 Sven Eric Niklander
Sven Eric Niklander Pablo Aránguiz
Pablo Aránguiz Fernando Faunes
Fernando Faunes René Martínez-Flores
René Martínez-Flores
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oral. Health , 12 October 2023
Sec. Oral Cancers
Volume 4 - 2023 | https://doi.org/10.3389/froh.2023.1285276
This article is part of the Research Topic Reviews in the Prevention and Early Detection of Oral Cancers View all 5 articles
The gradual accumulation and inadequate renewal of senescent cells over time drive organismal aging. Senescent cells undergo altered gene expression and release inflammatory mediators collectively termed the senescence-associated secretory phenotype (SASP), which significantly contributes to a spectrum of age-related disorders, including cancer. In the context of carcinogenesis, the SASP produced by senescent cells has been implicated in the promotion of epithelial cancers, including oral squamous cell carcinoma (OSCC), the most common form of oral cancer. Senescent cells within the tumor microenvironment release factors that amplify the growth and invasiveness of neighboring cancer cells. Senotherapeutics, including senolytics and senomorphics, emerge as promising modalities to target senescent cells and their associated inflammatory factors, thereby opening novel avenues for augmenting the efficacy of cancer treatments. Here, we review the general aspects of cellular senescence, focusing on the relation between senescence-related inflammation with cancer development. We also analyze the available evidence linking cellular senescence with OSCC, highlighting possible clinical applications.
The relation between organism aging and the development of different diseases (age-related diseases) is not new and is well accepted. Organism aging happens due to the accumulation and lack of renewal of aged cells across time (1). Cell aging or cellular senescence is a cellular state in which the growth capabilities of cells are irreversible lost in response to different stressors (2). An important feature of senescent cells is that they change their gene expression profile and develop an inflammatory secretome known as the senescence-associated secretory phenotype (SASP) (3, 4). The accumulation of senescent cells therefore can be detrimental, as the inflammatory factors secreted as part of the SASP can act on neighbor cells either predisposing, triggering or promoting the development of different diseases (5–7).
Many age-related diseases are influenced by the accumulation of senescent cells. This led to the development of new types of drugs, known as senotherapeutics. Senotherapeutics are drugs aimed to specifically target and eliminate senescent cells (senolytics) or to decrease the abundancy of inflammatory factors present in the SASP (senomorphics) (8). Although this field is relatively new, there are already human clinical trials reporting some of these drugs to be beneficial for the treatment of idiopathic pulmonary fibrosis (9) and many more are currently being held (https://clinicaltrials.gov/search?cond=cancer&intr=navitoclax).
There is robust evidence supporting stromal senescent cells as promoter of epithelial cancers of different origins. Recent studies have also suggested senescent cells to play a role in oral cancer squamous cell carcinoma (OSCC) development (10), progression (11) and therapy resistance (12). This could have an impact on the way we currently treat oral-precancerous lesions and OSCC. Here, we review the general aspects of cellular senescence, focusing on the relation between senescence-related inflammation with cancer development. We also analyze the available evidence linking cellular senescence with OSCC, highlighting possible clinical applications.
Senescence can be induced by different stimuli, including replicative stress, oxidative stress, oncogene signaling and DNA damage (13). Independent of the inductor, senescent cells acquire morphological alterations that differentiate them from non-senescent cells. There are 5 hallmarks of cellular senescence representing structural, epigenetic and signaling alterations: (i) chromatin reorganization, (ii) cell cycle arrest, (iii) metabolic adaptation, (iv) modifications of the lysosomal compartment and v) development of a secretory phenotype (SASP) (14, 15). There is no universal marker to identify senescent cells. The most used markers are the activity of senescent-associated β-galactosidase (SA-β-GAL), the expression of LaminB1, p16 and p21, the identification of senescence-associated heterochromatin foci (SAHFs) and the accumulation of DNA damage response (DDR) proteins, such as phosphorylated (γ) H2A. However, many others have been reported including the lack of expression of ki67 and the increased secretion of IL-6 and IL-8 SASP factors (15). None of the aforementioned features can be used by themselves to identify senescent cells, and a combination of two to four of these markers should be used (16).
Depending on the context, senescence can result in both beneficial and detrimental effects. In young individuals’ senescence contributes to tumor suppression and wound healing, primary by stopping the cell cycle and by secreting specific factors as part of the SASP (17–20). However, in older individuals or upon consistent and chronic damage, senescent cells accumulate in tissues. This contributes to tissue dysfunction, chronic inflammation, and age-related disorders, including cardiovascular diseases, fibrosis, diabetes, neurological disorders, and paradoxically cancer (21–23). These negatives effects are also attributed to the SASP. Due to these antagonistic effects, senescence is considered a double-edge sword in health and disease (24).
Whether a senescent cell that has irreversible exited the cell cycle can re-enter and become malignant is still under study (25, 26). Reversing senescence is not a common feature and is not the mechanism by which cellular senescence is considered to have pro-tumorigenic effects. The pro-tumorigenic effects of senescence are explained by the SASP. Neighbor non-cancerous senescent cells (or cancerous cells induced to senesce because of radiation or chemotherapy) secrete soluble factors that can be used by pre-malignant or malignant cells for their advantage, which is changing the current understanding of cancer biology. In fact, senescent cells have been recently proposed as an emerging hallmark of cancer with enabling/promoting capabilities (27).
Different experimental models have suggested that the composition of the SASP varies between cell types and the senescence inducer (28). Nevertheless, a recent report which compared the SASP after inducing senescence under two different modalities in 13 different cancer cell types found the expression of the SASP to be more influenced by the cell type rather than the senescence inductor (4). Although the composition of SASP is widely heterogenous and more than 50 different soluble factors might be overexpressed (3), there is substantial overlap among SASPs, with specific proteins being almost invariably, including IL-1α, IL-6, IL-8, MMP-1, MMP-3, MMP-10 (29).
Virtually every cell that senesce will develop a SASP, although there are reports from animal studies in which under specific conditions cell senesced without developing a secretory phenotype (19). Diverse signaling pathways are associated to induce and maintain the SASP, including: phosphoinositide-3-kinase (PI3K) (30), mammalian target of rapamycin (mTOR) (31–33), p38MAPK (34), STAT3 (35), GATA4 (36), cGAS/STING (37–39), IL-1 signaling pathway (40) and Rho-kinase (41). These pathways have one thing in common, to activate NF-kB and/or CEBPβ transcription factors as downstream effectors (42).
