 Thomas Duhen
Thomas Duhen Michael J. Gough
Michael J. Gough Rom S. Leidner
Rom S. Leidner Sasha E. Stanton
Sasha E. Stanton- Earle A. Chiles Research Institute, Providence Cancer Institute, Portland, OR, United States
The clinical response to cancer therapies involves the complex interplay between the systemic, tumoral, and stromal immune response as well as the direct impact of treatments on cancer cells. Each individual's immunological and cancer histories are different, and their carcinogen exposures may differ. This means that even though two patients with oral tumors may carry an identical mutation in TP53, they are likely to have different pre-existing immune responses to their tumors. These differences may arise due to their distinct accessory mutations, genetic backgrounds, and may relate to clinical factors including previous chemotherapy exposure and concurrent medical comorbidities. In isolation, their cancer cells may respond similarly to cancer therapy, but due to their baseline variability in pre-existing immune responses, patients can have different responses to identical therapies. In this review we discuss how the immune environment of tumors develops, the critical immune cell populations in advanced cancers, and how immune interventions can manipulate the immune environment of patients with pre-malignancies or advanced cancers to improve therapeutic outcomes.
Introduction
The local and systemic immune status of the patient plays a role in the response to conventional therapy for head and neck cancer (HNSCC). Therefore, it is critical to understand how the tumor develops both genetically and immunologically. It is in this context that this review examines the developing immune biology of HNSCC. While head and neck cancers can be found in the mucosal epithelium of the oral cavity, larynx, and pharynx, the oral cancers are predominantly associated with tobacco and alcohol use while tumors in the larynx and pharynx have been increasingly associated with HPV infection [1]. In addition, it is critical to understand whether the pre-existing immune environment can be manipulated to permit a more favorable response to treatment. From this perspective, the goal of immune manipulation is to render conventional therapies more effective in patients. Of course, it is sometimes possible to control tumors using only the immune system, but for the foreseeable future, immunotherapy will remain one element of treatment, as the fourth modality alongside some combination of the chemotherapy, surgery, and radiation therapy that is optimized to treat and cure HNSCC.
Evidence for immune involvement in treatment outcomes
A good example of the impact of immune cells on treatment outcome is that HNSCC patients with increased immune infiltrate, particularly of T cells, have improved overall and cancer free survival. This includes oral squamous cell carcinoma (OSCC) which is typically HPV negative (HPV-) and induced by carcinogen exposure (such as alcohol and cigarette exposure) [2, 3]. In 119 patients with HPV- OSCC, increased infiltrations of CD8 T cells at the invasive margin (IM) was associated with increased overall survival following treatment [2]. In 94 patients with OSCC, a higher density of CD3+ T cells in both the invasive margin (IM) and center of the tumor (CT) were associated with lower stage (T1 and T2 tumors) and increased CD8+ and CD4+ T cells in the invasive margin predicted lower risk of axillary metastases [3]. In a study of 328 OSCC in the TCGA, 7 features associated with improved prognosis, both disease free and overall survival (DFS and OS, respectively), included CD8 T cells in the IM, CD45 RO in IM, CD11b in IM and CT, CD20 in the CT, FOXP3 in the CT, and PD1 in the CT. These seven features predicted better DFS and OS than other clinical and pathologic features including gender, age, smoking status, alcohol use, grade, and stage with AUC 0.755 in the discovery cohort and 0.741 in the training cohort where all other features had AUC < 0.7 [4]. These data correlate well with the earlier multiparametric analysis of immune infiltrates by immunohistology [2], where the combination of a range of immune measures including the relative location of suppressive cells generated a cumulative suppression index that showed potential to predict patient outcome [2]. In each study, the patients were treated with conventional cancer therapies, therefore the response to surgery, radiation therapy, and chemotherapy is impacted by the immune environment of the tumor at presentation.
