 Vaibhav Sahni
Vaibhav Sahni Thomas E. Van Dyke
Thomas E. Van Dyke- 1Immunology and Infectious Disease, The Forsyth Institute, Cambridge, MA, United States
- 2Faculty of Medicine, Harvard University, Boston, MA, United States
Inflammation is a critical component in the pathophysiology of numerous disease processes, with most therapeutic modalities focusing on its inhibition in order to achieve treatment outcomes. The resolution of inflammation is a separate, distinct pathway that entails the reversal of the inflammatory process to a state of homoeostasis rather than selective inhibition of specific components of the inflammatory cascade. The discovery of specialized pro-resolving mediators (SPMs) resulted in a paradigm shift in our understanding of disease etiopathology. Periodontal disease, traditionally considered as one of microbial etiology, is now understood to be an inflammation-driven process associated with dysbiosis of the oral microbiome that may be modulated with SPMs to achieve therapeutic benefit.
Introduction
Historical perspective on inflammation
The medical principles of inflammation have been known since ancient times. Wounds and infections have always occupied a central space in the practice of medicine. Cornelius Celsus, a Roman physician is credited with being the first to explicitly define the clinical presentation of inflammation in 1 AD. He described four cardinal signs of inflammation: rubor (redness) and tumor (swelling) with calor (heat) and dolor (pain) while describing treatment procedures for chest pain in his treatise, De medicina (1). Along with other vascular changes specific to an acute inflammatory response, Waller and Cohnheim discovered the emigration of leukocytes from blood vessels, thereby establishing the physiological basis of the cardinal signs originally described by Celsus (1). Cohnheim further observed plasma leakage, vasodilatation and leukocyte emigration under a microscope. Cellular pathologie, authored by Rudolph Virchow, saw the addition of the disturbance of function (function laesa) as the fifth cardinal sign of inflammation (1). Virchow primarily contributed to the study of inflammation by establishing a cellular basis for the phenomenon, which was a paradigm shift from the more traditional view of humor imbalance. As such, function laesa is the only universal sign of inflammation, characterizing all its forms, while the original four were confined to be descriptive of only the acute phase.
1892 saw another milestone in the understanding of the inflammatory process when Elie Metchnikoff first described the process of phagocytosis and the cellular immunity theory. Metchnikoff stressed the favorable aspects of inflammation in his description of the role played by microphages (neutrophils) and macrophages in maintaining homoeostasis and host defense. Subsequently, Von Behring and Kitasato discovered diphtheria serum therapy, which led to the work of Ehrlich in developing the humoral immunity theory (1). Further development of the humoral immunity theory was provided by Bordet who discovered complement as an essential determinant of the immune response. Late in the 19th century, Pasteur and Koch established the germ theory of disease, which credited microbial insult as a major factor in the induction of an acute host inflammatory response.
Overview of inflammation
Inflammation may best be described as the innate response of an organism to an insult by noxious stimulants such as injury or infection (2). An acute inflammatory response induced by noxious stimuli at its most basic level in mammals involves accumulation of leukocytes and plasma (blood products) to the site of insult (2, 3). In the context of this review of inflammation in periodontal diseases, we focus on inflammation induced by microbial challenge.
The innate inflammatory response is triggered by innate immune receptors called pattern recognition receptors, such as NOD-like (NLRs) and Toll-like receptors (TLRs). The microbial challenge is initially recognized and mediated at a local level by mast cells and tissue macrophages that elaborate a wider inflammatory response, including the production of various mediators such as cytokines, chemokines, eicosanoids and vasoactive amines (1). This results in the production of localized exudate at the site of insult. The exudate is due to the leakage of otherwise vascular-confined elements such as leukocytes and plasma proteins, which accumulate in extravascular sites via postcapillary venules. The initial stimulus leads to an activation of the vascular endothelium, which selectively confines erythrocytes, while allowing neutrophils to exit the vascular compartment. This preferential selectivity is the result of an interaction of leukocyte integrins and selectins with receptors present on the vascular endothelial cells (4). Neutrophil chemotaxis to the site of infection is followed by their activation either directly via pathogen contact or indirectly through cytokine action. The neutrophils attempt to eliminate noxious agents, a process characterized by the production of reactive nitrogen and oxygen species (5) and lysosomal granule release. At this stage, collateral damage to healthy host tissue may occur since the neutrophil degranulation response is not confined (6). An acute inflammatory response is deemed successful if it results in the elimination of noxious agents as well as inflammatory cells with a return of tissue homeostasis.
Apart from pathogens, foreign bodies and autoimmune responses may also be a cause for the development of an inflammatory response. Unsuccessful attempts to eliminate foreign bodies or pathogens results in granuloma formation, which is basically an attempt by macrophages to “wall-off” the noxious agent to protect the host (2, 3).
Natural resolution of inflammation and specialized pro-resolving mediators
The acute inflammatory response may be divided into activation and resolution phases. Over time, evidence has accrued to suggest that the latter is not merely a passive process of chemoattractant and chemokine dilution which “fizzles out” after a while, but an active, programmed host response that “switches on” as the inflammatory lesion matures (7) that is mediated by a new class of eicosanoids collectively termed Specialized Proresolving Mediators (SPMs) (8–14). The discovery of this family of compounds introduced a fresh understanding of the inflammatory process, its development and indeed its resolution. Naturally, this led to a re-examination of previously established concepts surrounding disease etiology and treatment approaches. Building on previous knowledge, it is understood that the in vivo production of a mediator needs to be adequate to incite bioactions (11). The SPMs include resolvins, maresins, protectins and lipoxins that are derived from dietary Omega-3 polyunsaturated fatty acids (n-3 PUFA) or in the case of lipoxins, from endogenous arachidonic acid from cell membranes (15). Hence the concept of “good and laudable pus” which acknowledged the fact that pus contained the means to produce the resolution of inflammation. This family of compounds play an essential role in host defense by virtue of initiating the trafficking of leukocytes (16), a process known to be integral to the inflammatory response.
Upon challenge by a noxious stimulus, changes in blood flow, influx of neutrophils and edema are stimulated by prostaglandins and leukotrienes (17, 18). These processes manifest at a macro-level as the cardinal signs of inflammation. As the host inflammatory response builds up to this challenge, the pro-resolving mediators, maresins, protectins, resolvins and lipoxins along with their respective aspirin triggered (AT) variants undergo synthesis during the active phase of resolution (19, 20). It is imperative to recognize the synergy between the upregulation of pro-inflammatory elements of the response and the development of the resolution response itself.
The acute phase of inflammation may involve a microbial stimulus or one that is elicited because of the host's immune response to other forms of injury or insult. This is followed by local PUFA release from phospholipids found in cell membranes or their delivery to inflammatory sites by means of edema which would further result in the conversion of these PUFAs to specialized mediators (21).
Eicosanoids are produced from arachidonic acid within minutes of the initial insult (Figure 1). It is these leukotrienes and prostaglandins that signal neutrophils to arrive at the relevant site. Neutrophil availability is a function of LTD4 and LTC4 which impact vascular permeability, along with PGI2 and PGE2 which then affect blood flow (6, 22). As these products regulate neutrophil presence at the site of inflammation, the latter in turn tend to follow a transmigration gradient dictated by LTB4 (23). Once it is ensured that the right cells (neutrophils) are at the right place, neutrophil induction is mediated by these eicosanoids along with other immune system components such as the complement system, chemokines and cytokines (24, 25). It is relevant to understand these early events in the inflammatory phase since they are responsible for the subsequent lipid class-switching that occurs when the arachidonic acid metabolic system changes from leukotriene production to that of pro-resolving mediators such as lipoxins (26). If unencumbered, this process results in the natural resolution of inflammation wherein the acute phase proceeds to resolution (Figure 2).
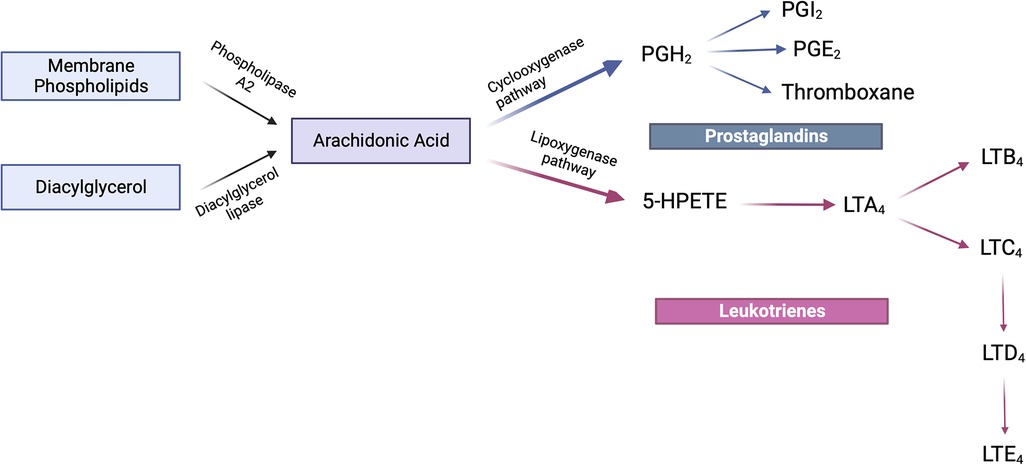
Figure 1. Arachidonic acid metabolism. Arachidonic acid is synthesized either from the action of phospholipase A2 upon membrane phospholipids or the diacylglycerol lipase-mediated conversion of diacylglycerol. The fate of this product is further determined by either the cyclooxygenase or lipoxygenase pathways. The cyclooxygenase-mediated pathway converts arachidonic acid to PGH2 which further elaborates the prostaglandins PGI2, PGE2 and thromboxanes. The lipoxygenase pathway leads to the formation of the intermediate 5-HPETE which is subsequently converted to LTA4. It is from LTA4 that LTB4 and LTC4 are formed of which the latter is converted to LTD4 which in turn forms LTE4. This family of compounds is termed leukotrienes. Created with BioRender.com.
