-OMe-SPS, and para-OMe-SPS, respectively. The crystal structures of the melt-cooled and stretched samples were determined by the quantitative analyses of the 2D-WAXD data (Wang et al., 2021; Tashiro, 2022). The meta- and para-OMe-SPS take the planar-zigzag chain conformations in the crystal lattice, similar to that of the SPS α and β forms. We call these crystalline forms of the OMe-SPS having the zigzag conformation the β form commonly. In the β form of the para-OMe-SPS, the bulky OMe groups jutting out of the main chain do not affect the chain conformation of the parent SPS chain of the zigzag form remarkably. On the other hand, the OMe groups located at the ortho-position of the rings are too bulky to keep the zigzag form and the trans-gauche exchange occurs to give the TTGG conformation even without any solvent. The meta-substitution is different from these two cases. The zigzag chains in the β form are relatively unstable and they transform to the TTGG conformation easily once when the solvent is supplied to the meta-sample.
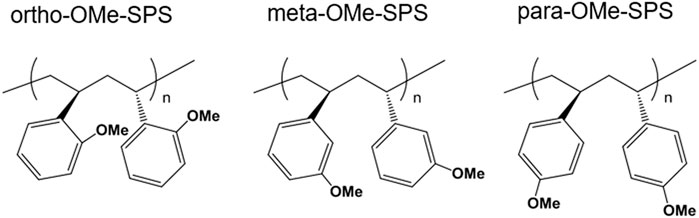
FIGURE 11. Chemical structures of three different OMe-SPS polymers.
The introduction of OMe groups modifies the solubility of the polymer remarkably. Different from the parent SPS case, these OMe-substituted SPS samples are easily soluble in such solvents as TOL and xylene of weak polarity, tetrahydrofuran (THF) of medium polarity, and chloroform of high polarity. The interactions of polymer chains with these solvents are strong for the ortho- and para-OMe-SPS, which are immediately solved even at room temperature and even in the rarefied solvent gas atmosphere. The meta-OMe-SPS behaves in a unique manner, which can form the solvent-complex in the rarefied THF or TOL atmosphere (Wang et al., 2022). The solvent molecules cause the large conformational change of the OMe-SPS skeletal chain from TTTT to TTGG form, as revealed in the remarkable changes of X-ray diffraction patterns and IR spectra (Wang et al., 2022). Figures 12 and 13 compare the crystal structures of the original meta-OMe-SPS (β form), the solvent complex (δ form) with TOL (or THF) and the γ form after extracting TOL (or THF) through the exchange with MeOH (Wang et al., 2022). Different from the case of SPS, the empty δ (δe) form cannot be formed stably, which transforms immediately to the γ form by the close packing of the alignments made of the TTGG chains (Figure 13). The heating of the solvent-complex (δ form) and the γ form does not cause the transition to the β form of the zigzag conformation, but they are melted finally, as known from the temperature-dependent X-ray diffraction measurement.
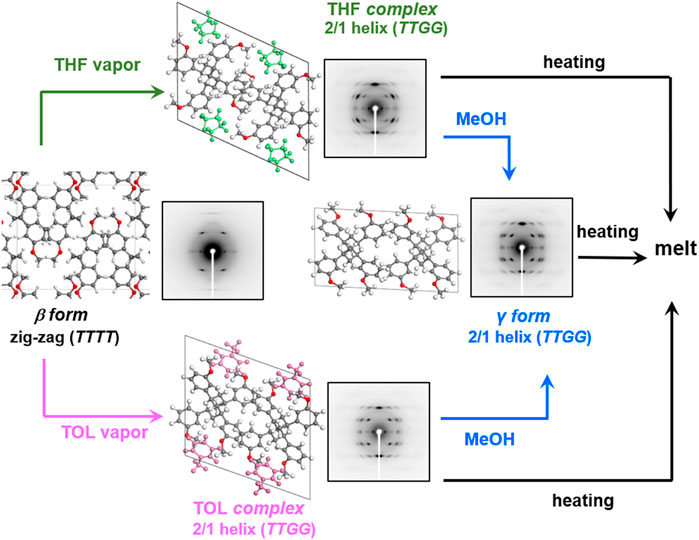
FIGURE 12. Solvent induced phase transition process of highly oriented and crystalline meta-OMe-SPS in presence of THF and TOL and subsequent MeOH washing (Reprinted with permission from Wang et al. (2022) Macromolecules, 2022, 55, 8222–8233 with some modification. Copyright 2022, American Chemical Society).
