 Flávia Caló Aquino Xavier1*
Flávia Caló Aquino Xavier1* Jamerson Carvalho Silva1
Jamerson Carvalho Silva1 Camila Oliveira Rodini2
Camila Oliveira Rodini2 Maria Fernanda Setubal Destro Rodrigues3
Maria Fernanda Setubal Destro Rodrigues3- 1Laboratory of Oral Surgical Pathology, School of Dentistry, Federal University of Bahia, Salvador, BA, Brazil
- 2Department of Biological Sciences, Bauru School of Dentistry, University of São Paulo, São Paulo, SP, Brazil
- 3Postgraduate Program in Biophotonics Applied to Health Sciences, Nove de Julho University (UNINOVE), São Paulo, SP, Brazil
Different mechanisms are involved in immune escape surveillance driven by Oral and Head and Neck Cancer Stem Cells (HNCSCs). The purpose of this review is to show the most current knowledge regarding the main impact of HNCSCs on tumor evasion through immunosuppression, CSCs phenotypes and environmental signals, highlighting strategies to overcome immune evasion. The main results drive the participation of cell surface receptors and secreted products and ligands, the crosstalk between cells, and genetic regulation. The reduction in CD8+ T cell recruitment and decreased effector of anti-PD-1 therapy by cells expressing BMI1 is a key event; Natural Killer cell ligands and cytokines needed for its activation and expansion are crucial to control tumor growth and to target CSCs by immunotherapy; CSCs expressing ALDH1 are related to increased expression of PD-L1, with a positive link between DNMT3b expression; CD276 expression in CSCs can act as a checkpoint inhibitor and together with Activator Protein 1 (AP-1) activation, they create continuous positive feedback that enables immune evasion by suppressing CD8+ T cells and prevent immune cell infiltration in head and neck cancer. These data demonstrate the relevance of the better understanding of the interaction between HNCSCs and immune cells in the tumor microenvironment. The ultimate clinical implication is to ground the choice of optimized targets and improve immune recognition for ongoing treatments as well as the response to approved immunotherapies.
Introduction
Cancer cells generally collapse the development of a specific antitumor immunity as a survival strategy, leading to “immune evasion mechanisms” that guarantee the success of tumor formation and progression [1]. Immune effector cells have cytotoxicity repressed in the tumor microenvironment due to different mechanisms driven by distinct cellular interactions and secreted factors [2]. In this context, recent studies have demonstrated that Cancer Stem Cells (CSCs) represent immune-privileged cells able to initiate tumor growth and mediate metastasis, tumor recurrence, and therapeutic resistance [3, 4].
CSCs are a non-immunogenic long-lived cell type that represents a relevant target cell population for mutations to occur until the development of an immune escape phenotype [5]. These cells survive all phases of the immune editing process, enabling them to efficiently modulate immune responses and avoid immune-mediated destruction [5]. CSCs can present a deficient expression of human leukocyte antigens (HLA)- A, B, and C and also antigen-processing machinery (APM) molecules in different types of cancer, which implicates in non-recognition by CD8+ T cells [4]. CSCs can avoid Natural Killer (NK) cytotoxicity by the low expression of ligands necessary for their activated state, like natural killer group 2D (NKG2D) ligands [4]. Therefore, the immune system defenses cannot defeat CSCs in the elimination phase. Consequently, these cells remain fully protected, achieving a dynamic balance between their quiescent state and augmented resistance to cell killing [6]. In this phase, CSCs acquire genetic and epigenetic alterations but are still niche confined and constrained by immune cells, which prevent the establishment of clinically relevant tumors [7]. However, less immunogenic and immunosuppressive CSCs clones emerge and expand in the equilibrium phase. When the immune system function is impaired by age, therapy, or disease, it uses this opportunity to divide itself. Asymmetric cellular division occurs during this process, and the recruitment of immunosuppressive cells to the tumor microenvironment (TME) favors rapid tumor growth [4].