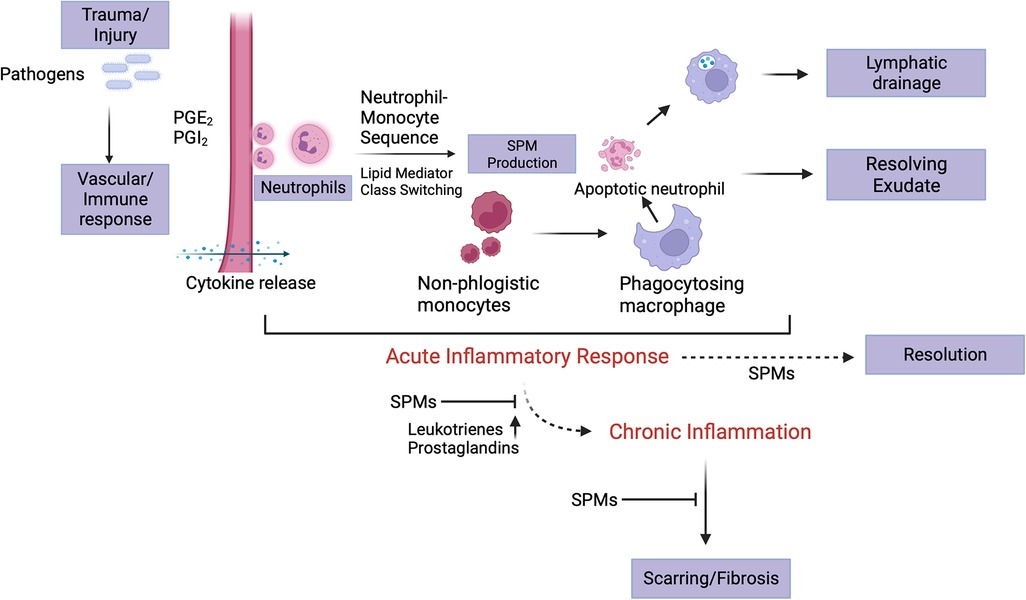
Figure 2. Acute inflammatory stage and its resolution. Eicosanoids play a crucial role in the initiation of inflammation subsequent to insult, which is manifest as the cardinal signs of inflammation. At the site of injury, trafficking of leukocytes forms part of the initial vascular response to the insult. Neutrophil migration is stimulated by LTB4, PGE2 and PGI2, the former being responsible for chemotaxis and adhesion while the latter two for vasodilation. An end to the acute phase of the inflammatory response is initiated by neutrophil-monocyte interaction that leads to a “class switch” in the lipid mediators (from eicosanoids to lipoxins). These lipoxins and then, resolvins signal non-phlogistic monocyte recruitment. The apoptotic neutrophils (“eat me” signals’) are subsequently cleared by the “big eater” macrophages in an SPM-mediated process termed efferocytosis. Once completed, this process paves the way for establishing homoeostasis. A failure in resolution may be marked by elevated levels of leukotrienes and prostaglandins, fibrosis and progression into the chronic phase. Created with BioRender.com.
The resolution response tends to get delayed in the event of an interruption with lipoxin production or of the availability of receptors for this family of compounds (27, 28). It is the acute inflammatory response that signals the development of the resolution phase. Essential fatty acids in the diet such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) supplement the endogenous arachidonic acid-based system where these n-3 PUFA have been demonstrated to enzymatically (LOX-dependent) produce SPMs (Figure 3).
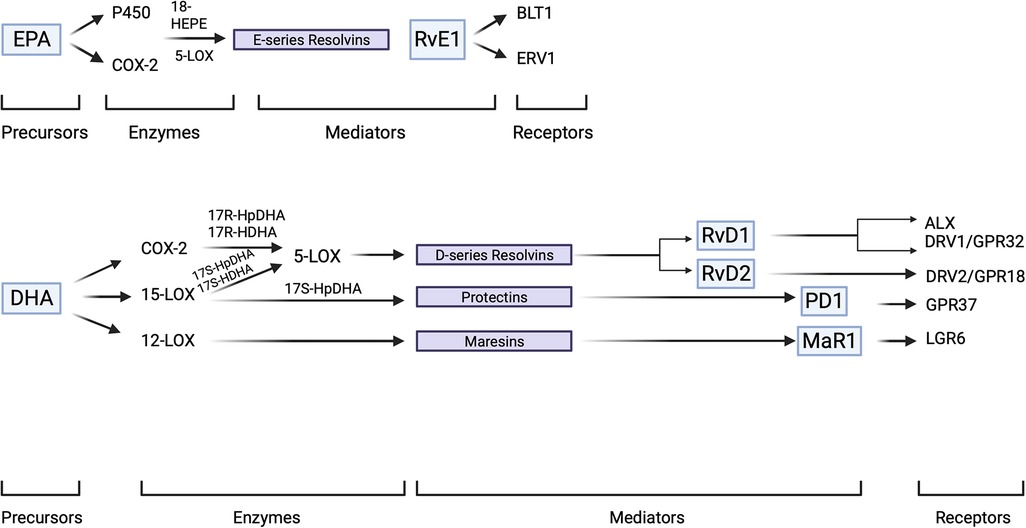
Figure 3. SPM overview. SPM precursors, EPA and DHA are enzymatically converted to SPMs which subsequently interact with their specific receptors to elaborate innate immune functions of a pro-resolving character. SPMs demonstrate stereoselectivity in their interaction with the corresponding GPCRs which result in the activation of intra-cellular signaling pathways. Created with BioRender.com.
Thus, the production of these stereoselective products has been demonstrated via multiple pathways, which can be broadly categorized as intrinsic and extrinsic. The former includes SPM synthesis within the host in the absence of an external stimulant whereas the latter necessitates an external influence to the process. The intrinsic mechanisms for SPM production are varied based on enzyme mediators. One of these involves native enzymes, such as 12-LOX (platelet-derived) and 5-LOX (leukocyte-derived or 15-LOX (eosinophil-, monocyte- or epithelial cell-derived) and 5-LOX (leukocyte-derived) to produce lipoxins (21, 29, 30).
The extrinsic mechanism involves the modification of native enzymes by other compounds, such as aspirin. Even though aspirin inhibits the production of prostaglandins, it does so through a unique mechanism that involves modification of COX2 to produce a new active enzyme; not merely blocking the activity of COX2. It does this through acetylation of COX2 yielding a product that is an active 15R-lipoxygenase (31). When arachidonic acid or n-3 PUFA interact with this new lipoxygenase followed by 5-LO actions, the product is a longer acting epimer of lipoxins called aspirin triggered or epi-lipoxins. Statins have a similar property wherein they nitrosylate COX2 yielding a 15R-LO product (31). Intrinsically, cytochrome P450 enzyme complex may also produce 15R-HETE in the absence of a stimulus from aspirin (30–32). SPMs are present in exudates. Resolving exudates are also comprised of n-3 fatty acid derived pro-resolving mediators, including protectins, resolvins and maresins.
Resolvins of the E- and D-series are derived enzymatically from EPA and DHA, respectively, via intrinsic mechanisms whereas, COX-2 and LOX activities acetylated by aspirin also lead to generation of aspirin triggered epimers in a fashion similar to 15-epi-lipoxins (10).
The autacoids produced from EPA via these enzymatic processes are termed E-series resolvins (RvE). Similarly, the derivatives of DHA were termed D-series resolvins (RvD). Another family of chemically active products derived from DHA and characterized by the presence of a conjugated triene double-bond are named protectins. The nomenclature of resolvins was adopted to signify their potent immunoregulatory and anti-inflammatory actions (10). Protectins or neuroprotectins (protectins derived from neural tissues) were named to signify the protective character of 10,17-docosatriene and its anti-inflammatory nature (11, 33). EPA derived compounds such as 18R-HEPA and 18R-HEPE have also been demonstrated to be present in resolving exudates (8). Transcellular synthesis of RvE1 in the vasculature, in the presence of aspirin, resulted in the formation of 18R-HEPE from EPA mediated by endothelial cell derived COX2 and further, leukocyte derived 5-LOX mediated RvE1 formation utilizing 18R-HEPE as the substrate (8, 34). The two major resolvin series derived from DHA include the RvD1-6 and their aspirin triggered isomers (AT-RvD1-6) (35).
17S-hydroperoxy-DHA derived from DHA through an enzymatic reaction mediated by 15-LOX and later by 5-LOX leads to the formation of the D-series of resolvins. In the case of their aspirin triggered epimers, 17R-HpDHA is derived from DHA through a mechanism mediated by COX-2 acetylated by aspirin also subsequently acts as a 5-LOX substrate for an eventual epimeric variant transformation.
Not just resolvins and protectins but maresins are also groups of additional compounds derived from DHA that have been observed in resolving exudates and exhibit pro-resolving and protective activities (11, 12). This is evidenced by the fact that 15-LOX mediated formation of protectin D1 from 17S-HpDHA and 12-LOX mediated maresin 1 (MaR1) formation from 14S-HpDHA have been observed at sites of inflammation (31, 36).
It is essential to understand how significantly the process of resolution differs from that of inhibition. Traditional therapeutic methodologies have focused on anti-inflammatory therapies, which are based on the concept of inhibiting the proinflammatory process. However, in doing so, they also cut-off the signals necessary to initiate and sustain the process of resolution. This is a critical caveat since the process of resolution is central to the favorable outcome of the inflammation cascade, since it reverses the inflammatory process and signals a return to homoeostasis and restoration of cellular structure. While anti-inflammatory strategies act to block enzymes or antagonize relevant receptors, the SPMs exert their activity in an agonistic manner through their interaction with their corresponding receptors. A G-protein coupled receptor, FPR2/ALX binds 15-epi-LXA4 and LXA4 with significant affinity. Other receptors for SPMs have also been identified such as GPR32 for LXA4 chemR23/CMKLR1/ERV1, BLT1 for RvE1, GPR32/DRV1, ALX/FPR2 for RvD1, GPR18/DRV2 for RvD2, GPR32 for RvD3, GPR37 for PD1 and LGR6 for MaR1 (37–39).