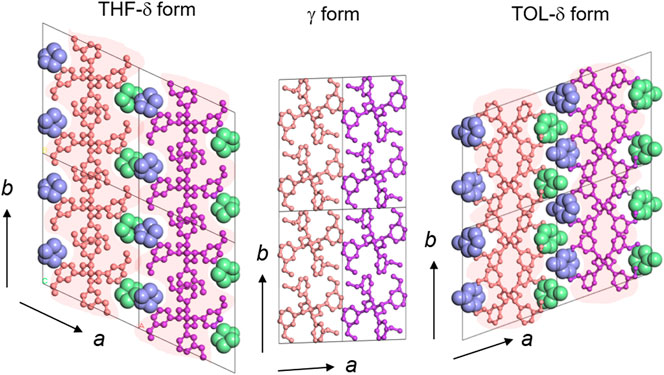
FIGURE 13. The packing modes of the solvent molecules in the vacant spaces produced by the surrounding TTGG molecular chains. Immediately after the evaporation of solvent molecules, the molecular chains are packed closely to generate the γ form. The chain conformation is kept unchanged in this process (Wang et al., 2022) (Reprinted with permission from Wang et al. (2022) Macromolecules, 2022, 55, 8222–8233 with some modification. Copyright 2022, American Chemical Society).
Another remarkable difference in the solvent complex formation phenomenon between the SPS and OMe-SPS cases is seen in the solid-to-solid phase transition. In the case of SPS, the oriented β form was not observed to transform to the complex when it is immersed in a solvent. Contrarily, the highly-crystalline meta-OMe-SPS shows the solvent-induced phase transition from the β form (TT conformation) to the δ form (TTGG conformation), just like the solid-to-solid phase transition although there might be another possibility of the dissolution of the original β form followed by the recrystallization to the δ form (Wang et al., 2022).
The solvent-complexation rate depends on the morphology of the OMe-SPS sample. The transition occurs relatively slowly when the unoriented crystalline sample is immersed in the solvent compared with the case of oriented sample. The kind of solvent species affects also this transition rate, as shown in Figures 14 and 15. In the case of THF, even the unoriented sample showed the transition from the β form to the δ form although the induction time (or the waiting time before the start of the complexation) was not very short. In the case of TOL, the oriented sample was found to show the transition to the oriented δ form, while the transition did not occur even for 200 h in the unoriented highly-crystalline sample. The effective size of THF molecule is smaller than TOL, making the movement in the sample easier for the former case.

FIGURE 14. (A) THF and (B) TOL vapor induced phase transition in unoriented/annealed meta-OMe-SPS.
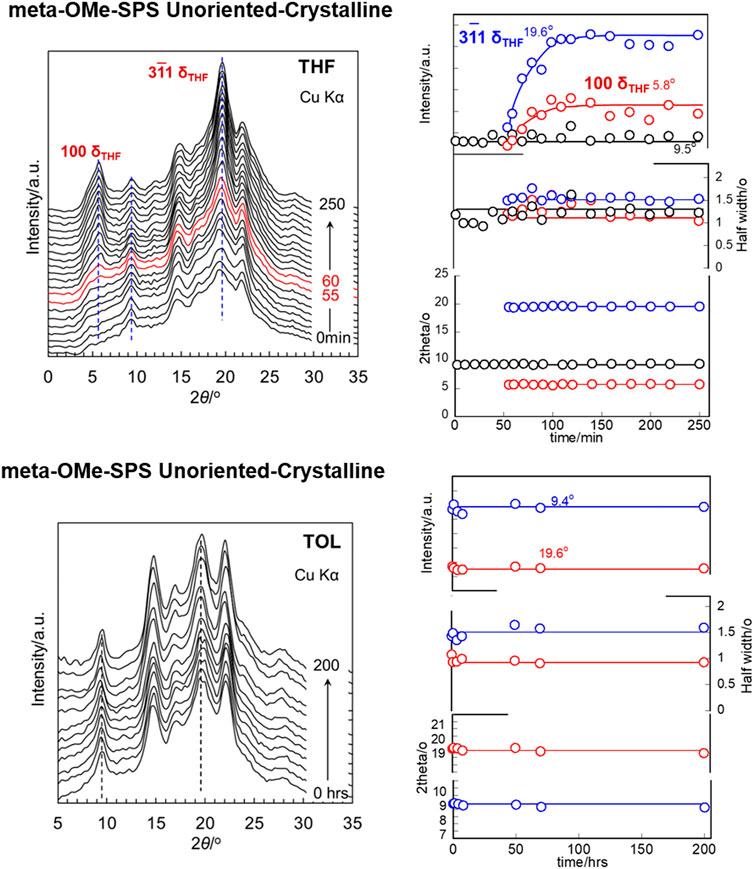
FIGURE 15. THF and TOL vapor induced phase transition in unoriented/annealed meta-OMe-SPS.
