 Thi Lan Anh Nguyen1,2*
Thi Lan Anh Nguyen1,2* Ha Thi Cam Dang2Jacco Koekkoek3
Ha Thi Cam Dang2Jacco Koekkoek3 Martin Braster1
Martin Braster1 John R. Parsons4
John R. Parsons4 Abraham Brouwer5,6
Abraham Brouwer5,6 Tjalf de Boer7
Tjalf de Boer7 Rob J. M. van Spanning1*
Rob J. M. van Spanning1*- 1Department of Molecular Cell Biology, Faculty of Science, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 2Institute of Biotechnology, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 3Department of Environment and Health, Faculty of Science, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 4Institute for Biodiversity and Ecosystem Dynamics, University of Amsterdam, Amsterdam, Netherlands
- 5BioDetection Systems, Amsterdam, Netherlands
- 6Department of Ecological Science, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 7MicroLife Solutions, Amsterdam, Netherlands
Four bacterial strains were isolated from enrichment cultures inoculated with soil from Bien Hoa military base in Vietnam contaminated with the herbicides 2,4-dichlorophenoxyacetate (2,4-D) and 2,4,5-trichlorophenoxyacetate (2,4,5-T). They were classified as Pseudomonas aeruginosa BT1 2.2, Sphingomonas histidinilytica BT1 5.2, Bordetella petrii BT1 9.2, and Achromobacter xylosoxidans BT1 10.2. All four were able to degrade 2,4-D and 2,4,5-T, but only the last three species used them as the sole sources of carbon and energy. Mass balance analyses suggest that between 33 and 46% of the carbon in the herbicides is incorporated into dry weight (DW). We obtained insight into their degradation pathways by the genomic analysis of these strains. A tfdCDEF gene cluster was found in A. xylosoxidans BT1 10.2 with amino acid sequences of their gene products showing high identity to those in B. petrii DSM12804. Bordetella petrii BT1 9.2 has a full complement of the tfdABCDEF genes. Surprisingly, the gene organization along with the amino acid sequences of the gene products are virtually identical to those of Cupriavidus pinatubonensis JMP134, referred to as type I tfd genes, and different from those of A. xylosoxidans BT1 10.2 and B. petrii DSM12804. We hypothesize that some of the genetic potential to degrade the herbicides has been recruited in recent mating events between these species and other members of the proteobacteria. This is the first report showing that B. petrii BT1 9.2 emerges as a key player in the degradation of 2,4-D.
Introduction
During the Vietnam War from 1961 to 1971, different types of herbicide and defoliant chemical, amongst which was Agent Orange, were used by the U.S. military as part of its chemical warfare program. Agent Orange is a mixture of equal parts of the herbicides 2,4-dichlorophenoxyacetate (2,4-D) and 2,4,5-trichlorophenoxyacetate (2,4,5-T) (Westing, 1984; Stellman et al., 2003). Many different types of bacteria isolated from different environments such as Cupriavidus pinatubonensis JMP 134 (Filer and Harker, 1997; Laemmli et al., 2000), Sphingomonas sp. TFD44 (Smejkal et al., 2001; Thiel et al., 2005), Achromobacter xylosoxidans subsp. denitrificans EST4002 (Vedler et al., 2004), Bradyrhizobium sp. HW13 (Kitagawa et al., 2002), Pseudomonas aeruginosa PAOlc, and Halomonas sp. EF43 (Kleinsteuber et al., 2001) can degrade 2,4-D. They have been isolated mainly from soils contaminated with herbicides (Ka et al., 1994; Tonso et al., 1995) and further genetic studies revealed their possession of key genes encoding the enzymes for 2,4-D degradation. These bacteria have been classified into two groups based on their phylogeny and genetic potential to express the 2,4-D-degrading enzymes (Serbent et al., 2019). Group I bacteria belong to the subdivision of Beta- and Gammaproteobacteria and include species from the genera Achromobacter, Burkholderia, Delftia, Halomonas, Pseudomonas, Ralstonia, Rhodoferax, and Variovorax (Hoffmann et al., 2003; Itoh et al., 2004). They usually have a full complement of a tfdABCDEF gene cluster for 2,4-D degradation. Group II bacteria include members of Alphaproteobacteria, closely related to the genera Sphingomonas and Bradyrhizobium (Kitagawa et al., 2002; Zharikova et al., 2018). Their organization of tfd genes is different from that of group I species and, importantly, Sphingomonas possesses a so-called cadABCD gene cluster encoding the subunits of a ring hydroxylating dioxygenase that replaces the function of TfdA (Kijima et al., 2018).
The first description of genes involved in the degradation of 2,4-D was based on studies on the Betaproteobacterium C. pinatubonensis (strain JMP 134/LMG 1197, previously termed Cupriavidus necator, Ralstonia eutropha, and originally Alcaligenes eutrophus), showing that it contained a so-called tfdABCDEF gene cluster expressing the enzymes for the degradation of 2,4-D (Don and Pemberton, 1981; Streber et al., 1987; Vallaeys et al., 1996). Subsequently, it was revealed that this species even had two copies of this gene cluster located adjacent to one another on plasmid pJP4 (Laemmli et al., 2000). The gene tfdA encodes an α-ketoglutarate-dependent 2,4-dichlorophenoxyacetate dioxygenase, which catalyses the initial cleavage of the acetate side chain of 2,4-D into 2,4-dichlorophenol (2,4-DCP) (Fukumori and Hausinger, 1993a,b). Subsequently, 2,4-dichlorophenol 6-monooxygenase which is encoded by tfdB converts 2,4-DCP into 3,5-dichlorocatechol (3,5-DCC). The substituted catechol is then sequentially degraded to 2-maleylacetate (2-MA) via an ortho-cleavage pathway by the enzymes encoded by the tfdCDEF genes, which include a chlorocatechol 1,2-dioxygenase, a dichloromuconate cycloisomerase, a carboxymethylenebutenolidase, and a maleylacetate reductase, successively (Liu and Chapman, 1984; Perkins et al., 1990; Laemmli et al., 2000). 2-Maleylacetate is then degraded to the succinyl moiety in succinyl-CoA and the acetyl moiety in acetyl-CoA in a sequence of reactions involving the products of the tfdF and pcaIJF genes, the last three of which encode a 3-oxoadipate CoA-transferase (PcaIJ) and a 3-oxoadipyl-CoA thiolase (PcaF), successively. Succinyl-CoA and acetyl-CoA may fuel the tricarboxylic acid (TCA) cycle. A list of relevant enzymes according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database is in the Supplementary Table 1.
In contrast to the diversity of 2,4-D-degrading bacteria, only a few aerobic 2,4,5-T degraders have been reported. The best known are members of genera Burkholderia (Kellogg et al., 1981; Danganan et al., 1994; Huong et al., 2007), Nocardioides (Golovleva et al., 1990), Sphingomonas (Huong et al., 2007), and Bradyrhizobium (Rice et al., 2005; Huong et al., 2007; Hayashi et al., 2016), all of which were capable of utilizing 2,4,5-T as the sole carbon and energy source. Burkholderia cepacia AC1100 has three gene clusters involved in the degradation of 2,4,5-T. The first one has tftAB genes encoding the two subunits of 2,4,5-trichlorophenoxyacetic acid oxygenase that converts 2,4,5-T to 2,4,5-trichlorophenol (2,4,5-TCP). The latter compound is further oxidized to 2,5-dichloro-p-benzoquinone (2,5-DCBQ) by a chlorophenol-4-monooxygenase encoded by the tftCD genes on the secend cluster (Danganan et al., 1994; Daubaras et al., 1995; Hübner et al., 1998; Zaborina et al., 1998; Gisi and Xun, 2003). An unknown factor then converts DCBQ into 2,5-dichlorohydroquinone (2,5-DCHQ). The third gene cluster, tftEFGH, encodes the enzymes for converting DCHQ to 3-oxoadipate (3-OXA). First, 2,5-dichlorohydroxyquinone reductive dehydrochlorinase (TftG) dechlorinates 2,5-DCHQ to chlorohydroquinone (CHQ) and then to hydroquinone (HQ) (Zaborina et al., 1998). Subsequent ring cleavage is then catalyzed by hydroxyquinone 1,2-dioxygenase (TftH) to yield 4-hydroxymuconic semialdehyde (4-HMSA) which then reacts to maleylacetate (2-MA). The latter is converted to 3-OXA by a maleylacetate reductase encoded by the tftE gene, an ortholog of tfdF. S. paucimobilis strain B90 has LinE and LinD proteins that differ from TftGH proteins, although their substrate and ultimate product are the same (Kumari et al., 2002). Here, the Tft and Tfd degradation pathways merge, ultimately resulting in the formation of succinyl-CoA and acetyl-CoA (Daubaras et al., 1995, 1996a). In yet another parallel pathway, 2,5-DCHQ is converted to 2-MA via the enzymes chlorophenol-4-monooxygenase (TftCD), 2-hydroxy-1,4-benzoquinone reductase (HbqR), and hydroxyquinol 1,2-dioxygenase (ChqB) (Ferraroni et al., 2005). These enzymes are found in for example Burkholderia and Nocardioides species (Daubaras et al., 1996b; Ferraroni et al., 2005; Takenaka et al., 2011).