Classically, the accumulation of genetic and epigenetic changes in target cells has been considered as the primary cause of carcinoma development (43). However, that simplistic view has changed, based on the finding that pre-cancerous epithelial lesions with “cancer-associate mutations” may never progress into cancer (44) with substantial evidence suggesting that the final trigger for developing the malignant phenotype could be a micro-environmental change (45, 46). The current knowledge supports the idea that solid tumors, such as carcinomas, are not just clonally evolved epithelial cells that have accumulated a critical number of cancer-predisposing mutations, but rather dysfunctional tissues where the mesenchymal component (stroma) plays an important role in the tumor pathogenesis (27), being even responsible for the acquisition of therapeutic resistance (47–50). The stromal component consists of a variety of cells, including fibroblasts, pericytes, endothelial cells, adipose cells, but as fibroblast are the most common stromal component within tissues, most of the studies focus on them (51). There is evidence sustaining senescent fibroblasts to facilitate tumorigenesis of epithelial cancers by generating a tumor permissive microenvironment, promoting the growth of malignant cells of breast (3), skin (52, 53), prostate (54), colon (30), gastric (55) and oral cancers (10). In addition to fibroblasts, other stromal cells (including pericytes, adipocytes, lymphocytes, among others) have also been implicated in the generation of a tumor permissive microenvironment (56, 57).
The mechanisms underlying these pro-tumorigenic effects are not entirely known. As the organism ages, the fibroblast renewal rate decreases (58). Senescence of immune cells also reduces the clearance of other senescent cells (59). Therefore, senescent cells accumulate in tissues, resulting in an aged tumor microenvironment (TME) with abundancy of SASP factors (1). It is hypothesized then, that this would generate a switch towards a more immunosuppressive immune infiltrate (60, 61), establishing chronic inflammation, generating a microenvironment prone for cancer formation and progression. The factors that accumulate in an aged TME include proteins able to remodelate the extracellular matrix (ECM), such as MMPs, plasminogen activator inhibitors (PAI1 and PAI2), tissue-type plasminogen activator (tPA) and the secretion of pro-inflammatory molecules and growth factors, such as CXCL1, CXCL8, CXCL2, IL-6, IL-1α and colony stimulating factors (CSFs) (62).
The TME is not the only source of senescent cells. Tumor cells themselves can be induced to senesce by chemotherapeutic drugs (23) or by radiation therapy (63), which is known as therapy induced senescence (TIS). TIS does not only affect cancerous cells, but also stromal cells, and is considered an off-target effect of cancer therapy (64). In theory, TIS of cancerous cells should be a desirable outcome of cancer treatment because even when the treatment is not able to eliminate all malignant cells, the remaining living cells are not able to proliferate. Nevertheless, cancerous TIS cells remain metabolically active and together with other stromal TIS cells are a source of chronic inflammation through the SASP, facilitating drug resistance (65), tumor relapse (66) and distant metastasis (67).
In human premalignancies, cellular senescence of the epithelial compartment is considered a tumor suppressor mechanism, as the acquisition of an indefinite replicative lifespan (one of the most important hallmarks of cancerous cells) depends on bypassing senescence via inactivation of p16INK4A and p53 (44, 68), the main pathways responsible for senescence induction and maintenance (13). There is evidence that senescent cells accumulate in vivo in human premalignancies, such as colonic adenomas (69), cervical intraepithelial neoplasia (70), Bowen's disease (71), ductal carcinoma in situ (72), actinic keratosis (73) and oral potentially malignant disorder (OPMD) (74–76). OPMD encompass a heterogenous group of disorders characterized by an increased risk for developing cancer (77). The global prevalence of OPMD has been estimated in 4.47%. Oral leukoplakia (OL) and oral submucous fibrosis (OSMF) are the most common disorders (78), with a malignant transformation rate of 9.5% and 5.2% respectively (79).
A recent study that analyzed 50 OL samples with and without dysplasia found p16 positive keratinocytes in all 50 lesions, which were not related to HPV infection (76). Similarly, another study analyzed senescent markers in 86 OL with different degrees of dysplasia and found that both γH2AX and p53 proteins increase progressively according to the severity of dysplasia (75). There are also reports that senescent cells and DNA damage accumulate in higher numbers in many human premalignancies compared to their corresponding malignancies (80–83). In OL with dysplasia, senescent markers cyclin D1, maspin, Rb, and p16INK4A have been found at higher levels compared to OSCC (74). All these data suggest that the epithelial senescence program prevents malignant transformation, which needs to be dismantled prior to cancer development.
Although senescence of the epithelial compartment has shown to be tumor suppressor, this does not mean that senescent epithelial cells are of no harm. In an OL with dysplasia, most, but not all dysplastic cells will senesce due to DNA damage response (DDR) due to oncogenic stress (oncogene induced senescence). But some cells will escape senescence and will become immortal, as has been shown in keratinocytes isolated from oral dysplasias (44). The acquisition of an immortal phenotype depends on p16 mutation or methylation, mutations or inactivation of p53 and reactivation of telomerase (44, 84). On the other side, dysplastic senescent keratinocytes will develop a SASP characterized by high levels of IL-6, IL-8 and IL-1α (cytokines with known oncogenic potential) (3, 85, 86) and other inflammatory and growth factors (87). These soluble factors can also induce paracrine senescence of surrounding normal keratinocytes, fibroblasts and other stromal cells (40), increasing the abundancy of SASP factors in the microenvironment. Thus, the “initiated” dysplastic keratinocytes that escaped senescence (immortal) will be exposed to an inflammatory pro tumorigenic microenvironment that can promote cancer development (46, 85) (Figure 1A). In fact, it has been recently shown that senescent mortal premalignant oral keratinocytes upregulate the expression of extracellular prostaglandins E1 and E2 (ePGE1 and 2), and that ePGE2, in conjunction with other SASP cytokines, are able to induce proliferation of immortal premalignant oral keratinocytes (88). If maintained over time, this could promote malignant transformation.
