 Alessandro Antonelli1,†
Alessandro Antonelli1,† Anna Martina Battaglia2,†
Anna Martina Battaglia2,† Alessandro Sacco2
Alessandro Sacco2 Lavinia Petriaggi2
Lavinia Petriaggi2 Emanuele Giorgio2
Emanuele Giorgio2 Selene Barone1
Selene Barone1 Flavia Biamonte2*
Flavia Biamonte2* Amerigo Giudice1
Amerigo Giudice1
- 1Department of Health Science, School of Dentistry, “Magna Graecia” University of Catanzaro, Catanzaro, Italy
- 2Laboratory of Biochemistry and Cellular Biology, Department of Experimental and Clinical Medicine, “Magna Graecia” University of Catanzaro, Catanzaro, Italy
Oral squamous cell carcinoma (OSCC) is an aggressive disease whose incomplete biological comprehension contributes to the inappropriate clinical management and poor prognosis. Thus, the identification of new promising molecular targets to treat OSCC is of paramount importance. Ferroptosis is a regulated cell death caused by the iron-dependent accumulation of reactive oxygen species and the consequent oxidative damage of lipid membranes. Over the last five years, a growing number of studies has reported that OSCC is sensitive to ferroptosis induction and that ferroptosis inducers exert a remarkable antitumor effect in OSCC, even in those displaying low response to common approaches, such as chemotherapy and radiotherapy. In addition, as ferroptosis is considered an immunogenic cell death, it may modulate the immune response against OSCC. In this review, we summarize the so far identified ferroptosis regulatory mechanisms and prognostic models based on ferroptosis-related genes in OSCC. In addition, we discuss the perspective of inducing ferroptosis as a novel strategy to directly treat OSCC or, alternatively, to improve sensitivity to other approaches. Finally, we integrate data emerging from the research studies, reviewed here, through in silico analysis and we provide a novel personal perspective on the potential interconnection between ferroptosis and autophagy in OSCC.
1 Introduction
Oral squamous cell carcinoma (OSCC) is an aggressive disease and one of the most prevalent head and neck malignancy (1). The pathogenesis of OSCC is a multistep process characterized by the accumulation of genetic and epigenetic events that lead to the dysregulation of oncogenic signaling pathways, such as the epidermal grow factor receptor (EGFR) (2), PI3K/AKT/mTOR (3), Wnt/β-catenin (4), and JAK/STAT (5) as well as impairment of suppressor pathways, such as TP53/RB and p16/Cyclin D1/Rb (6, 7), overall involved in the control of cell growth, proliferation, differentiation, and cell death (8). In addition, the role of tumor microenvironment (TME) in shaping oral cancer progression is increasingly recognized. TME is often an inflamed ecosystem characterized by high levels of oxidative stress and hypoxia due to exposure to risk factors, such as microbial infections and tobacco smoke (9–11). Oral TME is also populated by diverse cell types such as immune cells and stromal cells which crosstalk through autocrine–paracrine signaling pathways, thus controlling the proliferation and metastasis of tumor cells as well as hindering effective drug diffusion and immune cell invasion (12, 13). OSCC clinical management is still a serious challenge. It requires aggressive multimodality approaches, including surgery followed by radiotherapy alone or with chemotherapy. In addition, although targeted therapies, such as cetuximab against EGFR, and the pembrolizumab- and nivolumab-based immunotherapy, have been approved by the Food and Drug Administration (FDA), the prognosis is poor, over half of OSCC patients experience locoregional recurrence or metastasis, and the 5-year survival rate is still less than 50% (1–17). Besides, clinical and histopathological factors used in clinical practice to predict the prognosis of patients with oral cancer are not satisfactory yet. The tumor-node-metastasis (TNM) staging system is the most widely used for risk stratification. However, patients with the same TNM stage may show significantly different clinical outcomes and responses to treatment (18). This suggests that the diversity within cancer extends beyond clinical and pathological aspects, but rather is fundamentally driven by a still incompletely defined complex array of biological alterations.
Over the past decade, the growing understanding of the molecular mechanisms underlying the regulated cell deaths (RCDs), in particular the so-called non-apoptotic RCDs, such as autophagic cell-death (ACD), ferroptosis, necroptosis, and pyroptosis, has shed light on new key features of oral carcinogenesis. Moreover, it provided novel therapeutic targets and strategies to hit OSCC where defective apoptosis pathways often tip the balance in favor of tumor progression (19). Autophagy is a dynamic process during which cells digest organelles and proteins to meet their metabolic needs and is tightly regulated by autophagy-related genes (ATG) (20, 21). Autophagy acts as a “double edged sword” in OSCC by promoting either tumor progression or tumor suppression (22). To make few examples, autophagy can favor the early steps of oral carcinogenesis by protecting OSCC cells from nutrients and oxygen depletion caused by insufficient vascularization (23); besides, autophagy inhibition can enhance cisplatin sensitivity in OSCC cell lines (24). Conversely, in other studies, it has been demonstrated activating autophagy, i.e., by inhibiting mTOR pathway, suppresses tumor activity in OSCC (25). To make thing more complex, autophagy has been found to play crucial roles in ferroptosis execution through several mechanisms, such as ferritin degradation (ferritinophagy), which leads to intracellular iron accumulation, and degradation of lipid droplets (lipophagy), which causes lipid peroxidation, and mitophagy. The autophagy cargo receptor NCOA4 binds to ferritin and deliver it into lysosomes where it is degraded. Ferritin phagocytosis promotes iron release and accumulation in cytoplasm and causes ferroptosis in tumor cells (26, 27). The autophagic degradation of lipid droplets causes the release and accumulation of free fatty acids, which in turn promote lipid peroxidation and subsequent ferroptosis (28). As such, ferroptosis is now described as an autophagic cell death (28, 29).
Ferroptosis is an oxidative type of RCD driven by the iron-dependent accumulation of reactive oxygen species (ROS), followed by peroxidation of lipid membrane and mitochondrial dysfunction (26, 30, 31). A number of studies demonstrate that ferroptosis occurs in OSCC and it is mainly associated with the impairment of System Xc−/GSH/GPX4 antioxidant pathway. Notably, interfering with this pathway or with intracellular iron and ROS levels, either through genetic modification or the administration of natural and chemical compounds, have shown a remarkable therapeutic potential (29, 32–38). Pyroptosis and necroptosis represents two additional targets for OSCC treatment: however, studies are limited and both the biological mechanisms and the implications of these RCDs in OSCC are still poorly understood (19). Pyroptosis is mediated by caspase activation through either canonical or non-canonical pathways (39). Necroptosis is initiated by tumor necrosis factor (TNF) signal and is regulated by a necrosome complex composed by receptor-interacting kinases 1 and 3 (RIPK1 and RIPK3) and Mixed lineage kinase domain-like protein MLKL (40). Both, pyroptosis and necroptosis induce a lytic cell death accompanied by the release of pro-inflammatory cytokines (i.e., interleukin-1β) and damage-associated molecular patterns (DAMPs) (41). Considering, though, that inflammation has the potential to increase the risk of tumorigenesis, promote metastasis, and cause treatment failure, it suggested that pyroptosis and necroptosis may play a role in OSCC development and progression (19).
Since its first steps, ferroptosis research in OSCC highlighted that the pronounced imbalance between oxidative stress and antioxidant defenses makes this tumor a breeding ground for ferroptosis onset. Oral precancer and cancer are characterized by heightened levels of ROS and reactive nitrogen species (RNS), not only due to the increased metabolism driven by abnormal cell growth, but also by the overexpression of enzymes like nitric oxide synthase (NOS), cyclooxygenase (COX), and lipoxygenase (LOX), which are significant sources of both ROS and RNS. Furthermore, environmental factors such as tobacco smoke, alcohol, and pathogens infections exacerbate the production and the adverse effects of ROS and RNS (9–11, 42). Originally, overwhelming literature has extensively documented the pivotal role of oxidative stress in both initiating and promoting the multistep process of oral carcinogenesis. In agreement with this assumption, treatment interventions, such as the use of antioxidants, and even lifestyle interventions, such as quitting smoking and reducing alcohol consumption, has shown the potential to mitigate OSCC risk and improve treatment outcomes. However, a different current of thought suggests that in certain contexts antioxidants can protect cancer cells from oxidative damage, thus inhibiting cell death and promoting the survival and growth of oral cancer cells (43). In this last scenario, the concept of ferroptosis has stressed the hypothesis that exasperate the production of free radicals, rather than attenuate it, might represent an alternative approach to kill oral cancer cells. Therefore, achieving a comprehensive and in-depth understanding of the role of ferroptosis in OSCC is crucial for effectively guiding its application in OSCC treatment.
In this review, we summarize the so far identified ferroptosis regulatory mechanisms and prognostic models based on ferroptosis-related genes (FRGs) in OSCC. We also discuss the perspective of inducing ferroptosis as a novel strategy for OSCC treatment by describing the ferroptosis-based therapeutic interventions currently used to kill OSCC cells or to improve sensitivity to other therapeutic approaches. Furthermore, we integrate data emerging from the research studies reviewed here and we provide a novel personal perspective on the potential interconnection between ferroptosis and autophagy in OSCC that could offer “food for thought” for further studies. Considering that squamous cell carcinoma (SCC) constitutes most of oral cancer cases, the present review focuses on oral SCC (OSCC) affecting tongue, mouth floor, gingiva, and buccal mucosa.
1.1 Ferroptosis: core regulatory mechanisms and morphological features
First discovered by Dixon et al. in 2012, ferroptosis is a new form of RCD both biochemically and morphologically distinct from other forms of cell death. Ferroptosis is driven by the iron-dependent peroxidation of membrane phospholipids (PLs) containing polyunsaturated fatty acids (PUFA-PLs). When lipid peroxidation exceeds the buffering capability of antioxidant defense systems, the lethal accumulation of lipid peroxides on cellular membranes, and the subsequent membrane rupture, lead to ferroptotic cell death (Figure 1) (44–46).
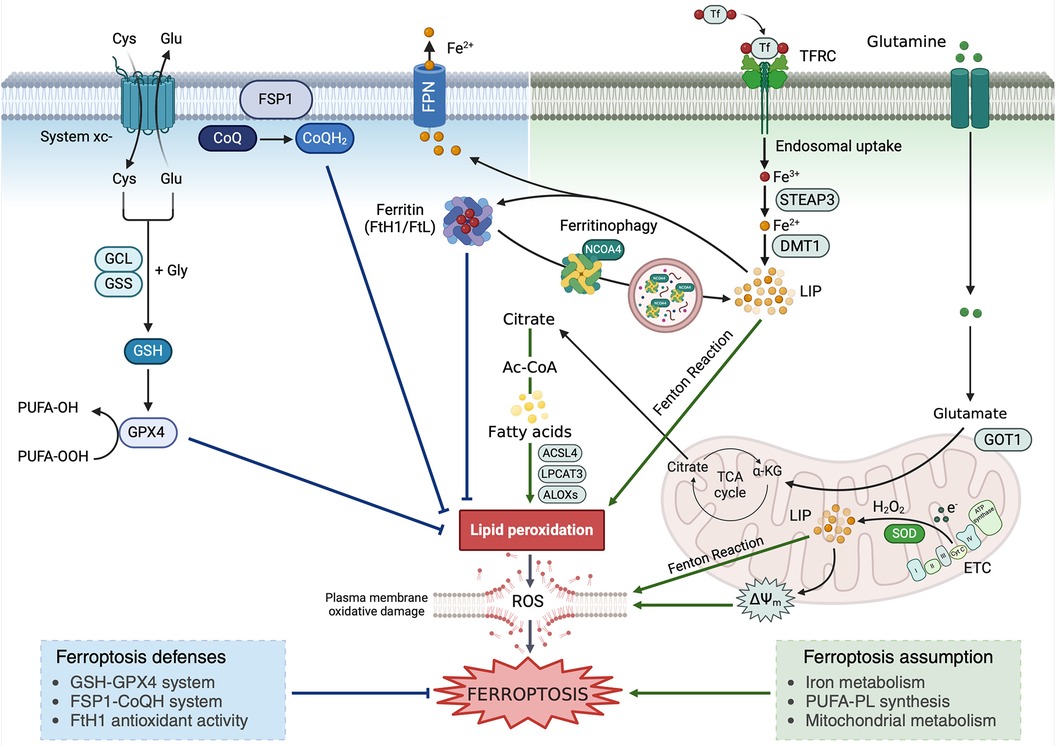
Figure 1. Ferroptosis occurrence relies on the antagonism between the promoting (right) and defense mechanisms (left). Graphic representation of the main ferroptosis-promoting events and ferroptosis defense mechanisms. Cys, cysteine; Glu, glutamate; Gly, glycine; GCL, glutamate cysteine ligase; GSS, glutathione synthetase; GSH, reduced glutathione; GPX4, glutathione peroxidase 4; PUFA, polyinsaturated fatty acids; CoQ, ubiquinone; CoQH2, ubiquinol; FTH1, ferritin heavy subunit 1; FTL, ferritin light subunit; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; NCOA4, nuclear receptor coactivator 4; DMT1, divalent metal trasposter 1; TFRC, transferrin receptor; Tf, transferrin; ALOX, arachidonate lipoxygenase; LIP, labile iron pool; TCA Cycle, tricarboxylic acids cycle; GOT1, glutamic-oxaloacetic transaminase 1; coA, coenzyme A; ETC, electron trasport chain; SOD, superoxide dismutase; ACSL4, acyl-coenzyme A (CoA) synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; ΔΨm, mitochondrial membrane potential.
Iron is a fundamental micronutrient playing a key role in a variety of cellular biological processes such as cell cycle, oxygen transport, electron transport chain (ETC)-mediated production of ATP, and various reactions involving iron-containing proteins, heme-containing proteins, iron–sulfur (Fe–S) cluster proteins, and iron-containing enzymes (47). Under physiological conditions, cells incorporate iron as ferric Fe3+ complexed with transferrin (Tf) via transferrin receptor-1 (TfR1). Afterward, Fe3+ dissociates from Tf and is reduced to ferrous iron (Fe2+), which can be transported to the cytosol via the divalent metal transporter l (DMT1), stored by ferritin, and exported into extracellular space by ferroportin (FPN) (48). When the intracellular amount of Fe2+ overwhelms the storage capacity of ferritin, it accumulates within the cytoplasm as labile iron pool (LIP), which participates in the Fenton and reactions to generate the hydroxyl radicals (49, 50). If ROS are not properly buffered by the cellular antioxidant machinery, they drive the non-enzymatic autoxidation of PUFA-PL. Alternatively, Fe3+ may act as an essential cofactor for arachidonate lipoxygenase (ALOX) enzymes responsible for the enzymatic lipid peroxidation (26, 31, 50–53). Therefore, dysregulation of proteins involved in iron metabolism, including the iron uptake TfR1, the iron storage protein ferritin, and the iron efflux protein FPN, can promote or suppress ferroptosis, by modulating the amount of intracellular LIP (54, 55). To make an example, the autophagic degradation of ferritin mediated by the nuclear receptor coactivator 4 (NCOA4), a phenomenon known as ferritinophagy, releases free iron into the cytoplasm thus promoting ferroptosis (26, 54). Conversely, the blockade of NCOA4 decreases the level of the LIP and suppresses ferroptosis (56).