In this way, the various such factors as the sample morphology, the interaction strength between the OMe groups and solvents etc. affect the complex formation rate of OMe-SPS sensitively. Similar observation was made for SPS itself as seen in Figures 8 and 9.
What we want to emphasize here is that, as mentioned above, the crystalline α and β phases of SPS do not transform into the solvent complex when the sample is immersed for a long time in the solvent. Once the polar OMe groups are introduced, the crystalline β form was found to transform to the δ form in the solvent vapor. This experimental result is important from the practical pint of view. By controlling the side groups of the phenyl rings, we might have a novel SPS derivative with the unexpectedly remarkable change of the functional characters.
3.2 Methyl- and halogen-substituted syndiotactic polystyrene
Another types of the SPS derivatives synthesized so far are methyl (Me)- and halogen-substituted SPS: the para-Me-SPS, the meta-Me-SPS, the para-chloro-SPS, and the para-fluoro-SPS (Dell’Isola et al., 1997; Petraccone et al., 1998; Loffredo et al., 2001; La Camera et al., 2001; Girolamo et al., 2003; Petraccone and Tarallo, 2004; Rosa et al., 1996; Petraccone et al., 2005; Galdi et al., 2009). For example, the para-Me-SPS exhibits the complicated phase transitions: the forms III, IV, and V having the planar zigzag chain conformation, and the forms I, II, and complex (δ) of the 2/1 helical chain conformation (Dell’Isola et al., 1997; De Rosa et al., 1996). The form IV is a mesomorphic form with planar zigzag chains. The oriented δ form was obtained by the exposure of form IV in the ortho-dichlorobenzene or THF vapor atmosphere, where the right-handed and left-handed chains are packed in the unit cell (δalpha). When the sample is treated in such different type of solvent vapor atmosphere as benzene or CS2, the δ form consisting of the helical chains with the same handedness is formed (δbeta). When cyclohexane or cyclohexanone are used in the solvent vapor treatment of the para-Me-SPS sample, the solvent complex with the c-axial length 11.7 Å was obtained, where the polymer chain takes the complicated conformation of T6G2T2G2 (Petraccone et al., 2005).
The SPS samples having the halogen atoms are expected to behave similarly to the case of OMe-SPS, since the halogen atoms may induce the polar bonds. For example, the amorphous para-chloro-SPS sample treated with para-chlorostyrene (p-CS) (Girolamo et al., 2003) and the para-fluoro-SPS sample treated with CHCl3, TOL, or p-difluorobenzene (Galdi et al., 2009) were reported to give the solvent/polymer complexes, although no detailed information was obtained about the structure.
4 Concluding remarks and perspectives
SPS and its derivatives exhibit a variety of solvent-induced crystallization and phase transition phenomena. They provide a very good platform to study the interactions between polymers and small molecules.
The small solvent molecules play a role as a kind of plasticizer. In many of polymer species, including PET, PLA, P3HT, etc., the polymer chains are activated in their thermal motions by the interactions with the solvent. This results in the easier crystallization even below the glass transition temperature. The typical cases have been discussed in the early section. However, the SPS chains are special cases. The SPS chains possess the phenyl rings as the side groups. The exquisite balance between the skeletal chain conformation and the phenyl rings creates the vacant spaces of flexible size and shape, into which the small solvent molecules can be stably entrapped with the relatively strong interactions with the polymer chains. In an extreme case, even n-alkane molecules with long size can be stored. Of course, the thermal effect enhances the motions of these solvent molecules and the empty δ form is produced when the interactions are weak. Not only the characteristic property of solvent molecules, but also the functional groups introduced as the side chains of the phenyl rings affect the solvent-induced crystallization and phase transition remarkably. At present, the studies of the effects of these functional groups on the solvent-induced transition have not yet been developed enough well. The present review might be a good stimulus for this research theme.