In the TME, CSCs interact with different cell types to favor its immune evasion [8]. Dendritic cell (DC) recruitment, maturation, and differentiation are impaired by CSCs mainly via increased secretion of TGF-β, which leads to the downregulation of CD80, CD86, and MHC class II molecules in DC, which are responsible for the co-stimulatory activity, as well as the development of PD-L+ DC that contributes to immunotherapy resistance [9]. CSCs positively correlate with T regulatory lymphocytes (Treg), a population of CD4+ T cells that contribute to tumor stemness and progression mainly via inhibiting effector T cells and secretion of IL-4, IL-10, IL-35, and TGF-β, cytokines types with an immunosuppressive function [10, 11]. Moreover, the recruitment of myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAM) into the TME by CSCs and their constant interaction, contribute not only to the establishment of an immunosuppressive TME and increased expression of PD-1 and PD-L1 by T lymphocytes and CSCs, respectively, but also promotes CSCs maintenance and survival via different mechanisms, including activation of mTOR, NF-κB, STAT3, and Src signaling pathways and secretion of different cytokines [12–14]. It is also essential to highlight that HLA-I or low expression of the APM by CSCs is another relevant mechanism by which they are poor targets for T cell-mediated immune response [15].
Head and neck squamous cell carcinoma (HNSCC) is the sixth-ranked worldwide most common cancer, characterized by very aggressive behavior and poor prognosis [16, 17]. Conventional treatment is associated with morbidity, toxicity, and discrete improvement in overall survival [18]. HNSCC is a heterogeneous group of malignancies with their origin from different anatomic subsites. In addition, they present a diversity of risk factors and a broad molecular profile, imposing difficulties in the study and treatment of these tumors [17]. HNSCC shows, among other solid tumors, one of the most inflamed TME and has a high tumor mutation burden, which may benefit from immunotherapy strategies [19]. Recently, the use of nivolumab and pembrolizumab (anti-PD-1 immune checkpoint inhibitors) in patients with recurrent HNSCC has demonstrated improved outcomes compared to standard therapy [20].
However, single-agent strategies in immunotherapy have caused either temporary or lasting responses only in a minor subset of HNSCC patients [21]. Mapping how head and neck cancers overcome immune surveillance within TME provides optimal strategies to better deal with this tolerance [21]. Therefore, understanding the cross-talk between HNCSC and the immune system is extremely important as these cells directly impact tumor development, progression, and response to therapy. Thus, this mini-review aims to present how HNCSCs contribute to immune evasion leading to immunosuppression and to the emergence of genetic and epigenetic genotypes with immune privilege and points out some strategies to overcome immune evasion.
HNCSCs and escape from the host immune surveillance
CSCs maintain a cross-talk with immune cells in the TME to promote an immunosuppressive milieu that allows tumor development as a result of escape from the host's immune surveillance [22]. However, the mechanisms displayed by CSCs that enable their survival under immune vigilance during HNSCC tumorigenesis and metastasis are not well-established [23]. Recent studies demonstrate a dual function of CSCs in the immune system. First, the outgrowth of these cells can elicit immune system responses to destroy them. Instead, immunoediting generates CSCs to survive even in immunocompetent patients or provide necessary conditions within the TME, allowing tumor progression [24].
Different mechanisms involved in immune escape driven by Oral and HNCSCs are summarized in Table 1 and Figure 1. Wang et al. [23] demonstrated that expression of CD276 and Activator Protein 1 (AP-1) created continuous positive feedback to enable immune evasion, self-renewal, and metastasis by CSCs in HNSCC. Miao et al. [35] reported that CSCs express CD80 to interact with T lymphocytes after Transforming Growth Factor-beta (TGF-β) impulse. They also showed that CD80 surface ligand on CSCs directly inhibited T cells cytotoxicity and mediated resistance to approaches with immunotherapies. Gong et al. [33] revealed that worse survival was related to a higher expression of MX1 (MX dynamin-like GTPase 1) and relied on the amount of CD8+ T-cells in HNSCC, including oral cavity cancers. In the same study, a cancer-specific IFN-I receptor (IFNAR1) provided a stemness state and the release of exosomes derived from CSCs carrying receptor ligands associated with immune checkpoint function.