There appears to be an overlap amongst some of the receptors in terms of binding compounds from the SPM family. A notable finding has been that of RvD1 exhibiting equipotent activity to LXA4 upon binding with ALX (37). Further, AT-RvD1, RvD3 and RvD1 all demonstrated high affinity binding to GPR32 (37). The understanding of these pathways is significant since disruption leads to impaired resolution that, in turn, leads an acute inflammatory lesion to progress into a chronic state (27, 40). This disruption in the resolution response may occur as a result of inadequacies in enzyme synthesis, receptor expression, essential PUFA deficiency or intra-cellular signaling. Even though the families of these immunoresolvents are structurally distinct, it is their common functional role in the resolution of inflammation that unites them as SPMs. Studies from experimental models have demonstrated these limit damage to tissues, promote healing, control the inflammatory response, decrease the interval to resolution and alleviate pain. These compounds do not act alone but are components of a larger resolution response of the body, which includes cytokines, annexin A1 protein, carbon monoxide and microRNAs and cyclin dependent kinase inhibitors (9, 41–44).
It was over a century ago that Metchnikoff noted that macrophagic clearance of neutrophils tended to resolve inflammation (16). As subsequent research identified the initiators of leukocyte recruitment to sites of insult, the thought process which evolved was to either block these signals or remove the initiators of inflammation in order to prevent inflammation. It was believed that the chemoattractants so involved, gradually undergo dilution and dissipation in a passive manner to end the inflammatory phase. It appears that the fundamental processes surrounding the resolution of inflammation, such as removal of apoptotic PMNs (polymorphonuclear neutrophils) and cessation of tissue-entry of PMNs are impacted by SPMs. This has been demonstrated in studies which have observed in resolving exudates, that cellular interactions expound signals to impair the influx of neutrophils and upregulate macrophagic clearance of these as well, both considered key processes for the resolution of inflammation.
It has been demonstrated that prostaglandins participate in the initial stages of inflammation impacting the production of pro-resolving mediators by stimulating mRNA translation for the responsible enzymes (26). This essentially is a “class-switching” of the lipid mediators to go from ones which are pro-inflammatory to ones which are pro-resolving in action. Temporal analyses of exudates point towards the biosynthesis of lipoxins subsequent to an early appearance of prostaglandins and leukotrienes. The appearance of lipoxins has been correlated with the resolution phase of inflammation (45). Macrophages of the M1-like and M2-like type have been observed to exhibit different responses to stimulation by pathogens and pharmacological inhibitors. The difference lies in the distinction between the lipid mediator profiles that are produced, which further characterizes the pro-resolving and inflammatory cell phenotypes. PGE2 and LTB4 are produced by M1-like macrophages and a simultaneous stimulation of the M2-like phenotype as a result of a challenge from S. aureus and E. coli (11, 46). From this, one may appreciate the significance of the lipid mediator class switch, which appears to impact the inflammatory cascade at numerous levels. It has also been observed that an inhibitor of microsomal prostaglandin E2 synthase-1 and 5-LOX selectively downregulates PG and LT production in the M1-like phenotype but enhances maresin and resolvin production in the M2-like variant (47).
Further credence to the extrinsic mechanism of SPM production can be evidenced by the observation that Resolvin E2, another bioactive member was identified when the common E-series precursor 18-HEPE was subjected to human recombinant 5-LOX (48). In general, SPMs being agonists have the capacity to signal important events in the resolution phase such as stimulating the clearance of apoptotic cells by macrophages and downregulating neutrophil infiltration (15). The pro-resolution and anti-inflammatory processes are not alike. The former involves agonists (SPMs) to halt the influx of neutrophils and stimulate non-phlogistic macrophage responses, these two cardinal features of resolution lead to a restoration of homoeostasis (49).
Every SPM has characteristic proresolving activity that is of significant importance to the resolution of inflammation. These include stopping or limiting neutrophil influx, cytokine/chemokine counter regulation, pain reduction and stimulation of nonphlogistic macrophage activity (50). SPMs act as agonists binding to receptors on multiple targets (neutrophils, macrophages, stromal cells) individually in order to bring about resolution. SPM production in humans is temporal with SPMs acting selectively by signaling resolution to control inflammation without being immunosuppressive (50, 51). In periodontal, kidney, eye and lung diseases, their actions have been demonstrated to be organ protective (50, 51).
SPMs, diurnal variation, and hormonal influence
SPM production demonstrates a diurnal pattern of regulation. A central mechanism requisite for the maintenance of resolution at the tissue level has been elucidated to involve the biosynthesis of pro-resolving mediators by acetylcholine (Ach) release. Ach levels have been demonstrated to be high in the early hours of the day in healthy volunteers. Blood from these volunteers when incubated with Ach demonstrated an upregulation of RvDn-3 DPA (52). Reduced plasma levels of RvDn-3 DPA in the early morning hours, essentially a loss of the diurnal regulation mechanism, has been postulated to contribute to the onset and propagation of cardiovascular disease (CVD) (52).
Uncovering the dysregulated circadian rhythmic production of pro-resolving mediators in various disease processes may aid in developing therapeutic strategies, especially in light of studies that have demonstrated the circadian rhythm regulated production of IL-6, TNF-alpha and Ly6chi monocytes (53, 54).
The role of hormonal regulation of SPMs has been demonstrated in a selective and rapid upregulation of SPMs characterized by a typical profile including RvD1 and RvD2 observed in mice administered parathyroid hormone (PTH), which triggers a pro-resolving circuit in the backdrop of bone remodeling (55).
Androgen DHT and estrogen have been demonstrated to downregulate the action of the pro-inflammatory LTB4. Indeed, a sex-based difference has been observed in the reaction of cultured conjunctival goblet cells to LXA4, with human male cells demonstrating a greater Ca2+ response (56). Similarly, in human tear samples, it was found that males demonstrated higher levels of RvD1 and RvD2 (57).
These findings suggest that future studies should further evaluate sex-based differences in SPM production and response also controlling for time of day.
The role of SPMs in infectious diseases
The inflammatory process has time and again been demonstrated to be a key underlying factor in the pathophysiology of numerous infectious disease processes. Naturally, a family of compounds that act to resolve this inflammation would potentially be of immense benefit in not only understanding the disease but in therapeutics as well. Recently, scientific literature has discussed the role SPMs may play in host modulation for several infectious diseases, which provides the scope for exploring a potentially novel therapeutic modality.
Inflammation has a central role to play in pneumonia, a highly prevalent disease of concern both at the community- and hospital acquired-levels. Typically, pneumonia elicits an acute response which is self-limiting in nature. In certain cases, this may progress into acute respiratory distress syndrome (ARDS) which is characterized by respiratory failure and hypoxemia (58). In cases of pneumonia induced by Escherichia coli, Lipoxin A4 (LXA4) has been observed to stimulate BCL-2-associated death promoter (BAD) phosphorylation and decrease the expression of myeloid cell leukemia sequence 1 (MCL1), which causes neutrophil apoptosis (58). RvE1 has also been observed to stimulate neutrophil apoptosis but via a different mechanism of activating caspases (58). In both the above situations, the severity associated with lung inflammation in its acute phase is blunted as a result of neutrophil death. Additionally, in a mouse model of pneumonia induced by E. coli, RvE1 has been demonstrated to decrease local pro-inflammatory cytokine production and promote bacterial clearance (59). It was shown that the actions of SPMs aid in E. coli clearance and the decreased levels of host inflammation, which enhanced survival (59). It is quite apparent that resolving the inflammatory process provided the added advantage of microbial clearance, which tends to act as the initiator in a number of disease processes.
Another infectious disease of bacterial etiology (but tick vector), Lyme disease, involves an inflammatory component that manifests after exposure to the initial etiological agent, which microbial in nature. Mouse models of Lyme disease have helped elucidate the systemic and local control of inflammation (60). In mice with an impaired SPM production mechanism, i.e., those deficient in 5-LOX, infection with Borrelia burgdorferi caused a similar pattern of arthritis development as it did in wild type counterparts, the key feature, however, was that the lack of resolvins and lipoxins impaired the ability of the host to resolve arthritis, giving rise to chronic disease, the inflammatory response of which persisted long after the causative agent was cleared (60).
Tuberculosis, in both its typical pulmonary and extra-pulmonary forms, is a disease of concern particularly in the developing world. In a pattern similar to other infectious diseases, it involves both a microbial and inflammatory component. The host response to Mycobacterium tuberculosis involves a balance between pro-inflammatory and pro-resolving mediators, which determines microbial clearance as well as pathogen induced inflammation (61). In a mouse model, it has been demonstrated that infection with Mycobacterium tuberculosis led to an increase in the levels of LTB4 (pro-inflammatory) and LXA4 (pro-resolving) with high levels of the latter being maintained during the chronic phase of the infection (61). Further, 5-LOX deficient animals have demonstrated increased survival with M. tuberculosis infections (61). This demonstrates the importance of the inflammatory response in determining disease outcomes. As was discussed previously, it is the inflammatory response which begets the generation of the resolution component. The M. tuberculosis strain has been related to lipoxin generation in the host which points towards SPMs effecting host modulation of the inflammatory response in tuberculosis infections (62). Either LXA4 or LTB4, if produced in excess, may result in a skewed response of the host to M. tuberculosis infection, which related to a dysregulation in the expression of tumor necrosis factor (TNF) (62).