In this review, we have not discussed the solvent effect on the higher-order structure consisting of the complicated crystalline and amorphous phases. As seen for PET and P3HT, the lamellar aggregation structure is more or less influenced by the application of the solvent. The control of the higher-order structure of crystalline polymers should be useful for the industrial development of the engineering polymer materials with the higher functional properties.
Author contributions
HW wrote the first draft of the manuscript; YM designed the manuscript structure; KT designed the manuscript structure and contributed to manuscript revision.
Funding
This research is supported by the National Natural Science Foundation of China (21704101) and the MEXT, Japan “Strategic Project to Support the Formation of Research Bases at Private Universities (2015–2019)”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Chatani, Y., Shimane, Y., Inagaki, T., Ijitsu, T., and Yukinari, T. (1993). Structural study on syndiotactic polystyrene. 2. Crystal structure of molecular compound with toluene. Polymer 34 (8), 1620–1624. doi:10.1016/0032-3861(93)90318-5
Chatani, Y., Shimane, Y., Ijitsu, T., and Yukinari, T. (1993). Structural study on syndiotactic polystyrene:3.Crystal structure of planar form I. Polymer 34 (3), 1625–1629. doi:10.1016/0032-3861(93)90319-6
Cornélis, H., Kander, R. G., and Martin, J. P. (1996). Solvent-induced crystallization of amorphous poly(ether ether ketone) by acetone. Polymer 37 (20), 4573–4578. doi:10.1016/0032-3861(96)00246-7
De Rosa, C., Guerra, G., Petraccone, V., and Corradini, P. (1991). Crystal structure of the α-form of syndiotactic polystyrene. Polym. J. 23, 1435–1442. doi:10.1295/polymj.23.1435
De Rosa, C., Guerra, G., Petraccone, V., and Pirozzi, B. (1997). Crystal structure of the emptied clathrate form (δe form) of syndiotactic polystyrene. Macromolecules 30 (14), 4147–4152. doi:10.1021/ma970061q
De Rosa, C., Petraccone, V., Guerra, G., and Manfredi, C. (1996). Polymorphism of syndiotactic poly(p-methylstyrene): Oriented samples. Polymer 37 (23), 5247–5253. doi:10.1016/0032-3861(96)00353-9
Dell’Isola, A., Floridi, G., Rizzo, P., Ruiz de Ballesreros, O., and Petraccone, V. (1997). On the clathrate forms of syndiotactic poly(p -methylstyrene). Macromol. Symp. 114, 243–249. doi:10.1002/masy.19971140132
Desai, A. B., and Wilkes, G. L. (1974). Solvent-induced crystallization of polyethylene terephthalate. J. Polym. Sci. C. Polym. Symp. 319 (46), 291–319. doi:10.1002/polc.5070460123
Duda, J. L., Ni, Y. C., and Vrentas, J. S. (1979). Toluene diffusion in molten polystyrene. J. Appl. Polym. Sci. 23 (3), 947–951. doi:10.1002/app.1979.070230329
Durning, C. J., Rebenfeld, L., Russel, W. B., and Weigmann, H. D. (1986). Solvent-induced crystallization. II. Morphology. J. Polym. Sci. B. Polym. Phys. 24 (6), 1341–1360. doi:10.1002/polb.1986.090240611
Galdi, N., Albunia, A. R., Oliva, L., and Guerra, G. (2009). Polymorphism of syndiotactic poly(p-fluoro-styrene). Polymer 50 (8), 1901–1907. doi:10.1016/j.polymer.2009.02.015
Girolamo, A. De, Mauro, D., Loffredo, F., Venditto, V., Longo, P., and Guerra, G. (2003). Polymorphic behavior of syndiotactic poly(p -chlorostyrene) and styrene/p-chlorostyrene cosyndiotactic random copolymers. Macromolecules 36 (20), 7577–7584. doi:10.1021/ma034678l