Table 1. The main immune evasion mechanisms and strategies to overcome immune evasion in oral and head and neck cancer stem cells.
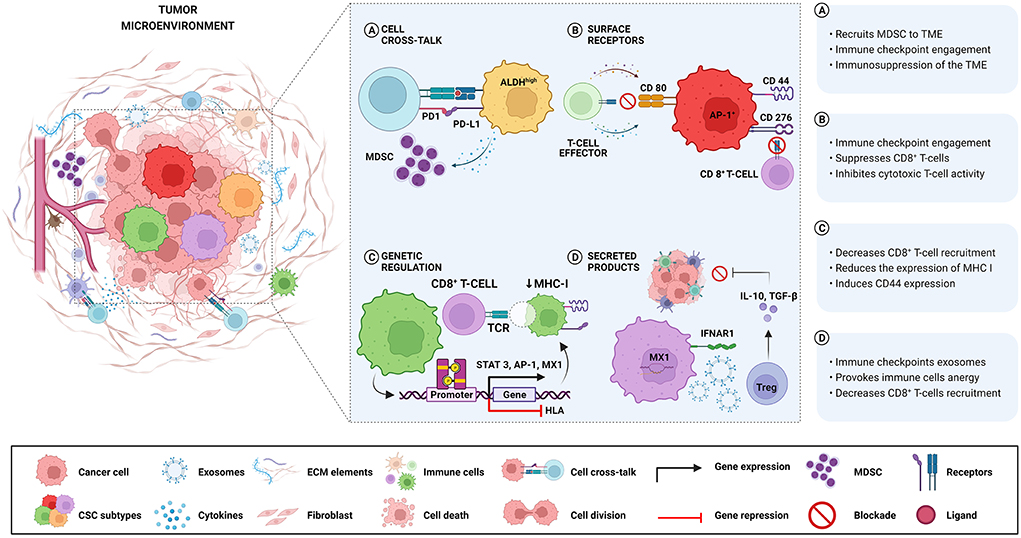
Figure 1. Different mechanisms involved in immune escape are driven by Oral and HNCSCs. (A) ALDHhigh-CSCs exhibit higher levels of PD-L1 and recruit MDSC with a suppressive role, causing negative regulation of immune responses in the TME. (B) CSCs expressing CD276 and AP-1 create a continuous positive feedback that enables immune evasion by suppressing CD8+ T cells. After TGF-β stimulation, CSCs express CD80 and inhibit T cell cytotoxicity leading to resistance to T cell immunotherapy. (C) The activation of the STAT3 pathway is related to the expression of PD-L1 in CD44+ cells, resulting in immune escape played by CSCs. Downregulation of HLA genes in CSCs decreases the expression of MHC class I causing non-recognition by T cells. (D) High expression of MX1 in CSCs decreases CD8+ T-cells infiltration concomitant with IFNAR1 expression associated with the release of exosomes containing immune checkpoint receptors. Noteworthy, the same CSCs can activate more than one immune avoidance mechanism.
A stemness profile has been linked to immune evasion. Cells with a CSCs phenotype undergoing EMT (Epithelial-Mesenchymal Transition) were correlated to ALDH1 activity [36]. Compared to ALDH negative cells, their positive counterparts demonstrated higher levels of PD-L1, with an increase after radiation treatment in OSCC [30]. Tsai et al. [30]. also evaluated the correlation between ALDH1 and MDSCs in OSCC. They reported that higher levels of PD-L1 were associated with tumors with ALDH1 positive expression in a combination of elevated levels of MDSCs. CD33+CD14+CD11b+HLA-DR− cells had their percentage increased in ALDH1+ tumors. Furthermore, epigenetic therapy by injecting DNA methyltransferase (DNMT) inhibitor 5-aza-2′-deoxycytidine (5-AZDC) attenuated the radiation-induced PD-L1 expression in tumors [30]. The authors proposed that MDSC recruitment and high PD-L1 in ALDH1+ tumors may be responsible for resistance to radiotherapy. Additionally, this could be reversed by agents with DNA hypomethylation effect as it knockdown CSC properties and radioresistance [30].