In this study, we isolated indigenous bacterial strains originally present in soil heavily contaminated with Agent Orange in Vietnam, all of which were dominant in enrichment cultures with 2,4-D and 2,4,5-T as the sole sources of carbon and energy, and contained the genes involved in the breakdown of these compounds. Here, we aimed at a more fundamental understanding of their physiological and genetic potential in that metabolic process. The approach in this study was to (i) to culture the indigenous strains under defined conditions with 2,4-D and 2,4,5-T as the sole carbon and energy sources, (ii) to monitor the degradation of these herbicides in time along with increases in cellular biomass, and (iii) to unravel their genome sequences to obtain insight in their metabolic potential. These efforts resulted in an integrative view of species and pathways involved in the degradation of these chlorinated phenoxyacetates.
Materials and Methods
Culturing Conditions and Isolation of Bacterial Strains
Herbicide contaminated soil for enrichment studies was collected from Bien Hoa airbase (10°58′14.3″ N 106°48′19.3″ E), Dong Nai Province, Vietnam (Nguyen et al., 2021). Ten grams of herbicide contaminated soil was added in basal salt medium (BS, 40 ml per culture) containing KH2PO4 0.5 g/l, (NH4)2SO4 0.25 g/l, MgSO4 0.2 g/l, CaCl2 0.5 g/l, and NaNO3 0.4 g/l (van der Zaan et al., 2012), and supplemented with 100 mg/l 2,4-D and 100 mg/l 2,4,5-T. Both compounds, more than 95% pure, were purchased from Sigma. Cultures were incubated in the dark on a rotary shaker at 30°C and 200 rpm (the first enrichment). After cultivation for 26 days, these cultures were then used to inoculate a second enrichment with 200 mg/l 2,4-D and 100 mg/l 2,4,5-T and further incubated for 25 days. Colony-forming units (CFUs) were isolated on nutrient agar (NA) at 30°C by serial dilution of the cultures at different time points after the start of the experiment. Nutrient agar medium contained 1.0 g peptone, 1.0 g meat extract, 0.5 g NaCl, and 1.5 g agar per 100 ml. Bacterial strains that were isolated as dominant CFUs were pre-cultured overnight in nutrient broth (NB) medium. The cells were harvested by centrifugation at 10,000 rpm for 5 min, washed two times with sterilized BS, and transferred into BS medium supplemented with 200 mg/l 2,4-D and 100 mg/l 2,4,5-T as inoculum for the degradation experiment under the same conditions as those for the enrichment cultures. A flask with dead cells, which were killed by heating the flask at 121°C during 15 min, amended with the herbicides was used as a control to display the effects of abiotic chemistry or denatured proteins. All experiments were independently performed in triplicate. Cell growth was determined by measuring the absorbance of cultures at 600 nm using a spectrophotometer (Multiskan GO, Thermo Scientific, U.S). We realize that the use of optical density values alone to detect growth of the bacterial cultures is less precise than other methods such as direct measurement of bacterial cell dry weight (DW) or flow cytometry. The concentrations of 2,4-D and 2,4,5-T in all culture samples were determined in time using tandem liquid chromatography-mass spectrometry (LC-MS/MS) (Aligent Technologies, 1200 Series). All experiments were performed according to the Biosafety Manual from the Vrije Universiteit Amsterdam entitled “Handling hazardous biological agents, genetically modified organisms and quarantine organisms” and approved by the biosafety officer of the same university.
Analysis of 2,4-D and 2,4,5-T by LC-MS/MS
The extraction and determination of the concentrations of 2,4-D and 2,4,5-T by LC-MS/MS along with the data analyses were performed exactly as described earlier (Nguyen et al., 2021).
DNA Isolation and 16S rRNA Gene Sequencing of Isolated Strains
The bacterial isolates were identified based on 16S rRNA gene sequence analysis. DNA of bacteria was extracted using the MoBio PowerSoil® DNA Isolation kit (Carlsbad, CA, USA) according to the manufacturer's instructions. Amplification was carried out with universal primers 8F (5′-AGAGTTTGATYMTGGCTCAG-3′) and 1512R (5′-ACGGYTACCTTGTTACGACTT-3′) as described previously (Weisburg et al., 1991). Reactions were performed in a Thermocycler (Biometra, Analytik Jena, Germany). PCR products were purified and sequenced by Macrogen Europe. These sequences were compared with known 16S rRNA gene sequences deposited in the GenBank database using the BLAST search at the National Center for Biotechnology Information (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Genomic DNA Extraction, Genome Sequencing, Assembly, and Annotation
Genomic DNA was extracted using the MoBio PowerSoil® DNA Isolation kit (Carlsbad, CA, USA). The quality and quantity of the extracted genomic DNA were checked by agarose gel electrophoresis and Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, cat. no. Q32851). Genome sequencing was carried out by BaseClear B.V (The Netherlands) according to their in-house protocol. Single-end or paired-end sequence reads were generated using the Illumina NovaSeq 6000 or MiSeq system. The sequences generated with the MiSeq system were performed under accreditation according to the scope of BaseClear b.v. (L457; NEN-EN-ISO/IEC 17025). When paired-end sequencing is being performed, the “Number of reads” noted in Supplementary Table 2 is referring to read pairs. FASTQ read sequence files were generated using bcl2fastq2 version 2.18. Initial quality assessment was based on data passing the Illumina Chastity filtering. Subsequently, reads containing PhiX control signal were removed using an in-house filtering protocol, and reads containing (partial) adapters were clipped (up to a minimum read length of 50 bp). The second quality assessment was based on the remaining reads using the FASTQC quality control tool version 0.11.5. Supplementary Table 3 shows essential data of the sequencing and assembly of the genomes of the four selected isolates.
The raw Illumina reads were used directly for genome assembly using SPAdes (version 3.13.0), which is a De Bruijn graph assembler for short reads with a read error correction (Bankevich et al., 2012). The following SPAdes settings were used: –careful, -kmer: 55,77,127. Assembly quality was determined using QUAST (Gurevich et al., 2013) where we determined the number of scaffolds, total genome length, and N50 as a measurement for genome completeness. We found that the removal of scaffolds smaller than 5,000 bps did not significantly impact the total assembled genome length. Consequently, these scaffolds were removed in order to better facilitate the annotation. The scaffold size-adjusted genomes were annotated using several programs and databases. Gene prediction and initial annotation were done using Prokka (Seemann, 2014). Predicted genes were further annotated using Interproscan (Pfam and hamap databases) and the CAZy carbohydrate activated enzyme database (all families) (Jones et al., 2014; Lombard et al., 2014). Signal peptides were predicted with SignalP and secondary metabolite clusters using AntiSMASH (Weber et al., 2015; Almagro Armenteros et al., 2019). The closest homologs were determined by BLAST analysis using the Clusters of Orthologs Groups (COG) database for each predicted gene separately and by taking the species with the most hits as closest species (Tatusov et al., 2000). The NCBI-annotated genomes of BT1 2.2 (PRJNA586721), BT1 5.2 (PRJNA586745), BT1 9.2 (PRJNA586752), and BT1 10.2 (PRJNA587043) were deposited in GenBank with their BioProject numbers listed in between brackets.
Phylogeny
The amino acid sequences of TftA homologs from the four isolated bacteria were compared with reference sequences available in the NCBI and KEGG databases. The sequences were aligned and a phylogenetic tree (Figure 3) was visualized using the MEGA version 7 software by the neighbor-joining method with 1,000 bootstrap replicates (Kumar et al., 2016).
Data Accessibility
The NCBI-annotated genomes of BT1 2.2 (PRJNA586721), BT1 5.2 (PRJNA586745), BT1 9.2 (PRJNA586752), and BT1 10.2 (PRJNA587043) that support the findings of this study were deposited in GenBank with their BioProject accession numbers listed in between brackets. Links to these data, initially for review only, are listed below. After acceptance of the manuscript, the data will become publicly available along with an assigned DOI number.