Gowd, E. B., Nair, S. S., and Ramesh, C. (2002). Crystalline transitions of the clathrate (δ) form of syndiotactic polystyrene during heating: Studies using high-temperature X-ray diffraction. Macromolecules 35 (22), 8509–8514. doi:10.1021/ma020640h
Gowd, E. B., Nair, S. S., Ramesh, C., and Tashiro, K. (2003). Studies on the clathrate (δ) form of syndiotactic polystyrene crystallized by different solvents using fourier transform infrared spectroscopy. Macromolecules 36 (19), 7388–7397. doi:10.1021/ma034935m
Gowd, E. B., Shibayama, N., and Tashiro, K. (2006). Structural changes in thermally induced phase transitions of uniaxially oriented δe form of syndiotactic polystyrene investigated by temperature-dependent measurements of X-ray fiber diagrams and polarized infrared spectra. Macromolecules 39 (24), 8412–8418. doi:10.1021/ma061659d
Gowd, E. B., Shibayama, N., and Tashiro, K. (2008). Structural correlation between crystal lattice and lamellar morphology in the phase transitions of uniaxially oriented syndiotactic polystyrene (δ and δe forms ) as revealed by simultaneous measurements of wide-angle and small-angle X-ray scatterings. Macromolecules 41 (7), 2541–2547. doi:10.1021/ma071759z
Gowd, E. B., and Tashiro, K. (2011). Effect of chain-length of n-alkane on solvent-induced crystallization and solvent exchange phenomenon in syndiotactic polystyrene. Polymer 52 (3), 822–829. doi:10.1016/j.polymer.2010.11.015
Gowd, E. B., and Tashiro, K. (2007). Effect of solvent molecules on phase transition phenomena of syndiotactic polystyrene. Macromolecules 40 (15), 5366–5371. doi:10.1021/ma070558s
Gowd, E. B., Tashiro, K., and Ramesh, C. (2008). Role of solvent molecules as a trigger for the crystal phase transition of syndiotactic polystyrene/solvent complex. Macromolecules 41 (24), 9814–9818. doi:10.1021/ma801742p
Gowd, E. B., Tashiro, K., and Ramesh, C. (2009). Structural phase transitions of syndiotactic polystyrene. Prog. Polym. Sci. 34, 280–315. doi:10.1016/j.progpolymsci.2008.11.002
Greis, O., Xu, Y., Asano, T., and Petermann, J. (1989). Morphology and structure of syndiotacticpolystyrene. Polymer 30 (4), 590–594. doi:10.1016/0032-3861(89)90140-7
Handa, Y. P., Zhang, Z., and Wong, B. (1997). Effect of compressed CO2 on phase transitions and polymorphism in syndiotactic polystyrene. Macromolecules 30 (26), 8499–8504. doi:10.1021/ma9712209
Immirzi, A., Candia, F. de, Iannelli, P., Zambelli, A., and Vittoria, V. (1988). Solvent-induced polymorphism in syndiotactic polystyrene. Makromol. Chem. Rapid Commun. 9, 761–764. doi:10.1002/marc.1988.030091108
Ishihara, N., Kuramoto, M., and Uoi, M. (1988). Stereospecific polymerization of styrene giving the syndiotactic polymer. Macromolecules 21 (12), 3356–3360. doi:10.1021/ma00190a003
Ishihara, N., Seiniya, T., Kuramoto, M., and Uoi, M. (1986). Crystalline syndiotactic polystyrene. Macromolecules 19 (9), 2464–2465. doi:10.1021/ma00163a027
Kaneko, F., and Sasaki, K. (2011). Crystalline complex of syndiotactic polystyrene with poly(ethylene glycol) dimethyl ethers. Macromol. Rapid Commun. 32 (13), 988–993. doi:10.1002/marc.201100136
Kaneko, F., Sasaki, K., Kashihara, N., and Okuyama, K. (2011). Complexation of syndiotactic polystyrene with crown ethers: 12-Crown-4, 15-crown-5 and 18-crown-6. Soft Mat. 9 (2–3), 107–123. doi:10.1080/1539445x.2011.552347