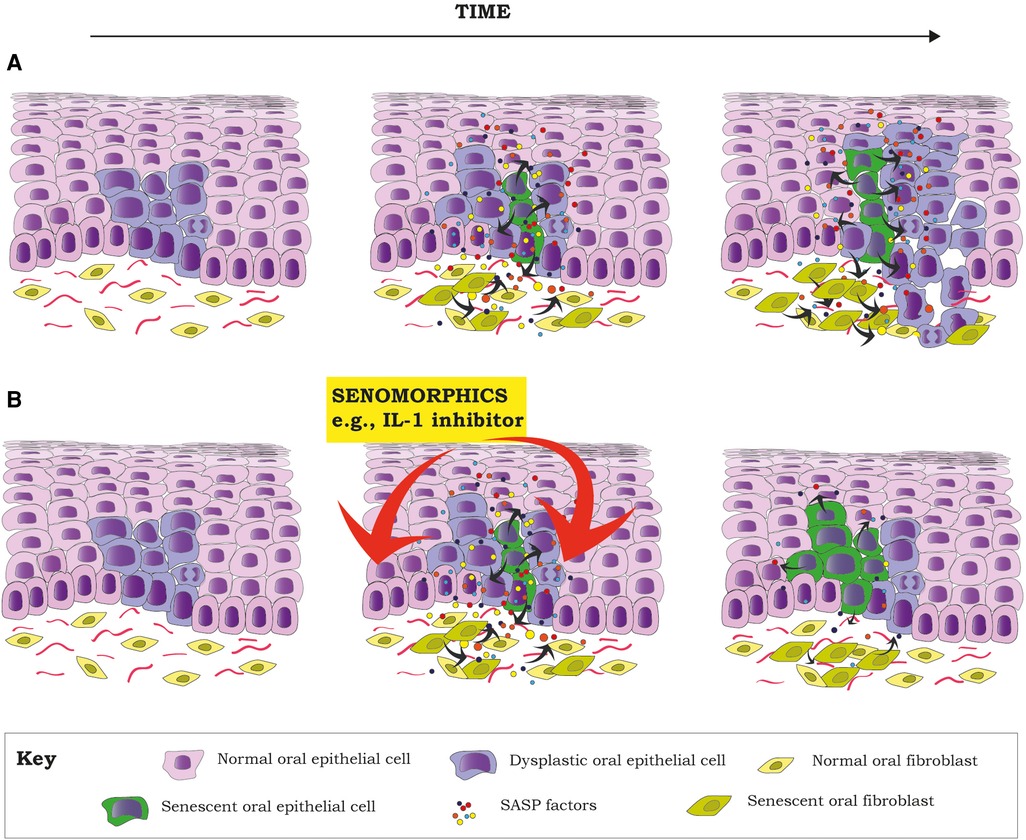
Figure 1. Senescent cells can create an environment for tumor promotion, by facilitating proliferation and the acquisition of a malignant phenotype of dysplastic cells (pre-malignant) through the secretion of different inflammatory factors as part of the SASP, a hallmark of senescent cells (A). Proposal of how the inhibition of different components of the SASP by senomorphics (e.g., IL-1 pathway) at the dysplasia state could decrease the SASP of senescent cells (both epithelial and stromal). This would potentially reduce the inflammatory component of the TME, reducing the chances of malignant transformation of already dysplastic cells (B). Red arrows mean inhibition.
While in the epithelial compartment senescence is mainly considered a tumor suppressor mechanism (although as mentioned above it could also act a tumor promoter), senescence of surrounding stromal cells, such as fibroblasts, is considered tumor promoting (68). In skin, senescent fibroblasts can promote carcinogenesis of keratinocytes via factors present in the SASP (53). For example, pre-malignant breast epithelial cells can become invasive and suffer epithelial-to-mesenchymal transition (EMT) after stimulation by SASP factors from surrounding senescent fibroblasts via a paracrine mechanism dependent of IL-6 and IL-8 (3). Pre-malignant non-tumorigenic human embryonic kidney cells, in the presence of SASP factors from senescent fibroblast, can also become tumorigenic with stem-like properties (89). Senescent prostatic fibroblasts are able to induce proliferation of prostate epithelial cells through a paracrine mechanism including hepatocyte growth factor (HGF), fibroblast growth factor 7 (FGF7) and amphiregulin (AREG) (54). GDF15, an essential factor from the SASP from senescent colonic fibroblasts, can promote malignant features (increased proliferation, migration, and invasion) in colon adenoma cell lines (30).
Whilst there is substantial evidence supporting a cancer promoting role of senescent fibroblast in different premalignancies, there is scarce information about their role in OPMD, being most studies performed in OSMF, in which cellular senescence has been proposed to have an essential role in the malignant transformation (90). OSMF is a progressive fibrotic disease caused by the chewing of areca nut characterized by a gradual reduction in the jaw opening (91) and an increased risk for oral cancer development (92). In the initial phases, reactive oxygen species (ROS) induce senescence of epithelial stem cells leading to epithelial atrophy (90, 93). This is followed by senescence of endothelial cells and fibroblasts (90), which reduces tissue vascularity generating hypoxia and fibrosis. Alkaloids and flavonoids from areca nut also stimulate fibroblasts to produce more collagen. This leads to more collagen deposition affecting deeper tissues as the disease progresses (46). As fibrosis increases, the epithelium atrophies due to senescence, and senescent fibroblasts accumulate in the stroma (90), which precedes the development of epithelial dysplasia (51). For the malignant transformation to happen, the dysplastic senescent epithelial cells must escape senescence, likely due to upregulation of LOLXL2 ad mucin 4 (94, 95). This will result in immortal dysplastic epithelial cells exposed to upregulated SASP factors secreted by senescent fibroblast, such as IL-1, IL-6, IL-8 and GRO-α/CXCL-1, which will increase ROS production, generating DSB, favoring cancer development (96).
In OSCC, most of the epithelial cancerous cells have escape senescence to become malignant through TP53 and p16INKA4 mutations, although a subset of cancerous cells has been reported to retain functional Tp53 or p16INKA4 (97, 98). Nevertheless, those mortal cancerous cells are expected to acquire further mutations to reach immortality or will disappear due to natural selection. Therefore, senescence of malignant keratinocytes is not naturally expected in OSCCs or should not have a significant effect in tumor behavior (unless the tumor is irradiated or treated with chemotherapeutic drugs). But as mentioned earlier, senescence of stromal components of the tumor can have deleterious effects. The most studied stromal cells in OSCCs are cancer-associated fibroblasts (CAF).