In contrast to monounsaturated fatty acids (MUFA), PUFA-PL are highly susceptible to peroxidation. Hence, their synthesis is a fundamental prerequisite for ferroptosis occurrence. PUFA-PL synthesis depends on the activity of two enzymes, acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). ACSL4 catalyzes the ligation of free PUFAs with CoA to generate PUFA-CoAs, which are subsequently re-esterified and incorporated into PLs by LPCAT3 to form PUFA-PLs. As mentioned before, the peroxidation of PUFA-PLs, then, is mainly a non-enzymatic autoxidation mechanism driven by the iron-dependent generation of ROS via Fenton reactions or, alternatively, a series of enzymatic reactions mediated by ALOXs (57). Inhibition of PUFA-PL synthesis, via inactivation of ACSL4 and LPCAT3, or suppression of ALOXs functions can block or attenuate ferroptosis (58). In contrast, inactivation of enzymes involved in MUFA-PL synthesis, such as stearoyl CoA desaturase 1 (SCD1) and ACSL3, sensitizes cancer cells to ferroptosis (59). One of the final products of lipid peroxidation is malonyldialdehyde (MDA) that contains two aldehyde groups that can react with thiol and amine groups of proteins, lipids, amino sugars, and nitrogenous bases of nucleic acids. Besides, they modify physical properties of cell membranes by increasing their permeability. This causes changes in electric potentials on both sides of the membrane, resulting in loss of integration of both the intracellular and the plasmatic membranes and inhibition of activity of membrane enzymes and carrier proteins (60).
Ferroptosis defense mechanisms involve cellular antioxidant systems that directly neutralize lipid peroxides. Ferroptosis defense systems can be divided into GPX4-dependent and GPX4-independent arms (61). The SLC7A11–GSH–GPX4 axis constitutes the major cellular system defending against ferroptosis. GSH is a thiol-containing tripeptide derived from glycine, glutamate, and cysteine, with cysteine being the rate-limiting precursor. Cancer cells absorb cysteine mainly through the antiporter System xc–-mediated uptake of cystine, followed by cystine reduction to cysteine in the cytosol. SLC7A11 is the transporter subunit in System xc−. GSH is the cofactor of GPX4, an antioxidant enzyme able to detoxify lipid hydroperoxides converting them to lipid alcohols (62). Genetic ablation or pharmacological inhibition of GPX4 induces unchecked lipid peroxidation and triggers potent ferroptosis in many cancer cell types (29, 35). Among the GPX4-independent defense mechanisms, the ferroptosis suppressor protein 1 (FSP1), localized on the plasma membrane, functions as a NAD(P)H-dependent oxidoreductase capable of reducing ubiquinone (CoQ) to ubiquinol (CoQH2), which in turns traps lipid peroxyl radicals, thereby suppressing ferroptosis (63, 64).
Finally, mitochondria are key organelles in ferroptosis execution as they represent the major source of ROS. During the activity of the ETC complexes I and III, the electron leakage generates superoxide (O2−) that is converted to hydrogen peroxide (H2O2) by superoxide dismutase (2 HO2 → O2 + H2O2). H2O2 can, then, react with LIP via Fenton reactions to generate hydroxyl radicals, which subsequently drive PUFA-PL peroxidation (65).
When observed through electron microscopy, ferroptosis presents with morphological features that make it unique compared to the other RCDs (Figure 2). Cells undergoing ferroptosis, indeed, do not show chromatin condensation, DNA fragmentation, apoptotic bodies or bubble-like protrusions as cells undergoing apoptosis and pyroptosis (66). Ferroptotic cells do not show organelle edema and content release with inflammatory response as cells undergoing necroptosis and pyroptosis (67). Ferroptotic cells appear as clear round cells with empty cytosol (the so-called ballooning phenotype) characterized by shrinkage of mitochondria with increased membrane density and vanishing mitochondrial cristae (26, 31). Ferroptotic cells may show cytoplasmic vacuolation and autophagosomes, which are typical characteristics of autophagy (68). However, the rapid vesicle turnover and the random distribution of vesicles within cells make the assessment of autophagosomes formation and degradation challenging (69).
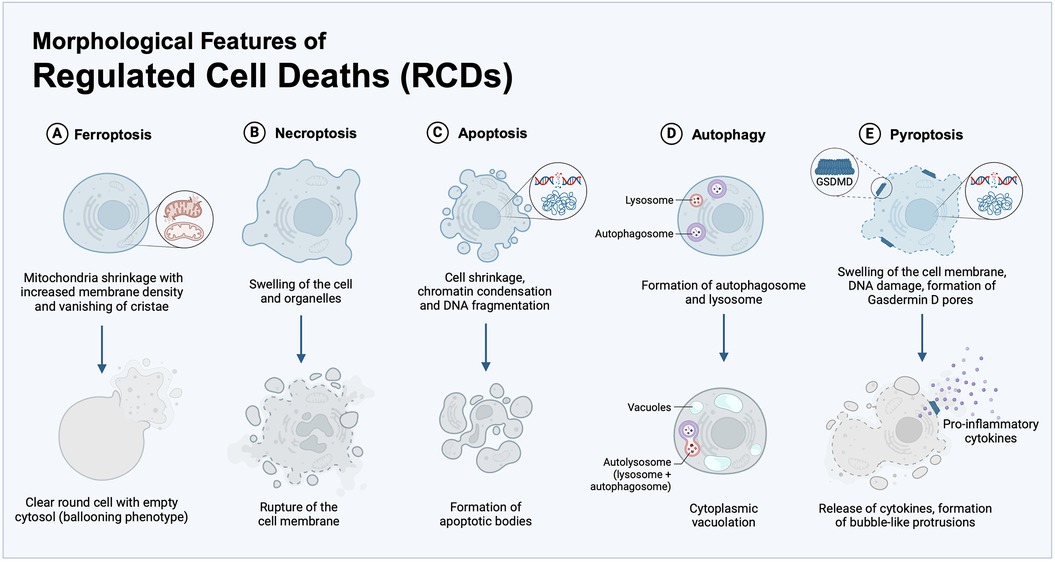
Figure 2. Morphological features of RCDs. Typical morphology of the main RCD processes, here including: (A) ferroptosis, identified by shrinkage of mitochondria with vanishing cristae and clear round cells with empty cytosol (ballooning phenotype); (B) necroptosis, characterized by membrane rupture and swelling of organelles, cellular collapse, and release of cellular contents; (C) apoptosis, defined by cell contraction, chromatin condensation, DNA fragmentation, and generation of apoptotic bodies; (D) autophagy, marked by the cytoplasmic vacuolation and autophagosome generation; (E) pyroptosis, that exhibits DNA damage and chromatin condensation, the formation of Gasdermin D (GSDMD) pores for secretion of inflammatory cytokines and bubble-like protrusions on the cellular membrane.
1.2 Overview of ferroptosis in cancer
Since its discovery in 2012, ferroptosis has sparked great interest in the cancer research community. In particular, targeting ferroptosis has provided new therapeutic opportunities in treating cancers that are refractory to conventional therapies (45, 70–72). Ferroptosis is implicated in tumor biology in multiple ways. First, it makes a cancer cell nutrient addiction, i.e., iron addiction, a targetable vulnerability (26, 55, 73). Plus, it may exert anticancer activity by interacting with several tumor suppressors (74). For instance, the major tumor suppressor p53, among other functions, binds to and downregulates the cystine transporter solute carrier family 7 member 11 (SLC7A11), a component of the cystine/glutamate antiporter, thus causing reduced glutathione (GSH) depletion, the consequent glutathione peroxidase 4 (GPX4) dysfunction and ferroptosis in breast cancer cells (75). On the contrary, other evidence indicates that ferroptosis signaling pathways may promote tumor growth and progression. In this regard, it has been demonstrated that ferroptosis-mediated oxidative stress can activate two key transcription factors involved in cancer development and progression, namely nuclear erythroid factor 2-related factor (NRF2) and hypoxia inducible factor 1 alpha (HIF1-α). When chronically activated, NRF2 supports cancer cells proliferation, metabolic reprogramming, and resistance to therapy (76, 77). HIF-1α enhances the transcription of genes involved in cell survival, angiogenesis, and metastasis (78).
As mentioned above, due to their metabolic requirements and oxidative burden, cancer cells may have a higher susceptibility to ferroptosis inducers (FINs). Over the last 5 years, several FINs have shown a great antitumor effect by targeting the tumor redox homeostasis, and their relative mechanisms of action have been elucidated. Today, the number of FINs is ever growing and can be classified as: (i) System Xc− inhibitors (Erastin, Sulfasalazine, Sorafenib), (ii) GSH depleters (FIN56), (iii) GPX4 inhibitors (RSL3), and (iv) iron metabolism modulators (Figure 3) (46, 79). In addition, a series of drugs already approved by FDA in chemotherapy, radiotherapy, immunotherapy, and targeted therapies, have been found to act also via ferroptosis induction or synergistically with FINs in different cancer types (Figure 4) (80, 81). The poly (ADP-ribose) polymerase (PARP) inhibitor olaparib promotes ferroptosis by suppressing SLC7A11 and, thus, synergizes with RSL3 in BRCA-proficient ovarian cancer cells (82). GPX4 inhibitors enhance the sensitivity of resistant triple-negative breast cancer (TNBC) cells to the EGFR-tyrosine kinase inhibitor gefitinib (83). However, certain tumor cells inevitably adopt mechanisms of ferroptosis evasion, such as heightening the antioxidant capacity as an adaptive response to increased ROS levels or, alternatively, overexpressing iron efflux pumps to avoid LIP accumulation. In these cases, the combination therapy of FINs with other therapeutic approaches represents a promising strategy to tackle resistance and preventing tumor recurrence (74).
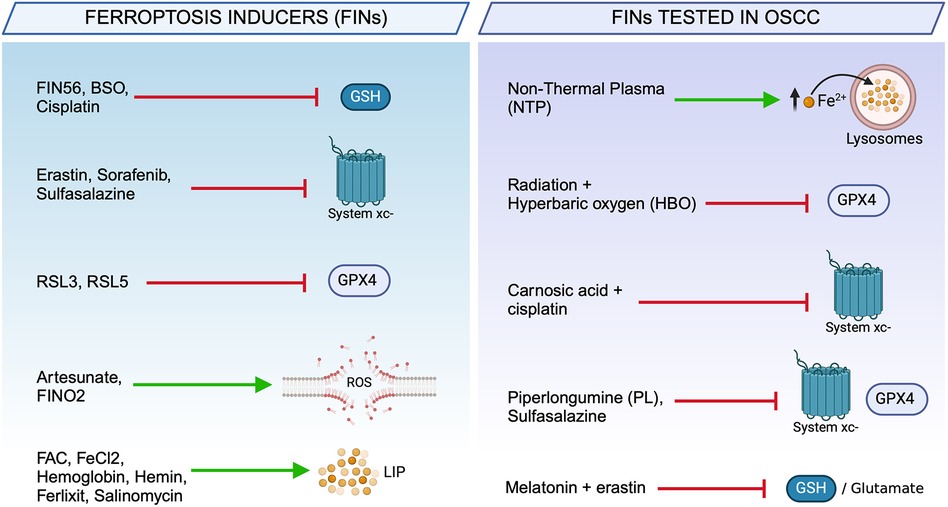
Figure 3. Chemical and natural compounds used to trigger ferroptosis. Ferroptosis inducers (FINs) used in cancer therapy classified according to their biochemical target (left). Ferroptosis-based therapeutic strategies used to trigger cell death in OSCC cells (right).
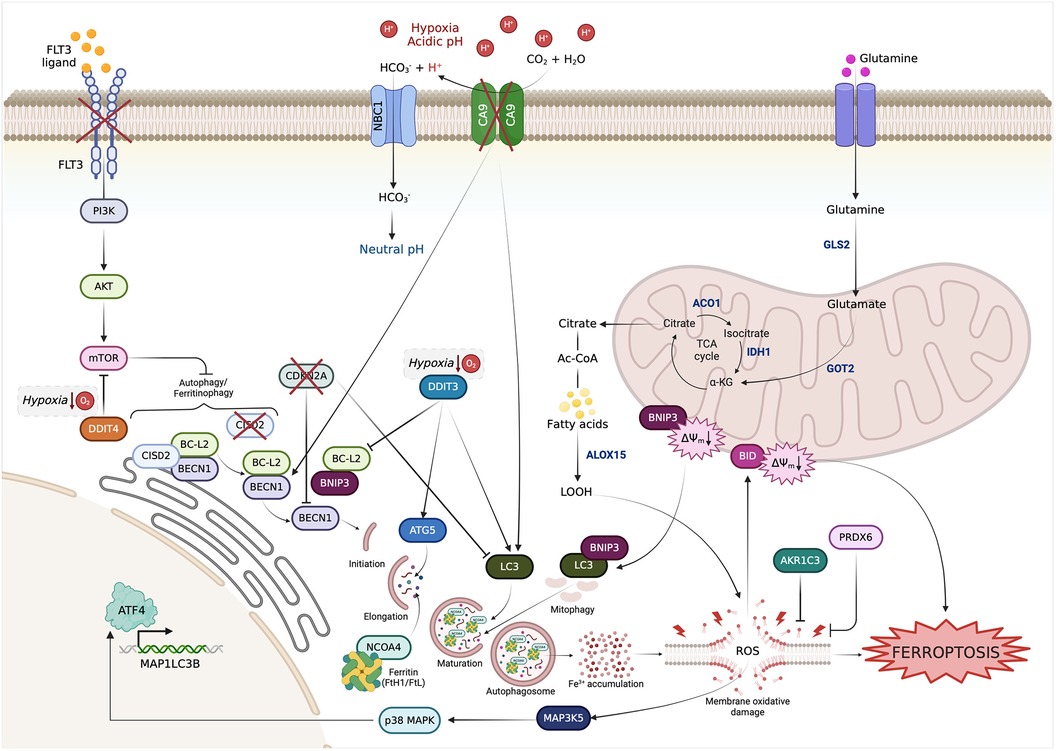
Figure 4. Interactions among the FRGs with prognostic value in OSCC based on autophagy. FRGs belonging to the prognostic models are involved in signaling pathways that converge on autophagy regulation.