Kaneko, F., Uda, Y., Kawaguchi, T., Ute, K., and Yamamuro, O. (2006). Structural and dynamical properties of N-alkane molecules in clathrate phase of syndiotactic polystyrene. Macromol. Symp. 242, 113–119. doi:10.1002/masy.200651017
La Camera, D., Petraccone, V., Artimagnella, S., and de Ballesteros, O. R. (2001). Crystal structure of the clathrate form of syndiotactic poly(p-methylstyrene) containing benzene. Macromolecules 34 (22), 7762–7766. doi:10.1021/ma010703g
Liu, W., Breault, B., and Brisson, J. (1995). Solvent induced crystallization of nylon-6I. J. Polym. Sci. B. Polym. Phys. 33, 619–627. doi:10.1002/polb.1995.090330412
Loffredo, F., Pranzo, A., Guerra, G., Venditto, V., and Longo, P. (2001). Clathrates with tetrahydrofuran of styrene-p-methyl styrene Co-syndiotactic copolymers. Macromol. Symp. 166, 165–172. doi:10.1002/1521-3900(200103)166:1<165::aid-masy165>3.0.co;2-r
Loffredo, F., Pranzo, A., Venditto, V., Longo, P., and Guerra, G. (2003). Clathrate phases of styrene/p-methylstyrene Co-syndiotactic copolymers. Macromol. Chem. Phys. 204 (5–6), 859–867. doi:10.1002/macp.200390052
Ma, W., Andersson, A., He, J., and Maurer, F. H. J. (2008). Free volume changes, crystallization, and crystal transition behavior of syndiotactic polystyrene in supercritical CO2 revealed by positron annihilation lifetime spectroscopy. Macromolecules 41 (14), 5307–5312. doi:10.1021/ma8009068
Ma, W., Yu, J., and He, J. (2004). Direct formation of γ form crystal of syndiotactic polystyrene from amorphous state in supercritical CO2. Macromolecules 37 (18), 6912–6917. doi:10.1021/ma0491715
Marubayashi, H., Asai, S., and Sumita, M. (2012). Complex crystal formation of poly(L-lactide) with solvent molecules. Macromolecules 45 (3), 1384–1397. doi:10.1021/ma202324g
Marubayashi, H., Asai, S., and Sumita, M. (2012). Crystal structures of poly(L-lactide)–CO2 complex and its emptied form. Polymer 53 (19), 4262–4271. doi:10.1016/j.polymer.2012.07.044
Marubayashi, H., Asai, S., and Sumita, M. (2013). Guest-induced crystal-to-crystal transitions of poly(L-lactide) complexes. J. Phys. Chem. B 117 (1), 385–397. doi:10.1021/jp308999t
Mensitieri, G., Venditto, V., and Guerra, G. (2003). Polymeric sensing films absorbing organic guests into a nanoporous host crystalline phase. Sensors Actuators B Chem. 92 (3), 255–261. doi:10.1016/s0925-4005(03)00273-9
Naddeo, C., Guadagno, L., Aciemo, D., Vittoria, V., Chimica, I., Salemo, U., et al. (1999). Studies of the γ ← α transition in syndiotactic polystyrene. Macromol. Symp. 138, 209–214. doi:10.1002/masy.19991380127
Ouyang, H., Lee, W. H., Ouyang, W., Shiue, S. T., and Wu, T. M. (2004). Solvent-induced crystallization in poly(ethylene terephthalate) during mass transport: Mechanism and boundary condition. Macromolecules 37 (20), 7719–7723. doi:10.1021/ma0400416
Ouyang, H., Lee, W. H., and Shih, M. C. (2002). Three stages of crystallization in poly(ethylene terephthalate) during mass transport. Macromolecules 35 (22), 8428–8432. doi:10.1021/ma020851m
Park, D., and Hong, J.-W. (1997). Solvent-induced crystallization and interaction parameter of the blends of bisphenol A polycarbonate and poly(phenyl methacrylate). Polym. J. 29 (12), 970–974. doi:10.1295/polymj.29.970