Opportunities to intervene to improve immune-guided prognosis
Despite this predictive and prognostic data, pre-treatment immune infiltrates do not currently alter the choice of conventional therapies given to HNSCC patients. Should we identify a patient predicted to have a poor response, we have two major options: Option 1 is to intensify treatment, or, if the predictive power is sufficiently strong and negative, perhaps palliation. Option 2 is to change the tumor so that the standard therapy becomes effective; for example, if there are few T cells in the tumor, can we not provide more? The reality is that option 2 reveals our limitations in understanding of the immune mechanisms that generate the pre-existing tumor environment at the time of treatment and in our capacity to meaningfully shape the contours of that environment for clinical benefit. There are interventions that can result in non-specific margination of T cells from the peripheral blood into peripheral tissues, such as systemic delivery of high dose IL-2. However, T cells specific for mutations in cancer are rare in the peripheral blood [5]. If the majority of T cells in the peripheral blood recognize CMV, EBV, or other common infectious agents, why would increasing the number of these cells in tumors impact patient outcome? As we will discuss, often the presence of T cell subpopulations that are enriched for tumor-specificity are more important than the infiltration of non-specific T cells into the tumor [6]. It seems rational that to meaningfully alter the outcome of a patient with poor T cell infiltration to the tumor, tumor specific T cells may need to be generated, expanded, and/or targeted to the tumor environment. Relatedly, the majority of T cells in HNSCC are found within the tumor stroma [2] rather than in contact with cancer cells, and these T cells may recirculate through the tumor environment without direct interactions with the nests of cancer cells [reviewed in Blair et al. [7]]. We may also need to consider local T cell support mechanisms such as local antigen presentation, and the differentiation signals that generate a resident tumor-specific T cell population in tumors. We should also consider the possibility that the T cells may not be the appropriate target that for manipulation to increase the accumulation of antigen-specific cells in the tumor. While this sounds counter-intuitive, tumors develop an array of immune regulatory populations that impact the ability of T cells to kill cancer cells that they recognize [reviewed in Medler et al. [8] and Tormoen et al. [9]]. Therapies that target macrophages and fibroblasts are outstanding as combination therapies in pre-clinical models, though to date they have not impacted clinical practice. These data mean that while sending a transient pulse of T cells to a tumor might increase overall infiltrates, this may not be impactful if they cannot engage with antigen, receive local cytokine support, or withstand local immune suppression that forms a critical part of the tumor stromal environment.
For example, fibroblasts that form part of the tumor stroma in HNSCC have been demonstrated to differentiate into suppressive phenotypes that associate with patient outcome [10]. While suppressive fibroblast populations were shown to impact T cell phenotypes ex vivo [10], cause and effect of suppressive fibroblast differentiation are difficult to separate in vivo, and targeting markers of specific fibroblast populations has not been impactful in our hands [11]. Fibroblast differentiation in tumors is regulated by a range of factors that also impact myeloid and T cell biology [12], and it seems likely that specific fibroblast phenotypes are reflective of broader immunoregulation of the tumor immune environment. For example, while TGFb is a potent modulator of macrophage and fibroblast phenotypes [13], genetic knockout of TGFb receptor components in different populations demonstrated that blocking TGFb responses only in T cells was sufficient to improve tumor control in pre-clinical models [14]. Similarly, cancer cells can dictate an immune environment that impacts multiple infiltrating cells via T cell responses. Murine HNSCC cancer cell lines selected for poor responsiveness to PD1 blockade exhibited downregulation of antigen processing and presentation, resulting in a shift from M1 to M2 macrophage differentiation and increased T reg infiltration [15]. In these models, T cell targeted interventions were sufficient to reshape the immune environment of the tumors and improve tumor control [15]. This is consistent with prior data demonstrating a reciprocal relationship between T cell and myeloid populations in tumors, and that T cell-specific immune interventions can reshape the tumor environment [16, 17]. Of course, not all immune interventions are sufficient to overcome the suppressive immune environment of tumors, and combination therapies are critical for patients that do not respond to monotherapies [18]. As we will discuss, specific immune environments of HNSCC are associated with improved outcome following conventional treatment of patients with modalities including surgery, chemotherapy, and radiation therapy [2, 3, 6, 19]. Similarly, there are immune environments that predict responses to immune therapies in many cancers, though at present few features have been validated as prognostic in HNSCC [20, 21]. We discuss how T cell targeted immune interventions can remodel the tumor immune environment such that even where they are not curative alone, they can potentially alter the response of cancer patients to conventional cancer therapies.