CAF are a poorly characterized heterogenous cell population with different subtypes, including activated myofibroblastic CAF (myCAF) and senescent fibroblasts (99, 100). There is substantial evidence that the accumulation of myCAF is associated with a poor prognosis in OSCC (83, 101), as myCAF support OSCC progression, tumor growth and invasion (99). Senescent oral fibroblasts have also been reported to have pro-tumorigenic effects in OSCC (10, 11, 100). They share an overlapping gene expression profile with myCAF (100), suggesting that these CAF phenotypes are closely related. Senescent fibroblasts are found in the stroma of OSCC in different quantities (74, 100) and have been reported to be in higher numbers than in normal oral mucosa and dysplastic oral lesions (10). Interestingly, the injection of small number of senescent cells into mice caused persistent physical dysfunction with shorter health and life span, indicating that the number of senescent cells is not important (102). Furthermore, the injection of senescent cells spread the senescence to host tissues (102), probably in a paracrine manner through components of the SASP (40, 103).
The accumulation of senescent fibroblast in OSCC is an early event unlikely to be due to replicative exhaustion, as some same age patients with OSCC have shown no senescent fibroblasts (10). Additionally, senescent fibroblasts also accumulate in OPMD in younger patients by a telomere-independent mechanism (93). Hassona et al. (2013) suggest that in OSCC, fibroblast senescence is caused by oxidative DNA damage. This would be induced by ROS produced by oral keratinocytes and fibroblasts from genetically unstable OSCC (developed from premalignancies with loss of TP53 and P16INK4A), but not genetically stable OSCC (developed from premalignancies with functional p53), in a TGF-β dependent manner (10). This is supported by the fact that keratinocytes from genetically unstable OSCC produce higher amounts of ROS and are deficient in antioxidant defenses, suggesting that p53 functionality is important in the regulation of ROS production (10). Recently, it has been also proposed that impairment of the autophagy process might be also responsible for the development of the senescent and myofibroblastic phenotypes in CAF (104).
Regardless the mechanism underlying the induction of fibroblast senescence, the SASP from senescent oral fibroblast has been shown to have pro-tumorigenic effects on malignant oral keratinocytes (10, 11, 68, 100). SASP factors MMP-2 (11), ROS, TGF-β (10), PGE2 and the miR-335/COX-2/PTEN axis (105) have been shown to induce dis-cohesion, EMT, migration and invasion of oral malignant keratinocytes (10, 11, 68, 100, 105). A recent paper also showed that both, early senescence and NF-kB-dependent SASP cytokines secreted by senescent OSCC cells induced to senesce by radiotherapy, are critical for radioresistance in OSCC in vivo (12).
Cellular aging is a relatively new research field that has developed during the last two decades and has gained increasing interest due to its relationship with organism aging and age-related disorders. Although initially cellular senescence was considered only as a potent anti-tumor mechanism, nowadays is also recognized as a potent tumor promoter. Consistent with this idea, senescent cells have been recently considered as an emerging hallmark of cancer with enabling characteristics (27) despite their well-known anti-tumorigenic functions.
Senescent cells might not only influence cancer behavior but also affect tolerance to cancer treatment, as the expression of a senescence marker in circulating T-cells before chemotherapy was associated with increased risk of chemotherapy-induced fatigue in humans. In addition, in vivo models in mice showed that eliminating TIS cells reduced short-and long-term effects of chemotherapeutic drugs, including cardiac dysfunction, physical activity, strength, bone marrow suppression and cancer recurrence (23).
This interest has led to the development of new drugs or to the finding of new uses for old drugs to specifically eliminate senescent cells or to target their SASP (senolytics and senomorphics respectively). This field, also known as senotherapeutics, has also emerged as new complimentary treatment alternative for some cancers.
Senolytic drugs consist usually of small molecule agents that target specifically anti-apoptotic pathways that are overexpressed in senescent cells as a pro-survival mechanism, but not in proliferating nor quiescent cells (13). There are different drugs targeting different components of the antiapoptotic pathways including: Navitoclax and ABT-737, both targeting the BCL-2 pathway (106, 107), Dasatinib, a tyrosine kinase inhibitor (108), AT-406, a regulator of anti-apoptotic proteins c-IAP2 and XIAP (109). These drugs have eliminated senescent cells in in vitro and in in vivo models improving clinical outcomes. For example, Navitoclax, has shown to eliminate ovarian and breast cancer TIS cells after PARP inhibitor therapy (110) and to induce tumor regression and improve therapeutic outcomes following conventional chemotherapy in mouse models (111). It has also shown to improve radiation-induced salivary gland hypofunction in irradiated mice by eliminating salivary gland senescent stem cells (112). Dasatinib, in combination with quercetin, reduces the population of senescent cells in mice attenuating adipose tissue inflammation, improving systemic metabolic function (108). The same combination of drugs has also been tested in clinical trials for the treatment of pulmonary fibrosis improving patient's physical function (9).
Senomorphics or SASP inhibitors are drugs aimed to decrease the pro-tumorigenic inflammatory component of the SASP, with the advantage of conserving pro-immunogenic functions of senescent cells, such as immunosurveillance. The main disadvantage of senomorphics over senolytics is that, as these drugs do not eliminate senescent cells, they might require long-term administration as their effect vanishes upon discontinuation (21). NF-κB is probably the most important signaling pathway mediating the inflammatory components of the SASP. Thus, most studies have explored the use of drugs targeting this transcription pathway or NF-κB-mediated cytokines, including metformin (113, 114), aventhramice C (115), Anakinra (IL-1 inhibitor) (116), Canakinumab (anti-IL-1β antibody) (117) or Simvastatin (IL-6 and IL-8 inhibitor) (118). Furthermore, the use of agents targeting other pathways such as Rho-kinase (Y27632) (41, 119), cGAS-STING (RU.521 (85), mTOR (Rapamycin) (31, 33), among many others, have also been assessed.
Inflammation seems to be of importance in OSCC development and progression (120). In solid tumors, the presence of senescent cells is one of the most important sources of inflammation. Therefore, targeting senescent cells to reduce inflammation seems a promising approach to find new treatment alternatives to improve treatment success. The increased expression of NF-κB signaling pathway (121, 122) and NF-κB-mediated cytokines (120), such as IL-1 in OPMD and OSCC, are reported to have impact in clinical outcomes (120, 123). IL-1 is considered a master regulator of the SASP and for the spread of paracrine senescence (40), and there is evidence from in vivo animal studies that IL-1 induces malignant transformation of oral precursor lesions and OSCC aggressiveness (124). Therefore, targeting senescent cells to reduce the overexpression of IL-1 with senolytics or senomorphics, in addition to surgical or chemotherapeutic treatment, could represent a novel treatment alternative for OPMDS and OSCC (Figure 1B). Although there are different clinical trials testing senolytics as single agents or in combination with other chemotherapeutic drugs for the treatment of different cancers, including lymphomas, melanoma, leukemia, lung, ovarian and prostate cancers (source: clinicaltrials.gov), more in vitro and in vivo animal studies are needed to support the use of senotherapeutics for OSCC treatment.