Furthermore, Prince et al. [29] used peripheral blood mononuclear cells (PBMCs) from HNSCC patients and demonstrated unique CSCs antigens in the ALDHhigh population cells isolated from the tumor specimen. The same research showed that DCs derived from PBMC and cultured in a preparation containing HNSCC ALDHhigh cells could elicit responses in autologous B and T lymphocytes. ALDHhigh CSCs induced antibodies and cytokine release and CTL activity. Hence, this strategy can guide CSC-DC vaccine production. Additionally, both humoral and cellular immunity against HNCSC was achievable, implying its potential for the treatment of HNSCC patients [29]. These findings were previously supported by Visus et al. [26] and indicate that CD8+ cells sensitized against ALDH1 positive HNCSC cells were able to target these cells and may contribute to tumor control. In fact, ALDH1A1-specific CD8+ T cells lysed ALDH+ cells, inhibiting tumor progression, and metastasis, and increasing the rate of survival of xenograft-bearing immunodeficient mice [26].
CD44 is another well-characterized marker associated with increased tumorigenesis, radioresistance, chemoresistance, and an immunosuppressive phenotype [28]. Lee et al. [28] showed that CD44+ cells were more immunosuppressive than their negative counterparts. This immunosuppression was partially switched when antibodies blocked the PD-1 receptor, suggesting a biologically and clinically relevant implication in PD-L1 expression between CD44+ and CD44− cells. A protein-coding gene known as STAT3 sustains a phosphorylated state in CD44+ cells and its blockade decreases the expression of PD-L1. Therefore, the STAT3 pathway may be related to the expression of PD-L1 in CD44+ cells, resulting in an immune escape played by CSCs [28].
Moreover, as CSCs decrease the expression of MHC class I to evade T cell recognition, they become susceptible to Natural Killer (NK) cells [37]. Thus, based on NK cells' capacity to target CSCs, Kaur et al. [31] proposed a new protocol describing a sustainable and durable expansion of NK cells stimulated by osteoclasts with cytotoxicity activity against oral squamous carcinoma stem cells (OSCSCs). Besides eliminating CSCs, the infusion with super-charged NK cells control tumor growth and induces stem-like differentiation in poorly differentiated tumors targeted by immunotherapy. Furthermore, an expansion of T cells was noted when co-cultured with DCs. In contrast, osteoclasts expand NK cells, suggesting that NK and T cells respond to different stimuli in the TME. These findings have a translational focus, facilitating future cancer immunotherapies. NK cells derived from HNSCC patients exhibited a distinct profile when compared to healthy patients, with lower cellular lyse ability and reduced secretion of cytokines. Moreover, ligands and crucial cytokines for NK activation and expansion were lower in OSCSCs [31]. Previously, Tseng et al. [25] demonstrated that NK cell-mediated cytotoxicity quickly targets OSCSCs compared to their differentiated counterpart. Co-cultures of CSCs with NK cells demonstrated increased IFN-γ and low levels of GM-CSF and interleukins (IL-6 and IL-8). Most importantly, OSCSCs expressed CD133 and CD44bright markers [25].