Pseudomonas aeruginosa strain: BT1 2.2: https://dataview.ncbi.nlm.nih.gov/object/PRJNA586721?reviewer=q7ko9k52t4a6uqlcrjdl3sdci2
Sphingomonas histidinilytica strain: BT1 5.2: https://dataview.ncbi.nlm.nih.gov/object/PRJNA586745?reviewer=i6f5dcie0oc5aamc0lu4h8g3ea
Bordetella petrii strain: BT 1 9.2: https://dataview.ncbi.nlm.nih.gov/object/PRJNA586752?reviewer=anls0i9ag2l5ehv9f3q7clktoi
Achromobacter xylosoxidans strain: BT1 10.2: https://dataview.ncbi.nlm.nih.gov/object/PRJNA587043?reviewer=ca8ottdc5qkfr9gdon77qj5ffi
Results
Isolation and Classification of 2,4-D- and 2,4,5-T-Degrading Bacteria
Fourteen different types of bacterial colony were isolated from herbicide contaminated soil communities enriched in cultures with 2,4-D and 2,4,5-T as the sole sources of carbon and energy. The colonies of these strains on NA plates varied in shape and color. The results of 16S rRNA gene sequencing indicated that the dominant isolates were from genera Rhodanobacter, Arthrobacter, Pseudomonas, Achromobacter, Sphingomonas, Bordetella, Bosea, Pedobacter, Sphingopyxis, Staphylococcus, and Ancylobacter (Table 1). Among these isolates, four Gram-negative strains that showed up as the most dominant species in the enrichments, Pseudomonas, Achromobacter, Sphingomonas, and Bordetella, were selected to study their herbicide biodegradation capacities and their corresponding genetic make-up. BLAST results showed that the 16S rRNA gene sequences of the isolates BT1 2.2, BT1 5.2, BT1 9.2, and BT1 10.2 were highly similar (>99% identity) to those of P. aeruginosa, S. histidinilytica., B. petrii, and A. xylosoxidans, respectively. Therefore, we classified them as P. aeruginosa BT1 2.2, S. histidinilytica BT1 5.2, B. petrii BT1 9.2, and A. xylosoxidans BT1 10.2, respectively.
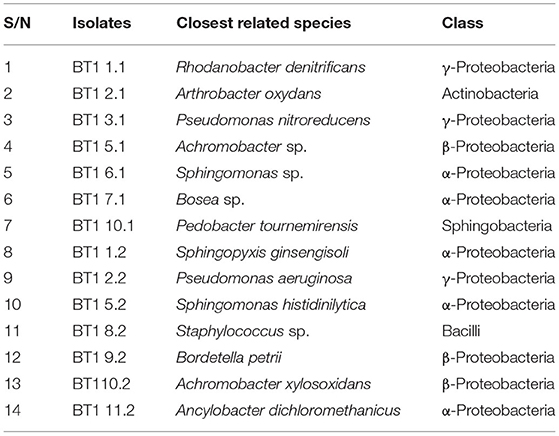
Table 1. List of bacterial isolates derived from the first and second enrichments (denoted by “1” and “2” as the last digit in the isolate names, respectively) of T1 soil communities on medium with herbicides.
Biodegradation of 2,4-D and 2,4,5-T by Four Strains Isolated From Enrichment Cultures
We set out a growth experiment where the isolates were cultured in basal salt medium supplemented with 2,4-D and 2,4,5-T as the sole sources of carbon and energy. We determined their growth curves (Figure 1A) and the removal of 2,4-D and 2,4,5-T in time (Figures 1B,C). Pseudomonas aeruginosa BT1 2.2 showed poor growth with a small increase in biomass after 7 days of cultivation, which subsequently decreased again after 10 days, most likely as a result of cell death and lysis. It did, however, show complete removal of 2,4-D and 2,4,5-T within 10 days of cultivation, but we have no data on the products that were formed. The other three strains showed substantial growth and complete removal of the herbicides within 10 days of cultivation. The fastest growers were B. petrii BT1 9.2 and A. xylosoxidans BT1 10.2, but the latter one had a lag time of 2 days before it started growing. Also, its final biomass was slightly less than that of B. petrii BT1 9.2. In both cases, maximum growth was reached after 4 days of cultivation. Sphingomonas histidinilytica BT1 5.2 needed a bit longer to reach its maximum growth yield. Due to its apparent slower growth rate, maximum biomass was obtained after 7 days, but the final yield was comparable to that of B. petrii BT1 9.2. The latter was the best performer concerning rates and yields of growth along with its ability to completely degrade 2,4-D and 2,4,5-T. To get a more comprehensive view of its capacities, we studied the responses of B. petrii BT1 9.2 in cultures with only 2,4-D or only 2,4,5-T and compared the growth data (Figure 2A) and removal of the herbicides (Figures 2B,C) with those from the culture with a mixture of both herbicides. The fastest growth and highest yields were observed in the cultures with only 2,4-D as the sole source of carbon and energy. Both growth rates and yields were slightly less in the mixture of the two compounds, and the poorest performance concerning rates and yield was observed with 2,4,5-T only. These growth properties coincided in part with the degradation rates of 2,4-D and 2,4,5-T. The former compound was removed within 2 days in the cultures with only 2,4-D, while it took 10 days in those with the mixture. Degradation of 2,4,5-T took 4 days of cultivation, while in the mixture this required 10 days.

Figure 1. Bacterial growth (A), degradation of 2,4-D (B), and degradation of 2,4,5-T (C) in mineral salt medium with a mixture of 2,4-D and 2,4,5-T inoculated with either of the four isolated species listed. The optical density (OD600 nm) at the start of each growth curve (0.32 for P. aeruginosa BT1 2.2, 0.26 for S. histidinilytica BT1 5.2, 0.32 for B. petrii BT1 9.2, and 0.39 for A. xylosoxidans BT1 10.2) was subtracted from those further on in these curves. Symbols are the means of three replicates. Results are shown as averages of triplicate cultures, with the error bars representing standard deviations.

Figure 2. Bacterial growth (A), degradation of 2,4-D (B), and degradation of 2,4,5-T (C) in mineral salt medium with either a mixture of 2,4-D and 2,4,5-T (mixture) or with only 2,4-D or 2,4,5-T as single herbicide inoculated with B. petrii BT1 9.2. Results are shown as averages of triplicate cultures, with the error bars representing standard deviations. The optical density (OD 600 nm) at the start of each growth curve (an average of about 0.32 for all three types of culture) was subtracted from those further on in these curves.
We were also interested to understand the stoichiometry of conversion of the herbicides into biomass. For that, we made use of a few relevant numbers related to Escherichia coli taken from the Bionumbers archive (Milo et al., 2010) and assumed similar values for our isolated strains. At an OD600 of 1, the number of E. coli cells is around 1012/L (Bionumbers ID 100985) with an average dry weight of 0.36 g/L (Bionumbers ID 109837). This is in the same order as when we approach these numbers from the average DW of an E. coli cell, which is 3.10−13 g DW (Bionumbers ID 100008), and, hence, 0.3 g/L DW in a culture with 1012 cells. For the actual ratio of biomass formation over the catabolic branch one has to consider the percentages carbon in 2,4-D and in that of the biomass, which are 43 and 49% (assuming a biomass formula C1H1.8O0.5N0.24; Bionumbers ID 101800), respectively. Using the value of 0.36 g DW/L for a culture with an OD600 of 1, we applied the calculations below. The increases in OD600 of B. petrii BT1 9.2, S. histidinilytica BT1 5.2, and A. xylosoxidans BT1 10.2 after degradation of a total of 300 mg/L herbicides 2,4-D and 2,4,5-T are 0.33, 0.29, and 0.23, respectively. These values correspond to 119, 104, and 83 mg DW/L, from which one could calculate the formation of 0.40, 0.35, and 0.28 mg DW/mg 2,4-D mineralized, respectively. When we consider the carbon content in the herbicides and the DW, these numbers are 0.46, 0.40, and 0.33 mg C in DW/ mg C in 2,4-D, respectively.
Our data are also in compliance with data from the literature. The group of Tiedje grew an isolated Bradyrhizobium strain HW13 in media with 2,4-D as the sole source of carbon and energy and the growth curve showed an increase in the optical density of around 0.09 (32.4 mg DW/L) upon full mineralization of 125 mg/L 2,4-D (Kamagata et al., 1997). This corresponds to 0.26 mg DW/mg 2,4-D mineralized. The value calculated from the growth curve of C. pinatubonensis JMP134 growing on 3-chlorobenzoate (Pérez-Pantoja et al., 2000; Plumeier et al., 2002) is 0.46 mg DW/mg 2,4-D mineralized. Both values are in the same range as those of our isolates.