Hence, both the pro-resolution and pro-inflammatory pathways seem to be important in determining host defense and outcomes of pathogen caused inflammation (63). It would appear that a therapeutic modality involving antibiotics to clear the pathogen along with SPMs to manage the immune response would be effective. This is further given credence by the finding that varying rates of infection were observed for human variants for the LTA4H and ALOX5 loci, both of which interfere with LXA4 and LTB4 production (64, 65).
Perhaps the most serious complication arising from a bacterial infection in its acute phase is that of sepsis. The host response in such cases tends to involve a rapidly progressing system-wide immune dysregulation which may result in shock (66). In a mouse cecal ligation puncture model, treatment of sepsis with LXA4 downregulated pro-inflammatory cytokine production and reduced gram-negative bacterial numbers, which resulted in improved survival (66, 67). RvD2, by means of downregulating the activity of nuclear factor-κB (NF-κB), regulates pro-inflammatory signals and thereby the overall inflammatory response at the systemic level (50, 59, 66, 67). As was demonstrated in a mouse model of sepsis, administration of RvD2 led to a significant decrease in cytokine production, particularly that of IL-10, IL-6 and interferon-α (IFN-α) (43). There was also reduced infiltration of leukocytes at the infection site (50). It is interesting to note that controlling the inflammatory response in such a manner brings about an overall reduction in bacterial counts both at the systemic and local levels along with an improvement in the overall survival of these animals (50).
Another consideration is one involving the timing of SPM administration. It was demonstrated in a mouse pneumosepsis model that treatment with LXA4 in the early stages limited the immune response since it reduced leukocyte infiltration. It was also noted that these mice had a deterioration in their survival rate along with impaired bacterial clearance (68). On the contrary, when LXA4 was utilized in the later stages of treatment, it demonstrated certain positive effects such as satisfactory infection clearance while blunting the protraction of the inflammatory response and thereby improving survival (69). By means of their activity affecting bacterial clearance and downregulation of the pathological inflammatory response, SPMs have been demonstrated to decrease antibiotic dosage requirements for clearing bacterial infections (67, 69). This finding assumes significance in the face of the growing global threat of antibiotic resistance.
SPMs also regulate the mechanisms by which certain viral pathogens interact with the host. In a comparison between influenza strains of varying virulence, it has been observed that the more virulent strains cause greater suppression of lipoxins and hence, an enhanced dissemination of the virus within the host (70). Protectin D1 levels have been observed to be downregulated by highly virulent influenza strains such as H5N1, which reinforces the inverse relationship between SPM activity and strain virulence (71). Apart from modifying the host immune response, protectin D1 and the isomeric protectin DX limit viral replication by scrambling the nuclear export mechanism (11, 45, 46). In fact, protectin D1 has been observed to exhibit better clinical outcomes in cases of influenza virus infections (72). Infected mice, treated with protectin D1 even as late as 48 h into the infection, reported an improvement in survival (72). This finding appears to be important when contemporary antiviral therapies seem to be declining in effectiveness (73). Infection with the respiratory syncytial virus (RSV), another respiratory pathogen of considerable relevance, is characterized by macrophage-driven bronchiolitis which is subsequently resolved by their alternatively activated counterparts (74). This may be explained by the fact that alternatively fated macrophages appear to have a relation to RSV mediated COX-2, RvE1 and LXA4 related protective action (74, 75).
Herpes Simplex Virus (HSV) infections tend to be particularly distressful and recurrent in their presentation with the potential to progress into states of morbidity. Ocular HSV infection presents a useful model to demonstrate control of the virus at the local level via a strong inflammatory response, the chronicity of which may have significant morbidity. In mice with ocular herpes, it was observed that treatment with topical RvE1 was associated with a reduced number of neutrophils, Th17 and Th1 cells in the cornea (50). The mechanisms for this were hypothesized to be multiple, including a downregulation of CD4+ T cells and neutrophil influx, increased anti-inflammatory mediator production and reduced production of pro-inflammatory mediators (76). One study in particular demonstrated that RvE1 may be utilized to regulate severity of lesions in immunopathologies of a viral origin (75). This essentially provides evidence of SPMs regulating the immune response initially elicited by microbial stimulus. The timely involvement of SPMs in the inflammatory cascade is crucial to the outcome of the inflammatory response.
Based on findings from a mouse model it has been suggested that lipoxin induction in response to stimulation by Toxoplasma gondii extract serves as a potent pathway for regulating the function of dendritic cells during the host innate immune response (77). Wild type mice exposed to T. gondii infection produced LXA4 during the early stages of the chronic infection, whereas mice deficient in 5-Lipooxygenase [5-LO(−/−)] exhibited shorter survival during the same time frame characterized by encephalitis (78). The mortality of these mice was attributable to greater levels of IFN-gamma and IL-12 and was prevented altogether by treatment with a stable analogue of LXA4 (78), again evidencing the essential role played by SPMs in determining the outcome of inflammation. Lipoxins have an alleged protective role in other parasitic infections such as those involving Plasmodium species, Angiostrongylus costaricensis and Trypanosoma cruzi (79–81).
Specialized proresolving mediators (SPMs) and the return to tissue homeostasis
Class-switching of lipid mediators from pro-inflammatory to anti-inflammatory or pro-resolution is imperative for initiation of the resolution phase of acute inflammation. This class-switch from prostaglandins and leukotrienes to pro-resolution elements leads to an inhibition of neutrophil recruitment in favor of monocytes, which remove cellular debris and set the stage for tissue remodeling (82). If the acute phase fails to enter the resolution phase, due to failure of production of adequate proresolving mediators or failure to eliminate the noxious stimulus, the inflammatory response becomes chronic and is characterized by persistence of inflammatory macrophages and T cells, which involves tertiary lymphoid tissue and granuloma formation (3, 51).
The inflammatory process and production of relevant mediators is temporal. Thus, along with the production of mediators to promote proinflammatory processes, there is an inherent feature of local termination of inflammation. These resolution pathways are activated by the proinflammatory pathways. Prostaglandins activate transcription of enzymes (12- and 15-lipoxygenases) that are required to produce lipoxins, resolvins, protectins and maresins that promote resolution of inflammation through specific receptors. There are multiple receptors for the various SPMs that have been reported. This interaction has been described as the alpha signaling the omega or the beginning signaling the end to indicate that the class-switch that occurs to produce molecules responsible for the resolution phase is initiated by the pro-inflammatory response of the initial stages.
The characterization of these compounds has changed the understanding of resolution of inflammation from the earlier belief that resolution was a passive process mediated by decay of proinflammatory mediators to an understanding that SPMs actively promote a resolution response. These new lipid mediator pathways open the door for novel therapeutics, since these lipids can be produced by complete organic synthesis, are small molecules (below 500 Da) and can be manufactured utilizing contemporary pharmaceutical methods. Both animal and human studies have revealed SPMs to be of benefit in the resolution of the inflammatory response, which is characteristic of several infectious diseases as well as periodontal disease. It is already known that such disease processes involve a microbial etiology that initiates the lesion in a susceptible host. In this review, we will explore the relationship that resolution of inflammation has with dysbiosis, for it is an interplay of these processes that eventually leads to a favorable therapeutic outcome.
SPMs in periodontal disease
Inflammation and periodontal disease
The etiology of periodontal disease is multifactorial, being both host-specific and polymicrobial. While microbial challenge may be the initiator of the disease process, the pathogenesis is inflammatory in nature (83, 84). The American Academy of Periodontology considers periodontitis as an inflammatory disease with bacterial initiation (9). Even though this definition represents a paradigm shift in the understanding of the pathogenesis of periodontal disease, there remains a lack of clarity regarding the relationship shared by the disease itself and the inflammatory response in that, it remains unclear whether the immune response preceded the dysbiosis or vice versa (85). There is evidence in literature to suggest that host immune responses tend to shape microbial communities in biofilms on mucosal surfaces (67). However, the extent to which bacterial biofilms play a role in triggering human disease is still under active investigation and debate (86–88).
Mucosal inflammatory lesions have been evidenced to occur as a result of the presence of biofilms at numerous locations in both animals and humans (86, 89). At the same time, in entities such as chronic rhinosinusitis and inflammatory bowel disease, even though biofilms have been detected on mucosal surfaces, their formation may be attributable to a deranged immune response upon stimulus from antigens of a non-microbial origin (90, 91). Hence, it would appear rational to argue that an immune response to non-microbial antigens may produce conducive environments for biofilm development. It is already known that microbes within biofilms behave differently when compared to their planktonic counterparts. Cells within biofilms tend to possess greater resistance to host defense mechanisms on account of an increase in biomass which impedes phagocytosis and extra-cellular polysaccharides that act as a physical barrier to antibodies, immune and complement systems (86). The biofilm need not even be extremely pathogenic, but an ineffective and hence, “frustrated” host immune response may end up causing collateral damage to surrounding tissues (86).
The primary etiologic agent of periodontitis is recognized as an insult originating from a pathogenic biofilm, but a specific bacterium or group of bacteria is not established as causing periodontitis (92, 93). Cross-sectional studies have implicated the “red-complex” bacteria, i.e., Porphyromonas gingivalis (P.g.), Treponema denticola (T.d.) and Tannerella forsythia (T.f.) to be associated with the presence of periodontitis, the causality for this is yet to be established in longitudinal studies (72). It is however, known that in susceptible hosts, the immune response to biofilm leads to a loss of tissue as part of the pathogenesis of periodontal disease (92).