PD-L1 has shown a variable expression across patients with HNSCC, although success occurs in a subset of patients treated with therapies based on checkpoint inhibitors [38]. Wang et al. [23] demonstrated that the host immune vigilance is overcome when CSCs express other ligands related to the immune checkpoint, such as CD276 (B7-H3). CD276, as a stem cell marker, was suitable to isolate CSCs once it was expressed over this subpopulation of cells from mouse and human HNSCC. CD8+ lymphocytes recovered the cytotoxic potential against CSCs when anti-CD276 antibodies were infiltrated, hindering the spread of cells to lymph nodes and the tumor progression in animal models of HNSCC. Using Next-Generation Sequencing (NGS) techniques to better characterize this mechanism, the results showed that CD276 is crucial for CSCs immune evasion, and blockade with anti-CD276 eliminates these cells in a CD8+ T cell-dependent manner, in addition to remodeling HNSCC heterogeneity and decreasing EMT [23].
Discussion
Even lymph nodes where high-density immune cells exist serve as niches for CSCs metastasizing in HNSCC [39]. This can be partly explained by the low immune response and augmented capacity to induce immunosuppression due to immunoediting imposed on tumor cells by effectors of the immune system [4]. Understanding the mechanisms involved in the interaction of Oral and HNCSCs and the immune system is extremely important and has been one of the most challenging questions in the new era of Immunology. These cells directly affect tumor development, progression, and response to therapy. The evolution of efficacious immunotherapies for head and neck cancer is based upon a deep knowledge of antitumor immunity and how these tumors evade immune surveillance. Despite the diversity of immune cells present in the tumor stroma, HNSSC developed many mechanisms by which antitumor immunity can be thwarted [21].
Tumoral heterogeneity has been supported by the presence and the maintenance of cancer cells with stemness ability. The CSC model proposed in many studies suggests that this specific subpopulation of cells within the tumor is responsible for disease progression and relapse after standard treatments [22]. The tumor progression requires different abilities by the subclones, which are present in cells that share stemness profile in Oral and HNSCC, giving a particular marker signature diversity that could result from its functional and molecular plasticity [40]. BMI1 is a crucial marker abnormally expressed in cells from HNSCC that shows self-renewal capacity [41]. The absence of the normal immune response on TME is majorly due to the lack of activated CD8+T cells. As previously reported, cytotoxic T cell activity is defeated by CSCs CD80+ and even resists adoptive cytotoxic T cell transfer (ACT)-based immunotherapy [35]. Wang et al. [23] also demonstrated that CD80 was expressed in BMI1+ CSCs, with decreased cytotoxic effect in lymphocytes. Additionally, Gong et al. [33] reported that CSCs signaling pathways simultaneously occur with an intrinsic activation of Interferon alpha and beta receptor subunit 1(IFNAR1) signaling, affecting the anti-tumor stimulation function of stimulator of interferon response cGAMP interactor 1 (STING1) on CD8+ lymphocytes by induction of a hypo-responsiveness status in HNSCC. Therefore, for properly targeting CSCs, detecting a panel of tumor-associated antigens specific to CSCs will amplify the potential of combinational therapies to target these cells effectively in HNSCC [24].
Furthermore, NK cells can significantly lyse CSCs, as shown by Jewett et al. [27]. When co-cultured with OSCSCs, an increase in NK cell cytotoxicity was noted compared with oral squamous carcinoma cells (OSCCs) in a more differentiated state. In addition, the tumor progression is allowed by the absence of effective responses of NK cells and the deficient stimulus for the expansion of CD8 lymphocytes [31]. Thus, continuous infusion of allogeneic NK cells in the TME may benefit patients with OSCC and is essential to controlling tumor growth [27, 31]. Noteworthy, IFN-γ secreted by NK cells induces differentiation of the CSCs, inducing the expression of MHC class I, essential to CD8+ T cell target function [31].