Pathways for Herbicide Degradation
We inspected the four genomes for their genetic potential to degrade 2,4-D and 2,4,5-T to benzoate and further on to succinyl-CoA and acetyl-CoA. To this end, we gathered the amino acid sequences of all the enzymes identified in the KEGG database as being involved in these metabolic pathways. A list of our selected enzymes along with their properties and some additional parameters is shown in Supplementary Table 1. The KEGG database amino acid sequences of the selected genes were then used to look for similar sequences in the genome sequences of our four isolates. The script for these analyses is described in the materials and methods section. The results of these analyses for each organism are listed in Supplementary Table 3. Percentages identity between the KEGG protein sequences and those expressed from the selected genomes are listed as well. The threshold for selection of the orthologous proteins was set at a minimum of 45% identity. Importantly, all four species showed (initial) degradation of 2,4-D and 2,4,5-T so they should at least have a tfdA gene to degrade 2,4-D into 2,4-DCP, and a tftAB gene set to encode the dioxygenase for converting 2,4,5-T into 2,4,5-TCP, or as yet unknown enzymes that could take over that role. A phylogenetic tree of TftA homologs from the four isolates is shown in Figure 3 along with the reference enzymes from the KEGG and NCBI databases. These reference enzymes also include CadA, BenA, and AntA as we had reason to believe that some of the homologs that we found group closer to these proteins than to the TftA group. As such we were able to judge the minimum potential of the four species to degrade 2,4-D and 2,4,5-T and their further metabolites ultimately to succinyl-CoA and acetyl-CoA. We first constructed the reference pathways for degradation of the herbicides using the KEGG database maps “chlorocyclohexane and chlorobenzene degradation” and “benzoate degradation.” A graphical representation of these pathways is shown in Figure 4. We then reconstructed the pathways in our four isolated species by identifying their sequences orthologous to those of the reference pathways (Supplementary Figure 1). Below is a further description of the four isolates with emphasis on their genetic potential to convert 2,4-D and 2,4,5-T.

Figure 3. Dendrogram showing the relatedness of relevant TftA homologous sequences of the four isolated species (names in light blue) along with reference protein sequences from KEGG and NCBI databases (names in black). Homologs include CadA, BenA, and AntA sequences from the KEGG database along with their best-matching counterparts in α-, β-, and γ-proteobacteria. The dendrogram was obtained by the neighbor-joining method using MEGA 7 based on sequence alignments calculated by CLUSTAL W using the default options. Bradyrhizobium TftA (WP_130222982.1): Bradyrhizobium sp. Leo121, Burkholderia TftA (AAB39767.1): Burkholderia cepacia, Gammaproteobacterial TftA (MSQ67289.1): Gammaproteobacterium, Rhodospirillales BenA (OYV39246.1): Rhodospirillales bacterium 20-64-7, Prolinoborus BenA (SPJ20396.1): Prolinoborus fasciculus, Acinetobacter BenA (P07769): Acinetobacter calcoaceticus, Niveispirillum AntA (WP_153203307.1): Niveispirillum sp. SYP-B3756, Vitreoscilla AntA (WP_058357594.1): Vitreoscilla massiliensis, Acinetobacter AntA (O85673): Acinetobacter baylyi ADP1, Sphingomonas CadA (YP_008494438.1): Sphingomonas sp. ERG5 and isolates Pseudomonas ORF 1030, ORF 4892, ORF 4898: P. aeruginosa BT1 2.2, Sphingomonas ORF 5533, ORF 1504, ORF 5379, ORF 1404: S. histidinilytica BT1 5.2, Bordetella ORF 2507: B. petrii BT1 9.2, Achromobacter ORF 2313, ORF 5061: A. xylosoxidans BT1 10.2.

Figure 4. KEGG pathways for the degradation of 2,4-D and 2,4,5-T. Genes encoding the enzymes in the pathways are listed in Supplementary Table 1.
Achromobacter xylosoxidans BT1 10.2 has its tfdCDEF genes clustered along with a regulatory tfdS gene located upstream and divergently transcribed from it (Figure 5A). Downstream are genes encoding the subunits of a ring hydroxylating dioxygenase most similar to a 2-aminobenzenesulfonate 2,3-dioxygenase first described in an Alcaligenes species (Mampel et al., 1999). The catalytic part is composed of AbsAa and AbsAb and the electron donor is the flavodoxin, AbsAc. This gene organization and corresponding amino acid sequences are almost identical to those of B. petrii DSM 12804. This high degree of identity may be the result of a recent gene transfer event between ancestors of these species. Its tfdB gene is located elsewhere on the genome (ORF2511; Supplementary Table 3). We did not find an obvious tfdA gene nor tftAB genes for the initial conversion of 2,4-D and 2,4,5-T, respectively. A likely candidate for tfdA is ORF 3265 (Supplementary Table 3, 35% identity with the KEGG reference protein from C. pinatubonensis). Those for tftAB are ORFs 2313 and 2314 (around 32% identities with their KEGG counterparts from B. cepacia). Other genes potentially involved in further degradation of 2,4,5-T breakdown products are hbqR and chqB expressing HbqR and ChqB for the conversion of 2-HBQ via BT to 2-MA. Yet, the genes encoding the enzymes to convert 2,4,5-T into HBQ were not identified in this species.

Figure 5. Organization of the tfd gene clusters of B. petrii BT1 9.2, A. xylosoxidans BT1 10.2, and S. histidinilytica BT1 5.2, along with those from C. pinatubonensis JMP134 and B. pertussis DSM 12804 (A), the tfd/cad gene cluster of S. histidinilytica BT1 5.2 along with that from Sphingomonas sp. ERG5 (B), and of the cat gene clusters of P. aeruginosa BT1 2.2 and S. histidinilytica BT1 5.2 along with those from R. erythropolis CCM2595, P. putida mt-2, and Burkholderia sp. TH2 (C). The arrows represent the transcription direction of the genes with their sizes reflecting their length in bp. P.a BT1 2.2: P. aeruginosa BT1 2.2, S.h BT1 5.2: S. histidinilytica BT1 5.2, B.p BT1 9.2: B. petrii BT1 9.2, A.x BT1 10.2: A. xylosoxidans BT1 10.2, C.p JMP134: C. pinatubonensis JMP134 (accession no. M35097.1 for the tfdI cluster and U16782.1 for the tfdII cluster), B.p DSM 12804: B. petrii DSM 12804 (accession no. AM902716.1), R.e CCM2595: R. erythropolis CCM2595 (accession no. FM995530.1), P.p mt-2: P. putida mt-2, B. sp. TH2: Burkholderia sp. TH2 (accession no. AB035483.1), S. sp. ERG5: Sphingomonas sp. ERG5 (accession no. KF494257.1).
Bordetella petrii BT1 9.2 has a full complement of tfdABCDEF genes encoding the enzymes for the sequential conversion of 2,4-D into 2-MA (Figure 5A). They are clustered and transcribed in the order tfdCDEFB with a lysR-type tfdS homolog upstream and divergently transcribed from that. A bit more downstream separated by four genes and with the transcription direction opposed to the other structural genes is the tfdA gene. One of the four genes sandwiched by the tfdA and remaining tfd genes is a gene encoding a PcaF-type enzyme responsible for cleavage of 3-oxoadipate-CoA into succinyl- and acetyl-CoA. The identity of the tfdBCDEF encoded proteins with the KEGG reference proteins from C. pinatubonensis JMP134 (accession no. M35097.1) is remarkably high, all of which ranging from 99 to 100%. Also, the gene organizations are identical suggestive of some kind of horizontal gene transfer between ancestors of these two Betaproteobacteria during recent evolution. We also compared the genetic organization of the isolated Vietnamese species with the one with its genome sequence deposited in the databases, B. petrii DSM 12804 (accession no. AM902716.1). Their gene organizations are different (Figure 5A). Also, the pairwise comparisons show that the identities are much lower than those between B. petrii BT1 9.2 and C. pinatubonensis (Supplementary Table 4). A second copy of the tfdA gene, not clustered with other tfd genes and with the expressed protein having 45% identity to the reference TfdA, was found elsewhere on the genome (ORF 3407). It has also the hbqR gene so it may be able to convert 2-HBQ to BT. However, it lacks reasonable homologs of enzymes to produce the substrate or to consume the product. Also, the hbqR gene is not clustered with other genes involved in 2,4,5-T degradation We did not find genes with high identity to tftAB genes but as this strain has the potential to perform the initial ring hydroxylating dioxygenase step, we assume that proteins related to the reference TftAB proteins are responsible for this reaction. Likely candidates to take over the role of TftA, all with identities to the reference TftA close to 30% are ORFs 524, 1633, 2507, and 3859. Their genes are all accompanied by homologs of tftB.