SN: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. PA: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. FF: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. RM: Conceptualization, Investigation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This research was funded by Fondo Núcleo DI-05-22/NUC, Universidad Andres Bello.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature. (2019) 571:183–92. doi: 10.1038/s41586-019-1365-2
2. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. (2015) 15:397–408. doi: 10.1038/nrc3960
3. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
4. Jochems F, Thijssen B, De Conti G, Jansen R, Pogacar Z, Groot K, et al. The cancer SENESCopedia: a delineation of cancer cell senescence. Cell Rep. (2021) 36:109441. doi: 10.1016/j.celrep.2021.109441
5. Chatsirisupachai K, Palmer D, Ferreira S, de Magalhães JP. A human tissue-specific transcriptomic analysis reveals a complex relationship between aging, cancer, and cellular senescence. Aging Cell. (2019) 18:e13041. doi: 10.1111/acel.13041
6. Lau V, Ramer L, Tremblay M. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun. (2023) 14:1670. doi: 10.1038/s41467-023-37304-3
7. de Yébenes VG, Briones AM, Martos-Folgado I, Mur SM, Oller J, Bilal F, et al. Aging-associated miR-217 aggravates atherosclerosis and promotes cardiovascular dysfunction. Arterioscler Thromb Vasc Biol. (2020) 40:2408–24. doi: 10.1161/ATVBAHA.120.314333
8. Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. Febs j. (2023) 290:1362–83. doi: 10.1111/febs.16350
9. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
10. Hassona Y, Cirillo N, Lim KP, Herman A, Mellone M, Thomas GJ, et al. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-beta. Carcinogenesis. (2013) 34:1286–95. doi: 10.1093/carcin/bgt035
11. Hassona Y, Cirillo N, Heesom K, Parkinson EK, Prime SS. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br J Cancer. (2014) 111:1230–7. doi: 10.1038/bjc.2014.438
12. Schoetz U, Klein D, Hess J, Shnayien S, Spoerl S, Orth M, et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis. (2021) 12:1162. doi: 10.1038/s41419-021-04454-5
13. Niklander SE, Lambert DW, Hunter KD. Senescent cells in cancer: wanted or unwanted citizens. Cells. (2021) 10:3315. doi: 10.3390/cells10123315
14. Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. (2016) 29:1–12. doi: 10.1016/j.arr.2016.05.003
15. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. Febs j. (2021) 288:56–80. doi: 10.1111/febs.15570
16. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. (2019) 19:439–53. doi: 10.1038/s41568-019-0156-2
17. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
18. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. (2013) 123:966–72. doi: 10.1172/JCI64098
19. Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. (2011) 286:36396–403. doi: 10.1074/jbc.M111.257071
20. Di Mitri D, Alimonti A. Non-cell-autonomous regulation of cellular senescence in cancer. Trends Cell Biol. (2016) 26:215–26. doi: 10.1016/j.tcb.2015.10.005
21. Schmitt CA, Wang B, Demaria M. Senescence and cancer—role and therapeutic opportunities. Nat Rev Clin Oncol. (2022) 19:619–36. doi: 10.1038/s41571-022-00668-4
22. Kasamatsu T, Awata-Shiraiwa M, Ishihara R, Murakami Y, Masuda Y, Gotoh N, et al. Sub-lethal doses of chemotherapeutic agents induce senescence in T cells and upregulation of PD-1 expression. Clin Exp Med. (2023) 6:2695–703.doi: 10.1007/s10238-023-01034-z
23. Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. (2017) 7:165–76. doi: 10.1158/2159-8290.CD-16-0241
24. Baker DJ, Narita M, Muñoz-Cánoves P. Cellular senescence: beneficial, harmful, and highly complex. FEBS J. (2023) 290:1156–60. doi: 10.1111/febs.16735
25. Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. (2005) 65:2795–803. doi: 10.1158/0008-5472.CAN-04-1270
26. Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem. (2003) 278:11731–4. doi: 10.1074/jbc.C300023200
27. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
28. Cuollo L, Antonangeli F, Santoni A, Soriani A. The senescence-associated secretory phenotype (SASP) in the challenging future of cancer therapy and age-related diseases. Biology (Basel. (2020) 9:485. doi: 10.3390/biology9120485
29. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
30. Guo Y, Ayers JL, Carter KT, Wang T, Maden SK, Edmond D, et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell. (2019) 18:e13013. doi: 10.1111/acel.13013
31. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR Regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. (2015) 17:1049–61. doi: 10.1038/ncb3195
32. van Vliet T, Varela-Eirin M, Wang B, Borghesan M, Brandenburg SM, Franzin R, et al. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol Cell. (2021) 9:2041–52. doi: 10.1016/j.molcel.2021.03.018
33. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. (2015) 17:1205–17. doi: 10.1038/ncb3225
34. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. (2011) 30:1536–48. doi: 10.1038/emboj.2011.69
35. Han X, Zhang T, Zhang X, Zhang R, Lao K, Mi Y, et al. AMPK Alleviates oxidative stress-induced premature senescence via inhibition of NF-κB/STAT3 axis-mediated positive feedback loop. Mech Ageing Dev. (2020) 191:111347. doi: 10.1016/j.mad.2020.111347
36. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. (2015) 349:aaa5612. doi: 10.1126/science.aaa5612
37. Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
38. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. (2017) 550:402–6. doi: 10.1038/nature24050
39. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. (2018) 9:1249. doi: 10.1038/s41467-018-03555-8
40. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. (2013) 15:978–90. doi: 10.1038/ncb2784