Long term impact of early intervention
It is reasonable to act early to regulate the immune environment of tumors. This is most clear in HPV positive (HPV+) cancers, where preventative vaccination is successfully blocking oral HPV infection [22], and therefore has the potential to block HPV-related tumorigenesis [23]. This preventative immunotherapy can be considered as one of a range of intervention points to direct immune responses to control HNSCC (Figure 1). If preventative measures are not possible, immune therapies can be targeted to early dysplasia to prevent further transformation (Figure 1). Finally, understanding the immune features that are prognostic in patient tumors can be used to guide interventions in high-risk patients to control tumor development. Even where these interventions fail to prevent malignancies, they may result in more treatable malignancies due to their impact on the immune environment of the developing tumor (Figure 1). Many immunotherapy candidates have proven ineffective as single agent cancer therapies, but may ultimately be used to induce the changes needed to alter the immune trajectory of conventional therapy. High quality immune monitoring combined with a deeper understanding of the key features of the tumor immune environment are needed to identify such agents. We review strategies to manipulate immunity during HNSCC tumorigenesis, the critical features of the tumor T cell environment that dictates outcome in HNSCC patients, how pre-existing immune environments impact conventional cancer therapy, and how we can alter the immune environment of tumors to support novel T cell therapies.
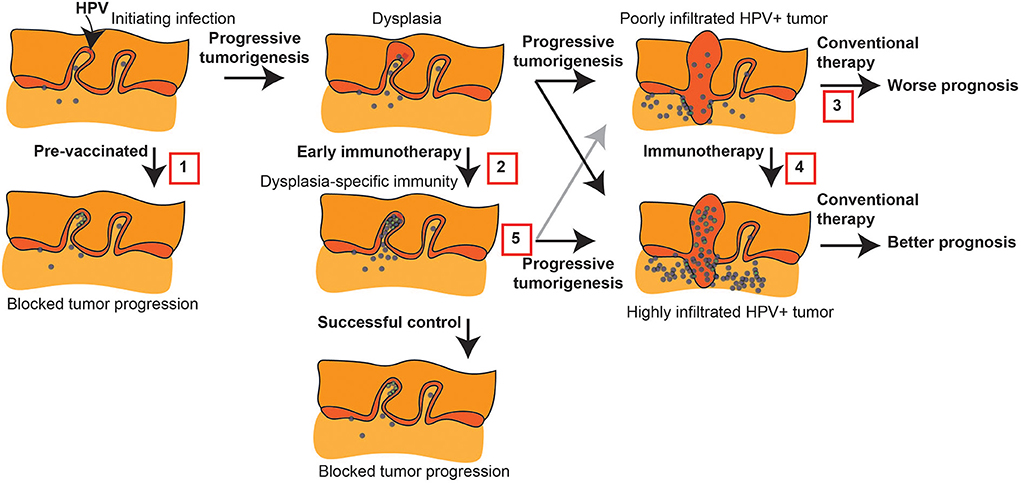
Figure 1. Immune intervention during tumorigenesis and prior to conventional therapy. HPV+ tumorigenesis provides a model to show potential interventions prior to conventional therapy for HNSCC that can impact outcome. 1. Protective HPV vaccination can prevent the initial transformation that can result from HPV infection. 2. Early immunotherapy of dysplasia can enhance HPV-specific responses or direct new immune responses to genes involved in progression to prevent further progression. 3. Malignant tumors can have a poor pre-existing immune response that is associated with poor prognosis following conventional therapy. 4. Immunotherapy can convert the tumor environment by directing immune responses to the tumor, and this has the potential to convert patients to an improved prognosis. 5. Even where early immunotherapy fails to prevent tumorigenesis, it may result in a tumor with a strong-pre-existing immune response that would be predicted to have a better prognosis following conventional therapy.