Sphingomonas histidinilytica BT1 5.2 has the genetic potential to express all enzymes for degradation of 2,4-D to succinyl-CoA and acetyl-CoA. Importantly, it does not have an apparent tfdA gene. The function of the tfdA gene product is likely taken over by a ring hydroxylating dioxygenase encoded by a cadABCD gene cluster that is sandwiched between the other tfd genes (Figure 5B). A similar gene organization is seen in Sphingomonas sp. TFD44 (accession no. AY598949.1), Sphingobium herbicidovorans MH (accession no. AJ628861.1), and Sphingomonas sp. ERG5 (accession no. KF494257.1) (Figure 4C) (Nielsen et al., 2013, 2017). Pairwise comparison of the gene products of the latter with those from our isolated strain indicates that they share a high degree of identity (more than 95%) both on the level of gene cluster organization and on the level of their amino acid sequences (Supplementary Table 4). Also, both clusters contain pcaIJF genes to make the enzymes for the conversion of 2-MA, the product of the Tfd proteins, into succinyl-CoA and acetyl-CoA, hence the potential of making a full pathway from 2,4-D to TCA-cycle intermediates. In addition to the gene cluster with tfd and cad genes, it has another one with copies of tfdCDEF genes on it, along with another set of pcaIJF genes. This one may be switched on for growth on chlorocatechols. Importantly, the suggested tfdD and tfdE genes in both clusters share a low identity with their counterparts from the KEGG reference proteins (in between 32 and 42%, see Supplementary Table 3). Nevertheless, we regard them as tfd genes because of their occurrence in the gene clusters with the complementary tfd genes. Its versatility in degrading chlorinated aromatics is further exemplified by the observation that it has a catABCD gene cluster under the apparent control of a catR regulatory gene (Figure 5C). Its gene organization looks quite similar to the one of P. aeruginosa BT1 2.2. Pairwise identities of the CatRABC proteins from both species are around 50% (Supplementary Table 4). Upstream of the cat gene cluster are two sets of genes that resemble those encoding the two subunits of the family of ring hydroxylating dioxygenases like AntAB, BenAB, and TftAB. It is tempting to speculate that these enzyme systems convert their as yet unknown aromatic substrates into catechol for further degradation to maleylacetate by the Cat proteins. It does not have obvious tftAB genes, yet it can convert it at least to 2,4,5-TCP. Hence another enzyme must be encoded by S. histidinilytica BT1 5.2. It might be that the CadABCD proteins can do that as they belong to the same family of ring-hydroxylating dioxygenases. Alternatively, that reaction may be carried out by the gene products of ORFs 5533 and 5532, which have around 30% identity with their counterparts TftA and TftB, respectively, from B. cepacia that serve as the KEGG database reference proteins.
Pseudomonas aeruginosa BT1 2.2 has catAB genes potentially involved in the 2,4-D breakdown. Their genes are clustered. A map of this gene cluster is in Figure 5C. A closer look at the gene cluster encompassing the catAB genes shows some interesting features as they make part of a typical catABC gene cluster, the proteins of which are involved in catechol degradation to 3-OXA. Transcribed in the same direction and sandwiched by the catB and catA genes is a gene homologous to catC and upstream but divergently transcribed a lysR-type regulatory gene possibly equivalent to catR (McFall et al., 1998; Nojiri et al., 2002). A similar gene organization is found in Pseudomonas putida mt-2 (Jiménez et al., 2014). Gene clusters composed of catRABC genes but organized differently are also found in R. erythropolis CCM2595 (accession no. FM995530.1) and Burkholderia sp. TH2 (accession no. AB035483.1). More upstream are two gene clusters encoding the subunits of ring-hydroxylating oxygenases, both of which under the apparent control of AraC-type regulators, their genes flanking these two gene clusters. The one most close to the cat gene cluster has two structural genes with the highest similarity with antABC genes to make an anthranilate 1,2-dioxygenase, AntAB as the catalytic core, and AntC as the flavodoxin-type electron donor. The one more downstream is most likely a benABCD gene cluster encoding a benzoate dioxygenase (BenABC) and a dihydroxy cyclohexadiene carboxylate dehydrogenase (BenD). Burkholderia sp. TH2 has also a set of benABCD genes adjacent to its cat gene cluster (Figure 5C). The phylogenetic tree with TftA homologs including AntA and BenA proteins shows their position in the sub-groups, which supports their suggested functions. Both enzyme systems, AntABC and BenABCD, convert their substrates, anthranilate, and benzoate, respectively, to catechol. Altogether, these findings suggest that P. aeruginosa BT1 2.2 is specialized in the degradation of benzoate and related compounds as it has ben and cat gene clusters for the sequential conversion of benzoate via catechol to 3-oxoadipate. Genes encoding PcaIJ and PcaF are found elsewhere on the genome for a further breakdown of 3-oxoadipate to succinyl-CoA and acetyl-CoA. It can convert 2,4-D at least into 2,4-DCP, and we speculate that this is achieved by the product of ORF3804, which has 38% identity with the KEGG reference tfdA from species C. pinatubonensis JMP134. An alternative gene set for tftAB genes might be ORFs 1030 and 1031. The protein from ORF 1030 has 30% identity with the KEGG reference TftA from B. cepacia AC1100.
Discussion
We have identified three species of proteobacteria as key players in the degradation of 2,4-D and 2,4,5-T in Vietnamese soil contaminated with herbicides, amongst which Agent Orange. They were identified as S. histidinilytica BT1 5.2, B. petrii BT1 9.2, and A. xylosoxidans BT1 10.2, the first is an Alphaproteobacterium, the latter two are Betaproteobacteria. All three were shown to grow and increase their numbers during enrichments on these herbicides, were isolated as dominant species at the end of culturing (Nguyen et al., 2021 manuscript in submission), and grew in batch cultures with 2,4-D and 2,4,5-T as the sole sources of carbon and energy. These were most likely fully metabolized by all three species to explain their conversion coefficients. Bordetella petrii BT1 9.2 catabolizes 54% of the herbicides to generate the ATP required to incorporate the cellular building blocks from the remaining 46% (maintenance not considered). Sphingomonas histidinilytica BT1 5.2, and A. xylosoxidans BT1 10.2 do that slightly less efficient. Importantly, we should note that deriving bacterial cell DW from measuring optical density at 600 nm alone is a less than precise method. The limitations of this are mentioned in materials and methods and may affect the results for the carbon utilization flux. Measuring bacterial cell dry weight directly would have given the carbon flux distribution with greater precision. Regardless of that, our physiological studies along with a detailed inspection of their genomes revealed indeed that they have the potential to express the key enzymes for the sequential degradation of 2,4-D to carbon dioxide. The pathways for degradation of 2,4,5-T in these three species are less clear.
The breakdown of 2.4-D by B. petrii and its use as the carbon and energy source is a novel finding as there are, to our knowledge, no studies that show the degradation of 2,4-D or 2,4,5-T by species of the genus Bordetella. There is only one report published that unravels its genome sequence, which displays genes involved in herbicide degradation, but these were not correlated to physiological studies (Gross et al., 2008). Like B. petrii BT1 9.2, also S. histidinilytica BT1 5.2 can grow on the herbicides and has a comparable set of tfd genes. A distinctive feature, however, is the observation that it does not have a tfdA gene to make an a-ketoglutaric acid-dependent 2,4-D dioxygenase. Instead, the tfdBCDEF genes in its cluster sandwich a set of four so-called cadABCD genes encoding the subunits of a ring hydroxylating dioxygenase that replaces the TfdA function. This seems to be quite common within the Alphaproteobacteria (Nielsen et al., 2013, 2017). In a previous study, we detected a tftA gene fragment using PCR with degenerate tftA primers (Nguyen et al., 2021 manuscript in submission). We now know that it does not have an obvious set of tftAB genes and that the PCR fragment is in fact from cadA. This result further underscores the overall view that members of the family of ring hydroxylating dioxygenases, such as TftA, BenA, and CadA, are quite similar to one another yet they have specific tasks in different reactions and pathways. The versatility of S. histidinilytica BT1 5.2 concerning the breakdown of the herbicides is further emphasized by the observation that it has a second gene cluster with tfdCDEF genes along with a regulatory lysR-type gene just as tfdS and again with pcaIJF genes. Also notable is the finding of a third gene cluster with catABCD genes and a regulatory catR gene, which would allow it to grow on catechols as well and emphasizes its versatility in growth on aromatic compounds. These findings were also reported in previous studies with Sphingomonas (Huong et al., 2007; Cycoń et al., 2011). Previously, S. herbicidovorans MH was reported to degrade the herbicide phenoxyalkanoic acid (Kohler, 1999; Müller et al., 2004), but this strain could not degrade 2,4,5-T (Kohler, 1999). Just like B. petrii BT1 9.2 and S. histidinilytica BT1 5.2, also A. xylosoxidans BT1 10.2 can grow on 2,4-D and use it as the sole source of carbon and energy. Its genetic potential, however, is less clear as it does have tfdBCDEF genes along with pcaIJF genes, but we could not identify a tfdA homolog with more than 45% identity to the reference homolog in the KEGG database. To explain the disappearance of 2,4-D and growth of A. xylosoxidans BT1 10.2 in the culture, we assume that it carries a gene encoding an alternative enzyme or that it makes use of a TfdA homolog with a lower identity.