Traditionally, therapeutics have revolved around the biofilm spectrum of disease, generally aimed at removing the aetiologic factor. It has been acknowledged that the bacteria being dealt with are present in large numbers and are spread across a multitude of sites (94). Moreover, these exist as biofilm-based communities which aid in their defense against antimicrobials and host response (94). Additionally, the microorganisms possess a formidable potential for multiplication and attachment to both like and unlike surfaces (94). Naturally, these factors hinder traditional therapeutics involving biofilm removal strategies, an imperfect treatment modality. At best, traditional periodontal therapies provide some benefit impeding or ceasing the progress of periodontal disease (94). These methods are, however, not always successful and may require adjunctive or alternative therapeutic measures (94). Antimicrobial therapies for the treatment of periodontal disease may include physical debridement, ones aimed at killing or altering the metabolism of the microorganisms in question or ones which alter the environment in which these microorganisms exist (94). Treatment modalities based on mechanical methods require accessibility to the biofilms to be rendered effectively. Fortunately, dental biofilms are quite accessible and are amenable to attempts at mechanical debridement. Given their ability to resist antimicrobial agents and host response, it would be logical to initially attempt a physical means of control for these microorganisms.
No doubt, mechanical therapy is able to remove a vast majority of microorganisms, however, the enormous multiplicative potential of bacteria allows these numbers to return to roughly baseline numbers within 3 months of therapy (94). There is also evidence in literature to suggest a return to baseline total counts within 4–8 days of therapy (95, 96). Since there appears to be no singularly isolated microorganism identified as the etiological agent for periodontal disease and in light of the fact that traditional therapeutic measures fall short of providing satisfactory results, let alone a “cure”, it would be logical to examine the relationship between inflammation and dysbiosis.
Periodontitis entails a significant increase in pro-inflammatory mediators, particularly prostaglandin E2 (PGE2) and leukotriene B4 (LTB4), which are generally considered lipid mediators related to early stages of the host response (97). Gingival Crevicular Fluid (GCF) has been reported to have increased exudative flow rates in states of active inflammation and has also been utilized as an analyte of interest being representative of the local and systemic inflammatory response. Increased GCF levels of pro-inflammatory markers such as PGE2, in periodontal disease can be correlated with disease activity and severity along with being potentially utilized to predict future disease progression (97). Another example of the relationship between the immune and microbial spectrum of disease pathophysiology is provided by in vitro studies that have demonstrated an increase in COX-2 activity as a result of stimulation from periodontopathogens (98).
As has already been discussed, neutrophils act as the “first responders” to sites of inflammation and form a crucial component in determining the progress of the lesion. Initially, it was demonstrated that neutrophils isolated from patients with periodontal disease, particularly those with the localized aggressive variant (as classified at the time), produced greater levels of LXA4 when compared to controls (98). Subsequently, in vivo priming of neutrophils and increased LXA4 levels were demonstrated in patients with asthma, a non-infectious inflammatory condition (99). Similar outcomes were demonstrated in vitro via cytokine induction (100). From such data it was argued that lesions of chronic periodontal disease featured a hyperactive neutrophil population which resulted in activating lipoxin production pathways. It is interesting to note that either due to dysfunctional receptor binding or insufficient quantities of lipoxins, the inflammatory phase was not resolved if a natural production of resolvins was relied upon (101). That being said, adequate quantities of lipoxins were protective against tissue damage in periodontal disease induced by bacteria (101). This was evidenced in a rabbit model which revealed that rabbits overexpressing 15-LO had decreased recruitment of neutrophils and in turn, lower levels of potentially tissue harming enzymes being released (101).
Further evidence as to the role of lipoxins in protecting against damage caused by periodontal disease is provided from findings in patients of localized aggressive periodontitis in whom LO activity appeared to be somewhat aberrant in nature (98, 102). The platelets of such patients featured an increase in the surface expression of P-selectin while the monocytes and neutrophils expressed an upregulation in CD18 expression (102). This was correlated with greater platelet-monocyte and platelet-neutrophils aggregations in the whole blood of these patients (102). These findings point towards the role of SPMs in protecting the host against bacterial-induced periodontal disease.
The pathogenesis of periodontitis may be regarded as being a result of the robust and protracted host immune response to a microbial challenge presented by organisms such as P. gingivalis and Actinobacillus spp. (103). The control of periodontal disease also attenuates the immune response to disease at a systemic level, which is characterized by blunted systemic inflammation as represented by reduced CRP levels and reduced interactions between neutrophils and platelets (103–105). It has been demonstrated in a rabbit model of ligature and P. gingivalis induced periodontal disease, that topical application of RvE1 to control inflammation leads to a reversal of changes in the biofilm associated with disease and essentially constituted a resolution of dysbiosis with P. gingivalis elimination (103). SPM usage does not have an identifiable side effect profile and long term, or recurrent usage does not increase susceptibility to infection. Since SPMs function to resolve the acute phase of inflammation, their utilization as therapeutic agents in the initial or early stages of periodontal disease provides a benefit (104).
Topical treatment with RvE1 has been demonstrated to sharply diminish the inflammation related to increased activation of neutrophils in the periodontium, elucidating a role for this class of lipid mediators to modify in vivo, local inflammatory processes presenting as a result of microbial challenge (106). RvE1 was also demonstrated to directly inhibit osteoclast induced resorption of bone, which was expressed as the ability of RvE1 treatment to prevent the appearance of TRAP+ cells in the bone (106). These findings demonstrate a more favorable outcome of inflammation when pro-resolution treatment strategies were employed instead of the traditional anti-inflammation regime.
Inflammation and dysbiosis
Inflammation was initially described as possessing certain cardinal signs. These may be considered the result of endogenous and exogenous chemical mediators released as a result of insult. Microbial peptides may act as exogenous mediators which serve as chemoattractants to signal neutrophil infiltration at the site of insult, where these may act by phagocytosing cell debris and microorganisms (107). Phagosomes within neutrophils fuse with lysosomal granules to mature into phagolysosomes that contain enzymes and reactive oxygen species to process the entrapped debris and microorganisms (107). At this stage, the initial inflammatory response can be viewed as one which is protective in nature and its timely resolution would ensure that it is self-limiting as well (107).
A failure of timely resolution of inflammation due to any number of factors would generally mean that the acute inflammatory episode would progress into a more protracted phase of chronic inflammation (108). This is likely to happen in situations where biofilms are involved wherein the microbial community is able to mount a sufficient defense against the host response leading to collateral damage (108). A great deal of theoretical knowledge surrounding the concept of dysbiosis in periodontal disease is based out of experience with research of the gut microbiome. With the complexity of biofilm structure and the potential multitude of host responses it may elicit at various levels, it would not be completely unfair to hypothesize that no one particular microorganism or “keystone” pathogen would be responsible for the initiation of disease. Indeed, there is no such evidence in literature to support such a finding (108). This adds further credence to the role of the inflammatory process in the pathogenesis of periodontal disease. Further, it is recognized that bacteria involved in disease initiation are generally commensal in nature and the so called periodontopathogens form a very minor portion of the biofilm in its early stages (109).
The ecological plaque hypothesis was the first to recognize that dysbiosis of the microbiome would be the result of a persistent and excessive inflammatory response along with pocket formation, which essentially alters the environment in which microorganisms grow by exerting selective pressure upon other specific microorganisms (110). The microbial community pertaining to the periodontium remains generally stable in a state of health while maintaining a dynamic equilibrium (108). Gingivitis may be considered as representative of homoeostasis in that it tends to remain a stable inflammatory lesion (108). It is a failure of timely resolution of inflammation that slides into a chronic phase leading to a loss of tissue (108). Bacteria associated with periodontal disease increase in significant numbers with the progress of the lesion (108). Similarly, a shift to healthy state also involves a change in the composition of the bacterial community (108, 111, 112).
While there is no doubt that polymicrobial insult causes gingivitis, whether the disease progresses is determined by the host response to this insult. This assumes particular importance when one considers the wealth of evidence implicating the host response as the factor responsible for driving tissue destruction (113, 114). Recent literature has suggested that the host immune response may dictate biofilm composition (103). Essentially, this implies that the inflammatory response in turn causes an alteration of the biofilm microenvironment and leads to specific selection of microorganisms. From this, one could infer that inflammation in the periodontal pocket would precede the overgrowth of certain periodontopathogens such as P. gingivalis and Tannerella forsythia at this site (115). Naturally, the initiating factor of disease falls into question. Microbial diversity appears to be stable in both health and severe forms of periodontal disease, whereas it is in shallow pockets that a greater diversity of microorganisms has been observed. A longitudinal study by Tanner et al. demonstrated that there appeared to be no particular microorganisms that preceded the occurrence of attachment loss. They found that gingival inflammation was the only reliable predictor of attachment loss (115).
Accumulating evidence of these concepts over the years has rendered the initial assumption of specific periodontopathogens causing disease as invalid. Coming back full circle to the ecological plaque hypothesis, one may view periodontal disease pathogenesis as the result of plaque accumulation which elicits an inflammatory response that in turn alters the microenvironment of the biofilm, specifically selecting for certain “pathogens” to overgrow, which further magnify the inflammatory response and enter a cyclical process of disease progression. Indeed, it has been demonstrated in a rabbit model that the progression and even onset of P.g induced periodontitis was prevented by Resolvin E1 (RvE1) by greater than 95% (106).
An overview of SPMs, their precursors, target cells and cell-specific activity is provided (Table 1).