Manipulating immunity during tumorigenesis
Immune environments during tumorigenic progression
As discussed above, increased immune infiltrate, particularly of T cells, are associated with improved overall and cancer free survival in patients with HNSCC [2, 3]. However, T cell infiltrates are only one feature of a complex pattern of infiltrating cells that can also impact outcome [2, 4]. The immune system's recognition of the tumor develops early in oral dysplasia and leukoplakia. In a study evaluating RNAseq for 19 patients with normal mucosa, dysplasia, and invasive tumor, immune gene expression was highest in oral dysplasia. In oral dysplasia the cytotoxic effector cell genes (including granzyme A and B) had the highest expression while in advanced disease there was highest expression of chronic inflammation (macrophages, neutrophils, and Th2 T cells) [24, 25]. There is both a developing innate and adaptive immune response starting in dysplasia that may be important in determining whether the pre-malignant lesion will progress to invasive disease. HNSCC has 4 subtypes: classical, basal, atypical, and mesenchymal that are associated with different clinical outcomes [26]. There are also two subtypes of oral dysplasia: the immune subtype is associated with an inflamed immune environment with increased CD8 T cells, monocytic dendritic cells, and macrophages and the classical subtype associated with EGFR overexpression and increased loss of heterozygosity. It is this second classical subtype, not the immune subtype, that is typically associated with progression to invasive disease and the genetic instability associated with carcinogen exposure; however, there are no specific differences in smoking or alcohol exposure between the two subtypes [27]. In progression from oral dysplasia to HNSCC, regulatory T cells, associated with immunosuppression and cancer progression, increase in proportion in the immune infiltrate [28, 29]. Oral leukoplakia is a very early oral lesion with ~3% risk of malignant transformation while proliferative leukoplakia has ~10% risk of malignant transformation. In 58 patients (29 with localized leukoplakia and 29 with proliferative leukoplakia) assessed using Nanostring digital spatial profiling, proliferative leukoplakia shows increased immune infiltrate over localized leukoplakia [30]. The proliferative leukoplakia (regardless of the degree of dysplasia) had increased expression of immune activation markers including cytotoxic T cells, B cells, and NK cells as well as increased stromal PDL1 expression [30]. In HNSCC there has been an association of FOXP3+ CD4+ T cells with improved prognosis [31, 32]. In the 4-nitroquinoline 1-oxide (4-NQO) carcinogen model of HNSCC, transiently knocking out FOXP3 cells in mice that express FOXP3 under a human diphtheria receptor caused increased T cell infiltrate in the tumor but 2.5 times increase in progression to HNSCC [33]. These data demonstrate that the immune environment of dysplasia is less immunosuppressive than invasive disease and suggest a potential pre-malignant use of immune therapies in oral dysplasia, as discussed below.
As could be expected with increased immune recognition with progressive oral lesions, increased immune checkpoint expression can be seen starting in dysplasia and increasing in invasive cancer. In leukoplakia, using the combined positive score assessment of PDL1 expression, high PDL1 expression predicted lower 5-year cancer free survival, with 70% of tumors expressing low PDL1 and 37% expressing high PDL1 [30]. While 93% (26/27) of the local leukoplakia had low PDL1, only 21% (6/28) proliferative leukoplakia tumors had low PDL1 [30]. In the 4-NQO mouse model of oral tumorigenesis, treatment with systemic anti-PD-1 immune checkpoint inhibition prevented progression [34]. Similarly, in the K14-Cre mutTP53fl/fl transgenic mouse model of HNSCC treated with oral 4NQO, local delivery of anti-PD-1 immune checkpoint inhibition loaded on a hydrogel prevented progression to HNSCC [35]. Based on these data, there are several clinical trials ongoing to determine whether immune checkpoint inhibitor therapy could prevent invasive disease in patients with dysplastic lesions: NCT03692325 (nivolumab), NCT03603223 (pembrolizumab), and NCT04504552 (avelumab).