We noticed also the disappearance of 2,4,5-T in all of the cultures, yet we are unaware of the fate of its metabolites. We did not recognize in either of the species a full suite of genes encoding the key enzymes for its sequential degradation. Perhaps as yet unknown enzymes may be recruited for 2,4,5-T breakdown and bacterial growth as we did see some increase in biomass of B. petrii BT1 9.2 growing on 2,4,5-T. It is more likely, however, that at least the initial step in the degradation of 2,4,5-T is catalyzed by a member of the TftA family, but with a relatively low identity with the KEGG counterpart. Indeed, we show that pairwise identities indicate that the orthologous enzymes in some cases substantially differ from one species to another. Hence, the KEGG proteins may not identify those that have such low identity, yet can carry out the same reaction. This is particularly true for the TfdD and TfdE proteins. If one looks at the pairwise identities of the type I and type II proteins of C. pinatubonensis for example then it is noteworthy that they share only 15% (TfdE) and 35% (TfdD) identities, respectively (Laemmli et al., 2000). Hence, we could use this observation of low identity as a valid argument that S. histidinilytica BT1 5.2 may well have counterparts of the tfdD and tfdE genes from C. pinatubonensis, although they share an identity of <45%.
Not all dominant species isolated from the enrichment cultures were able to use the herbicides as the sources of carbon and energy. Pseudomonas aeruginosa BT1 2.2 has no obvious set of tfd genes, which explains its poor growth on the herbicides. This is in contrast with other studies, which showed that Pseudomonas species were able to biodegrade 2,4-D (Marrón-Montiel et al., 2006; Yang et al., 2017). Pseudomonas aeruginosa BT1 2.2 does have catAB genes, though, but these are clustered with a catC gene and a regulatory catR gene, suggestive for a role in catechol metabolism. That would make sense as the same cluster is preceded by benABCD and antABC gene clusters, allowing it to convert benzoate and anthranilate, respectively, to catechol. A possible reason for its dominance in the enrichment cultures may be that it feeds on metabolites of the herbicides in the enrichment cultures. Alternatively, it manages to survive by feeding on the contents of lysed cells. As such, the enrichment culture may be regarded as a food web with different trophic levels ranging from primary consumers to scavengers.
We further noticed high degrees of similarities of gene cluster organizations along with the amino acid sequences of their gene products (more than 95%) between those of B. petrii BT1 9.2 and C. pinatubonensis JMP134 (gene cluster I) on the one hand and those of B. petrii DSM12804 and A. xylosoxidans BT1 10.2 on the other hand. That observation suggests recent events of horizontal gene transfer between members of these Betaproteobacteria. Surprisingly, the cluster and its gene products from B. petrii that we isolated from Vietnamese soils are different from the ones that were already deposited in the database (B. petrii DSM 12804). Apparently, the exchanges between B. petrii species and their mating partners were independent events. The gene clusters from C. pinatubonensis and B. petrii DSM 12804 are on plasmids, but we have no information about the genetic location of the tfd clusters from our isolates as we have only a set of contigs. Since the identities are so high, we suggest that gene exchange events occurred quite recently and that these events supported growth on chlorinated aromatics such as 2,4-D and 2,4,5-T, which were produced by the American Chemical Paint Company only after the 1940s. As a consequence, it now allows B. petrii BT1 9.2 and C. pinatubonensis JMP134 to degrade 2,4-D to 2-MA as they have a full complement of the tfdABCDEF genes. The clusters from A. xylosoxidans BT1 10.2 and B. petrii DSM 12804 have only tfdCDEF genes, hence their products serve to convert (chlorinated) catechol into 2-MA. Catechols are much older than the phenoxyacetates as they are also formed during wood fires and breakdown of other aromatics (Baldwin et al., 1994; Tao et al., 2004). Hence these clusters might become useful via genetic transfer in species that already have some TfdA and TfdB activity to boost the 2,4-D degrading pathway. Altogether, our integrative systems ecology approach where we combined physiology with genomics and downstream analyses resulted in a more fundamental insight into the degradation of 2,4-D and 2,4,5-T, the species involved in that, and the types of the pathway that they use. Such an approach may easily be applied to other types of bioremediation as well.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA586721; https://www.ncbi.nlm.nih.gov/genbank/, PRJNA586745; https://www.ncbi.nlm.nih.gov/genbank/, PRJNA586752; https://www.ncbi.nlm.nih.gov/genbank/, PRJNA587043.
Author Contributions
TLAN designed and performed the research, analyzed the data, prepared figures and tables, authored or reviewed drafts of the paper, and approved the final draft. HD delivered soil material, reviewed drafts of the paper, and approved the final draft. JK determined the concentration of the herbicides, approved the final draft. MB assisted in the laboratory experiments, reviewed the draft, and approved the final draft. JP gave suggestions, reviewed the draft, and approved the final draft. TB analyzed and annotated the genome of the isolates, reviewed the draft, and approved the final draft. AB reviewed drafts of the paper and approved the final draft. RS designed the experiments, analyzed the data, prepared figures and tables, authored or reviewed drafts of the paper, and approved the final draft. All authors contributed to the article and approved the submitted version.
Funding
This research was financially supported by a BE Basic Foundation-FES grant (grant no: FES0905) from the Dutch Ministry of Economic Affairs and by a grant from the Ministry of Science and Technology of Vietnam (MOST, project code 826/QD-BKHCN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Brett Olivier and Chrats Melkonian for their assistance in data management.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frsc.2021.692018/full#supplementary-material
Supplementary Figure 1. Reconstruction of the pathways for 2,4-D and 2,4,5-T degradation in our four isolated species by identifying their sequences orthologous to those of the reference pathways found at the KEGG database. These orthologous sequences, which share more than 45% identity, are marked with a blue circle in each map. Orthologous genes that make part of more extended clusters with complementary tfd genes are marked with a thick blue circle, those not clustered with cognate genes are marked with a thin blue circle. Steps in the pathways predicted from the 2,4-D and 2,4,5-T degradation experiments are marked with yellow circles. Thick green circles in the S. histidinilytica map are suggested orthologs with an identity lower than 45%, but making part of a gene cluster with complementary tfd genes. Enzymes that are not marked are absent as judged by the absence of the allocated gene from the corresponding species.
Supplementary Table 1. Enzymes in KEGG pathways for degradation of 2,4-D and 2,4,5-T along with relevant characteristics.
Supplementary Table 2. Statistics of sequencing after demultiplexing and filtering.
Supplementary Table 3. Genes encoding KEGG enzymes for degradation of 2,4-D and 2,4,5-T in the four isolated species. Those with an identity more than 45% with the homolog from the KEGG database are marked.
Supplementary Table 4. Pairwise identities between tfd enzymes of the isolated species and of related species. Identities more than 50 and 95% are in orange and red, respectively.
References
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Baldwin, I. T., Staszak-Kozinski, L., and Davidson, R. (1994). Up in smoke: I. Smoke-derived germination cues for postfire annual, Nicotiana attenuata torr. Ex. Watson. J. Chem. Ecol. 20, 2345–2371.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Cycoń, M., Zmijowska, A., and Piotrowska-Seget, Z. (2011). Biodegradation kinetics of 2,4-D by bacterial strains isolated from soil. Cent. Eur. J. Biol. 6, 188–198. doi: 10.2478/s11535-011-0005-0
Danganan, C. E., Ye, R. W., Daubaras, D. L., Xun, L., and Chakrabarty, A. M. (1994). Nucleotide sequence and functional analysis of the genes encoding 2,4,5-trichlorophenoxyacetic acid oxygenase in Pseudomonas cepacia AC1100. Appl. Environ. Microbiol. 60, 4100–4106.
Daubaras, D. L., Danganan, C. E., Hübner, A., Ye, R. W., Hendrickson, W., and Chakrabarty, A. M. (1996a). Biodegradation of 2,4,5-trichlorophenoxyacetic acid by Burkholderia cepacia strain AC1100: evolutionary insight. Gene 179, 1–8.
Daubaras, D. L., Hershberger, C. D., Kitano, K., and Chakrabarty, A. M. (1995). Sequence analysis of a gene cluster involved in metabolism of 2,4,5-trichlorophenoxyacetic acid by Burkholderia cepacia AC1100. Appl. Environ. Microbiol. 61, 1279–1289.
Daubaras, D. L., Saido, K., and Chakrabarty, A. M. (1996b). Purification of hydroxyquinol 1,2-dioxygenase and maleylacetate reductase: the lower pathway of 2,4,5-trichlorophenoxyacetic acid metabolism by Burkholderia cepacia AC1100. Appl. Environ. Microbiol. 62, 4276–4279.
Don, R. H., and Pemberton, J. M. (1981). Properties of six pesticide degradation plasmids isolated from Alcaligenes paradoxus and Alcaligenes eutrophus. J. Bacteriol. 145, 681–686.