Little is known about whether immunotherapy against immune checkpoints could target CSCs [23]. Studies of PD-L1 (also known as CD274) expression in CSCs have yielded contradictory results in HNSCC [42], indicating that CSCs targeted with anti-PD/PD-L1 antibodies may not be as affected as thought, and other immune checkpoints can be involved. Wang et al. [23] recently showed that CSCs expressing CD276 in HNSCC orchestrate immune vigilance toward tumor initiation, progression, and metastasis. CD276 hindrance CD8+ T cells anti-tumor effects were improved with the elimination of CSCs by anti-CD276 immunotherapy, thus inhibiting tumor growth and spread. Importantly, CD276 blockade significantly inhibited lymph node metastasis of HNSCC, enhancing anti-tumor immunity [23]. Although CD276 was found to be upregulated in HNSCC, the molecular mechanism controlling CD276 expression remains unclear [23].
Targeting CSCs markers in HNSCC by PD1 blockade immunotherapy promotes CD8+ cell infiltration and improves cisplatin response [23, 34]. The anti-PD therapies applied to patients with HNSCC depend on the activated pathway, but in some cases, immune-related adverse events can occur, leading to treatment failure [1]. Since CSCs cause some of these events, identifying specific defects in the antitumor immune response and combining different target therapy approaches may improve treatment [1, 32, 34]. In this context, it is important to better characterize the CSCs markers in HNSCC to define optimized targets and improve ongoing treatments, especially OSCC immunotherapy. Following this idea, Tsai et al. [30] analyzed the link between ALDH1, PD-L1, and circulating MDSCs by FACS. These authors revealed ALDH1-positive tumors with high levels of circulating MDSCs, significantly incremented after radiotherapy. Furthermore, ALDH1-expressing tumor cells had higher PD-L1 expression, which was enhanced by radiation [30]. Wang et al. [23] also demonstrated in HNSCC an association between BMI1+ and CD276high CSCs in the invasive tumor front, supporting that this phenotype of CSC in invasive niches might be controlled by molecular mechanisms.
In summary, in head and neck cancer, NK and T cells respond to different stimuli provided by the CSCs and other stromal cells on the TME and usually have limited anti-tumoral activity due to immunosuppression. The absence of the normal immune response on the TME is mainly due to the lack of activated CD8+ T cells and low expression of ligands and cytokines needed for NK cell activation, cytotoxicity, and expansion. These findings highlight the relevance of multimodality therapies that disrupt the TME to release NK and T cells in head and neck cancer from an unfavorable immune condition. As the CSCs are relevant actors in tumor development and progression and show immune privileges in the TME, combining different therapy approaches targeting CSCs and immunotherapy may contribute to achieving better clinical results in head and neck cancer patients.
Author contributions
FX, JS, CR, and MR contributed to the conception and design of the study. FX and JS organized the database. FX wrote the first draft of the manuscript. JS, CR, and MR wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Acknowledgments
Figure 1 was created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sanmamed MF, Chen LA. Paradigm shift in cancer immunotherapy: from enhancement to normalization [published correction appears in Cell. 2019 Jan 24; 176(3):677.]. Cell. (2018) 175:313–26. doi: 10.1016/j.cell.2018.09.035
2. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. (2015) 35(Suppl):S185–98. doi: 10.1016/j.semcancer.2015.03.004
3. Sultan M, Coyle KM, Vidovic D, Thomas ML, Gujar S, Marcato P, et al. Hide-and-seek: the interplay between cancer stem cells and the immune system. Carcinogenesis. (2017) 38:107–18. doi: 10.1093/carcin/bgw115