Myeloid targets in tumorigenesis
While much of the literature has focused on the changes in adaptive immune response during progression from dysplasia to invasive disease, the innate immune response also plays a role in HNSCC development. In evaluating the immune infiltrate in the epithelium and stroma between untreated oral mucosa, dysplasia, and invasive disease in the 4NQO mouse model by RNA sequencing, the largest differences in immune infiltrate were between normal tissue and dysplasia levels of M2 macrophages (CD163+ macrophages) in both the epithelium and stroma. In normal tissue M2 macrophages were 19.1% of the immune population but in dysplasia they were 52.6%. Based on the mouse studies, an M2 macrophage gene signature was then evaluated in patients. In 86 patients with oral leukoplakia, the M2 macrophage signature was associated with improved cancer free survival [36]. The role of macrophages in progression has not been consistent in the literature because in a second study of 45 patients with oral epithelia precursor lesions and 82 patients with OSCC, the presence of M2 macrophages in precursor lesions were associated with risk of progression to OSCC [37]. The presence of another innate population, dendritic cells (DC), also is important in the tumor immune environment. DC are exclusively responsible for cross-presentation of tumor-associated antigens to CD8 T cells and can integrate innate immune signals and CD4 T cell responses to maximize tumor-specific CD8 T cell expansion. When comparing 48 oral dysplasia and 50 OSCC patients, there was a decrease in mature CD83+ Langerhans DC in the OSCC samples as compared to the oral dysplasia samples. In contrast, there was an increased plasmacytoid DC infiltrate in the OSCC as compared to dysplasia. The decreased CD83+ DC were associated with a decrease in CD8+ T cell infiltrate in the OSCC suggesting that the reduction in mature DC could result in lesser T cell recognition of the tumor and thus allowing immune escape [38]. The developing type II innate immune response and decreased mature antigen presenting cells found in the invasive cancer suggest that it is better able to escape immune detection as compared to dysplasia.
Immune interventions to prevent tumorigenesis
With the high risk of recurrence of dysplastic lesions and risk of progression to invasive disease, vaccine strategies have been evaluated for HNSCC prevention. The vaccine strategies are separated into HPV-positive dysplasia and HPV-negative dysplasia. The HPV vaccines have been shown to prevent invasive cervical cancer in patients with cervical dysplasia and studies are currently ongoing to evaluate prevention of HNSCC in patients with HPV-positive oral dysplasia [39]. However, HPV-negative dysplasia does not benefit from this prevention therapy as there are no viral antigens to target. Tumor associated antigens including CEA, antigens identified by SEREX screens, or cancer testis antigens have been evaluated as target antigens for HPV-negative HNSCC [40]. In pre-clinical modeling, DC were pulsed with pre-malignant cell lysate and then infused in 4-NQO treated mice with oral dysplasia. The DC vaccine induced a pro-inflammatory immune response and the mice developed significantly fewer invasive lesions as compared to control mice (p < 0.001) [41].
In addition to direct immune targets such as PD1 as discussed above [34, 35], the cancer-targeted interventions intended to block the progression to malignancy may exploit immune mechanisms. Overexpression of EGFR can predict progression to malignancy in pre-malignant oral lesions [42]. The monoclonal anti-EGFR antibody, cetuximab, given with erlotinib for secondary prevention in patients with stage I/II HNSCC (NCT00400374) showed 71% pathologic complete response at the time of surgery, but long-term recurrence has not been reported and this combination does have considerable toxicities [43]. When erlotinib was evaluated in a randomized trial against placebo in high grade oral dysplasia (EPOC trial), there was no difference in oral cancer free survival. Despite this, there is still significant clinical and pre-clinical efforts to identify therapies for dysplasia to prevent invasive disease and improve clinical outcomes.