Petraccone, V., Esposito, G., Tarallo, O., and Caporaso, L. (2005). A new clathrate class of syndiotactic poly(p-methylstyrene) with a different chain conformation. Macromolecules 38 (13), 5668–5674. doi:10.1021/ma050257n
Petraccone, V., La Camera, D., Pirozzi, B., Rizzo, P., and De Rosa, C. (1998). Crystal structure of the clathrate form of syndiotactic poly(p-methylstyrene) containing tetrahydrofuran. Macromolecules 31 (17), 5830–5836. doi:10.1021/ma9718964
Petraccone, V., and Tarallo, O. (2004). Comparative analysis of the clathrate forms of syndiotactic polystyrene, poly (p-Methylstyrene) and poly (m-Methylstyrene). Macromol. Symp. 213, 385–394. doi:10.1002/masy.200450934
Praveena, N. M., Shaiju, P., Amal Raj, R. B., and Gowd, E. B. (2022). Infrared bands to distinguish amorphous, meso and crystalline phases of poly(lactide)s: Crystallization and phase transition pathways of amorphous, meso and co-crystal phases of poly(ʟ-lactide) in the heating process. Polymer 240 (1), 124495. doi:10.1016/j.polymer.2021.124495
Praveena, N. M., Virat, G., Krishnan, V. G., and Gowd, E. B. (2022). Stereocomplex formation and hierarchical structural changes during heating of supramolecular gels obtained by polylactide racemic blends. Polymer 241 (15), 124530. doi:10.1016/j.polymer.2022.124530
Rizzo, P., Albunia, A. R., and Guerra, G. (2005). Polymorphism of syndiotactic polystyrene: Gamma phase crystallization induced by bulky non-guest solvents. Polymer 46 (23), 9549–9554. doi:10.1016/j.polymer.2005.07.063
Rizzo, P., Ianniello, G., Venditto, V., Tarallo, O., and Guerra, G. (2015). Poly(L-Lactic acid): Uniplanar orientation in cocrystalline films and structure of the cocrystalline form with cyclopentanone. Macromolecules 48 (20), 7513–7520. doi:10.1021/acs.macromol.5b00908
Rosa, C. De, Petraccone, V., Guerra, G., Manfredi, C., Chimica, D., Federico, N., et al. (1996). Polymorphism of syndiotactic poly(p-methylstyrene ): Oriented samples. Polymer 23, 5247–5253. doi:10.1016/0032-3861(96)00353-9
Shaiju, P., Murthy, N. S., and Gowd, E. B. (2016). Molecular, crystalline, and lamellar length-scale changes in the poly(L-lactide) (PLLA) during cyclopentanone (CPO) desorption in PLLA/CPO cocrystals. Macromolecules 49 (1), 224–233. doi:10.1021/acs.macromol.5b02425
Sheldon, R. P., and Blakey, P. R. (1962). Liquid-induced crystallization in polymers. Nature 195 (4837), 172–173. doi:10.1038/195172a0
Tarallo, O., Petraccone, V., R. Albunia, A., Daniel, C., and Guerra, G. (2010). Monoclinic and triclinic δ -clathrates of syndiotactic polystyrene. Macromolecules 43 (20), 8549–8558. doi:10.1021/ma1013513
Tarallo, O., Schiavone, M. M., and Petraccone, V. (2011). Structural characterization of the δ-clathrate forms of syndiotactic polystyrene with n-alkanes. Polymer 52 (6), 1426–1435. doi:10.1016/j.polymer.2011.01.041
Tashiro, K., Shimada, S., and Nakatani, T. (2006). Structural change of polymers investigated by the rapid-2D-IR microscopic imaging method. Polym. Repr. Soc. Polym. Sci. Jpn., 812. doi:10.1007/978-981-15-9562-2
Tashiro, K. (2005). Structural evolution process in solvent-induced crystallization phenomenon of syndiotactic polystyrene. Macromol. Symp. 222, 115–120. doi:10.1002/masy.200550413
Tashiro, K. (2022). Structural science of crystalline polymers: Basic concepts and practices. Singapore: Springer.