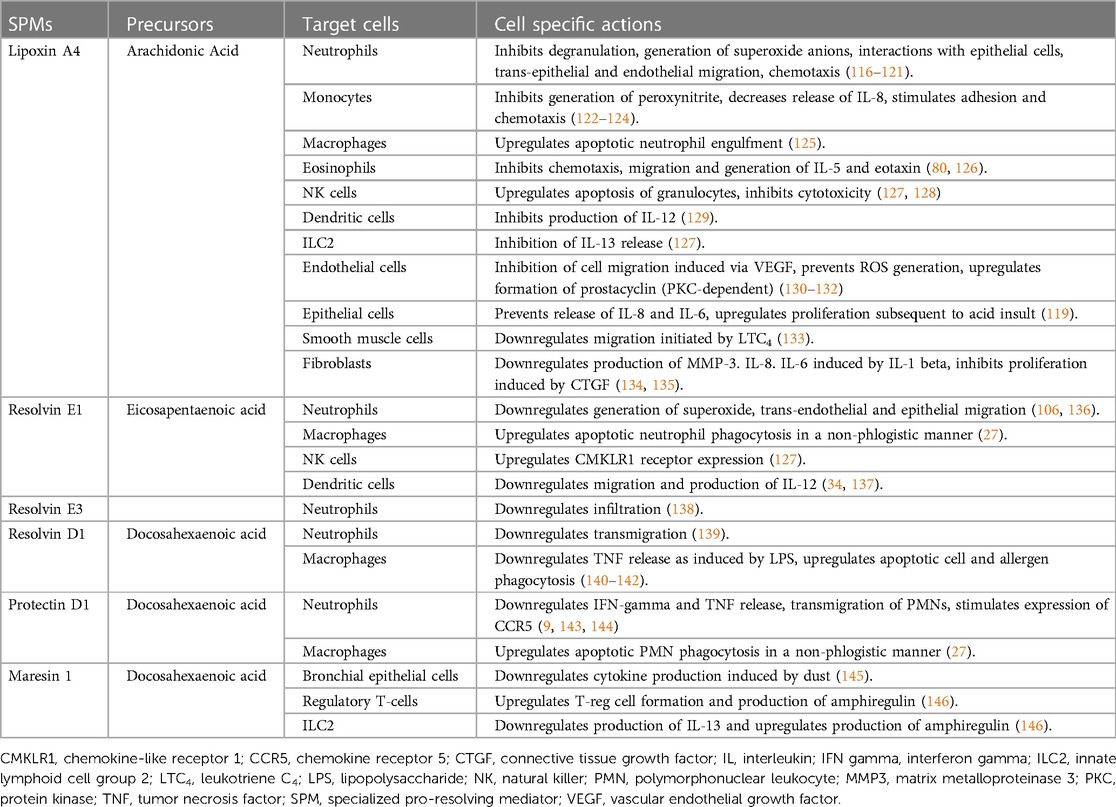
Table 1. An overview of SPMs, their precursors, target cells and cell-specific activity.
Conclusion
In summary, it would appear that SPMs do not act alone via a purely immunological pathway but exert effects on dysbiosis as well. This assumes significant importance in terms of our understanding of periodontal disease, its etiopathology as well as treatment. It would be logical to argue that periodontal disease must be treated as an immunological imbalance created as a result of microbial insult and not just an isolated case of the latter infecting host tissues to cause the disease. This is evidenced quite clearly in the increased diversity and microbial load in periodontal disease when compared to exogenous infections. Clearly, there appears to be immunological factors at play which modify the bacterial diversity and numbers in periodontal disease. With such an understanding, it would be more appropriate to state that Periodontitis is an inflammatory disease initiated by bacteria.
Author contributions
VS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review and editing. TD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
Supported in part by NIH, NIDCR grants DE025020 and DE032406.
Conflict of interest
TD is a named as inventor on Forsyth owned patents that have been licensed for development. He is a founder of Nocendra, Inc., a Forsyth spin out company developing SPMs for treatment of periodontitis.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. (2010) 140:771–6. doi: 10.1016/j.cell.2010.03.006
3. Kumar V, Cotran RS, Robbins SL. Robbins basic pathology. London, United Kingdom: Saunders (2003).
4. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
5. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
7. Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. (2003) 339:166–72. doi: 10.1126/science.1230720
8. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with anti-inflammatory actions generated from omega-3 fatty acids via cyclooxygenase2-nonsteroidal anti-inflammatory drugs and transcellular processing. J Exp Med. (2002) 192:1197–204. doi: 10.1084/jem.192.8.1197
9. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. (2005) 174:4345–55. doi: 10.4049/jimmunol.174.7.4345
10. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. (2002) 196:1025–37. doi: 10.1084/jem.20020760
11. HongS GK, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. (2003) 278:14677–87. doi: 10.1074/jbc.M300218200
12. Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. (2009) 206:15–23. doi: 10.1084/jem.20081880
13. Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med. (2015) 21:1071–5. doi: 10.1038/nm.3911
14. Dalli J, Colas RA, Serhan CN. Novel n-3 immunoresolvents: structures and actions. Sci Rep. (2013) 3:1940. doi: 10.1038/srep01940
15. Maderna P, Godson C. Lipoxins: resolutionary road. Br J Pharmacol. (2009) 158:947–59. doi: 10.1111/j.1476-5381.2009.00386.x
16. Cotran RS, Kumar V, Collins T. Robbins pathologic basis of disease. Philadelphia: W.B. Saunders (1999).
17. Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. (1987) 237:1171–6. doi: 10.1126/science.2820055
18. Samuelsson B. Role of basic science in the development of new medicines: examples from the eicosanoid field. J Biol Chem. (2012) 287:10070–80. doi: 10.1074/jbc.X112.351437
19. Serhan CN. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol. (2004) 122:305–21. doi: 10.1007/s00418-004-0695-8
20. Serhan CN, Chiang N. Resolution phase lipid mediators of inflammation: agonists of resolution. Curr Opin Pharmacol. (2013) 13:632–40. doi: 10.1016/j.coph.2013.05.012
21. Serhan CN, Sheppard KA. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J Clin Invest. (1990) 85:772–80. doi: 10.1172/JCI114503
22. Badr KF, DeBoer DK, Schwartzberg M, Serhan CN. Lipoxin A4 antagonizes cellular and in vivo actions of leukotriene D4 in rat glomerular mesangial cells: evidence for competition at a common receptor. Proc Natl Acad Sci U S A. (1989) 86:3438–42. doi: 10.1073/pnas.86.9.3438
23. Malawista SE, de Boisfleury Chevance A, van Damme J, Serhan CN. Tonic inhibition of chemotaxis in human plasma. Proc Natl Acad Sci U S A. (2008) 105:17949–54. doi: 10.1073/pnas.0802572105
24. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. (2012) 11:633–52. doi: 10.1038/nrd3800
25. Mizgerd JP. Acute lower respiratory tract infection. New Engl J Med. (2008) 358:716–27. doi: 10.1056/NEJMra074111
26. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. (2001) 2:612–9. doi: 10.1038/89759
27. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation- resolution programmes. Nature. (2007) 447:869–74. doi: 10.1038/nature05877
28. Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. (2010) 184:2611–9. doi: 10.4049/jimmunol.0903526
29. Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J Clin Invest. (1993) 92:1572–9. doi: 10.1172/JCI116738
30. Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A. (1984) 81:5335–9. doi: 10.1073/pnas.81.17.5335
31. Chiang N, Takano T, Clish CB, Petasis NA, Tai HH, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (ATL) generation by human leukocytes and murine peritonitis exudates: development of a specific 15-epi-LXA4 ELISA. J Pharmacol Exp Ther. (1998) 287:779–90.9808710
32. Claria J, Lee MH, Serhan CN. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol Med. (1996) 2:583–96. doi: 10.1007/BF03401642
33. Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci U S A. (2004) 101:8491–96. doi: 10.1073/pnas.0402531101
34. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. (2005) 201:713–22. doi: 10.1084/jem.20042031
35. Serhan CN, Arita M, Hong S, Gotlinger K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids. (2004) 39:1125–32. doi: 10.1007/s11745-004-1339-7
36. Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. (2011) 111:5922–43. doi: 10.1021/cr100396c
37. Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee C, Yang R, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci U S A. (2010) 107:1660–5. doi: 10.1073/pnas.0907342107
38. Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med. (2015) 212:1203–17. doi: 10.1084/jem.20150225
39. Lavy M, Gauttier V, Poirier N, Barillé-Nion S, Blanquart C. Specialized pro-resolving mediators mitigate cancer-related inflammation: role of tumor-associated macrophages and therapeutic opportunities. Front Immunol. (2021) 12:702785. doi: 10.3389/fimmu.2021.702785
40. Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, et al. Defective lipoxin-mediated anti- inflammatory activity in the cystic fibrosis airway. Nat Immunol. (2004) 5:388–92. doi: 10.1038/ni1056
41. Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. (2009) 9:62–70. doi: 10.1038/nri2470
42. Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits. FASEB J. (2011) 25:544–60. doi: 10.1096/fj.10-169599
43. Shinohara M, Kibi M, Riley IR, Chiang N, Dalli J, Kraft BD, et al. Cell-cell interactions and bronchoconstrictor eicosanoid reduction with inhaled carbon monoxide and resolvin D1. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L746–57. doi: 10.1152/ajplung.00166.2014
44. Leitch AE, Cd L, Marwick JA, Duffin R, Haslett C, Rossi AG. Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ. (2012) 19:1950–61. doi: 10.1038/cdd.2012.80
45. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. (2018) 128:2657–69. doi: 10.1172/JCI97943
46. Werz O, Gerstmeier J, Libreros S, De la Rosa X, Werner M, Norris PC, et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun. (2018) 9:59. doi: 10.1038/s41467-017-02538-5
47. Cheung SY, Werner M, Esposito L, Troisi F, Cantone V, Liening S, et al. Discovery of a benzenesulfonamide-based dual inhibitor of microsomal prostaglandin E(2) synthase-1 and 5- lipoxygenase that favorably modulates lipid mediator biosynthesis in inflammation. Eur J Med Chem. (2018) 156:815–30. doi: 10.1016/j.ejmech.2018.07.031
48. Tjonahen E, Oh S, Siegelman J, Elangovan S, Percarpio K, Hong S, et al. Resolvin E2: identification and anti-inflammatory actions: pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem Biol. (2006) 13:1193–202. doi: 10.1016/j.chembiol.2006.09.011
49. Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, et al. Anti-inflammatory action of neuroprotection D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. (2006) 176:1848–59. doi: 10.4049/jimmunol.176.3.1848
50. Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. (2009) 461:1287–91. doi: 10.1038/nature08541
51. Serhan CN. Resolution phases of inflammation: novel endogenous anti-inflammatory and pro- resolving lipid mediators and pathways. Annu Rev Immunol. (2007) 25:101–37. doi: 10.1146/annurev.immunol.25.022106.141647
52. Colas RA, Souza PR, Walker ME, Burton M, Zasłona Z, Curtis AM, et al. Impaired production and diurnal regulation of vascular RvDn-3 DPA increase systemic inflammation and cardiovascular disease. Circ Res. (2018) 122:855–63. doi: 10.1161/CIRCRESAHA.117.312472
53. Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk HD, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci U S A. (2009) 106:21407–12. doi: 10.1073/pnas.0906361106
54. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. (2013) 341:1483–8. doi: 10.1126/science.1240636
55. McCauley LK, Dalli J, Koh AJ, Chiang N, Serhan CN. Cutting edge: parathyroid hormone facilitates macrophage efferocytosis in bone marrow via proresolving mediators resolvin D1 and resolvin D2. J Immunol. (2014) 193:26–9. doi: 10.4049/jimmunol.1301945
56. Yang M, Fjærvoll HK, Fjærvoll KA, Wang NH, Utheim TP, Serhan CN, et al. Sex-based differences in conjunctival goblet cell responses to pro-inflammatory and pro-resolving mediators. Sci Rep. (2022) 12:16305. doi: 10.1038/s41598-022-20177-9
57. English JT, Norris PC, Hodges RR, Dartt DA, Serhan CN. Identification and profiling of specialized pro-resolving mediators in human tears by lipid mediator metabolomics. Prostaglandins Leukot Essent Fatty Acids. (2017) 117:17–27. doi: 10.1016/j.plefa.2017.01.004
58. El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. (2009) 180:311–9. doi: 10.1164/rccm.200810-1601OC
59. Seki H, Fukunaga K, Arita M, Arai H, Nakanishi H, Taguchi R, et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol. (2010) 184:836–43. doi: 10.4049/jimmunol.0901809
60. Blaho VA, Zhang Y, Hughes-Hanks JM, Brown CR. 5-Lipoxygenase-deficient mice infected with Borrelia burgdorferi develop persistent arthritis. J Immunol. (2011) 186:3076–84. doi: 10.4049/jimmunol.1003473
61. Bafica A, Scanga CA, Serhan CN, Machado F, White S, Sher A, et al. Host control of mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest. (2005) 115:1601–6. doi: 10.1172/JCI23949
62. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. (2012) 148:434–46. doi: 10.1016/j.cell.2011.12.023
63. Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol. (2016) 16:51–67. doi: 10.1038/nri.2015.4
64. Tobin DM, Vary JC Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. (2012) 140:717–30. doi: 10.1016/j.cell.2010.02.013
65. Herb F, Thye T, Nieman S, Browne EL, Chinbuah MA, Gyapong J, et al. ALOX5 variants associated with susceptibility to human pulmonary tuberculosis. Hum Mol Genet. (2008) 17:1052–60. doi: 10.1093/hmg/ddm378
66. Walker J, Dichter E, Lacorte G, Kerner D, Spur B, Rodriguez A, et al. Lipoxin a4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock. (2011) 36:410–6. doi: 10.1097/SHK.0b013e31822798c1
67. Ueda T, Fukunaga K, Seki H, Miyata J, Arita M, Miyasho T, et al. Combination therapy of 15-epi-lipoxin A4 with antibiotics protects mice from Escherichia coli-induced sepsis. Crit Care Med. (2014) 42:e288–95. doi: 10.1097/CCM.0000000000000162
68. Sordi R, Menezes-de-Lima O, Horewicz V, Scheschowitsch K, Santos LF, Assreuy J. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int Immunopharmacol. (2013) 17:283–92. doi: 10.1016/j.intimp.2013.06.010
69. Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. (2012) 484:524–8. doi: 10.1038/nature11042
70. Cilloniz C, Pantin-Jackwood MJ, Ni C, Goodman AG, Peng X, Proll SC, et al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J Virol. (2010) 84:7613–24. doi: 10.1128/JVI.00553-10
71. Tam VC, Quehenberger O, Oshansky CM, Suen R, Armando AM, Treuting PM, et al. Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell. (2013) 154:213–27. doi: 10.1016/j.cell.2013.05.052
72. Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. (2013) 153:112–25. doi: 10.1016/j.cell.2013.02.027
73. Ng S, Cowling BJ, Fang VJ, Chang KH, Ip DKM, Cheng CKY, et al. Effects of oseltamivir treatment on duration of clinical illness and viral shedding and household transmission of influenza virus. Clin Infect Dis. (2010) 50:707–14. doi: 10.1086/650458
74. Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JCG, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4-, and IFN-β-dependent. Mucosal Immunol. (2010) 3:291–300. doi: 10.1038/mi.2010.6
75. Richardson JY, Ottolini MG, Pletneva L, Boukhvalova M, Zhang S, Vogel SN, et al. Respiratory syncytial virus (RSV) infection induces cyclooxygenase 2: a potential target for RSV therapy. J Immunol. (2005) 174:4356–64. doi: 10.4049/jimmunol.174.7.4356
76. Rajasagi NK, Reddy PB, Suryawanshi A, Mulik S, Gjorstrup P, Rouse BT. Controlling herpes simplex virus-induced ocular inflammatory lesions with the lipid-derived mediator resolvin E1. J Immunol. (2011) 186(3):1735–46. doi: 10.4049/jimmunol.1003456
77. Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med. (1997) 186:1819–29. doi: 10.1084/jem.186.11.1819
78. Aliberti J, Serhan C, Sher A. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in toxoplasma gondii infection. J Exp Med. (2002) 196:1253–62. doi: 10.1084/jem.20021183
79. Shryock N, McBerry C, Gonzalez RMS, Janes S, Costa FTM, Aliberti J, et al. Lipoxin A4 and 15-epi-lipoxin A4 protect against experimental cerebral malaria by inhibiting IL-12/IFN-γ in the brain. PLoS One. (2013) 8:e61882. doi: 10.1371/journal.pone.0061882
80. Bandeira-Melo C, Serra MF, Diaz BL, Cordeiro RS, Silva PM, Lenzi HL, et al. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in angiostrongylus costaricensis- infected rats: relationship with concurrent eosinophilia. J Immunol. (2000) 164:1029–36. doi: 10.4049/jimmunol.164.2.1029
81. Molina-Berrios A, Campos-Estrada C, Henriquez N, Faúndez M, Torres G, Castillo C, et al. Protective role of acetylsalicylic acid in experimental trypanosoma cruzi infection: evidence of a 15-epi-lipoxin A4-mediated effect. PLoS Negl Trop Dis. (2013) 7:e2173. doi: 10.1371/journal.pntd.0002173
82. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. (2005) 6:1191–7. doi: 10.1038/ni1276
83. Van Dyke TE. Inflammation and periodontal diseases: a reappraisal. J Periodontol. (2008) 79(8 Suppl):1501–2. doi: 10.1902/jop.2008.080279
84. Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol. (2008) 79(8 Suppl):1601–8. doi: 10.1902/jop.2008.080173
85. Dongari-Bagtzoglou A. Mucosal biofilms: challenges and future directions. Expert Rev Anti Infect Ther. (2008) 6:141–4. doi: 10.1586/14787210.6.2.141
86. Macfarlane S, Dillon JF. Microbial biofilms in the human gastrointestinal tract. J Appl Microbiol. (2007) 102(5):1187–96. doi: 10.1111/j.1365-2672.2007.03287.x
87. Akiyama H, Morizane S, Yamasaki O, Oono T, Iwatsuki K. Assessment of Streptococcus pyogenes microcolony formation in infected skin by confocal laser scanning microscopy. J Dermatol Sci. (2003) 32:193–9. doi: 10.1016/S0923-1811(03)00096-3
88. Post JC, Hiller NL, Nistico L, Stoodley P, Ehrlich GD. The role of biofilms in otolaryngologic infections: update 2007. Curr Opin Otolaryngol Head Neck Surg. (2007) 15:347–51. doi: 10.1097/MOO.0b013e3282b97327
89. Hall-Stoodley L, Hu FZ, Gieseke A, Nistico L, Nguyen D, Hayes J, et al. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. J Am Med Assoc. (2006) 296(2):202–11. doi: 10.1001/jama.296.2.202
90. Coticchia J, Zuliani G, Coleman C, Carron M, Gurrola J 2nd, Haupert M, et al. Biofilm surface area in the pediatric nasopharynx: chronic rhinosinusitis vs obstructive sleep apnea. Arch Otolaryngol Head Neck Surg. (2007) 133(2):110–4. doi: 10.1001/archotol.133.2.110
91. Swidinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. (2007) 43(7):3380–9. doi: 10.1128/JCM.43.7.3380-3389.2005
92. Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res. (2003) 82:82–90. doi: 10.1177/154405910308200202
93. Van Dyke TE. Control of inflammation and periodontitis. Periodontol 2000. (2007) 45:158–66. doi: 10.1111/j.1600-0757.2007.00229.x
94. Socransky SS, Haffajee AD. Dental biofilms: difficult therapeutic targets. Periodontol 2000. (2002) 28:12–55. doi: 10.1034/j.1600-0757.2002.280102.x
95. Sharaway AM, Sabharwal K, Socransky SS, Lobene RR. A quantitative study of plaque and calculus formation in normal and periodontally involved mouths. J Periodontol. (1966) 37:495–501. doi: 10.1902/jop.1966.37.6.495
96. Furuichi Y, Lindhe J, Ramberg P, Volpe AR. Patterns of de novo plaque formation in the human dentition. J Clin Periodontol. (1992) 19:423–33. doi: 10.1111/j.1600-051X.1992.tb00673.x
97. Offenbacher S, Odle BM, Van Dyke TE. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J Periodontal Res. (1986) 21:101–12. doi: 10.1111/j.1600-0765.1986.tb01443.x
98. Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. (2000) 39:4761–8. doi: 10.1021/bi992551b
99. Chavis C, Vachier I, Chanez P, Bousquet J, Godard P. 5(S),15(S)-dihy- droxyeicosatetraenoic acid and lipoxin generation in human poly- morphonuclear cells: dual specificity of 5-lipoxygenase towards endogenous and exogenous precursors. J Exp Med. (1996) 183:1633–43. doi: 10.1084/jem.183.4.1633