Ferroptosis plays a versatile and complex role of in the crosstalk between tumor cells and TME. Ferroptotic cells, indeed, release DAMPs that recruit and activate immune cells such as macrophages, initiating an antitumor immune response (84). Moreover, lipid peroxidation may also increase T cell-mediated cytotoxicity against tumor cells (85). Simultaneously, mediators released by immune cells also have a crucial impact on regulating the susceptibility of cancer cells to ferroptosis. Notably, the occurrence of ferroptosis in tumor infiltrating immune cells affect their survival and immunomodulatory function, ultimately reprograming tumor progression in the TME (86).
2 Literature search strategy and characteristics of the selected studies
To discover the eligible studies reporting the roles of ferroptosis in OSCC, the survey of the literature was conducted on 4 life-science databases: MEDLINE (PubMed), Scopus, Web of Science, and Google Scholar. The timeframe of searching was spanned from the inception of these databases to 2023. In each database, the searching strategy used the following keywords: “Ferroptosis”, “Oral squamous cell carcinoma”, “Oral cancer”, “Oral carcinoma”, and “OSCC”. Original research articles, i.e., in vitro, in vivo, and in silico studies were selected. Conversely, articles that were either unavailable in English language or unavailable as full text were excluded. The preliminary screening was assessed based on titles and the evaluation of abstracts, followed by the analysis of the entire texts. The key features of these studies, including the first author's name, the country where the studies were conducted, the publication year of the article, the type of study, materials and methods, and the main findings are summarized in Table 1. Briefly, the selected studies were conducted in 4 different countries (n = 14 in China, n = 1 in Japan, n = 1 in Taiwan, n = 1 in Japan and China, and n = 1 in Japan and Australia) and published between 2019 and 2023. The methodological approach of these studies was either experimental research (n = 11, 61%) or bioinformatics analysis (n = 7, 39%). Among the experimental research studies, n = 7 were based exclusively on in vitro assays in OSCC cells and n = 4 combined in vitro and in vivo assays, the latter performed with xenograft murine models or OSCC patients-derived tissue specimens. Most of the in silico studies used molecular and clinical data extracted from online dataset such as The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and International Cancer Genome Consortium (ICGC).
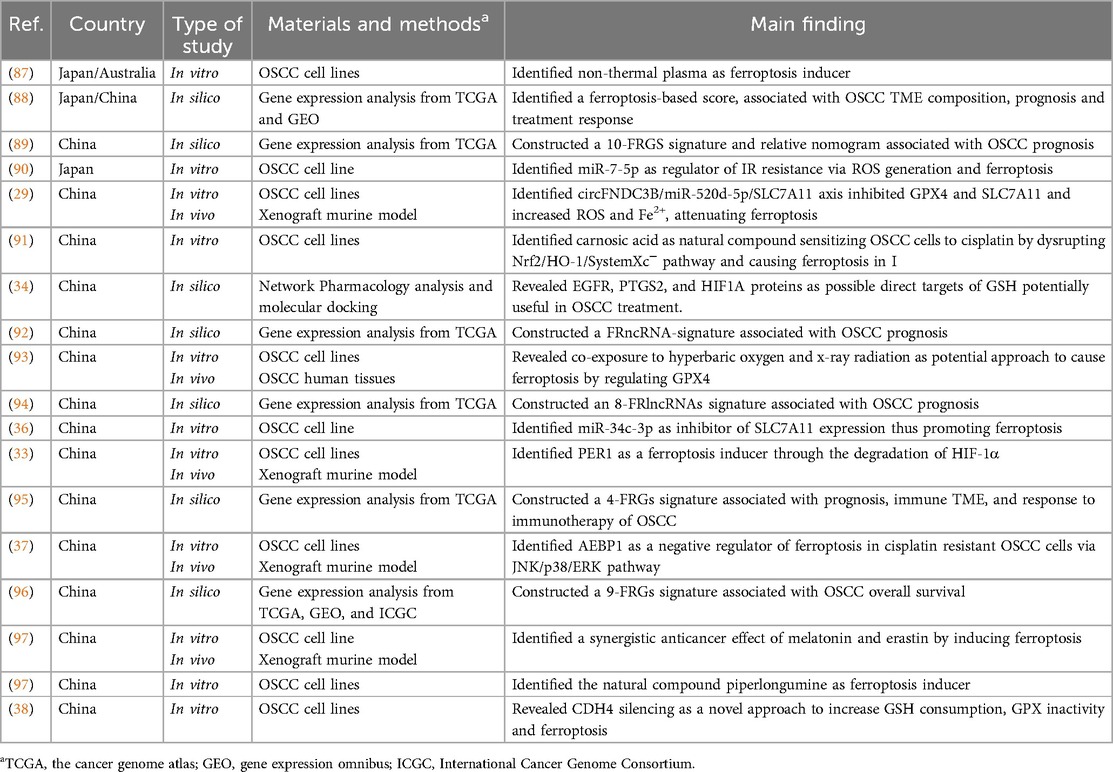
Table 1. Main characteristics of studies on ferroptosis and OSCC included in the review.
3 Targets of ferroptosis in OSCC
Over the past five years, a growing number of in vitro and in vivo studies have demonstrated that ferroptosis opens a new avenue for OSCC treatment, essentially by interfering with iron metabolism and redox homeostasis of OSCC cells. Moreover, in silico analysis have led to the development of different prognostic models and/or scores based on ferroptosis related genes (FRGs) that hold the promise to direct personalized treatments in the future.
3.1 Core regulatory mechanisms of ferroptosis in OSCC
The research landscape of ferroptosis and its implication in OSCC moved its first steps around five years ago. Knowledge on the core regulatory mechanisms driving ferroptosis in OSCC is summarized in Table 2. In the study by Zhou, Q. et al., it has been demonstrated that treatment with the ferroptosis inducer (FIN) sulfasalazine (SSZ) causes the suppression of the transcriptional factor adipocyte enhancer-binding protein 1 (AEBP1) and thus the downregulation of its target genes GPX4 and SLC7A11. Consequently, OSCC cells are unable to buffer lipid peroxides caused by the accumulation of intracellular free iron and ultimately undergo ferroptosis (37). According to Yang, Y. et al., the forced overexpression of PER1 causes ferroptosis in OSCC cells (33). PER1 is a core circadian clock gene normally expressed at low levels in OSCC tissues compared to adjacent healthy counterparts. When overexpressed in OSCC cells, by using expression vector transduction, it binds to and degrade HIF-1α (33) that has been recently considered a ferroptosis suppressor in several in vivo tumor models (98). HIF-1α, indeed, may inhibit ferroptosis by promoting the lactate production and TME acidification, which is associated with ferroptosis repression, or may enhance the transcription of the glutamate transporter SLC1A1, thus promoting cystine uptake, GSH synthesis and ferroptosis resistance (99). In OSCC, Yang, Y. et al. demonstrated that PER1 overexpression causes the degradation of HIF-1α, which in turn, is associated with reduced expression of SLC7A11, the consequent inhibition of the System xc−/GSH/GPX4 axis, the accumulation of ROS, and ferroptotic cell death (33). R-cadherin (CDH4) is a cell adhesion protein belonging to the cadherins family. Xie, J. et al. found that CDH4 is commonly overexpressed in OSCC, where it participates in epithelial-to-mesenchymal transition (EMT), invasion, and metastasis (38). Notably, CDH4 knockdown raises the consumption rate of GSH, thus causing the suppression of GPX4 activity, the accumulation of lipid peroxides, and ferroptosis (38). In support of the key role of GSH in ferroptosis occurrence in OSCC cells, a complex network pharmacology analysis, recently demonstrated that targeting GSH dramatically reduces OSCC survival by causing the dysregulation of 14 ferroptosis target molecules, namely EGFR, PTGS2, HIF-1α, VEGFA, TFRC, SLC2A1, CAV1, CDKN2A, SLC3A2, IFNG, NOX4, DDIT4, CA9, and DUSP1. Among these, the molecular docking analysis highlighted that GSH shows a strong binding affinity to EGFR, PTGS2, and HIF-1α (34). The main role of HIF-1α as a ferroptosis repressor has been described above. EGFR regulates ferroptosis by interacting with mTOR and MAPK signaling pathways in ovarian and lung cancer as well as hepatocellular carcinoma and glioblastoma (100–103). Notably, the overexpression of EGFR and the downstream recruitment and activation of oncogenic molecules, including MAPK, PI3 K/AKT, and JAK/STAT3, is one of the principal events involved in the control of differentiation, proliferation, angiogenesis, and metastasis of OSCC (104, 105). The prostaglandin-endoperoxide synthase (PTGS2) encodes for the cyclooxygenase-2 (COX-2) enzyme devoted to the biosynthesis of endoperoxides, such as prostaglandins (PGs), in response to a variety of inflammatory stimuli (106). In oral cancer, COX-2 enzyme regulates the biosynthesis of prostaglandin E2 (PGE2), which in turn induces the vascular endothelial growth factor (VEGF) expression in endothelial cells and angiogenesis (107); besides, high expression levels of COX2 are associated with lymph node metastasis (108–110). Since endogenous endoperoxides, such as PTGS2, have been found as natural triggers for ferroptosis, PTGS2 is now considered a biomarker of ferroptosis in several cancer types, including colorectal cancer and gastric cancer (111, 112). In OSCC, the relationship among GSH, HIF-1α, EGFR, PTGS2, and ferroptosis is still limited to a molecular docking analysis and needs to be further investigated in future studies.

Table 2. Regulatory mechanisms of ferroptosis in OSCC.
3.2 Non-coding RNA modulate ferroptosis in OSCC
Non-coding RNAs (NcRNAs) are a group of non-coding transcripts with limited protein-coding potential that regulate many cell life activities by controlling gene expression and protein synthesis. Three major classes of functional ncRNAs are (i) short microRNAs (miRNAs), (ii) long ncRNAs (lncRNAs), and (iii) circular RNAs (circRNAs) (113). NcRNAs are frequently deregulated in cancer and are widely considered biomarkers in both tumor diagnosis and prognosis. In addition, ncRNAs are tightly involved in the pathogenesis of cancer by affecting tumor cell proliferation, tumor metabolism, angiogenesis, metastasis, and drug resistance. Thus, targeting ncRNAs represents a promising therapeutic option to treat cancer and improve patients’ outcome (52, 114–118). As researchers have deepened their interest in ferroptosis, a growing number of non-coding (nc) RNAs has been proven to regulate, either positively or negatively, ferroptosis or to impact the propensity of tumor cells to undergo ferroptosis (119, 120).
In OSCC, Sun, K. et al., found that miR-34c-3p, directly targets SLC7A11 and reduces cysteine intake, thus impairing GSH production and GPX4 antioxidant activity. Consequently, OSCC cells undergo ferroptosis (36). Based on this study, miR-34c may function as a tumor suppressor in OSCC. miR-34c has previously been counted among the microRNAs involved in the regulation of head and neck cancer (HNSCC). Several studies report that miR-34c exerts a suppressive function on the growth and invasiveness of HNSCC and that low levels of miR-34c correlate with worse overall survival (OS) in patients with HNSCC (121, 122). Other studies, instead, show that miR-34c is upregulated in HNSCC tumor tissue compared to neighboring healthy tissues and that overexpression of miR-34c promotes the proliferation, migration, and invasion of OSCC cells in vitro (123, 124).
In addition to direct regulation of ferroptotic key players, miRNAs may indirectly regulate this cell death process via interaction with other ncRNAs, such as lncRNAs and circRNAs. CircRNAs are single-stranded ncRNAs that form circular conformations through non-canonical splicing or back-splicing events (125). Many circRNAs can act as microRNA sponges to relieve the inhibitory effect of miRNAs on their target genes and increase the expression level of corresponding genes (126). In the study of OSCC, Yang, J. et al. demonstrated that circFNDC3B acts as a molecular sponge for miR-520d-5p and, thus, relieves its inhibitory effect on SLC7A11 target gene. As a result, SLC7A11 is overexpressed. Consistently with these findings, Yang, J et al. observed that high levels of circFNDC3B correlate with a low susceptibility to ferroptosis of OSCC cells (29). Consistently, Chen et al. previously found that circFNDC3B is highly expressed in OSCC tissues and that circFNDC3B promotes proliferation, migration, and invasion of OSCC cells by targeting miR-1322/MED1 axis (127). Notably, these two studies agree with the cancer-promoting role of circFNDC3B in OSCC and propose two different mechanisms of action that need to be further investigated.
3.3 Ferroptosis and prognosis in OSCC
During the last decades, many efforts have been made to identify markers potentially correlated with OSCC outcome. As a result, several factors ranging from clinical and histopathological features to genetic and molecular alterations have been described. However, their use is still limited and only few of them are applied in the routine clinical practice (128).
Among the clinicopathological markers, tumor staging, angiogenesis, tumor budding, and perineural invasion are the most described. Tumor staging is typically correlated with prognosis. In OSCC, clinical stages II-III-IV significantly correlate with a worse survival. Indeed, while the 5-year survival rate for stage I oral cancer patients is around 80%, it breaks down to 53% for those with stage IV disease (129). Angiogenesis, quantified as number of blood vessels present in specific areas of the tumor, is significantly associated with poor prognosis, as it correlates with tumor size, nodal metastasis and relapse (130). Tumor budding is present in around 35% OSCC and, when present, correlates shorter overall survival and lymph node metastasis (131, 132). Perineural invasion (PNI), generally detected in around 18% of OSCC patients, correlates with poor prognosis (133, 134). More recently, specifically after the approval of pembrolizumab and nivolumab for treating recurrent or metastatic oral cancer, the overall assessment of tumor-infiltrating immune cells has emerged as a hot target for the prognostication of OSCC. The presence of tumor-infiltrating lymphocytes (TILs) in the invasive front of oral cancer has been reported as a promising biomarker for a better prognosis (135, 136). Hadler-Olsen E and Wirsing AM indicated that CD163+ tumor-associated macrophages (TAMs) and CD57+ natural killer (NK) cells were the most promising predictors of a better survival in OSCC patients (137). The potential role of CD163+ TAMs and CD57+ NK cells in predicting OSCC prognosis was also found by Huang, Z. et al., together with CD8+ TILs, CD45RO+ TILs and CD68+ TAMs (138).