Ferraroni, M., Seifert, J., Travkin, V. M., Thiel, M., Kaschabek, S., Scozzafava, A., et al. (2005). Crystal structure of the hydroxyquinol 1,2-dioxygenase from Nocardioides simplex 3E, a key enzyme involved in polychlorinated aromatics biodegradation. J. Biol. Chem. 280, 21144–21154. doi: 10.1074/jbc.M500666200
Filer, K., and Harker, A. R. (1997). Identification of the inducing agent of the 2,4-dichlorophenoxyacetic acid pathway encoded by plasmid pJP4. Appl. Environ. Microbiol. 63, 317–320.
Fukumori, F., and Hausinger, R. P. (1993a). Alcaligenes eutrophus JMP134 “2,4-dichlorophenoxyacetate monooxygenase” is an alpha-ketoglutarate-dependent dioxygenase. J. Bacteriol. 175, 2083–2086. doi: 10.1128/jb.175.7.2083-2086.1993
Fukumori, F., and Hausinger, R. P. (1993b). Purification and characterization of 2,4-dichlorophenoxyacetate/alpha-ketoglutarate dioxygenase. J. Biol. Chem. 268, 24311–24317.
Gisi, M. R., and Xun, L. (2003). Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:flavin adenine dinucleotide oxidoreductase (TftC) of Burkholderia cepacia AC1100. J. Bacteriol. 185, 2786–2792. doi: 10.1128/jb.185.9.2786-2792.2003
Golovleva, L. A., Pertsova, R. N., Evtushenko, L. I., and Baskunov, B. P. (1990). Degradation of 2,4,5-Trichlorophenoxyacetic acid by a Nocardioides simplex culture. Biodegradation 1, 263–271. doi: 10.1007/BF00119763
Gross, R., Guzman, C. A., Sebaihia, M., dos Santos, V. A., Pieper, D. H., Koebnik, R., et al. (2008). The missing link: Bordetella petrii is endowed with both the metabolic versatility of environmental bacteria and virulence traits of pathogenic Bordetellae. BMC Genomics 9, 449. doi: 10.1186/1471-2164-9-449
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hayashi, S., Sano, T., Suyama, K., and Itoh, K. (2016). 2,4-Dichlorophenoxyacetic acid (2,4-D)- and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T)-degrading gene cluster in the soybean root-nodulating bacterium Bradyrhizobium elkanii USDA94. Microbiol Res 188–189, 62–71. doi: 10.1016/j.micres.2016.04.014
Hoffmann, D., Kleinsteuber, S., Müller, R. H., and Babel, W. (2003). A transposon encoding the complete 2,4-dichlorophenoxyacetic acid degradation pathway in the alkalitolerant strain Delftia acidovorans P4a. Microbiology 149, 2545–2556. doi: 10.1099/mic.0.26260-0
Hübner, A., Danganan, C. E., Xun, L., Chakrabarty, A. M., and Hendrickson, W. (1998). Genes for 2,4,5-trichlorophenoxyacetic acid metabolism in Burkholderia cepacia AC1100: characterization of the tftC and tftD genes and locations of the tft operons on multiple replicons. Appl. Environ. Microbiol. 64, 2086–2093.
Huong, N. L., Itoh, K., and Suyama, K. (2007). Diversity of 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T)-degrading bacteria in Vietnamese soils. Microbes Environ. 22, 243–256. doi: 10.1264/jsme2.22.243
Itoh, K., Tashiro, Y., Uobe, K., Kamagata, Y., Suyama, K., and Yamamoto, H. (2004). Root nodule Bradyrhizobium spp. harbor tfdAalpha and cadA, homologous with genes encoding 2,4-dichlorophenoxyacetic acid-degrading proteins. Appl. Environ. Microbiol. 70, 2110–2118. doi: 10.1128/aem.70.4.2110-2118.2004
Jiménez, J. I., Pérez-Pantoja, D., Chavarría, M., Díaz, E., and de Lorenzo, V. (2014). A second chromosomal copy of the catA gene endows Pseudomonas putida mt-2 with an enzymatic safety valve for excess of catechol. Environ. Microbiol. 16, 1767–1778. doi: 10.1111/1462-2920.12361
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Ka, J. O., Holben, W. E., and Tiedje, J. M. (1994). Genetic and phenotypic diversity of 2,4-dichlorophenoxyacetic acid (2,4-D)-degrading bacteria isolated from 2,4-D-treated field soils. Appl. Environ. Microbiol. 60, 1106–1106.
Kamagata, Y., Fulthorpe, R. R., Tamura, K., Takami, H., Forney, L. J., and Tiedje, J. M. (1997). Pristine environments harbor a new group of oligotrophic 2,4-dichlorophenoxyacetic acid-degrading bacteria. Appl. Environ. Microbiol. 63, 2266–2272. doi: 10.1128/aem.63.6.2266-2272.1997
Kellogg, S. T., Chatterjee, D. K., and Chakrabarty, A. M. (1981). Plasmid-assisted molecular breeding: new technique for enhanced biodegradation of persistent toxic chemicals. Science. 214, 1133–1135. doi: 10.1126/science.7302584
Kijima, K., Mita, H., Kawakami, M., and Amada, K. (2018). Role of CadC and CadD in the 2,4-dichlorophenoxyacetic acid oxygenase system of Sphingomonas agrestis 58-1. J. Biosci. Bioeng. 125, 649–653. doi: 10.1016/j.jbiosc.2018.01.003
Kitagawa, W., Takami, S., Miyauchi, K., Masai, E., Kamagata, Y., Tiedje, J. M., et al. (2002). Novel 2,4-dichlorophenoxyacetic acid degradation genes from oligotrophic Bradyrhizobium sp. strain HW13 isolated from a pristine environment. J Bacteriol, 184(2), 509–518. doi: 10.1128/jb.184.2.509-518.2002
Kleinsteuber, S., Müller, R. H., and Babel, W. (2001). Expression of the 2,4-D degradative pathway of pJP4 in an alkaliphilic, moderately halophilic soda lake isolate, Halomonas sp. EF43. Extremophiles 5, 375–384. doi: 10.1007/s007920100202
Kohler, H. P. E. (1999). Sphingomonas herbicidovorans MH: a versatile phenoxyalkanoic acid herbicide degrader. J. Ind. Microbiol. Biotechnol. 23, 336–340. doi: 10.1038/sj/jim/2900751
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kumari, R., Subudhi, S., Suar, M., Dhingra, G., Raina, V., Dogra, C., et al. (2002). Cloning and characterization of lin genes responsible for the degradation of Hexachlorocyclohexane isomers by Sphingomonas paucimobilis strain B90. Appl. Environ. Microbiol. 68, 6021–6028. doi: 10.1128/aem.68.12.6021-6028.2002
Laemmli, C. M., Leveau, J. H., Zehnder, A. J., and van der Meer, J. R. (2000). Characterization of a second tfd gene cluster for chlorophenol and chlorocatechol metabolism on plasmid pJP4 in Ralstonia eutropha JMP134(pJP4). J. Bacteriol. 182, 4165–4172. doi: 10.1128/jb.182.15.4165-4172.2000
Liu, T., and Chapman, P. J. (1984). Purification and properties of a plasmid-encoded 2,4-dichlorophenol hydroxylase. FEBS Lett. 173, 314–318. doi: 10.1016/0014-5793(84)80797-8
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–495. doi: 10.1093/nar/gkt1178
Mampel, J., Ruff, J., Junker, F., and Cook, A. M. (1999). The oxygenase component of the2-aminobenzenesulfonate dioxygenase system from Alcaligenes sp. strain O-1. The GenBank accession number for the sequence reported in this paper is AF109074. Microbiology 145, 3255–3264. doi: 10.1099/00221287-145-11-3255
Marrón-Montiel, E., Ruiz-Ordaz, N., Rubio-Granados, C., Juárez-Ramírez, C., and Galíndez-Mayer, C. J. (2006). 2,4-D-degrading bacterial consortium: isolation, kinetic characterization in batch and continuous culture and application for bioaugmenting an activated sludge microbial community. Process Biochem. 41, 1521–1528. doi: 10.1016/j.procbio.2006.02.012
McFall, S. M., Chugani, S. A., and Chakrabarty, A. M. (1998). Transcriptional activation of the catechol and chlorocatechol operons: variations on a theme. Gene 223, 257–267. doi: 10.1016/s0378-1119(98)00366-7
Milo, R., Jorgensen, P., Moran, U., Weber, G., and Springer, M. (2010). BioNumbers–the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–D753. doi: 10.1093/nar/gkp889
Müller, T. A., Byrde, S. M., Werlen, C., van der Meer, J. R., and Kohler, H.-P. E. (2004). Genetic analysis of phenoxyalkanoic acid degradation in Sphingomonas herbicidovorans MH. Appl. Environ. Microbiol. 70, 6066. doi: 10.1128/AEM.70.10.6066-6075.2004