4. Galassi C, Musella M, Manduca N, Maccafeo E, Sistigu A. The immune privilege of cancer stem cells: a key to understanding tumor immune escape and therapy failure. Cells. (2021) 10:2361. doi: 10.3390/cells10092361
5. Lei MML, Lee TKW. Cancer stem cells: emerging key players in immune evasion of cancers. Front Cell Dev Biol. (2021) 9:692940. doi: 10.3389/fcell.2021.692940
6. Bruttel VS, Wischhusen J. Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front Immunol. (2014) 5:360. doi: 10.3389/fimmu.2014.00360
7. Bhatia A, Kumar Y. Cancer stem cells and tumor immunoediting: putting two and two together. Expert Rev Clin Immunol. (2016) 12:605–7. doi: 10.1586/1744666X.2016.1159133
8. Tsuchiya H, Shiota G. Immune evasion by cancer stem cells. Regen Ther. (2021) 17:20–33. doi: 10.1016/j.reth.2021.02.006
9. Zhong M, Zhong C, Cui W, Wang G, Zheng G, Li L, et al. Induction of tolerogenic dendritic cells by activated TGF-β/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer. (2019) 19:439. doi: 10.1186/s12885-019-5670-9
10. Oh E, Hong J, Yun CO. Regulatory T cells induce metastasis by increasing Tgf-β and enhancing the epithelial–mesenchymal transition. Cells. (2019) 8:1387. doi: 10.3390/cells8111387
11. Liu S, Zhang C, Wang B, Zhang H, Qin G, Li C, et al. Regulatory T cells promote glioma cell stemness through TGF-β-NF-κB-IL6-STAT3 signaling. Cancer Immunol Immunother. (2021) 70:2601–16. doi: 10.1007/s00262-021-02872-0
12. Jinushi M. Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic microenvironments. Oncoimmunology. (2014) 3:e28862. doi: 10.4161/onci.28862
13. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. (2014) 352:160–8. doi: 10.1016/j.canlet.2014.05.008
14. Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. (2016) 18:632–44. doi: 10.1038/ncb3355
15. Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of cancer-initiating cells. BMC Cancer. (2018) 18:469. doi: 10.1186/s12885-018-4389-3
16. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, et al. Cancer statistics for the year 2020: An overview. Int J Cancer. (2021) 149:778–89. doi: 10.1002/ijc.33588
17. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR, et al. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. (2020) 6:92. doi: 10.1038/s41572-020-00224-3
18. Szturz P, Cristina V, Herrera Gómez RG, Bourhis J, Simon C, Vermorken JB, et al. Cisplatin eligibility issues and alternative regimens in locoregionally advanced head and neck cancer: recommendations for clinical practice. Front Oncol. (2019) 9:464. doi: 10.3389/fonc.2019.00464
19. Ferris RL. Immunology and immunotherapy of head and neck cancer. J Clin Oncol. (2015) 33:3293–304. doi: 10.1200/JCO.2015.61.1509
20. Cohen EEW, Bell RB, Bifulco CB, Burtness B, Gillison ML, Harrington KJ, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J Immunother Cancer. (2019) 7:184. doi: 10.1186/s40425-019-0662-5
21. Horton JD, Knochelmann HM, Day TA, Paulos CM, Neskey DM. Immune evasion by head and neck cancer: foundations for combination therapy. Trends Cancer. (2019) 5:208–32. doi: 10.1016/j.trecan.2019.02.007
22. Wilczyński JR, Wilczyński M, Paradowska E. Cancer stem cells in ovarian cancer-a source of tumor success and a challenging target for novel therapies. Int J Mol Sci. (2022) 23:2496. doi: 10.3390/ijms23052496
23. Wang C, Li Y, Jia L, Kim JK, Li J, Deng P, et al. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell. (2021) 28:1597–613.e7. doi: 10.1016/j.stem.2021.04.011
24. Qian X, Ma C, Nie X, Lu J, Lenarz M, Kaufmann AM, et al. Biology and immunology of cancer stem(-like) cells in head and neck cancer. Crit Rev Oncol Hematol. (2015) 95:337–45. doi: 10.1016/j.critrevonc.2015.03.009
25. Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A, et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS ONE. (2010) 5:e11590. doi: 10.1371/journal.pone.0011590