41. Simay Demir YD, Ozdemir A, Sucularli C, Benhur E, Ark M. The implication of ROCK 2 as a potential senotherapeutic target via the suppression of the harmful effects of the SASP: do senescent cancer cells really engulf the other cells? Cell Signal. (2021) 84:110007. doi: 10.1016/j.cellsig.2021.110007
42. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
43. Goruppi S, Dotto GP. Mesenchymal stroma: primary determinant and therapeutic target for epithelial cancer. Trends Cell Biol. (2013) 23:593–602. doi: 10.1016/j.tcb.2013.08.006
44. McGregor F, Muntoni A, Fleming J, Brown J, Felix DH, MacDonald DG, et al. Molecular changes associated with oral dysplasia progression and acquisition of immortality: potential for its reversal by 5-azacytidine. Cancer Res. (2002) 62:4757–66.12183435
45. Prime SS, Cirillo N, Cheong SC, Prime MS, Parkinson EK. Targeting the genetic landscape of oral potentially malignant disorders has the potential as a preventative strategy in oral cancer. Cancer Lett. (2021) 518:102–14. doi: 10.1016/j.canlet.2021.05.025
46. Parkinson EK, Prime SS. Oral senescence: from molecular biology to clinical research. Front Dent Med. (2022) 3:822397. doi: 10.3389/fdmed.2022.822397
47. Liu H, Zhao H, Sun Y. Tumor microenvironment and cellular senescence: understanding therapeutic resistance and harnessing strategies. Semin Cancer Biol. (2022) 86:769–81. doi: 10.1016/j.semcancer.2021.11.004
48. Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. (2012) 18:1359–68. doi: 10.1038/nm.2890
49. Chen F, Long Q, Fu D, Zhu D, Ji Y, Han L, et al. Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat Commun. (2018) 9:4315. doi: 10.1038/s41467-018-06860-4
50. Xu Q, Long Q, Zhu D, Fu D, Zhang B, Han L, et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell. (2019) 18:e13027. doi: 10.1111/acel.13027
51. Prime SS, Cirillo N, Hassona Y, Lambert DW, Paterson IC, Mellone M, et al. Fibroblast activation and senescence in oral cancer. J Oral Pathol Medicine. (2016) 2:82–8. doi: 10.1111/jop.12456
52. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. (2001) 98:12072–7. doi: 10.1073/pnas.211053698
53. Malaquin N, Vercamer C, Bouali F, Martien S, Deruy E, Wernert N, et al. Senescent fibroblasts enhance early skin carcinogenic events via a paracrine MMP-PAR-1 axis. PLoS One. (2013) 8:e63607. doi: 10.1371/journal.pone.0063607
54. Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. (2006) 66:794–802. doi: 10.1158/0008-5472.CAN-05-1716
55. Yasuda T, Koiwa M, Yonemura A, Miyake K, Kariya R, Kubota S, et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep. (2021) 34:108779. doi: 10.1016/j.celrep.2021.108779
56. Xu M, Zhang T, Xia R, Wei Y, Wei X. Targeting the tumor stroma for cancer therapy. Mol Cancer. (2022) 21:208. doi: 10.1186/s12943-022-01670-1
57. Gabai Y, Assouline B, Ben-Porath I. Senescent stromal cells: roles in the tumor microenvironment. Trends Cancer. (2023) 9:28–41. doi: 10.1016/j.trecan.2022.09.002
58. Varani J, Dame MK, Rittie L, Fligiel SE, Kang S, Fisher GJ, et al. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. (2006) 168:1861–8. doi: 10.2353/ajpath.2006.051302
59. Teissier T, Boulanger E, Cox LS. Interconnections between inflammageing and immunosenescence during ageing. Cells. (2022) 11:359. doi: 10.3390/cells11030359
60. Sharma S, Dominguez AL, Lustgarten J. High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J Immunol. (2006) 177:8348–55. doi: 10.4049/jimmunol.177.12.8348
61. Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, et al. Blood CD33(+)HLA-DR(-) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol. (2013) 93:633–7. doi: 10.1189/jlb.0912461
62. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. (2020) 20:89–106. doi: 10.1038/s41568-019-0222-9
63. Kumar S, Suman S, Fornace AJ, Datta K. Intestinal stem cells acquire premature senescence and senescence associated secretory phenotype concurrent with persistent DNA damage after heavy ion radiation in mice. Aging (Albany NY. (2019) 11:4145–58. doi: 10.18632/aging.102043
64. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. (2010) 102:1536–46. doi: 10.1093/jnci/djq364
65. Kehagias P, Kindt N, Krayem M, Najem A, Agostini G, Acedo Reina E, et al. Regorafenib induces senescence and epithelial-mesenchymal transition in colorectal cancer to promote drug resistance. Cells. (2022) 11:3663. doi: 10.3390/cells11223663
66. Fletcher-Sananikone E, Kanji S, Tomimatsu N, Di Cristofaro LFM, Kollipara RK, Saha D, et al. Elimination of radiation-induced senescence in the brain tumor microenvironment attenuates glioblastoma recurrence. Cancer Res. (2021) 81:5935–47. doi: 10.1158/0008-5472.CAN-21-0752
67. Guccini I, Revandkar A, D’Ambrosio M, Colucci M, Pasquini E, Mosole S, et al. Senescence reprogramming by TIMP1 deficiency promotes prostate cancer metastasis. Cancer Cell. (2021) 39:68–82.e9. doi: 10.1016/j.ccell.2020.10.012
68. Parkinson EK, James EL, Prime SS. Senescence-derived extracellular molecules as modulators of oral cancer development: a mini-review. Gerontology. (2016) 62:417–24. doi: 10.1159/000440954
69. Kriegl L, Neumann J, Vieth M, Greten FR, Reu S, Jung A, et al. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol. (2011) 24:1015–22. doi: 10.1038/modpathol.2011.43
70. Zhang Y, Guo L, Xing P, Chen Y, Li F, Zhu W, et al. Increased expression of oncogene-induced senescence markers during cervical squamous cell cancer development. Int J Clin Exp Pathol. (2014) 7:8911–6.25674264
71. Oh CW, Penneys N. P27 and mib1 expression in actinic keratosis, bowen disease, and squamous cell carcinoma. Am J Dermatopathol. (2004) 26:22–6. doi: 10.1097/00000372-200402000-00004
72. Pare R, Soon PS, Shah A, Lee CS. Differential expression of senescence tumour markers and its implications on survival outcomes of breast cancer patients. PLoS One. (2019) 14:e0214604. doi: 10.1371/journal.pone.0214604