These data demonstrate that the immune environment of advanced cancers is developed through tumorigenesis and revealed at diagnosis. There are multiple strategies that can be incorporated to modify the contours of the immune environment during progression, which may mitigate subsequent emergence of advanced disease. Even where tumors still progress, it remains possible that those cancers that emerge may have a distinct immune profile which may alter their response to conventional cancer therapies. While it is difficult to identify tumors early, it is hopeful that the lessons from immunotherapy of pre-malignant disease may be applied to change the environment of patients with advanced cancers.
T cell environment of responders
Tumor-specific T cell subsets in HNSCC
As introduced above, T cell infiltration in the tumor environment is associated with better OS in oral cancers. However, initial studies did not characterize the T cell compartment to identify which T cell populations responsible for tumor growth control. Most studies have broadly focused on CD8 T cells and their role in the response against cancer. This is due in part to their capacity to recognize and kill tumor cells that present tumor antigen-derived peptides on MHC class I molecules. In oral cancers the presence of CD8 T cells at the tumor site has been correlated with better OS and improved response to check-point inhibitors. However, the CD8 T cell infiltrate is heterogenous and we have recently shown that only a fraction of the tumor-infiltrating (TIL) CD8 T cells from HNSCC patients were specific for tumor antigens [6]. Tumor-reactive CD8 T cells that co-express the surface molecules CD39 and CD103 could efficiently kill autologous tumor cells after in vitro expansion [6, 44]. CD8 T cells with a similar phenotype were also observed in other tumor histologies such as melanoma, breast cancer, colorectal cancer, and non-small cell lung cancer [6, 45]. Expression of CD103 and CD69 and lack of CCR7 expression by CD39+CD103+ CD8 TIL favors their tumor residency [6, 44] and this is highlighted by little overlap between the TCR repertoire of CD39+CD103+CD8 TIL and circulating memory CD8 T cells [6]. In addition, an increased frequency of CD39+CD103+ CD8 cells among total tumor infiltrating CD8 T cells at time of surgery correlated with a better OS in a cohort of HNSCC patients [6]. Together, these results imply that CD39+CD103+CD8 T cells in the tumor are involved in the anti-tumor immune response. Therefore, gaining insight about the development requirements, maintenance, and cellular interactions of these cells in the tumor might provide new avenues for the treatments for cancer. In addition, understanding the changes in this CD8 T cell compartment between dysplasia and invasive cancer would provide valuable information for future interventions.
A recent study in HPV-positive HNSCC patients helps demonstrate the regulation of these cells [46]. Using tetramers to isolate HPV-reactive CD8 T cells, which all expressed PD-1, the investigators found that HPV-specific T cells were composed of three transcriptionally distinct cell clusters: a stem-like cluster; a transitory cluster; and a terminally differentiated cluster [46]. While the stem-like subset was characterized by the expression of TCF7 and IL7R, the transitory subset expressed PRDM1 and IFNG, and the terminally differentiated subset expressed HAVCR2, GZMA, GZMB, and PRF1. Following cell sorting and in-vitro peptide stimulation, cells from the stem-like subset but not the terminally differentiated subset were able to proliferate and differentiate into effector-like CD8 T cells [46]. Cells from the other subset maintained their terminally differentiated phenotype. Thus, an efficient anti-tumor immune response might require controlling the balance between those three subsets to maintain a pool of effector CD8 T cells capable of recognizing and lysing tumor cells. It will be important to determine whether a similar pathway exists for non-viral tumor antigens such as neoantigen-reactive CD8 T cells and understand if this mechanism is influenced by the affinity of a TCR for its cognate epitope.