Among the biological markers used to further assess the prognosis of OSCC patients, HPV infection represents the main predictor of a better prognosis in terms of a higher overall survival rate, a better treatment response, a reduced disease progression risk and recurrence. As such, HPV protein p16 is one of the most investigated prognostic biomarkers of OSCC (139, 140). Additional biomarkers bring insights into the identification of more aggressive OSCC, which may benefit from more specific therapeutic approaches, although most of them do not show a clinical utility (128). To make an example, FAM3C (the family with sequence similarity 3 member C) is upregulated in OSCC compared to healthy mucosa and epithelial dysplasia, and patients with a higher FAM3C expression are more likely to have a poor prognosis. In addition, the expression of FAM3C correlates with the expression of immune checkpoints such as PD-L1, VISTA, and B7-H4, EMT marker Slug, and the cancer stem cells (CSC) markers SOX2 and ALDH1 (141).
Currently, data supporting the relationship between ferroptosis and prognosis in OSCC are mostly based on in silico analyses. During the last 4 years, five different research groups, drawing from The Cancer Genome Atlas (TCGA) database, found that numerous ferroptosis-related genes (FRGs) and ferroptosis-related lncRNAs (frlncRNAs) were differentially expressed in OSCC tissues compared to healthy mucosa (89, 92, 94–96), thus further supporting the involvement of ferroptosis in the molecular pathogenesis of OSCC. Then, each of these five research groups established a distinct risk model, based on the expression of FRGs or frlncRNAs, able to cluster OSCC patients in “high risk” and “low risk” groups characterized respectively by a bad and a good prognosis, in terms of overall survival (OS) (Table 3). First, Li, H. et al., constructed a prognostic signature composed by 10 FRGs, namely Autophagy Related 5 (ATG5), BH3 Interacting Domain Death Agonist (BID), Aconitase 1 (ACO1), Glutamic-Oxaloacetic Transaminase 1 (GOT1), Aldo-Keto Reductase Family 1 Member C3 (AKR1C3), Glutaminase 2 (GLS2), Arachidonate 15-Lipoxygenase (ALOX15), Synthesis Of Cytochrome C Oxidase 2 (SCO2), Microtubule Associated Protein 1 Light Chain 3 Alpha (MAP1LC3A), Mitogen-Activated Protein Kinase Kinase Kinase 5 (MAP3K5) (89). Later, Fan et al. identified nine prognostic FRGs; three of them, ALOX15, ATG5 and MAP1LC3A, were in common with the results by Li, H. et al. (89), the other 6 include CDGSH Iron Sulfur Domain 2 (CISD2), DNA Damage Inducible Transcript 4 (DDIT4), Carbonic Anhydrase 9 (CA9), Beclin 1 (BECN1), BCL2 Interacting Protein 3 (BNIP3), and Peroxiredoxin 5 (PRDX5) (96). Then, Yin, G. et al., found 5 FRGs with prognostic value for OSCC patients: the above mentioned BNIP3, together with Ferritin Heavy Chain 1 (FTH1), Fms Related Receptor Tyrosine Kinase 3 (FLT3), Cyclin Dependent Kinase Inhibitor 2A (CDKN2A), and DNA Damage Inducible Transcript 3 (DDIT3) (95). Two further prognostic models for OSCC patients were constructed based on frlncRNAs. Some of them, namely AC099850.3, AC090246.1, AL512274.1, and Myocardial Infarction Associated Transcript (MIAT) were common between the two models (92, 94). The prognostic model developed by Qiu, L. et al., also included Firre Intergenic Repeating RNA Element (FIRRE), Long Intergenic Non-Protein Coding RNA 1305 (LINC01305), AC079921.2, and Long Intergenic Non-Protein Coding RNA 524 (LINC00524) (94) while that of Li, T. et al., also contained ALMS1 Intronic Transcript 1 (ALMS1-IT1), AC021087.4, and HOTARM1 as risk factors and STARD4 Antisense RNA 1 (STARD4-AS1) as protection factor (92). To date, these data show a promising link between ferroptosis and prognosis of OSCC. In future studies, the predictive value of these prognostic models needs to be verified by additional clinical data and the inherent mechanism of these FRGs in OSCC needs to be unveiled.
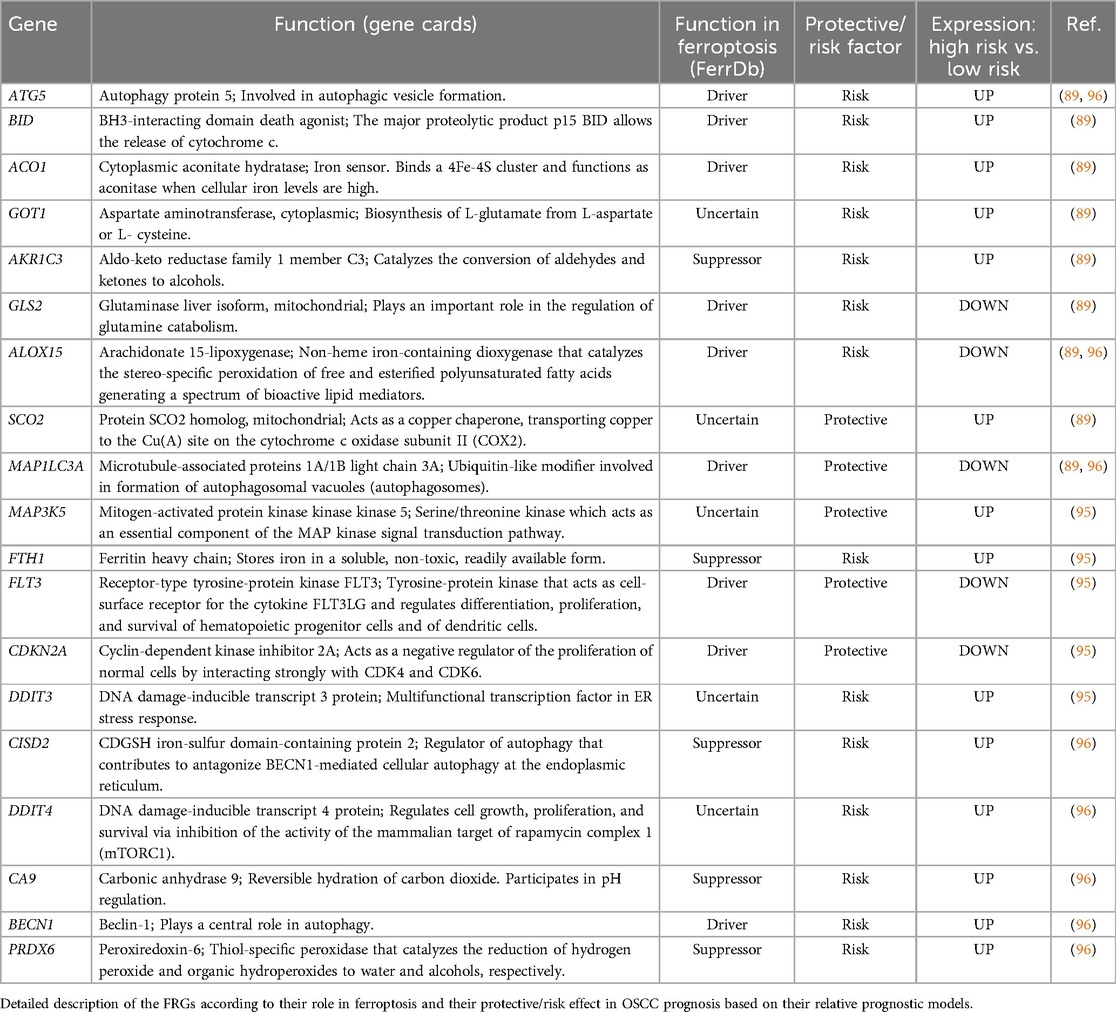
Table 3. List of FRGs with prognostic value in OSCC.
3.4 Ferroptosis-related prognostic signatures and oral tumor immunity
Due to environmental pressures, such as alteration of microbiome, HPV infection, and stromal remodeling, OSCC is characterized by extensive infiltration of immune cells, including tumor-infiltrating lymphocytes (TILs) [T cells, B cells and natural killer (NK) cells] and myeloid lineage cells (macrophages, neutrophils, dendritic cells, and myeloid-derived suppressor cells) (1, 142). However, OSCC shows the ability to evade immune surveillance through a variety of strategies, some of them specific for OSCC and others shared with other solid tumors (1). First, tobacco smoke and alcohol promote a high mutational rate in human leukocyte antigen (HLA) genes that are critical for OSCC to evade immune surveillance (143). OSCC cells can directly enhance the expression of inhibitory molecules such as (i) programmed death ligand-1 (PD-L1), which interacts with programmed death-1 (PD-1) on T cells to suppress their activity, or (ii) cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), which interacts with CD80/86 on antigen-presenting cells (APCs) to block the differentiation of naïve T cells (144–147). Alternatively, OSCC cells can inhibit both innate and adaptive immune response by releasing immune-inhibitory cytokines, such as tumor growth factor-β (TGF-β) and interleukin-6 (IL-6), or suppressing the production of immune-activating cytokines, such as interleukin-2 (IL-2) (148, 149).
Like for other forms of RCD, ferroptosis plays critical roles in the regulation of antitumor immunity (150). First, ferroptosis can be considered an immunogenic cell death (ICD) as cells undergoing ferroptosis often release damage-associated molecular patterns (DAMPs) that modulate cellular immune response (151). Cancer cells dying from ferroptosis may release high-mobility group box 1 (HMGB1) and adenosine triphosphate (ATP), which in turn stimulate the macrophage-mediated production of tumor necrosis factor α (TNFα), thus causing inflammation (152). Pancreatic cancer cells undergoing ferroptosis release mutant KRASG12D proteins that are uptaken by macrophages, which in turn respond by adopting a pro-tumorigenic M2 phenotype (153). Immune cells resident within the TME, from their side, can undergo ferroptosis thus affecting the antitumor immune response. For instance, inducing ferroptosis in Tregs, by suppressing GPX4, leads to the production of the proinflammatory interleukin-1β (IL-1β), which in turn activates T helper 17 (TH17) response and enhances antitumor immunity (154). Alternatively, immune cells resident within the TME trigger ferroptosis in tumor cells. Interferon gamma (IFNγ), released by CD8+ T cells, downregulates the expression of the two subunits of the System Xc−, SLC3A2 and SLC7A11, thus promoting lipid peroxidation and ferroptosis in ovarian cancer and melanoma cells (85).
In the study of oral cancer Gu, W. et al., highlighted that OSCC tissues characterized by high levels of ferroptosis driver genes showed a TME that they defined as “immune-inflamed”, since characterized by high expression levels of immune inhibitory molecules, such as PD-L1 and CTLA-4, and high infiltration of immune cells. Notably, this group of OSCC patients exhibited clinical features associated with a better prognosis, i.e., low-grade, early-stage tumors, no lymph node metastasis, and good response to immune checkpoints (ICIs)-based immunotherapy (88). Besides, it is worth noting that each of the five signatures described above, independently from their composition, was characterized by FRGs or frncRNAs functionally involved, among others, in the modulation of immune response according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. Indeed, a further analysis of the TME of OSCC patients belonging to the “high-risk” group with poor prognosis highlighted that these patients were characterized by a TME poor of APCs and relative functions, especially immature dendritic cells (iDCs and B cells), and by a low percentage of immune effector cells including CD8+T cells and NK cells (89, 92, 94–96). Considering that, when present, iDCs and B cells favor the immune activity against OSCC (85) and that, CD8+T and NK cells kill tumor cells and, thus, prevent OSCC growth and metastasis by remodeling of the oral TME via IFN-γ and TNF-α (155), the authors concluded that the bad prognosis of the “high-risk” OSCC patients was, in some way, associated with the impairment of the antitumor immune response.
Overall, these data highlight that ferroptosis-based multigene signatures may provide a valid tool to predict both the immune status and prognosis of OSCC patients. However, so far, these data lack experimental validation and, necessarily, need a complete molecular characterization before being used as prognostic biomarkers or targets for novel therapies.
3.5 Targeting ferroptosis to treat OSCC
OSCC requires a multidisciplinary approach including surgical intervention followed, if necessary, by postoperative radiation or chemotherapy. In most cases, surgery is the first-line treatment for oral carcinomas. In advanced cases, postoperative radiation, chemoradiation, oncogene-targeted therapy, and immunotherapy may be administered (1). However, aggressive OSCC have a poor prognosis, with limited improvements in survival over many decades (18, 136, 156).
Given the role of oxidative stress in carcinogenesis of OSCC, both small molecules and natural compounds targeting the redox balance have become appealing targets for intervention in this field. Treatment with polyenolpyrrole auxarconjugatin B causes DNA damage and apoptosis via generation of intracellular ROS in oral squamous cell carcinoma xenografts (157). Cisplatin-resistant oral cancer cells exposed to curcumin nanoparticles show increased production of ROS in association with increased levels of apoptotic proteins, such as caspase 9, cytochrome c, and apoptotic protease activating factor 1 (Apaf-1) (158). Xanthorrhizol, a natural sesquiterpenoid, induces apoptosis via ROS-mediated p38 MAP kinase and JNK activation (159).
Although still limited, experimental evidence indicates that targeting ferroptosis represents a powerful approach to kill OSCC cells. Non-thermal plasma (NTP) causes ferroptotic cell death in OSCC cells (87). NTP is a body temperature ionized gas consisting of electrons, ions, neutral atoms, and radicals (160). Originally, NTP was found to cause oxidative stress and apoptosis in many cancer types, including OSCC (161, 162). In 2019, Sato, K. et al., demonstrated for the first time that NTP causes ferroptosis in OSCC cells through the accumulation of iron within lysosomes, the production of mitochondrial ROS, and lipid peroxidation (87). According to Sato, K. et al., the use of NTP to trigger ferroptosis in OSCC cells showed two advantages. First, the ferroptosis-mediated cytotoxic effect of NTP was less harmful to normal tissues; indeed, they found that NTP selectively killed OSCC cells without affecting the surrounding normal fibroblasts. The biological reason underlying this difference was that tumor cells are generally characterized by higher intracellular iron amount and oxidative stress compared to non-tumor cells (87, 163). Besides, due to the accessibility of the oral cavity, clinical application of NTP would be easier at this site compared to other organs (87).
In addition to chemical drugs, also natural compounds promote ferroptosis in OSCC. The natural product piperlongumine (PL), a natural alkaloid extracted from pepper, causes ferroptosis in OSCC cells (164). PL shows a cytotoxic effect in many cancer types, by causing oxidative stress, cell cycle arrest and autophagy (165, 166). According to Wang, Z.K. et al., PL triggers ferroptosis in OSCC cells by reducing the expression of SLC7A11 and GPX4 and increasing lipid peroxidation. In non-tumor cells, instead, PL seems to act as herbal medicine with very little toxicity (164).