100. Fiore S, Serhan CN. Formation of lipoxins and leukotrienes during receptor- mediated interactions of human platelets and recombinant human granulocyte/ macrophage colony-stimulating factor-primed neutrophils. J Exp Med. (1990) 172:1451–7. doi: 10.1084/jem.172.5.1451
101. Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. (2003) 171:6856–65. doi: 10.4049/jimmunol.171.12.6856
102. Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by resolvin E1. PloS One. (2011) 6:e24422. doi: 10.1371/journal.pone.0024422
103. Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. (2007) 179(10):7021–9. doi: 10.4049/jimmunol.179.10.7021
104. Hasturk H, Schulte F, Martins M, Sherzai H, Floros C, Cugini M, et al. Safety and preliminary efficacy of a novel host-modulatory therapy for reducing gingival inflammation. Front Immunol. (2021) 12:704163. doi: 10.3389/fimmu.2021.704163
105. Borgeson E, Lönn J, Bergström I, Brodin VP, Ramström S, Nayeri F, et al. Lipoxin A4 inhibits porphyromonas gingivalis-induced aggregation and reactive oxygen species production by modulating neutrophil-platelet interaction and CD11b expression. Infect Immun. (2011) 79:1489–97. doi: 10.1128/IAI.00777-10
106. Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, et al. Rve1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J. (2006) 20:401–3. doi: 10.1096/fj.05-4724fje
107. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8(5):349–61. doi: 10.1038/nri2294
108. Singh VP, Proctor SD, Willing BP. Koch’s postulates, microbial dysbiosis and inflammatory bowel disease. Clin Microbiol Infect. (2016) 22:594–9. doi: 10.1016/j.cmi.2016.04.018
109. Kirst ME, Li EC, Alfant B, Chi YY, Walker C, Magnusson I, et al. Dysbiosis and alterations in predicted functions of the subgingival microbiome in chronic periodontitis. Appl Environ Microbiol. (2015) 81:783–93. doi: 10.1128/AEM.02712-14
110. Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. (1994) 8(2):263–71. doi: 10.1177/08959374940080022001
111. Riep B, Edesi-Neuss L, Claessen F, Skarabis H, Ehmke B, Flemmig TF, et al. Are putative periodontal pathogens reliable diagnostic markers? J Clin Microbiol. (2009) 47:1705–11. doi: 10.1128/JCM.01387-08
112. Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol. (2006) 44:3665–73. doi: 10.1128/JCM.00317-06
113. Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000. (1997) 14:9–11. doi: 10.1111/j.1600-0757.1997.tb00189.x
114. Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. (2018) 16:745–59. doi: 10.1038/s41579-018-0089-x
115. Tanner ACR, Kent R Jr, Kanasi E, Lu SC, Paster BJ, Sonis ST, et al. Clinical characteristics and microbiota of progressing slight chronic periodontitis in adults. J Clin Periodontol. (2007) 34:917–30. doi: 10.1111/j.1600-051X.2007.01126.x
116. Papayianni A, Serhan CN, Brady HR. Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells. J Immunol. (1996) 156:2264–72. doi: 10.4049/jimmunol.156.6.2264
117. Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR, et al. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. (1995) 34:14609–15. doi: 10.1021/bi00044a041
118. Levy BD, Fokin VV, Clark JM, Wakelam MJ, Petasis NA, Serhan CN. Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: a ‘stop’ signaling switch for aspirin-triggered lipoxin A4. FASEB J. (1999) 13:903–11. doi: 10.1096/fasebj.13.8.903
119. Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A4 regulates bronchial epithelial cell responses to acid injury. Am J Pathol. (2006) 168:1064–72. doi: 10.2353/ajpath.2006.051056
120. Colgan SP, Serhan CN, Parkos CA, Delp-Archer C, Madara JL. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J Clin Invest. (1993) 92:75–82. doi: 10.1172/JCI116601
121. Gewirtz AT, Fokin VV, Petasis NA, Serhan CN, Madara JL. LXA4, aspirin-triggered 15-epi-LXA4, and their analogs selectively downregulate PMN azurophilic degranulation. Am J Physiol. (1999) 276:C988–94. doi: 10.1152/ajpcell.1999.276.4.C988
122. Bonnans C, Vachier I, Chavis C, Godard P, Bousquet J, Chanez P. Lipoxins are potential endogenous anti-inflammatory mediators in asthma. Am J Respir Crit Care Med. (2002) 165:1531–5. doi: 10.1164/rccm.200201-053OC
123. Jozsef L, Zouki C, Petasis NA, Serhan CN, Filep JG. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-κB and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc Natl Acad Sci U S A. (2002) 99:13266–71. doi: 10.1073/pnas.202296999
124. Maddox JF, Serhan CN. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: selective inactivation by dehydrogenation and reduction. J Exp Med. (1996) 183:137–46. doi: 10.1084/jem.183.1.137
125. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. (2000) 164:1663–7. doi: 10.4049/jimmunol.164.4.1663
126. Soyombo O, Spur BW, Lee TH. Effects of lipoxin A4 on chemotaxis and degranulation of human eosinophils stimulated by platelet-activating factor and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Allergy. (1994) 49:230–4. doi: 10.1111/j.1398-9995.1994.tb02654.x
127. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. (2013) 5:174ra26. doi: 10.1126/scitranslmed.3004812
128. Ramstedt U, Serhan CN, Nicolaou KC, Webber SE, Wigzell H, Samuelsson B. Lipoxin A-induced inhibition of human natural killer cell cytotoxicity: studies on stereospecificity of inhibition and mode of action. J Immunol. (1987) 138:266–70. doi: 10.4049/jimmunol.138.1.266
129. Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat Immunol. (2002) 3:76–82. doi: 10.1038/ni745
130. Cezar- de-Mello PF, Nascimento-Silva V, Villela CG, Fierro IM. Aspirin-triggered lipoxin A4 inhibition of VEGF-induced endothelial cell migration involves actin polymerization and focal adhesion assembly. Oncogene. (2006) 25:122–9. doi: 10.1038/sj.onc.1209002
131. Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Fierro IM. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb Haemostasis. (2007) 97:88–98. doi: 10.1160/TH06-06-0315
132. Brezinski ME, Gimbrone MA Jr, Nicolaou KC, Serhan CN. Lipoxins stimulate prostacyclin generation by human endothelial cells. FEBS Lett. (1989) 245:167–72. doi: 10.1016/0014-5793(89)80214-5
133. Parameswaran K, Radford K, Fanat A, Stephen J, Bonnans C, Levy BD, et al. Modulation of human airway smooth muscle migration by lipid mediators and th-2 cytokines. Am J Respir Cell Mol Biol. (2007) 37:240–7. doi: 10.1165/rcmb.2006-0172OC
134. Wu SH, Wu XH, Lu C, Dong L, Chen ZQ. Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol. (2006) 34:65–72. doi: 10.1165/rcmb.2005-0184OC
135. Sodin-Semrl S, Taddeo B, Tseng D, Varga J, Fiore S. Lipoxin A4 inhibits IL-1β-induced IL-6, IL-8, and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J Immunol. (2000) 164:2660–6. doi: 10.4049/jimmunol.164.5.2660
136. Campbell EL, Louis NA, Tomassetti SE, Canny GO, Arita M, Serhan CN, et al. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. (2007) 21:3162–70. doi: 10.1096/fj.07-8473com
137. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U S A. (2005) 102:7671–6. doi: 10.1073/pnas.0409271102
138. Isobe Y, Arita M, Matsueda S, Iwamoto R, Fujihara T, Nakanishi H, et al. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J Biol Chem. (2012) 287:10525–34. doi: 10.1074/jbc.M112.340612
139. Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. (2007) 282:9323–34. doi: 10.1074/jbc.M609212200
140. Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, et al. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol. (2012) 189:1983–91. doi: 10.4049/jimmunol.1101665
141. Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. (2012) 120:e60–72. doi: 10.1182/blood-2012-04-423525
142. Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol. (2006) 177:5902–11. doi: 10.4049/jimmunol.177.9.5902
143. Ariel A, Fredman G, Sun Y, Kantarci A, Van Dyke TE, Luster AD, et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. (2006) 7:1209–16. doi: 10.1038/ni1392
144. Ariel A, Li P, Wang W, Tang W, Fredman G, Hong S, et al. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J Biol Chem. (2005) 280:43079–86. doi: 10.1074/jbc.M509796200
145. Nordgren TM, Heires AJ, Wyatt TA, Poole JA, LeVan TD, Cerutis DR, et al. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir Res. (2013) 14:51. doi: 10.1186/1465-9921-14-51
Keywords: inflammation, resolution of inflammation, periodontal disease, SPM, resolvins, lipoxins, protectins and maresins, oral health
Citation: Sahni V and Van Dyke TE (2023) Immunomodulation of periodontitis with SPMs. Front. Oral. Health 4:1288722. doi: 10.3389/froh.2023.1288722
Received: 4 September 2023; Accepted: 9 October 2023;
Published: 20 October 2023.
Edited by:
Jôice Dias Corrêa, Pontifical Catholic University of Minas Gerais, BrazilReviewed by:
Fernanda Rocha, University of Florida, United StatesFranco Cavalla, University of Chile, Chile
Josefine Hirschfeld, University of Birmingham, United Kingdom
© 2023 Sahni and Van Dyke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas E. Van Dyke dHZhbmR5a2VAZm9yc3l0aC5vcmc=