26. Visus C, Wang Y, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8? T cells. Clin Cancer Res. (2011) 17:6174–84. doi: 10.1158/1078-0432.CCR-11-1111
27. Jewett A, Tseng HC, Arasteh A, Saadat S, Christensen RE, Cacalano NA, et al. Natural killer cells preferentially target cancer stem cells; role of monocytes in protection against NK cell mediated lysis of cancer stem cells. Curr Drug Deliv. (2012) 9:5–16. doi: 10.2174/156720112798375989
28. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, et al. CD44+ Cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res. (2016) 22:3571–81. doi: 10.1158/1078-0432.CCR-15-2665
29. Prince MEP, Zhou L, Moyer JS, Tao H, Lu L, Owen J, et al. Evaluation of the immunogenicity of ALDH(high) human head and neck squamous cell carcinoma cancer stem cells in vitro. Oral Oncol. (2016) 59:30–42. doi: 10.1016/j.oraloncology.2016.05.013
30. Tsai MS, Chen WC, Lai CH, Chen YY, Chen MF. Epigenetic therapy regulates the expression of ALDH1 and immunologic response: relevance to the prognosis of oral cancer. Oral Oncol. (2017) 73:88–96. doi: 10.1016/j.oraloncology.2017.08.007
31. Kaur K, Cook J, Park SH, Topchyan P, Kozlowska A, Ohanian N, et al. Novel strategy to expand super-charged NK cells with significant potential to lyse and differentiate cancer stem cells: differences in NK expansion and function between healthy and cancer patients. Front Immunol. (2017) 8:297. doi: 10.3389/fimmu.2017.00297
32. Jia L, Zhang W, Wang CY. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. (2020) 27:238–53.e6. doi: 10.1016/j.stem.2020.06.022
33. Gong W, Donnelly CR, Heath BR, Bellile E, Donnelly LA, Taner HF, et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology. (2021) 10:1997385. doi: 10.1080/2162402X.2021.1997385
34. Jia L, Wang Y, Wang C. circFAT1 promotes cancer stemness and immune evasion by promoting STAT3 activation. Adv Sci. (2021) 8:2003376. doi: 10.1002/advs.202003376
35. Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour MS, et al. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell. (2019) 177:1172–86.e14. doi: 10.1016/j.cell.2019.03.025
36. Qian X, Wagner S, Ma C, Coordes A, Gekeler J, Klussmann JP, et al. Prognostic significance of ALDH1A1-positive cancer stem cells in patients with locally advanced, metastasized head and neck squamous cell carcinoma. J Cancer Res Clin Oncol. (2014) 140:1151–8. doi: 10.1007/s00432-014-1685-4
37. Shokouhifar A, Firouzi J, Nouri M, Sarab GA, Ebrahimi MNK. cell upraise in the dark world of cancer stem cells. Cancer Cell Int. (2021) 21:682. doi: 10.1186/s12935-021-02400-1
38. Ferris RL, Blumenschein GJr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
39. Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y, et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J Clin Invest. (2016) 126:169–80. doi: 10.1172/JCI82066
40. Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, et al. The immune landscape of viral and carcinogen-driven head and neck cancer. Immunity. (2020) 52:183–99.e9. doi: 10.1016/j.immuni.2019.11.014
41. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. (2012) 30:372–8. doi: 10.1002/stem.1035
Keywords: head and neck squamous cell carcinoma (HNSCC), cancer stem cell (CSC), immune surveillance, immune evasion, immunotherapy
Citation: Xavier FCA, Silva JC, Rodini CO and Rodrigues MFSD (2022) Mechanisms of immune evasion by head and neck cancer stem cells. Front. Oral. Health 3:957310. doi: 10.3389/froh.2022.957310
Received: 30 May 2022; Accepted: 14 July 2022;
Published: 02 August 2022.
Edited by:
Babak Baban, Augusta University, United StatesReviewed by:
Évila Salles, Augusta University, United StatesCopyright © 2022 Xavier, Silva, Rodini and Rodrigues. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flávia Caló Aquino Xavier, ZmNhbG9AdWZiYS5icg==