Ferroptosis induction has also shown the potential to enhance or restore sensitivity of OSCC cells to other therapeutic strategies. As mentioned before, radiotherapy is the first line of treatment for most OSCC patients (89, 167). Radiotherapy is typically administered postoperatively and depends on variables such as the primary tumor size, positive surgical margins, and the presence of perineural, lymphatic, and vascular invasion. In addition, chemotherapy has recently become a popular adjunct treatment for locally advanced OSCC. Even though chemotherapy is not considered a curative treatment for oral carcinomas, it can be administered prior to surgery or in conjunction with irradiation before or after surgery. Adjuvant chemotherapy and radiotherapy are becoming standard remedies for advanced oral cancers. Chemotherapy and radiotherapy cause apoptosis of OSCC cells, among others, by causing an oxidative stress injury (168). However, the clinical efficiency of both therapeutic approaches is often limited by the development of chemo- or radio-resistance, determined by the dysregulation of apoptotic pathways (1, 169, 170). In 2022, Liu, J. et al. found that exposure to hyperbaric oxygen (HBO) enhances the sensitivity to radiation therapy of OSCC cells by expanding the oxidative damage caused by the sole treatment with radiotherapy (93). Previously, it has been shown that OSCC cells undergo apoptosis following HBO, perhaps via activation of the MAP kinase pathway (171). According to Liu, J. et al., the molecular basis of the synergistic effect of HBO and radiotherapy does not rely on apoptosis induction, but rather on ferroptosis. HBO, indeed, inhibits GPX4 expression and causes lipid peroxidation (93). In parallel, Han, L., et al. found that carnosic acid, a polyphenolic abietane diterpene derived from rosemary, sensitizes resistant OSCC cells to cisplatin by causing ferroptosis (91). Carnosic acid has previously shown cytotoxic effects in several cancers and to enhance the therapeutic efficacy of tamoxifen in estrogen receptor (ER) positive breast cancer (172, 173). Han, L., et al. demonstrated that carnosic acid suppresses the antioxidant pathway mediated by nuclear factor E2 related factor 2 (Nrf2/NFE2L2) and its downstream target System Xc−, thus leading to ROS accumulation, lipid peroxidation, and ferroptosis in cisplatin-resistant OSCC cells. Notably, in this study, carnosic acid seemed to preserve normal oral keratinocytes.
More recently, melatonin was found to synergize with erastin to trigger ferroptosis in OSCC cells by reducing the intracellular levels of GSH and increasing those of lipid ROS (97). Although generally used for its anti-inflammatory and immune system regulatory properties (174), melatonin has shown to induce apoptosis and autophagy in cancer cells (175, 176). In the study by Wang, C., et al. the combinatory treatment with erastin and melatonin markedly reduced the OSCC size in vivo, without obvious systemic side effects (97).
4 Discussion
Since its discovery in 2012, ferroptosis has emerged as an “Achille's heel” particularly in certain cancers, whose pathways related to redox homeostasis, iron and lipid metabolisms are dysregulated as part of the molecular pathogenesis. Hence, targeting this vulnerability, through the use of FINs, has provided a new potential antitumor therapeutic approach) (44–46). During the last five years, an increasing amount of compelling evidence has indicated that ferroptosis-regulatory pathways are activated or disrupted during oral carcinogenesis. Compared to tumors, such as melanoma, lung cancer, breast cancer, and colorectal cancer, whose relationship with ferroptosis has been under investigation from longer, the vast majority of studies in OSCC indicate that ferroptosis occurs mainly through the inhibition of the antioxidant pathways driven by the System Xc−/GSH/GPX4 axis and the reprogramming of cytoplasmic iron metabolism progression (19). Data demonstrating the contribution of tumor metabolism, such as glucose and glutamine metabolism, but also that of mitochondrial iron and of mevalonate, the role of Coenzyme Q in the protection against ferroptosis, or the role of major oncogenic signaling pathways in the regulation of ferroptosis, such as AMPK pathways and Hippo pathway, are still missing.
As the understanding of ferroptosis-regulating pathways in OSCC has caught on, new druggable targets and new ferroptosis modulators have emerged as likely to trigger oral cancer cell death. In this regard, System Xc−/GSH/GPX4 proteins synthesis and activity appear as the most targeted ferroptosis regulators both in vitro and in vivo, thus confirming a prominent role of this pathway in OSCC progression (33, 34, 37, 38). Few studies show that the epigenetic machinery of OSCC may have an appreciable role in the regulation of ferroptosis target genes, either through the direct activity of miRNAs or the competitive regulatory mechanism of endogenous RNA mediated by circRNAs (29, 36). Furthermore, three important information arises from the pharmacological studies assessing the effects of FINs in OSCC. First, the array of compounds able to trigger ferroptosis in OSCC is not limited to chemical drugs but also natural compounds seem to have considerable research value. Second, OSCC cells appear in some cases more sensitive to ferroptosis than their corresponding normal oral epithelial cells in vitro. These data highlight the existence of appropriate therapeutic windows that would allow selective ferroptosis induction in OSCC while sparing normal tissues. Third, the use of ferroptosis inducing compounds in combination with other anticancer therapies, such as chemo-radiotherapy, leads to the induction of mixed-type cell deaths that enhances tumor suppression and counteract resistance of OSCC cells to chemo-radiotherapy (87, 91, 93, 97, 164). Even in this context, ferroptosis research in OSCC lags behind other tumors, where various modalities (i.e., immunotherapy and targeted therapy), selected Food and Drug Administration-approved compounds (i.e., statins and artemisinin), and also dietary interventions (i.e., the inclusion of arachidonic acid and the restriction in methionine and cysteine) have garnered recognition for their potential in inducing or sensitizing cancer cells to ferroptosis.
Most of the factors used in clinical practice to predict prognosis in OSCC are based on clinical and histological characteristics. However, OSCC patients displaying clinical and pathological similarities may show different outcomes. Hence, clinicopathological factors alone are not sufficient to efficiently predict prognosis and exploring new prognostic models from a biological perspective appears mandatory (128). Recently, prognostic models based on ferroptosis have provided a valid tool to predict OS in different cancers. In this regard, five FRGs- or frlncRNAs- signatures have been found to correlate with prognosis of OSCC patients in silico. Interestingly, these ferroptosis-related signatures agree that the poor prognosis of OSCC patients might result from the dysregulation of immune status. In addition to providing a list of new potential prognostic biomarkers, that need to be further validated in larger cohort of patients, this finding puts the ground for future in-depth analysis of the relationship between ferroptosis and antitumor immune response in OSCC and opens up promising horizons for the application of immunotherapy in combination with FINs in this disease (89, 92, 94–96).
Ferroptosis research in cancer has shown that there is crosstalk between ferroptosis and other RCD pathways as it involves similar gene mutations and protein alterations (19). In this review, we have noticed that the vast majority of FRGs with prognostic values in OSCC are functionally involved in autophagy (Figure 4). ATG5, MAP1LC3A, BECN1 are the main drivers of phagophore formation and maturation and are considered autophagy-related biomarkers able to predict prognosis in OSCC (177). To sustain autophagy flux, transcriptional upregulation of genes encoding for proteins involved in this process is crucial. In this regard, the transcription factors DDIT3 and DDIT4 are key players. Under hypoxic conditions, DDIT3 promotes the transcriptional upregulation of MAP1LC3A and ATG5 (178). DDIT3 can also stimulate autophagosome formation through downregulation of BCL2 expression. Indeed, when present, BCL2 binds to BECN1 and hinders the generation of VPS34 complex I and, thus, the phosphorylation of phosphatidylinositol-3 (PI3) on autophagic membranes (178). BNIP3 is an outer mitochondrial membrane (OMM) protein, belonging to the BCL2-family, that directly interacts with MAP1LC3 to mediate mitochondrial autophagy. Under hypoxic conditions, BNIP3 is induced and localized to the mitochondria, where it triggers loss of membrane potential, increases ROS production, and causes mitophagy. BNIP3 can regulate autophagic cell death also by competing with BECN1 for binding to BCL2 (179). In OSCC, BNIP3 has been found to drive mitophagy and to sensitize OSCC cells to cisplatin treatment (180). CISD2, also known as nutrient-deprivation autophagy factor-1, binds to BCL2 at the endoplasmic reticulum (ER), contributes to the interaction of BCL2 with BECN1 thereby hindering formation of the BECN1-PIK3C3 autophagosome-initiating complex in response to nutrient stress (181). MAP3K5 promotes autophagy partly converging into DDIT3-mediated pathways. Briefly, in response to ER stress signals, the ERN1 (Endoplasmic reticulum to nucleus signaling 1) induces MAP3K5 to activate MAPK/p38 (mitogen-activated protein kinase), which in turn phosphorylate BCL2 (182). DDIT4 expression increases under the hypoxic condition and causes or sensitizes cancer cells to autophagy through the inhibition of mTOR signaling pathway (183). Notably, the mTOR pathway is altered in around 30% of oral cancer, where it modulates proliferation, invasion, angiogenesis, migration, and metabolism of OSCC cells. Hence, targeting mTOR exerts a great antitumor activity in OSCC and relative clinical trials show encouraging results (184). Besides, DDIT4 has been previously found upregulated in oral cancer and associated with a bad prognosis in terms of advanced TNM stage, higher tumor mutational burden (TMB) and low immune score and infiltrations (185, 186). CA9 is a hypoxia-inducible gene that is up-regulated in tumor cells undergoing oxygen and nutrient deprivation (187). In Hela cervical cancer cells, it has been found that treatment with CA9 inhibitors causes the overexpression MAP1LC3A and BECN1 and triggers autophagy (188). mTOR is activated, among others, by the cytokine receptor FLT3. In acute myeloid leukemia cells, the gain-of-functions mutations of FLT3 promotes cell survival, at least in part, through the activation of mTOR. Notably, the use of FLT3 inhibitors, i.e., sorafenib, triggers autophagy in AML cells by blocking the mTOR signaling pathway (189, 190). The pro-apoptotic BID has never been directly observed in autophagy execution. However, a comprehensive bioinformatics analysis identified BID as belonging to an autophagy-related multigene signature correlated with overall survival in OSCC (191). CDKN2A is a tumor suppressor gene as it prevents phosphorylation of Retinoblastoma (Rb) protein and inhibits cell cycle progression and cell proliferation. CDKN2A is mutated with a loss of function and deletions in more than 80% of OSCC. Plus, low expression of CDKN2A is associated with decrease in overall survival and recurrence in OSCC patients (192). Notably, CDKN2A silencing promoted autophagy and upregulated the autophagy markers LC3II and BECN1 in endometrial cancer (193). Overall, in light of these observations, we suggest giving importance to the autophagic flux during ferroptosis execution also in OSCC and we open up a new point of reflection on new possible targetable regulatory molecules.
In conclusion, we believe that ferroptosis represents a novel piece of the intricate puzzle of oral cancer biology and also a new glimmer of hope for oral cancer treatment. Nevertheless, there are still many research gaps that need to be filled to certainly bring ferroptosis among the new promising opportunities for OSCC cancer treatment. First, the translation of therapeutic interventions targeting ferroptosis vulnerabilities into clinical applications necessitates the identification and validation of OSCC patient populations whose cancers exhibit heightened susceptibility to ferroptosis, based on insights derived from cell line and preclinical studies. These studies will be necessary to unravel the differences in ferroptosis execution in diverse OSCC subtypes, to discern whether these difference stem from variances in cellular or cancer contexts, or in mutation profiles, or in features of TME. In this last regard, efforts will be required to more in-depth understand ferroptosis function in immune cell populations. Subsequently, rigorous clinical trials are needed to assess the efficacy of FINs in OSCC patients, to establish their safety when combined with other ferroptosis–sensitizing treatments, and to assess their ability to replicate preclinical successes in overcoming therapeutic resistance. Equally important will be the development of robust biomarkers or metabolomic profiles for predicting and monitoring tumor responsiveness to FINs. The establishment of reliable tools for detecting these biomarkers also in biofluids, such as saliva, is paramount to this effort.