Tashiro, K., Yoshioka, A., and Hashida, T. (2003). Microscopically-viewed structural changes in solvent-induced phase transitions of synthetic polymers. Macromol. Symp. 203, 13–26. doi:10.1002/masy.200351302
Tremel, K., and Ludwigs, S. (2014). P3HT revisited – from molecular scale to solar cell devices. P3HT Revisit. Mol. Scale Sol. Cell. Devices, 26, 39
Uda, Y., Kaneko, F., and Kawaguchi, T. (2004). Selective guest uptake from solvent mixtures in the clathrate phase of syndiotactic polystyrene. Macromol. Rapid Commun. 25 (22), 1900–1904. doi:10.1002/marc.200400328
Vittoria, V., De Candia, F., Iannelli, P., and Immirzi, A. (1988). Solvent-induced crystallization of glassy syndiotactic polystyrene. Makromol. Chem. Rapid Commun. 9, 765–769. doi:10.1002/marc.1988.030091109
Wang, H., Liu, D., Cui, D., Men, Y., and Tashiro, K. (2022). Crystal structures of syndiotactic polymethoxystyrene-solvent complexes derived by X-ray diffraction data analysis and computer simulation. Macromolecules 55 (18), 8222–8233. doi:10.1021/acs.macromol.2c01290
Wang, H., Liu, D., Cui, D., Men, Y., and Tashiro, K. (2021). Effect of methoxy side groups on the crystal structures of a series of syndiotactic polymethoxystyrenes as studied by the X-ray diffraction data analysis. Macromolecules 54 (4), 1881–1893. doi:10.1021/acs.macromol.0c02577
Wang, H., and Tashiro, K. (2022). “Structrues and phase transitions of PLA and its related polymers,” in Poly(lactic acid): Synthesis, structures, properties, processing, applications, and end of life (John Wiley & Sons).
Wang, H., Wu, C. J., Cui, D. M., and Men, Y. F. (2018). Equilibrium crystallization temperature of syndiotactic polystyrene γ form. Chin. J. Polym. Sci. 36 (6), 749–755. doi:10.1007/s10118-018-2059-1
Waywood, W. J., and Durning, C. J. (1987). Solvent-induced crystallization of a compatible polymer blend. Polym. Eng. Sci. 27 (17), 1265–1274. doi:10.1002/pen.760271702
Woo, E. M., Sun, Y. S., and Yang, C. (2001). Polymorphism , thermal behavior , and crystal stability in syndiotactic polystyrene vs . Its miscible blends. Prog. Polym. Sci. 26, 945–983. doi:10.1016/s0079-6700(01)00010-7
Yoshioka, A., and Tashiro, K. (2003). Polymer-solvent interactions in crystalline δ form of syndiotactic polystyrene viewed from the solvent-exchange process in the δ form and the solvent evaporation phenomenon in the thermally induced δ-γ phase transition. Macromolecules 36 (10), 3593–3600. doi:10.1021/ma021774y
Yoshioka, A., and Tashiro, K. (2004). Solvent effect on the glass transition temperature of syndiotactic polystyrene viewed from time-resolved measurements of infrared spectra at the various temperatures and its simulation by molecular dynamics calculation. Macromolecules 37 (2), 467–472. doi:10.1021/ma035505z
Yoshioka, A., and Tashiro, K. (2003). Thermally- and solvent-induced crystallization kinetics of syndiotactic polystyrene viewed from time-resolved measurements of infrared spectra at the various temperatures (1) estimation of glass transition temperature shifted by solvent absorption. Polymer 44 (21), 6681–6688. doi:10.1016/s0032-3861(03)00696-7
Keywords: syndiotactic polystyrene, solvent induced crystallization, solvent induced phase transition, solvent induced aggregate structure, X-ray diffraction, crystal structure
Citation: Wang H, Men Y and Tashiro K (2022) Solvent-induced crystallization and phase-transition phenomena in syndiotactic polystyrene and its relatives. Front. Soft. Matter 2:1041872. doi: 10.3389/frsfm.2022.1041872
Received: 11 September 2022; Accepted: 12 October 2022;
Published: 26 October 2022.
Edited by:
Hsin-Lung Chen, National Tsing Hua University, TaiwanReviewed by:
Erathimmanna Bhoje Gowd, National Institute for Interdisciplinary Science and Technology (CSIR), IndiaPaola Rizzo, University of Salerno, Italy
Copyright © 2022 Wang, Men and Tashiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai Wang, aGFpd2FuZ0BkbHV0LmVkdS5jbg==; Kohji Tashiro, a3Rhc2hpcm9AYWljaGlzci5qcA==