73. Hida Y, Kubo Y, Arase S. Activation of fibroblast growth factor receptor 3 and oncogene-induced senescence in skin tumours. Br J Dermatol. (2009) 160:1258–63. doi: 10.1111/j.1365-2133.2009.09068.x
74. Bascones-Martínez A, López-Durán M, Cano-Sánchez J, Sánchez-Verde L, Díez-Rodríguez A, Aguirre-Echebarría P, et al. Differences in the expression of five senescence markers in oral cancer, oral leukoplakia and control samples in humans. Oncol Lett. (2012) 3:1319–25. doi: 10.3892/ol.2012.649
75. Leung EY, McMahon JD, McLellan DR, Syyed N, McCarthy CE, Nixon C, et al. DNA Damage marker phosphorylated histone H2AX is a potential predictive marker for progression of epithelial dysplasia of the oral cavity. Histopathology. (2017) 71:522–8. doi: 10.1111/his.13260
76. Tomo S, Biss SP, Crivelini MM, de Oliveira SHP, Biasoli É R, Tjioe KC, et al. High p16(INK4a) immunoexpression is not HPV dependent in oral leukoplakia. Arch Oral Biol. (2020) 115:104738. doi: 10.1016/j.archoralbio.2020.104738
77. Warnakulasuriya S, Kujan O, Aguirre-Urizar JM, Bagan JV, González-Moles M, Kerr AR, et al. Oral potentially malignant disorders: a consensus report from an international seminar on nomenclature and classification, convened by the WHO collaborating centre for oral cancer. Oral Dis. (2021) 27:1862–80. doi: 10.1111/odi.13704
78. Mello FW, Miguel AFP, Dutra KL, Porporatti AL, Warnakulasuriya S, Guerra ENS, et al. Prevalence of oral potentially malignant disorders: a systematic review and meta-analysis. J Oral Pathol Med. (2018) 47:633–40. doi: 10.1111/jop.12726
79. Iocca O, Sollecito TP, Alawi F, Weinstein GS, Newman JG, De Virgilio A, et al. Potentially malignant disorders of the oral cavity and oral dysplasia: a systematic review and meta-analysis of malignant transformation rate by subtype. Head Neck. (2020) 42:539–55. doi: 10.1002/hed.26006
80. Parkinson EK. Senescence as a modulator of oral squamous cell carcinoma development. Oral Oncol. (2010) 46:840–53. doi: 10.1016/j.oraloncology.2009.09.009
81. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. (2007) 8:729–40. doi: 10.1038/nrm2233
82. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. (2005) 436:642. doi: 10.1038/436642a
83. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. (2005) 436:660–5. doi: 10.1038/nature03841
84. McGregor F, Wagner E, Felix D, Soutar D, Parkinson K, Harrison PR. Inappropriate retinoic acid receptor-beta expression in oral dysplasias: correlation with acquisition of the immortal phenotype. Cancer Res. (1997) 57:3886–9.9307265
85. Niklander SE, Crane HL, Darda L, Lambert DW, Hunter KD. The role of icIL-1RA in keratinocyte senescence and development of the senescence-associated secretory phenotype. J Cell Sci. (2021) 134:1–16. doi: 10.1242/jcs.252080
86. Ortiz-Montero P, Londono-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. (2017) 15:17. doi: 10.1186/s12964-017-0172-3
87. Schwartz RE, Shokhirev MN, Andrade LR, Gutkind JS, Iglesias-Bartolome R, Shadel GS. Insights into epithelial cell senescence from transcriptome and secretome analysis of human oral keratinocytes. Aging (Albany NY). (2021) 13:4747–77. doi: 10.18632/aging.202658
88. Karen-Ng LP, Ahmad US, Gomes L, Hunter KD, Wan H, Hagi-Pavli E, et al. Extracellular prostaglandins E1 and E2 and inflammatory cytokines are regulated by the senescence program in potentially premalignant oral keratinocytes. Cancers (Basel. (2022) 14:2636. doi: 10.3390/cancers14112636
89. Castro-Vega LJ, Jouravleva K, Ortiz-Montero P, Liu WY, Galeano JL, Romero M, et al. The senescent microenvironment promotes the emergence of heterogeneous cancer stem-like cells. Carcinogenesis. (2015) 36:1180–92. doi: 10.1093/carcin/bgv101
90. Sharma M, Hunter KD, Fonseca FP, Radhakrishnan R. Emerging role of cellular senescence in the pathogenesis of oral submucous fibrosis and its malignant transformation. Head Neck. (2021) 43:3153–64. doi: 10.1002/hed.26805
91. Sharma M, Radhakrishnan R. Limited mouth opening in oral submucous fibrosis: reasons, ramifications, and remedies. J Oral Pathol Med. (2017) 46:424–30. doi: 10.1111/jop.12513
92. Sharma M, Shetty SS, Radhakrishnan R. Oral submucous fibrosis as an overhealing wound: implications in malignant transformation. Recent Pat Anticancer Drug Discov. (2018) 13:272–91. doi: 10.2174/1574892813666180227103147
93. Pitiyage GN, Slijepcevic P, Gabrani A, Chianea YG, Lim KP, Prime SS, et al. Senescent mesenchymal cells accumulate in human fibrosis by a telomere-independent mechanism and ameliorate fibrosis through matrix metalloproteinases. J Pathol. (2011) 223:604–17. doi: 10.1002/path.2839
94. Macha MA, Rachagani S, Pai P, Gupta S, Lydiatt WM, Smith RB, et al. MUC4 Regulates cellular senescence in head and neck squamous cell carcinoma through p16/Rb pathway. Oncogene. (2015) 34:1698–708. doi: 10.1038/onc.2014.102
95. Wiel C, Augert A, Vincent DF, Gitenay D, Vindrieux D, Le Calvé B, et al. Lysyl oxidase activity regulates oncogenic stress response and tumorigenesis. Cell Death Dis. (2013) 4:e855. doi: 10.1038/cddis.2013.382
96. Illeperuma RP, Kim DK, Park YJ, Son HK, Kim JY, Kim J, et al. Areca nut exposure increases secretion of tumor-promoting cytokines in gingival fibroblasts that trigger DNA damage in oral keratinocytes. Int J Cancer. (2015) 137:2545–57. doi: 10.1002/ijc.29636
97. Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. (1981) 41:1657–63.7214336
98. Hunter KD, Thurlow JK, Fleming J, Drake PJ, Vass JK, Kalna G, et al. Divergent routes to oral cancer. Cancer Res. (2006) 66:7405–13. doi: 10.1158/0008-5472.CAN-06-0186