Author contributions
AA: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. AB: Data curation, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. AS: Supervision, Validation, Visualization, Writing – review & editing. LP: Supervision, Validation, Visualization, Writing – review & editing. EG: Supervision, Validation, Visualization, Writing – review & editing. SB: Software, Visualization, Writing – review & editing. FB: Conceptualization, Data curation, Formal Analysis, Investigation, Project administration, Resources, Validation, Writing – original draft, Writing – review & editing. AG: Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Figures were created by using BioRender (www.biorender.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tan Y, Wang Z, Xu M, Li B, Huang Z, Qin S, et al. Oral squamous cell carcinomas: state of the field and emerging directions. Int J Oral Sci. (2023) 15(1):1–23. doi: 10.1038/s41368-023-00249-w
2. Goffin JR, Zbuk K. Epidermal growth factor receptor: pathway, therapies, and pipeline. Clin Ther. (2013) 35:1282–303. doi: 10.1016/j.clinthera.2013.08.007
3. Aggarwal S, John S, Sapra L, Sharma SC, Das SN. Targeted disruption of PI3K/akt/mTOR signaling pathway, via PI3K inhibitors, promotes growth inhibitory effects in oral cancer cells. Cancer Chemother Pharmacol. (2019) 83:451–61. doi: 10.1007/S00280-018-3746-X
4. Liu F, Millar SE. Wnt/β-catenin signaling in oral tissue development and disease. J Dent Res. (2010) 89:318. doi: 10.1177/0022034510363373
5. Huang JS, Yao CJ, Chuang SE, Yeh CT, Lee LM, Chen RM, et al. Honokiol inhibits sphere formation and xenograft growth of oral cancer side population cells accompanied with JAK/STAT signaling pathway suppression and apoptosis induction. BMC Cancer. (2016) 16. doi: 10.1186/S12885-016-2265-6
6. Vijayalakshmi N, Selvaluxmi G, Majhi U, Rajkumar T. Alterations found in pl6/rb/cyclin D1 pathway in the dysplastic and malignant cervical epithelium. Oncol Res. (2007) 16:527–33. doi: 10.3727/096504007783438367
7. Zhang J, Chen T, Yang X, Cheng H, Spath SS, Clavijo PE, et al. Attenuated TRAF3 fosters activation of alternative NF-κB and reduced expression of antiviral interferon, TP53, and RB to promote HPV-positive head and neck cancers. Cancer Res. (2018) 78:4613–26. doi: 10.1158/0008-5472.CAN-17-0642
8. Nikitakis NG, Pentenero M, Georgaki M, Poh CF, Peterson DE, Edwards P, et al. Molecular markers associated with development and progression of potentially premalignant oral epithelial lesions: current knowledge and future implications. Oral Surg Oral Med Oral Pathol Oral Radiol. (2018) 125:650–69. doi: 10.1016/J.OOOO.2018.03.012
9. Korde S, Sridharan G, Gadbail A, Poornima V. Nitric oxide and oral cancer: a review. Oral Oncol. (2012) 48:475–83. doi: 10.1016/J.ORALONCOLOGY.2012.01.003
10. Choudhari SK, Chaudhary M, Gadbail AR, Sharma A, Tekade S. Oxidative and antioxidative mechanisms in oral cancer and precancer: a review. Oral Oncol. (2014) 50:10–8. doi: 10.1016/J.ORALONCOLOGY.2013.09.011
11. Kesarwala A, Krishna M, Mitchell J. Oxidative stress in oral diseases. Oral Dis. (2016) 22:9. doi: 10.1111/ODI.12300
12. Elmusrati A, Wang J, Wang CY. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma. Int J Oral Sci. (2021) 13(1):1–11. doi: 10.1038/s41368-021-00131-7
13. Liu C, Wang M, Zhang H, Li C, Zhang T, Liu H, et al. Tumor microenvironment and immunotherapy of oral cancer. Eur J Med Res. (2022) 27(1):1–19. doi: 10.1186/S40001-022-00835-4
14. Pai SI, Zandberg DP, Strome SE. The role of antagonists of the PD-1:pD-L1/PD-L2 axis in head and neck cancer treatment. Oral Oncol. (2016) 61:152–8. doi: 10.1016/J.ORALONCOLOGY.2016.08.001
15. Liu T, David M, Ellis O, Low TH, Palme CE, Clark J, et al. Treatment for oral squamous cell carcinoma: impact of surgeon volume on survival. Oral Oncol. (2019) 96:60–5. doi: 10.1016/J.ORALONCOLOGY.2019.06.030
16. Szturz P, Vermorken JB. Management of recurrent and metastatic oral cavity cancer: raising the bar a step higher. Oral Oncol. (2020) 101. doi: 10.1016/J.ORALONCOLOGY.2019.104492
17. Yamakawa N, Umeda M, Yoshii Y, Mitsudo K, Noguchi M, Kusukawa J, et al. Multicenter retrospective study of nivolumab for recurrent/metastatic oral squamous cell carcinoma. Oral Dis. (2022) 30:247–58. doi: 10.1111/ODI.14471
18. Zanoni DK, Montero PH, Migliacci JC, Shah JP, Wong RJ, Ganly I, et al. Survival outcomes after treatment of cancer of the oral cavity (1985–2015). Oral Oncol. (2019) 90:115–21. doi: 10.1016/J.ORALONCOLOGY.2019.02.001
19. Siquara da Rocha LO, de Morais EF, de Oliveira LQR, Barbosa AV, Lambert DW, Gurgel Rocha CA, et al. Exploring beyond common cell death pathways in oral cancer: a systematic review. Biology (Basel). (2024) 13:103–52. doi: 10.3390/BIOLOGY13020103
20. Patil S, Rao RS, Raj AT. Dual role of autophagy in oral cancer. J Int Oral Health. (2015) 7:i. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4479785/ (Accessed July 7, 2023).
21. Peña-Oyarzún D, Reyes M, Hernández-Cáceres MP, Kretschmar C, Morselli E, Ramirez-Sarmiento CA, et al. Role of autophagy in the microenvironment of oral squamous cell carcinoma. Front Oncol. (2020) 10:602661. doi: 10.3389/FONC.2020.602661
22. Abd El-Aziz YS, Leck LYW, Jansson PJ, Sahni S. Emerging role of autophagy in the development and progression of oral squamous cell carcinoma. Cancers (Basel). (2021) 13:6152. doi: 10.3390/CANCERS13246152
23. Nomura H, Uzawa K, Yamano Y, Fushimi K, Ishigami T, Kouzu Y, et al. Overexpression and altered subcellular localization of autophagy-related 16-like 1 in human oral squamous-cell carcinoma: correlation with lymphovascular invasion and lymph-node metastasis. Hum Pathol. (2009) 40:83–91. doi: 10.1016/J.HUMPATH.2008.06.018
24. Hu H, Li B, Wang J, Tan Y, Xu M, Xu W, et al. New advances into cisplatin resistance in head and neck squamous carcinoma: mechanisms and therapeutic aspects. Biomed Pharmacother. (2023) 163:114778–89. doi: 10.1016/J.BIOPHA.2023.114778
25. Xie J, Li Q, Ding X, Gao Y. Targeting mTOR by CZ415 inhibits head and neck squamous cell carcinoma cells. Cell Physiol Biochem. (2018) 46:676–86. doi: 10.1159/000488724
26. Battaglia AM, Sacco A, Perrotta ID, Faniello MC, Scalise M, Torella D, et al. Iron administration overcomes resistance to erastin-mediated ferroptosis in ovarian cancer cells. Front Oncol. (2022) 12. doi: 10.3389/FONC.2022.868351
27. Liu MZ, Kong N, Zhang GY, Xu Q, Xu Y, Ke P, et al. The critical role of ferritinophagy in human disease. Front Pharmacol. (2022) 13. doi: 10.3389/FPHAR.2022.933732
28. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2020) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
29. Yang J, Cao XH, Luan KF, Huang YD. Circular RNA FNDC3B protects oral squamous cell carcinoma cells from ferroptosis and contributes to the malignant progression by regulating miR-520d-5p/SLC7A11 axis. Front Oncol. (2021) 11. doi: 10.3389/FONC.2021.672724
30. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. (2019) 12:1–16. doi: 10.1186/S13045-019-0720-Y
31. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the “iron maiden” cell death. Cells. (2020) 9. doi: 10.3390/cells9061505
32. Fukuda M, Ogasawara Y, Hayashi H, Okuyama A, Shiono J, Inoue K, et al. Down-regulation of glutathione peroxidase 4 in oral cancer inhibits tumor growth through SREBP1 signaling. Anticancer Res. (2021) 41:1785–92. doi: 10.21873/ANTICANRES.14944
33. Yang Y, Tang H, Zheng J, Yang K. The PER1/HIF-1alpha negative feedback loop promotes ferroptosis and inhibits tumor progression in oral squamous cell carcinoma. Transl Oncol. (2022) 18:1936–5233. doi: 10.1016/J.TRANON.2022.101360
34. Huang C, Zhan L. Network pharmacology identifies therapeutic targets and the mechanisms of glutathione action in ferroptosis occurring in oral cancer. Front Pharmacol. (2022) 13:851540. doi: 10.3389/FPHAR.2022.851540/BIBTEX
35. Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG, Gao LC. System Xc -/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. (2022) 13. doi: 10.3389/FPHAR.2022.910292
36. Sun K, Ren W, Li S, Zheng J, Huang Y, Zhi K, et al. MiR-34c-3p upregulates erastin-induced ferroptosis to inhibit proliferation in oral squamous cell carcinomas by targeting SLC7A11. Pathol Res Pract. (2022) 231. doi: 10.1016/J.PRP.2022.153778
37. Zhou Q, Wang X, Zhang Y, Wang L, Chen Z. Inhibition of AEBP1 predisposes cisplatin-resistant oral cancer cells to ferroptosis. BMC Oral Health. (2022) 22. doi: 10.1186/S12903-022-02503-9
38. Xie J, Lan T, Zheng DL, Ding LC, Lu YG. CDH4 inhibits ferroptosis in oral squamous cell carcinoma cells. BMC Oral Health. (2023) 23. doi: 10.1186/S12903-023-03046-3
39. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. (2001) 9:113–4. doi: 10.1016/S0966-842X(00)01936-3
40. Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. (2018) 15. doi: 10.1186/S12974-018-1235-0
41. Gao J, Xiong A, Liu J, Li X, Wang J, Zhang L, et al. PANoptosis: bridging apoptosis, pyroptosis, and necroptosis in cancer progression and treatment. Cancer Gene Ther. (2024) 31(7):970–83. doi: 10.1038/s41417-024-00765-9
42. Frohwitter G, Zimmermann OL, Kreutzer K, Doll C, Rendenbach CM, Dommisch H, et al. Oxidative and nitrosative stress in oral squamous cell carcinoma. Cells Tissues Organs. (2020) 209:120–7. doi: 10.1159/000508705
43. Cockfield JA, Schafer ZT. Antioxidant defenses: a context-specific vulnerability of cancer cells. Cancers (Basel). (2019) 11. doi: 10.3390/CANCERS11081208
44. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/J.CELL.2012.03.042
45. Gao J, Luo T, Wang J. Gene interfered-ferroptosis therapy for cancers. Nat Commun. (2021) 12(1):1–16. doi: 10.1038/s41467-021-25632-1
46. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. (2022) 22(7):381–96. doi: 10.1038/s41568-022-00459-0
47. Yiannikourides A, Latunde-Dada GO. A short review of iron metabolism and pathophysiology of iron disorders. Medicines. (2019) 6:85. doi: 10.3390/MEDICINES6030085
48. Kosman DJ. A holistic view of mammalian (vertebrate) cellular iron uptake. Metallomics. (2020) 12:1323. doi: 10.1039/D0MT00065E
49. Di Sanzo M, Cozzolino F, Battaglia AM, Aversa I, Monaco V, Sacco A, et al. Ferritin heavy chain binds peroxiredoxin 6 and inhibits cell proliferation and migration. Int J Mol Sci. (2022) 23. doi: 10.3390/IJMS232112987/S1
50. Scicchitano S, Vecchio E, Battaglia AM, Oliverio M, Nardi M, Procopio A, et al. The double-edged sword of oleuropein in ovarian cancer cells: from antioxidant functions to cytotoxic effects. Int J Mol Sci. (2023) 24. doi: 10.3390/IJMS24010842
51. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. (2016) 26:165. doi: 10.1016/J.TCB.2015.10.014
52. Zolea F, Battaglia AM, Chiarella E, Malanga D, De Marco C, Bond HM, et al. Ferritin heavy subunit silencing blocks the erythroid commitment of K562 cells via miR-150 up-regulation and GATA-1 repression. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18102167
53. Sacco A, Battaglia AM, Botta C, Aversa I, Mancuso S, Costanzo F, et al. Iron metabolism in the tumor microenvironment— implications for anti-cancer immune response. Cells. (2021) 10. doi: 10.3390/cells10020303
54. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. (2020) 8:590226. doi: 10.3389/FCELL.2020.590226/BIBTEX
55. Battaglia AM, Sacco A, Vecchio E, Scicchitano S, Petriaggi L, Giorgio E, et al. Iron affects the sphere-forming ability of ovarian cancer cells in non-adherent culture conditions. Front Cell Dev Biol. (2023) 11. doi: 10.3389/FCELL.2023.1272667
56. Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. (2021) 12. doi: 10.1038/S41467-021-24859-2
57. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen Species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. (2019) 2019. doi: 10.1155/2019/5080843
58. Gan B. ACSL4, PUFA, and ferroptosis: new arsenal in anti-tumor immunity. Signal Transduct Target Ther. (2022) 7. doi: 10.1038/S41392-022-01004-Z
59. Lei G, Zhuang L, Gan B. mTORC1 and ferroptosis: regulatory mechanisms and therapeutic potential. Bioessays. (2021) 43. doi: 10.1002/BIES.202100093
60. Ratajczak-Wrona W, Jablonska E, Antonowicz B, Dziemianczyk D, Grabowska SZ. Levels of biological markers of nitric oxide in serum of patients with squamous cell carcinoma of the oral cavity. Int J Oral Sci. (2013) 5(3):141–5. doi: 10.1038/ijos.2013.59
61. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. (2020) 8. doi: 10.3389/FCELL.2020.586578
62. Hua Y, Yang S, Zhang Y, Li J, Wang M, Yeerkenbieke P, et al. Modulating ferroptosis sensitivity: environmental and cellular targets within the tumor microenvironment. J Exp Clin Cancer Res. (2024) 43(1):1–21. doi: 10.1186/S13046-023-02925-5
63. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/S41586-019-1707-0
64. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/S41586-021-03539-7
65. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–363.e3. doi: 10.1016/J.MOLCEL.2018.10.042
66. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6. doi: 10.1038/S41392-021-00507-5
67. Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. (2021) 7(1):1–10. doi: 10.1038/s41420-021-00579-w