Nguyen, T. L. A., Dang, H. T. C., Koekkoek, J., Dat, T. T. H., Braster, M., Brandt, B. W., et al. (2021). Correlating biodegradation kinetics of 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T) to the dynamics of microbial communities originating from soil in Vietnam contaminated with herbicides. Front. Sustain. Cities 3:692012. doi: 10.3389/frsc.2021.692012
Nielsen, T. K., Rasmussen, M., Demanèche, S., Cecillon, S., Vogel, T. M., and Hansen, L. H. (2017). Evolution of Sphingomonad gene clusters related to pesticide catabolism revealed by genome sequence and mobilomics of Sphingobium herbicidovorans MH. Genome Biol. Evol. 9, 2477–2490. doi: 10.1093/gbe/evx185
Nielsen, T. K., Xu, Z., Gözdereliler, E., Aamand, J., Hansen, L. H., and Sørensen, S. R. (2013). Novel insight into the genetic context of the cadAB genes from a 4-chloro-2-methylphenoxyacetic acid-degrading Sphingomonas. PLoS ONE 8, e83346. doi: 10.1371/journal.pone.0083346
Nojiri, H., Maeda, K., Sekiguchi, H., Urata, M., Shintani, M., Yoshida, T., et al. (2002). Organization and transcriptional characterization of catechol degradation genes involved in carbazole degradation by Pseudomonas resinovorans strain CA10. Biosci. Biotechnol. Biochem. 66, 897–901. doi: 10.1271/bbb.66.897
Pérez-Pantoja, D., Guzmán, L., Manzano, M., Pieper, D. H., and González, B. (2000). Role of tfdC(I)D(I)E(I)F(I) and tfdD(II)C(II)E(II)F(II) gene modules in catabolism of 3-chlorobenzoate by Ralstonia eutropha JMP134(pJP4). Appl. Environ. Microbiol. 66, 1602–1608. doi: 10.1128/aem.66.4.1602-1608.2000
Perkins, E. J., Gordon, M. P., Caceres, O., and Lurquin, P. F. (1990). Organization and sequence analysis of the 2,4-dichlorophenol hydroxylase and dichlorocatechol oxidative operons of plasmid pJP4. J. Bacteriol. 172, 2351–2359. doi: 10.1128/jb.172.5.2351-2359.1990
Plumeier, I., Pérez-Pantoja, D., Heim, S., González, B., and Pieper, D. H. (2002). Importance of different tfd genes for degradation of chloroaromatics by Ralstonia eutropha JMP134. J. Bacteriol. 184, 4054–4064. doi: 10.1128/jb.184.15.4054-4064.2002
Rice, J. F., Menn, F. M., Hay, A. G., Sanseverino, J., and Sayler, G. S. (2005). Natural selection for 2,4,5-trichlorophenoxyacetic acid mineralizing bacteria in agent orange contaminated soil. Biodegradation 16, 501–512. doi: 10.1007/s10532-004-6186-8
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Serbent, M. P., Rebelo, A. M., Pinheiro, A., Giongo, A., and Tavares, L. B. B. (2019). Biological agents for 2,4-dichlorophenoxyacetic acid herbicide degradation. Appl. Microbiol. Biotechnol. 103, 5065–5078. doi: 10.1007/s00253-019-09838-4
Smejkal, C. W., Vallaeys, T., Burton, S. K., and Lappin-Scott, H. M. (2001). A rapid method to screen degradation ability in chlorophenoxyalkanoic acid herbicide-degrading bacteria. Lett. Appl. Microbiol. 32, 273–277. doi: 10.1046/j.1472-765x.2001.00900.x
Stellman, J. M., Stellman, S. D., Christian, R., Weber, T., and Tomasallo, C. (2003). The extent and patterns of usage of Agent Orange and other herbicides in Vietnam. Nature 422, 681–687. doi: 10.1038/nature01537
Streber, W. R., Timmis, K. N., and Zenk, M. H. (1987). Analysis, cloning, and high-level expression of 2,4-dichlorophenoxyacetate monooxygenase gene tfdA of Alcaligenes eutrophus JMP134. J. Bacteriol. 169, 2950–2955. doi: 10.1128/jb.169.7.2950-2955.1987
Takenaka, S., Koshiya, J., Okugawa, S., Takata, A., Murakami, S., and Aoki, K. (2011). Fe-superoxide dismutase and 2-hydroxy-1,4-benzoquinone reductase preclude the auto-oxidation step in 4-aminophenol metabolism by Burkholderia sp. strain AK-5. Biodegradation 22, 1–11. doi: 10.1007/s10532-010-9369-5
Tao, Y., Fishman, A., Bentley, W. E., and Wood, T. K. (2004). Oxidation of benzene to phenol, catechol, and 1,2,3-trihydroxybenzene by toluene 4-monooxygenase of Pseudomonas mendocina KR1 and toluene 3-monooxygenase of Ralstonia pickettii PKO1. Appl. Environ. Microbiol. 70, 3814–3820. doi: 10.1128/aem.70.7.3814-3820.2004
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Thiel, M., Kaschabek, S. R., Gröning, J., Mau, M., and Schlömann, M. (2005). Two unusual chlorocatechol catabolic gene clusters in Sphingomonas sp. TFD44. Arch. Microbiol. 183, 80–94. doi: 10.1007/s00203-004-0748-3
Tonso, N. L., Matheson, V. G., and Holben, W. E. (1995). Polyphasic characterization of a suite of bacterial isolates capable of degrading 2,4-D. Microb. Ecol. 30, 3–24. doi: 10.1007/bf00184510
Vallaeys, T., Fulthorpe, R. R., Wright, A. M., and Soulas, G. (1996). The metabolic pathway of 2,4-dichlorophenoxyacetic acid degradation involves different families of tfdA and tfdB genes according to PCR-RFLP analysis. FEMS Microbiol. Ecol. 20, 163–172. doi: 10.1016/0168-6496(96)00027-X
van der Zaan, B. M., Saia, F. T., Stams, A. J., Plugge, C. M., de Vos, W. M., Smidt, H., et al. (2012). Anaerobic benzene degradation under denitrifying conditions: Peptococcaceae as dominant benzene degraders and evidence for a syntrophic process. Environ. Microbiol. 14, 1171–1181. doi: 10.1111/j.1462-2920.2012.02697.x
Vedler, E., Vahter, M., and Heinaru, A. (2004). The completely sequenced plasmid pEST4011 contains a novel IncP1 backbone and a catabolic transposon harboring tfd genes for 2,4-dichlorophenoxyacetic acid degradation. J. Bacteriol. 186, 7161–7174. doi: 10.1128/jb.186.21.7161-7174.2004
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Westing, A. H. (1984). “Herbicides in war: past and present,” in Herbicides in War: The Long-Term Ecological and Human Consequence, eds A. H. Westing and Stockholm International Peace Research Institute (London; Philadelphia, PA: Taylor and Francis), 3–24.
Yang, Z., Xu, X., Dai, M., Wang, L., Shi, X., and Guo, R. (2017). Rapid degradation of 2,4-dichlorophenoxyacetic acid facilitated by acetate under methanogenic condition. Bioresour. Technol. 232, 146–151. doi: 10.1016/j.biortech.2017.01.069
Zaborina, O., Daubaras, D. L., Zago, A., Xun, L., Saido, K., Klem, T., et al. (1998). Novel pathway for conversion of chlorohydroxyquinol to maleylacetate in Burkholderia cepacia AC1100. J. Bacteriol. 180, 4667–4675.
Keywords: bioremediation, Agent Orange, tfd gene clusters, horizontal gene transfer (HGT), chlorophenoxy acid herbicides
Citation: Nguyen TLA, Dang HTC, Koekkoek J, Braster M, Parsons JR, Brouwer A, de Boer T and van Spanning RJM (2021) Species and Metabolic Pathways Involved in Bioremediation of Vietnamese Soil From Bien Hoa Airbase Contaminated With Herbicides. Front. Sustain. Cities 3:692018. doi: 10.3389/frsc.2021.692018
Received: 07 April 2021; Accepted: 29 October 2021;
Published: 26 November 2021.
Edited by:
David Iain McBride, University of Otago, New ZealandReviewed by:
Paunita Iuliana Boanca, University of Agricultural Sciences and Veterinary Medicine of Cluj-Napoca, RomaniaGregor Steinhorn, Unitec Institute of Technology, New Zealand
Copyright © 2021 Nguyen, Dang, Koekkoek, Braster, Parsons, Brouwer, de Boer and van Spanning. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rob J. M. van Spanning, cm9iLnZhbi5zcGFubmluZ0B2dS5ubA==; Thi Lan Anh Nguyen, dC5sLmEubmd1eWVuQHZ1Lm5s; bGFuYW5oZG44NkBnbWFpbC5jb20=