Role of CD4 T cell help
Even though HPV-driven oral cancers have a better prognosis than HPV-negative tumors, in our experience there is no significant difference in the frequency of CD39+CD103+CD8 T cells among total CD8 T cells infiltrating the tumor between those two groups of patients (unpublished data). This observation suggests that other components of the immune environment influence the biology of tumor-reactive CD8 T cells. Indeed, it is known that CD8 T cells need to receive signals from CD4 helper T (Th) cells to become fully licensed to kill target cells [47]. Strong infiltration by CD4 T cells in oropharyngeal squamous cell carcinoma (OPSCC) was associated with lower T stage, improved disease specific survival and prolonged overall survival supporting a role for these cells in the anti-tumor immune response [48, 49]. However, these data did not address the complexity of the CD4 compartment and the role of the distinct CD4 T cell subsets in this effect. Similar to their frequency in the peripheral blood, CD4 T cells account for a large portion of the T cells in the tumor. Among the CD4 T cells infiltrating HNSCC, 30–40% of the cells are Treg cells [50], involved in regulating immune responses, preventing immune pathologies, and thus limit anti-tumor immune responses. In breast cancer and NSCLC, CCR8 expression identifies a subset of highly immunosuppressive Treg cells which also express CD39 and ICOS [51, 52]. The remainder of the intratumoral CD4 T cells include distinct cell subsets specialized for immune responses against different immunological insults and orchestrating both cellular and humoral immune responses to infectious agents and cancer [53]. Each subset is characterized by a master transcription factor and the secretion of hallmark cytokines. However, there is only scant data identifying the Th composition in oral cancers. Most of the TIL CD4 Th cells have an effector/memory phenotype. Among those, some express high levels of the activation/exhaustion markers PD-1, HLA-DR, CD39, and CTLA-4 [50, 54, 55]. In contrast to the CD8 compartment, CD103+ CD4 Th cells are rare [19, 55], suggesting distinct requirements between CD4 and CD8 T cells for CD103 expression.
The comparison of HPV+ and HPV-HNSCC tumors by single cell RNAseq analysis has shown differences in the CD4 compartment [54]. HPV+ tumors showed an enrichment in cells with a follicular CD4 Th (Tfh) cells or Th1 gene signatures, whereas HPV-negative tumors were composed mostly of effector or effector/memory CD4 Th cells [54]. The observed difference regarding Tfh cell signature reflects the higher frequency of B cells in HPV+ HNSCC as compared to HPV- HNSCC [54, 56, 57]. Alternatively, the data could be explained by the different anatomical locations between those two types of HNSCC, where HPV+ HNSCC exists as part of the oral ring of secondary lymphoid organs. In a mouse model of NSCLC, the presence of both neoantigen-reactive CD4 Th cells and B cells in the tumor was shown to be beneficial for the anti-tumor immune response [58]. Furthermore, interactions between those two cell types were necessary for the generation of IL-21-producing Tfh cells that promoted anti-tumor immunity by enhancing CD8 T cell effector functions [58]. Tfh-like CD4 T cells characterized by the secretion of the B cell-attracting CXCL13 have also been identified in breast cancer and their frequency correlates with better outcome [58]. More recently, we reported the presence of a subset of CD4 Th cells that co-express PD-1 and ICOS, a phenotype reminiscent of Tfh cells, in the tumor microenvironment (TME) of HNSCC tumors [19]. Those cells also expressed the transcription factor BCL6 and secreted IL-21 and CXCL13. More importantly, PD-1+ICOS+ CD4 Th cells were enriched in T cells recognizing tumor-associated antigens such as HPV but also tumor specific neoantigens. Future studies are needed to confirm whether PD-1+ICOS+ CD4 Th cells interact with B cells and CD8 T cells in the TME. Such experiments are important to distinguish the true function of CD4 Tfh and B cells in HNSCC, since they may be a secondary feature of tumor immune biology. Tertiary lymphoid follicles have been detected in a wide array of cancers [59], including poorly-immune responsive tumors [60]. Their formation has been linked to inflammatory conditions in multiple disease and may be a feature of inflammatory tumors rather than a cause of anti-tumor