68. Zhou Y, Shen Y, Chen C, Sui X, Yang J, Wang L, et al. The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer? Cancer Biol Med. (2019) 16:630. doi: 10.20892/J.ISSN.2095-3941.2019.0158
69. Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. (2003) 9:65. doi: 10.1007/bf03402040
70. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. (2020) 27:420–35. doi: 10.1016/J.CHEMBIOL.2020.02.005
71. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. (2022) 185:2401–21. doi: 10.1016/J.CELL.2022.06.003
72. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. (2022) 21. doi: 10.1186/S12943-022-01530-Y
73. Battaglia AM, Sacco A, Aversa I, Santamaria G, Palmieri C, Botta C, et al. Iron-mediated oxidative stress induces PD-L1 expression via activation of c-myc in lung adenocarcinoma. Front Cell Dev Biol. (2023) 11:1208485. doi: 10.3389/FCELL.2023.1208485/BIBTEX
74. Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, et al. Ferroptosis in cancer: from molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. (2024) 9(1):1–30. doi: 10.1038/s41392-024-01769-5
75. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520(7545):57–62. doi: 10.1038/nature14344
76. Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. (2013) 53:401. doi: 10.1146/ANNUREV-PHARMTOX-011112-140320
77. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/HEP.28251
78. Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-inducible factors and cancer. Curr Sleep Med Rep. (2017) 3:1. doi: 10.1007/S40675-017-0062-7
79. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Annu Rev Cancer Biol. (2019) 3:35–54. doi: 10.1146/ANNUREV-CANCERBIO-030518-055844
80. Balihodzic A, Prinz F, Dengler MA, Calin GA, Jost PJ, Pichler M. Non-coding RNAs and ferroptosis: potential implications for cancer therapy. Cell Death Differ. (2022) 29:1094. doi: 10.1038/S41418-022-00998-X
81. Du Y, Guo Z. Recent progress in ferroptosis: inducers and inhibitors. Cell Death Discov. (2022) 8. doi: 10.1038/S41420-022-01297-7
82. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. (2021) 42. doi: 10.1016/J.REDOX.2021.101928
83. Song X, Wang X, Liu Z, Yu Z. Role of GPX4-mediated ferroptosis in the sensitivity of triple negative breast cancer cells to gefitinib. Front Oncol. (2020) 10. doi: 10.3389/FONC.2020.597434
84. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature. (2019) 572(7769):402–6. doi: 10.1038/s41586-019-1426-6
85. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–5. doi: 10.1038/S41586-019-1170-Y
86. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8+ T cells. Cancer Immunol Res. (2021) 9:184–99. doi: 10.1158/2326-6066.CIR-20-0384
87. Sato K, Shi L, Ito F, Ohara Y, Motooka Y, Tanaka H, et al. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic fe(II)-dependent manner. J Clin Biochem Nutr. (2019) 65:8–15. doi: 10.3164/JCBN.18-91
88. Gu W, Kim M, Wang L, Yang Z, Nakajima T, Tsushima Y. Multi-omics analysis of ferroptosis regulation patterns and characterization of tumor microenvironment in patients with oral squamous cell carcinoma. Int J Biol Sci. (2021) 17:3476–92. doi: 10.7150/IJBS.61441
89. Li H, Zhang X, Yi C, He Y, Chen X, Zhao W, et al. Ferroptosis-related gene signature predicts the prognosis in oral squamous cell carcinoma patients. BMC Cancer. (2021) 21. doi: 10.1186/S12885-021-08478-0
90. Tomita K, Nagasawa T, Kuwahara Y, Torii S, Igarashi K, Roudkenar MH, et al. MiR-7-5p is involved in ferroptosis signaling and radioresistance thru the generation of ROS in radioresistant HeLa and SAS cell lines. Int J Mol Sci. (2021) 22:8300. doi: 10.3390/IJMS22158300
91. Han L, Li L, Wu G. Induction of ferroptosis by carnosic acid-mediated inactivation of Nrf2/HO-1 potentiates cisplatin responsiveness in OSCC cells. Mol Cell Probes. (2022) 64. doi: 10.1016/J.MCP.2022.101821
92. Li T, Wang Y, Xiang X, Chen C. Development and validation of a ferroptosis-related lncRNAs prognosis model in oral squamous cell carcinoma. Front Genet. (2022) 13. doi: 10.3389/FGENE.2022.847940
93. Liu J, An W, Zhao Q, Liu Z, Jiang Y, Li H, et al. Hyperbaric oxygen enhances x-ray induced ferroptosis in oral squamous cell carcinoma cells. Oral Dis. (2022) 30:116–27. doi: 10.1111/ODI.14461
94. Qiu L, Tao A, Liu F, Ge X, Li C. Potential prognostic value of a eight ferroptosis-related lncRNAs model and the correlative immune activity in oral squamous cell carcinoma. BMC Genom Data. (2022) 23. doi: 10.1186/S12863-022-01097-Z
95. Yin J, Fu J, Zhao Y, Xu J, Chen C, Zheng L, et al. Comprehensive analysis of the significance of ferroptosis-related genes in the prognosis and immunotherapy of oral squamous cell carcinoma. Bioinform Biol Insights. (2022) 16:1–13. doi: 10.1177/11779322221115548
96. Fan X, Zhong Y, Yuan F, Zhang L, Cai Y, Liao L. A ferroptosis-related prognostic model with excellent clinical performance based on the exploration of the mechanism of oral squamous cell carcinoma progression. Sci Rep. (2023) 13. doi: 10.1038/S41598-023-27676-3
97. Wang L, Wang C, Li X, Tao Z, Zhu W, Su Y, et al. Melatonin and erastin emerge synergistic anti-tumor effects on oral squamous cell carcinoma by inducing apoptosis, ferroptosis, and inhibiting autophagy through promoting ROS. Cell Mol Biol Lett. (2023) 28. doi: 10.1186/S11658-023-00449-6
98. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. (2013) 123:3664. doi: 10.1172/JCI67230
99. Yang Z, Su W, Wei X, Qu S, Zhao D, Zhou J, et al. HIF-1α drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. (2023) 42. doi: 10.1016/J.CELREP.2023.112945
100. Kadioglu O, Saeed MEM, Mahmoud N, Azawi S, Mrasek K, Liehr T, et al. Identification of novel drug resistance mechanisms by genomic and transcriptomic profiling of glioblastoma cells with mutation-activated EGFR. Life Sci. (2021) 284:119601. doi: 10.1016/J.LFS.2021.119601
101. Ni J, Chen K, Zhang J, Zhang X. Inhibition of GPX4 or mTOR overcomes resistance to lapatinib via promoting ferroptosis in NSCLC cells. Biochem Biophys Res Commun. (2021) 567:154–60. doi: 10.1016/J.BBRC.2021.06.051
102. Sun J, Zhou C, Zhao Y, Zhang X, Chen W, Zhou Q, et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. (2021) 41. doi: 10.1016/J.REDOX.2021.101942
103. Li HW, Liu MB, Jiang X, Song T, Feng SX, Wu JY, et al. GALNT14 Regulates ferroptosis and apoptosis of ovarian cancer through the EGFR/mTOR pathway. Future Oncol. (2022) 18:149–61. doi: 10.2217/FON-2021-0883/ASSET/IMAGES/LARGE/FIGURE8.JPEG
104. Benchekroun MT, Saintigny P, Thomas SM, El-Naggar AK, Papadimitrakopoulou V, Ren H, et al. Epidermal growth factor receptor expression and gene copy number in the risk of oral cancer. Cancer Prev Res (Phila). (2010) 3:800. doi: 10.1158/1940-6207.CAPR-09-0163
105. Rehmani HS, Issaeva N. EGFR in head and neck squamous cell carcinoma: exploring possibilities of novel drug combinations. Ann Transl Med. (2020) 8:813. doi: 10.21037/ATM.2020.04.07
106. Gómez-Valenzuela F, Escobar E, Pérez-Tomás R, Montecinos VP. The inflammatory profile of the tumor microenvironment, orchestrated by cyclooxygenase-2, promotes epithelial-mesenchymal transition. Front Oncol. (2021) 11:686792. doi: 10.3389/FONC.2021.686792/BIBTEX
107. Kamal MV, Damerla RR, Dikhit PS, Kumar NA. Prostaglandin-endoperoxide synthase 2 (PTGS2) gene expression and its association with genes regulating the VEGF signaling pathway in head and neck squamous cell carcinoma. J Oral Biol Craniofac Res. (2023) 13:567–74. doi: 10.1016/J.JOBCR.2023.07.002
108. Gallo O, Masini E, Bianchi B, Bruschini L, Paglierani M, Franchi A. Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Hum Pathol. (2002) 33:708–14. doi: 10.1053/hupa.2002.125376
109. Kyzas PA, Stefanou D, Agnantis NJ. COX-2 expression correlates with VEGF-C and lymph node metastases in patients with head and neck squamous cell carcinoma. Mod Pathol. (2005) 18:153–60. doi: 10.1038/MODPATHOL.3800244
110. Kono M, Watanabe M, Abukawa H, Hasegawa O, Satomi T, Chikazu D. Cyclo-oxygenase-2 expression is associated with vascular endothelial growth factor C expression and lymph node metastasis in oral squamous cell carcinoma. J Oral Maxillofac Surg. (2013) 71:1694–702. doi: 10.1016/J.JOMS.2013.04.015
111. Guan Z, Chen J, Li X, Dong N. Tanshinone IIA induces ferroptosis in gastric cancer cells through p53-mediated SLC7A11 down-regulation. Biosci Rep. (2020) 40:20201807. doi: 10.1042/BSR20201807/226033
112. Zhao X, Chen F. Propofol induces the ferroptosis of colorectal cancer cells by downregulating STAT3 expression. Oncol Lett. (2021) 22. doi: 10.3892/OL.2021.13028
113. Sandberg K, Samson WK, Ji H. Decoding noncoding RNA: the long and short of it. Circ Res. (2013) 113:240. doi: 10.1161/CIRCRESAHA.113.301865
114. Biamonte F, Zolea F, Santamaria G, Battaglia AM, Cuda G, Costanzo F. Human haematological and epithelial tumor-derived cell lines express distinct patterns of onco-microRNAs. Cell Mol Biol. (2017) 63:75–85. doi: 10.14715/cmb/2017.63.11.14
115. Biamonte F, Battaglia AM, Zolea F, Oliveira DM, Aversa I, Santamaria G, et al. Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis. (2018) 9. doi: 10.1038/s41419-018-1216-3
116. Biamonte F, Santamaria G, Sacco A, Perrone FM, Di Cello A, Battaglia AM, et al. MicroRNA let-7g acts as tumor suppressor and predictive biomarker for chemoresistance in human epithelial ovarian cancer. Sci Rep. (2019) 9. doi: 10.1038/s41598-019-42221-x
117. Grillone K, Riillo C, Riillo C, Scionti F, Rocca R, Rocca R, et al. Non-coding RNAs in cancer: platforms and strategies for investigating the genomic “dark matter.”. J Exp Clin Cancer Res. (2020) 39(1):1–19. doi: 10.1186/S13046-020-01622-X
118. Lv Y, Lv Y, Wang Z, Yuan K, Zeng Y. Noncoding RNAs as sensors of tumor microenvironmental stress. J Exp Clin Cancer Res. (2022) 41(1):1–20. doi: 10.1186/S13046-022-02433-Y
119. Hsieh PL, Chao SC, Chu PM, Yu CC. Regulation of ferroptosis by non-coding RNAs in head and neck cancers. Int J Mol Sci. (2022) 23:3142. doi: 10.3390/IJMS23063142
120. Zuo YB, Zhang YF, Zhang R, Tian JW, Lv XB, Li R, et al. Ferroptosis in cancer progression: role of noncoding RNAs. Int J Biol Sci. (2022) 18:1829. doi: 10.7150/IJBS.66917
121. Liu M, Wu H, Liu T, Li Y, Wang F, Wan H, et al. Regulation of the cell cycle gene, BTG2, by miR-21 in human laryngeal carcinoma. Cell Res. (2009) 19:828–37. doi: 10.1038/CR.2009.72
122. Re M, Çeka A, Rubini C, Ferrante L, Zizzi A, Gioacchini FM, et al. MicroRNA-34c-5p is related to recurrence in laryngeal squamous cell carcinoma. Laryngoscope. (2015) 125:E306–12. doi: 10.1002/LARY.25475
123. Severino P, Brüggemann H, Andreghetto FM, Camps C, Klingbeil MFG, de Pereira WO, et al. MicroRNA expression profile in head and neck cancer: hOX-cluster embedded microRNA-196a and microRNA-10b dysregulation implicated in cell proliferation. BMC Cancer. (2013) 13:533. doi: 10.1186/1471-2407-13-533
124. Shen Y, Sun C, Zhao B, Guo H, Li J, Xia Y, et al. miR-34c-5p mediates the cellular malignant behaviors of oral squamous cell carcinoma through targeted binding of TRIM29. Ann Transl Med. (2021) 9:1537. doi: 10.21037/ATM-21-4679
125. Zhang Y, Liang W, Zhang P, Chen J, Qian H, Zhang X, et al. Circular RNAs: emerging cancer biomarkers and targets. J Exp Clin Cancer Res. (2017) 36(1):1–13. doi: 10.1186/S13046-017-0624-Z
126. Panda AC. Circular RNAs act as miRNA sponges. Adv Exp Med Biol. (2018) 1087:67–79. doi: 10.1007/978-981-13-1426-1_6
127. Chen X, Kong D, Deng J, Mo F, Liang J. Overexpression of circFNDC3B promotes the progression of oral tongue squamous cell carcinoma through the miR-1322/MED1 axis. Head Neck. (2022) 44:2417–27. doi: 10.1002/HED.27152
128. de Oliveira LQR, Almangush A, Al-Samadi A, Salo T, Coletta RD. Prognostic markers for oral cancer: an overview of the current status and directions for future research. J Oral Pathol Med. (2023) 52:288–93. doi: 10.1111/JOP.13406
129. Rylands J, Lowe D, Rogers SN. Outcomes by area of residence deprivation in a cohort of oral cancer patients: survival, health-related quality of life, and place of death. Oral Oncol. (2016) 52:30–6. doi: 10.1016/J.ORALONCOLOGY.2015.10.017
130. Bagan J, Sarrion G, Jimenez Y. Oral cancer: clinical features. Oral Oncol. (2010) 46:414–7. doi: 10.1016/J.ORALONCOLOGY.2010.03.009
131. Seki M, Sano T, Yokoo S, Oyama T. Tumour budding evaluated in biopsy specimens is a useful predictor of prognosis in patients with cN0 early stage oral squamous cell carcinoma. Histopathology. (2017) 70:869–79. doi: 10.1111/HIS.13144
132. Almangush A, Pirinen M, Heikkinen I, Mäkitie AA, Salo T, Leivo I. Tumour budding in oral squamous cell carcinoma: a meta-analysis. Br J Cancer. (2018) 118:577–86. doi: 10.1038/BJC.2017.425
133. Alkhadar H, Macluskey M, White S, Ellis I. Perineural invasion in oral squamous cell carcinoma: incidence, prognostic impact and molecular insight. J Oral Pathol Med. (2020) 49:994–1003. doi: 10.1111/JOP.13069
134. Caponio VCA, Troiano G, Togni L, Zhurakivska K, Santarelli A, Laino L, et al. Pattern and localization of perineural invasion predict poor survival in oral tongue carcinoma. Oral Dis. (2023) 29:411–22. doi: 10.1111/ODI.13900