99. Bienkowska KJ, Hanley CJ, Thomas GJ. Cancer-associated fibroblasts in oral cancer: a current perspective on function and potential for therapeutic targeting. Front Oral Health. (2021) 2:686337. doi: 10.3389/froh.2021.686337
100. Mellone M, Hanley CJ, Thirdborough S, Mellows T, Garcia E, Woo J, et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging (Albany NY). (2016) 9:114–32. doi: 10.18632/aging.101127
101. Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, et al. Stromal features are predictive of disease mortality in oral cancer patients. J Pathol. (2011) 223:470–81. doi: 10.1002/path.2830
102. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
103. Borghesan M, Fafian-Labora J, Eleftheriadou O, Carpintero-Fernandez P, Paez-Ribes M, Vizcay-Barrena G, et al. Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. (2019) 27:3956–3971.e6. doi: 10.1016/j.celrep.2019.05.095
104. Tan ML, Parkinson EK, Yap LF, Paterson IC. Autophagy is deregulated in cancer-associated fibroblasts from oral cancer and is stimulated during the induction of fibroblast senescence by TGF-β1. Sci Rep. (2021) 11:584. doi: 10.1038/s41598-020-79789-8
105. Kabir TD, Leigh RJ, Tasena H, Mellone M, Coletta RD, Parkinson EK, et al. A miR-335/COX-2/PTEN axis regulates the secretory phenotype of senescent cancer-associated fibroblasts. Aging (Albany NY). (2016) 8:1608–35. doi: 10.18632/aging.100987
106. Ritschka B, Knauer-Meyer T, Gonçalves DS, Mas A, Plassat JL, Durik M, et al. The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. (2020) 34:489–94. doi: 10.1101/gad.332643.119
107. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. (2016) 15:428–35. doi: 10.1111/acel.12445
108. Islam MT, Tuday E, Allen S, Kim J, Trott DW, Holland WL, et al. Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age. Aging Cell. (2023) 22:e13767. doi: 10.1111/acel.13767
109. Peilin W, Songsong T, Chengyu Z, Zhi C, Chunhui M, Yinxian Y, et al. Directed elimination of senescent cells attenuates development of osteoarthritis by inhibition of c-IAP and XIAP. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2618–32. doi: 10.1016/j.bbadis.2019.05.017
110. Fleury H, Malaquin N, Tu V, Gilbert S, Martinez A, Olivier MA, et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun. (2019) 10:2556. doi: 10.1038/s41467-019-10460-1
111. Saleh T, Carpenter VJ, Tyutyunyk-Massey L, Murray G, Leverson JD, Souers AJ, et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L) -BAX interaction. Mol Oncol. (2020) 14:2504–19. doi: 10.1002/1878-0261.12761
112. Peng X, Wu Y, Brouwer U, van Vliet T, Wang B, Demaria M, et al. Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis. (2020) 11:854. doi: 10.1038/s41419-020-03074-9
113. Nguyen TT, Ung TT, Li S, Lian S, Xia Y, Park SY, et al. Metformin inhibits lithocholic acid-induced interleukin 8 upregulation in colorectal cancer cells by suppressing ROS production and NF-kB activity. Sci Rep. (2019) 9:2003. doi: 10.1038/s41598-019-38778-2
114. Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. (2013) 4:2192. doi: 10.1038/ncomms3192
115. Lim JS, Lee DY, Kim HS, Park SC, Park JT, Kim HS, et al. Identification of a novel senomorphic agent, avenanthramide C, via the suppression of the senescence-associated secretory phenotype. Mech Ageing Dev. (2020) 192:111355. doi: 10.1016/j.mad.2020.111355
116. Lau L, Porciuncula A, Yu A, Iwakura Y, David G. Uncoupling the senescence-associated secretory phenotype from cell cycle exit via interleukin-1 inactivation unveils its protumorigenic role. Mol Cell Biol. (2019) 39:1–18. doi: 10.1128/MCB.00586-18
117. Schenk KM, Reuss JE, Choquette K, Spira AI. A review of canakinumab and its therapeutic potential for non-small cell lung cancer. Anticancer Drugs. (2019) 30:879–85. doi: 10.1097/CAD.0000000000000832
118. Sakoda K, Yamamoto M, Negishi Y, Liao JK, Node K, Izumi Y. Simvastatin decreases IL-6 and IL-8 production in epithelial cells. J Dent Res. (2006) 85:520–3. doi: 10.1177/154405910608500608
119. Niklander S, Bandaru D, Lambert DW, Hunter KD. ROCK Inhibition modulates the senescence-associated secretory phenotype (SASP) in oral keratinocytes. FEBS Open Bio. (2020) 10:2740–9. doi: 10.1002/2211-5463.13012
120. Niklander SE. Inflammatory mediators in oral cancer: pathogenic mechanisms and diagnostic potential. Front Oral Health. (2021) 2:642238. doi: 10.3389/froh.2021.642238
121. Wu D, Wu P, Zhao L, Huang L, Zhang Z, Zhao S, et al. NF-κB Expression and outcomes in solid tumors: a systematic review and meta-analysis. Medicine (Baltimore). (2015) 94:e1687. doi: 10.1097/MD.0000000000001687
122. Gu Y, Liu H, Kong F, Ye J, Jia X, Zhang Z, et al. miR-22/KAT6B axis is a chemotherapeutic determiner via regulation of PI3k-Akt-NF-kB pathway in tongue squamous cell carcinoma. J Exp Clin Cancer Res. (2018) 37:164. doi: 10.1186/s13046-018-0834-z
123. Niklander SE, Murdoch C, Hunter KD. IL-1/IL-1R signaling in head and neck cancer. Front Oral Health. (2021) 2:722676. doi: 10.3389/froh.2021.722676
Keywords: cellular senescence, oral cancer, mouth neoplasm, oral potentially malignant disorder, SASP, senotherapeutics
Citation: Niklander SE, Aránguiz P, Faunes F and Martínez-Flores R (2023) Aging and oral squamous cell carcinoma development: the role of cellular senescence. Front. Oral. Health 4:1285276. doi: 10.3389/froh.2023.1285276
Received: 29 August 2023; Accepted: 29 September 2023;
Published: 12 October 2023.
Edited by:
Abhimanyu Kumar Jha, Sharda University, IndiaReviewed by:
Elin Hadler-Olsen, UiT The Arctic University of Norway, Norway© 2023 Niklander, Aránguiz, Faunes and Martínez-Flores. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Eric Niklander c3Zlbi5uaWtsYW5kZXJAdW5hYi5jbA== René Martínez-Flores cmVuZS5tYXJ0JiN4MDBFRDtuZXpAdW5hYi5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.