135. Heikkinen I, Bello IO, Wahab A, Hagström J, Haglund C, Coletta RD, et al. Assessment of tumor-infiltrating lymphocytes predicts the behavior of early-stage oral tongue cancer. Am J Surg Pathol. (2019) 43:1392–6. doi: 10.1097/PAS.0000000000001323
136. Almangush A, Mäkitie AA, Triantafyllou A, de Bree R, Strojan P, Rinaldo A, et al. Staging and grading of oral squamous cell carcinoma: an update. Oral Oncol. (2020) 107. doi: 10.1016/J.ORALONCOLOGY.2020.104799
137. Hadler-Olsen E, Wirsing AM. Tissue-infiltrating immune cells as prognostic markers in oral squamous cell carcinoma: a systematic review and meta-analysis. Br J Cancer. (2019) 120:714–27. doi: 10.1038/S41416-019-0409-6
138. Huang Z, Xie N, Liu H, Wan Y, Zhu Y, Zhang M, et al. The prognostic role of tumour-infiltrating lymphocytes in oral squamous cell carcinoma: a meta-analysis. J Oral Pathol Med. (2019) 48:788–98. doi: 10.1111/JOP.12927
139. Fakhry C, D’Souza G. Discussing the diagnosis of HPV-OSCC: common questions and answers. Oral Oncol. (2013) 49:863. doi: 10.1016/J.ORALONCOLOGY.2013.06.002
140. Wang F, Zhang H, Xue Y, Wen J, Zhou J, Yang X, et al. A systematic investigation of the association between HPV and the clinicopathological parameters and prognosis of oral and oropharyngeal squamous cell carcinomas. Cancer Med. (2017) 6:910–7. doi: 10.1002/CAM4.1045
141. Wu CC, Xiao Y, Li H, Mao L, Deng WW, Yu GT, et al. Overexpression of FAM3C is associated with poor prognosis in oral squamous cell carcinoma. Pathol Res Pract. (2019) 215:772–8. doi: 10.1016/J.PRP.2019.01.019
142. Peltanova B, Raudenska M, Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol Cancer. (2019) 18. doi: 10.1186/S12943-019-0983-5
143. de Ruiter EJ, Ooft ML, Devriese LA, Willems SM. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: a systematic review and meta-analysis. Oncoimmunology. (2017) 6. doi: 10.1080/2162402X.2017.1356148
144. Finn OJ. A believer’s overview of cancer immunosurveillance and immunotherapy. J Immunol. (2018) 200:385–91. doi: 10.4049/JIMMUNOL.1701302
145. Lenouvel D, González-Moles MÁ, Talbaoui A, Ramos-García P, González-Ruiz L, Ruiz-Ávila I, et al. An update of knowledge on PD-L1 in head and neck cancers: physiologic, prognostic and therapeutic perspectives. Oral Dis. (2020) 26:511–26. doi: 10.1111/ODI.13088
146. Mei Z, Huang J, Qiao B, Lam AK. Immune checkpoint pathways in immunotherapy for head and neck squamous cell carcinoma. Int J Oral Sci. (2020) 12(1):1–9. doi: 10.1038/s41368-020-0084-8
147. Venkatesiah SS, Augustine D, Mishra D, Gujjar N, Haragannavar VC, Awan KH, et al. Immunology of oral squamous cell carcinoma—a comprehensive insight with recent concepts. Life. (2022) 12. doi: 10.3390/LIFE12111807
148. Wolf GT, Fee WE, Dolan RW, Moyer JS, Kaplan MJ, Spring PM, et al. Novel neoadjuvant immunotherapy regimen safety and survival in head and neck squamous cell cancer. Head Neck. (2011) 33:1666–74. doi: 10.1002/HED.21660
149. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: mDSCs, TAMs, and tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol. (2016) 58:59–70. doi: 10.1016/J.ORALONCOLOGY.2016.05.002
150. Gu X, Liu Y, Dai X, Yang YG, Zhang X. Deciphering the potential roles of ferroptosis in regulating tumor immunity and tumor immunotherapy. Front Immunol. (2023) 14. doi: 10.3389/FIMMU.2023.1137107
151. Legrand AJ, Konstantinou M, Goode EF, Meier P. The diversification of cell death and immunity: memento Mori. Mol Cell. (2019) 76:232–42. doi: 10.1016/J.MOLCEL.2019.09.006
152. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/J.BBRC.2019.01.090
153. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. (2020) 16:2069–83. doi: 10.1080/15548627.2020.1714209
154. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain treg cell activation and suppression of antitumor immunity. Cell Rep. (2021) 35. doi: 10.1016/J.CELREP.2021.109235
155. Jewett A, Kos J, Fong Y, Ko MW, Safaei T, Perišić Nanut M, et al. NK cells shape pancreatic and oral tumor microenvironments; role in inhibition of tumor growth and metastasis. Semin Cancer Biol. (2018) 53:178–88. doi: 10.1016/J.SEMCANCER.2018.08.001
156. Yete S, Saranath D. MicroRNAs in oral cancer: biomarkers with clinical potential. Oral Oncol. (2020) 110. doi: 10.1016/J.ORALONCOLOGY.2020.105002
157. Hua KF, Liao PC, Fang Z, Yang FL, Yang YL, Chen YL, et al. Generation of reactive oxygen Species by polyenylpyrroles derivatives causes DNA damage leading to G2/M arrest and apoptosis in human oral squamous cell carcinoma cells. PLoS One. (2013) 8:67603. doi: 10.1371/JOURNAL.PONE.0067603
158. Chang PY, Peng SF, Lee CY, Lu CC, Tsai SC, Shieh TM, et al. Curcumin-loaded nanoparticles induce apoptotic cell death through regulation of the function of MDR1 and reactive oxygen species in cisplatin-resistant CAR human oral cancer cells. Int J Oncol. (2013) 43:1141–50. doi: 10.3892/IJO.2013.2050/HTML
159. Kim JY, An JM, Chung WY, Park KK, Hwang JK, Kim DS, et al. Xanthorrhizol induces apoptosis through ROS-mediated MAPK activation in human oral squamous cell carcinoma cells and inhibits DMBA-induced oral carcinogenesis in hamsters. Phytother Res. (2013) 27:493–8. doi: 10.1002/PTR.4746
160. Moreau M, Orange N, Feuilloley MGJ. Non-thermal plasma technologies: new tools for bio-decontamination. Biotechnol Adv. (2008) 26:610–7. doi: 10.1016/J.BIOTECHADV.2008.08.001
161. Vandamme M, Robert E, Lerondel S, Sarron V, Ries D, Dozias S, et al. ROS implication in a new antitumor strategy based on non-thermal plasma. Int J Cancer. (2012) 130:2185–94. doi: 10.1002/IJC.26252
162. Chang JW, Kang SU, Shin YS, Kim KI, Seo SJ, Yang SS, et al. Non-thermal atmospheric pressure plasma induces apoptosis in oral cavity squamous cell carcinoma: involvement of DNA-damage-triggering sub-G(1) arrest via the ATM/p53 pathway. Arch Biochem Biophys. (2014) 545:133–40. doi: 10.1016/J.ABB.2014.01.022
163. Shi L, Ito F, Wang Y, Okazaki Y, Tanaka H, Mizuno M, et al. Non-thermal plasma induces a stress response in mesothelioma cells resulting in increased endocytosis, lysosome biogenesis and autophagy. Free Radic Biol Med. (2017) 108:904–17. doi: 10.1016/J.FREERADBIOMED.2017.04.368
164. Wang Z-Q, Li Y-Q, Wang D-Y, Shen Y-Q, Rd MN. Natural product piperlongumine inhibits proliferation of oral squamous carcinoma cells by inducing ferroptosis and inhibiting intracellular antioxidant capacity. Transl Cancer Res. (2021) 12:2911–22. doi: 10.21037/TCR-22-1494
165. Piska K, Gunia-Krzyżak A, Koczurkiewicz P, Wójcik-Pszczoła K, Pękala E. Piperlongumine (piplartine) as a lead compound for anticancer agents - synthesis and properties of analogues: a mini-review. Eur J Med Chem. (2018) 156:13–20. doi: 10.1016/J.EJMECH.2018.06.057
166. Lv F, Deng M, Bai J, Zou D, Wang J, Li H, et al. Piperlongumine inhibits head and neck squamous cell carcinoma proliferation by docking to akt. Phytother Res. (2020) 34:3345–58. doi: 10.1002/PTR.6788
167. Melo-Alvim C, Neves ME, Santos JL, Abrunhosa-Branquinho AN, Barroso T, Costa L, et al. Radiotherapy, chemotherapy and immunotherapy—current practice and future perspectives for recurrent/metastatic oral cavity squamous cell carcinoma. Diagnostics. (2023) 13. doi: 10.3390/DIAGNOSTICS13010099
168. Contant C, Rouabhia M, Loubaki L, Chandad F, Semlali A. Anethole induces anti-oral cancer activity by triggering apoptosis, autophagy and oxidative stress and by modulation of multiple signaling pathways. Sci Rep. (2021) 11:13087. doi: 10.1038/S41598-021-92456-W
169. Usman S, Jamal A, The MT, Waseem A. Major molecular signaling pathways in oral cancer associated with therapeutic resistance. Front Oral Health. (2020) 1:603160. doi: 10.3389/FROH.2020.603160
170. Bos T, Ratti JA, Harada H. Targeting stress-response pathways and therapeutic resistance in head and neck cancer. Front Oral Health. (2021) 2:676643. doi: 10.3389/FROH.2021.676643/BIBTEX
171. Spiegelberg L, Djasim UM, Van Neck HW, Wolvius EB, Van Der Wal KG. Hyperbaric oxygen therapy in the management of radiation-induced injury in the head and neck region: a review of the literature. J Oral Maxillofac Surg. (2010) 68:1732–9. doi: 10.1016/J.JOMS.2010.02.040
172. Han N, Zhou Q, Huang Q, Liu K. Carnosic acid cooperates with tamoxifen to induce apoptosis associated with caspase-3 activation in breast cancer cells in vitro and in vivo. Biomed Pharmacother. (2017) 89:827–37. doi: 10.1016/J.BIOPHA.2017.01.084
173. El-Huneidi W, Bajbouj K, Muhammad JS, Vinod A, Shafarin J, Khoder G, et al. Carnosic acid induces apoptosis and inhibits akt/mTOR signaling in human gastric cancer cell lines. Pharmaceuticals. (2021) 14:230. doi: 10.3390/PH14030230
174. Slominski AT, Hardeland R, Zmijewski MA, Slominski RM, Reiter RJ, Paus R. Melatonin: a cutaneous perspective on its production, metabolism, and functions. J Invest Dermatol. (2018) 138:490–9. doi: 10.1016/J.JID.2017.10.025
175. Li Y, Li S, Zhou Y, Meng X, Zhang JJ, Xu DP, et al. Melatonin for the prevention and treatment of cancer. Oncotarget. (2017) 8:39896–921. doi: 10.18632/ONCOTARGET.16379
176. Mehrzadi S, Pourhanifeh MH, Mirzaei A, Moradian F, Hosseinzadeh A. An updated review of mechanistic potentials of melatonin against cancer: pivotal roles in angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Cancer Cell Int. (2021) 21:188. doi: 10.1186/S12935-021-01892-1
177. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. (2019) 26:605. doi: 10.1038/S41418-018-0252-Y
178. Rashid HO, Yadav RK, Kim HR, Chae HJ. ER Stress: autophagy induction, inhibition and selection. Autophagy. (2015) 11:1956. doi: 10.1080/15548627.2015.1091141
179. Burton TR, Gibson SB. The role of bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. (2009) 16(4):515–23. doi: 10.1038/cdd.2008.185
180. Shaw JJP, Boyer TL, Venner E, Beck PJ, Slamowitz T, Caste T, et al. Inhibition of lysosomal function mitigates protective mitophagy and augments ceramide nanoliposome-induced cell death in head and neck squamous cell carcinoma. Mol Cancer Ther. (2020) 19:2621–33. doi: 10.1158/1535-7163.MCT-20-0182
181. Chang NC, Nguyen M, Shore GC. BCL2-CISD2: an ER complex at the nexus of autophagy and calcium homeostasis? Autophagy. (2012) 8:856–7. doi: 10.4161/AUTO.20054
182. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. (2000) 103:239–52. doi: 10.1016/S0092-8674(00)00116-1
183. Zhang Y, Liu L, Hou X, Zhang Z, Zhou X, Gao W. Role of autophagy mediated by AMPK/DDiT4/mTOR axis in HT22 cells under oxygen and glucose deprivation/reoxygenation. ACS Omega. (2023) 8:9221–9. doi: 10.1021/ACSOMEGA.2C07280
184. Su YC, Lee WC, Wang CC, Yeh SA, Chen WH, Chen PJ. Targeting PI3K/AKT/mTOR signaling pathway as a radiosensitization in head and neck squamous cell carcinomas. Int J Mol Sci. (2022) 23. doi: 10.3390/IJMS232415749
185. Han Y, Wang X, Xia K, Su T. A novel defined hypoxia-related gene signature to predict the prognosis of oral squamous cell carcinoma. Ann Transl Med. (2021) 9:1565. doi: 10.21037/ATM-21-4990
186. Zhang Z, Zhu H, Zhao C, Liu D, Luo J, Ying Y, et al. DDIT4 promotes malignancy of head and neck squamous cell carcinoma. Mol Carcinog. (2023) 62:332–47. doi: 10.1002/MC.23489
187. Pastorek J, Pastorekova S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol. (2015) 31:52–64. doi: 10.1016/J.SEMCANCER.2014.08.002
188. Temiz E, Koyuncu I, Durgun M, Caglayan M, Gonel A, Güler EM, et al. Inhibition of carbonic anhydrase IX promotes apoptosis through intracellular pH level alterations in cervical cancer cells. Int J Mol Sci. (2021) 22:6098. doi: 10.3390/IJMS22116098
189. Koschade SE, Klann K, Shaid S, Vick B, Stratmann JA, Thölken M, et al. Translatome proteomics identifies autophagy as a resistance mechanism to on-target FLT3 inhibitors in acute myeloid leukemia. Leukemia. (2022) 36(10):2396–407. doi: 10.1038/s41375-022-01678-y
190. Grant S. Disrupting autophagy in FLT3-mutant acute myeloid leukemia. Haematologica. (2023) 108:1458. doi: 10.3324/HAEMATOL.2022.282054
191. Huang G, Lu Z, Rao Y, Gao H, Lv X. Screening and identification of autophagy-related biomarkers for oral squamous cell carcinoma (OSCC) via integrated bioinformatics analysis. J Cell Mol Med. (2021) 25:4444. doi: 10.1111/JCMM.16512
192. Padhi SS, Roy S, Kar M, Saha A, Roy S, Adhya A, et al. Role of CDKN2A/p16 expression in the prognostication of oral squamous cell carcinoma. Oral Oncol. (2017) 73:27–35. doi: 10.1016/J.ORALONCOLOGY.2017.07.030
Keywords: oral squamous cell carcinoma, ferroptosis, autophagy, prognosis, signature, ferroptosis inducers
Citation: Antonelli A, Battaglia AM, Sacco A, Petriaggi L, Giorgio E, Barone S, Biamonte F and Giudice A (2024) Ferroptosis and oral squamous cell carcinoma: connecting the dots to move forward. Front. Oral. Health 5:1461022. doi: 10.3389/froh.2024.1461022
Received: 7 July 2024; Accepted: 12 August 2024;
Published: 4 September 2024.
Edited by:
Ricardo D. Coletta, Campinas State University, BrazilReviewed by:
Daniel Peña-Oyarzun, San Sebastián University, ChileEverton Freitas De Morais, State University of Campinas, Brazil
Copyright: © 2024 Antonelli, Battaglia, Sacco, Petriaggi, Giorgio, Barone, Biamonte and Giudice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flavia Biamonte, ZmxhdmlhLmJpYW1vbnRlQHVuaWN6Lml0
†These authors share first authorship