immunity. It is unclear whether T cells in the tertiary lymphoid structures can emigrate into the tumor, or must follow lymphoid drainage and rely on recirculation to re-enter the tumor environment. Spatial analysis of the TME will be necessary to confirm the cellular interactions of CD4 Tfh and CD8 in human tumor specimens. As discussed earlier, multiparametric analysis of immune infiltrates by immunohistology [2], demonstrated that the relative location of CD8 T cells in relation to CD4 Treg formed part of a cumulative suppression index that showed potential to predict HNSCC patient outcome [2]. Interestingly, in patients with pancreatic cancer, tumors with tertiary lymphoid structures were less enriched for tumor-infiltrating CD103+ cells [60], suggesting that the biology driving expansion of CD4 Tfh in tumor follicular structures may be different from that driving Trm. Within the tumor, T cells are generally enriched in the tumor stroma, and less frequent in cancer cell nests [61–63]. Cells with the Trm phenotype are enriched in cancer cell nests rather than the tumor stroma in breast cancer patients [63], while stem-like CD8 T cells expressing the Tcf1 marker were located in the stroma and absent from cancer cell nests in melanoma patients [64]. Further studies are needed in HNSCC to confirm these data, but these data suggest that Trm exist near cancer cells, while tumor specific T cells with other phenotypes recirculate through the tumor stroma [7]. Along with spatial information, it will also be important to understand the molecular signals required to induce the recruitment and differentiation of CD4 Th cells into Tfh-like CD4 T cells in the tumor environment. Finally, it will be essential to determine if the Tfh-like CD4 T cells can directly recognize tumor antigens in HNSCC patients, since this will directly relate to the specificity of B cells that they support and the CD8 T cells that they might help. These data indicate that specific T cell subtypes in HNSCC impact patient survival. Given our new tools to identify and characterize tumor-specific T cells through a combination of epitope prediction, functional analysis, and single cell RNASeq, we can anticipate rapid advances in our understanding of the critical tumor antigen-reactive T cells in HNSCC.
Impact of immunotherapy on pre-existing or new responses to tumor antigens
In vivo initiation of immunity
Classical responses to exogenous infection, for example immunity to an infecting virus, depends on an efficient transition from innate to adaptive immunity. Initial infection of HPV into basal cells in the tonsillar epithelia, can result in innate immune detection of the invading virus (Figure 2A). These innate signals can result in local immune activation, such as STING-mediated nucleic acid sensing resulting in type I IFN production [65]. Innate immune responses can result in local dendritic cell maturation and trafficking to draining lymph nodes to initiate anti-viral responses and activate the vascular endothelia to encourage infiltration of T cells to the infection site (Figure 2A). Successful antiviral immunity can result in destruction of infected cells. Unfortunately, HPV has a series of evolutionary adaptations to block various aspects of innate and adaptive detection [65–67] permitting immune escape. These immune evasion mechanisms can allow tumor initiation and limit immune responses in tumors (Figure 2B). While there can be tumor-specific T cells in pre-malignancies and malignant tumors, there are many ways that the tumor environment can suppress adequate anti-tumor immune function, including expression of checkpoint regulators, suppressive macrophages and Treg (Figure 2B) and these features can support tumor growth despite the existence of tumor-specific T cells. As we will discuss, interventions that form the basis for current immunotherapies focus on releasing existing cells from checkpoint inhibition, deplete or reprogram suppressive cells, and generate local inflammation to increase immune function in the tumor (Figure 2B). However, in patients with poor pre-existing immunity to their tumor, alternative immune interventions such as adoptive transfer or vaccination may be necessary to provide new cells to target the cancer cell. Removal of a negative feature, such as immune suppression, can only have impact if there are pre-existing anti-tumor immune responses waiting to become active.
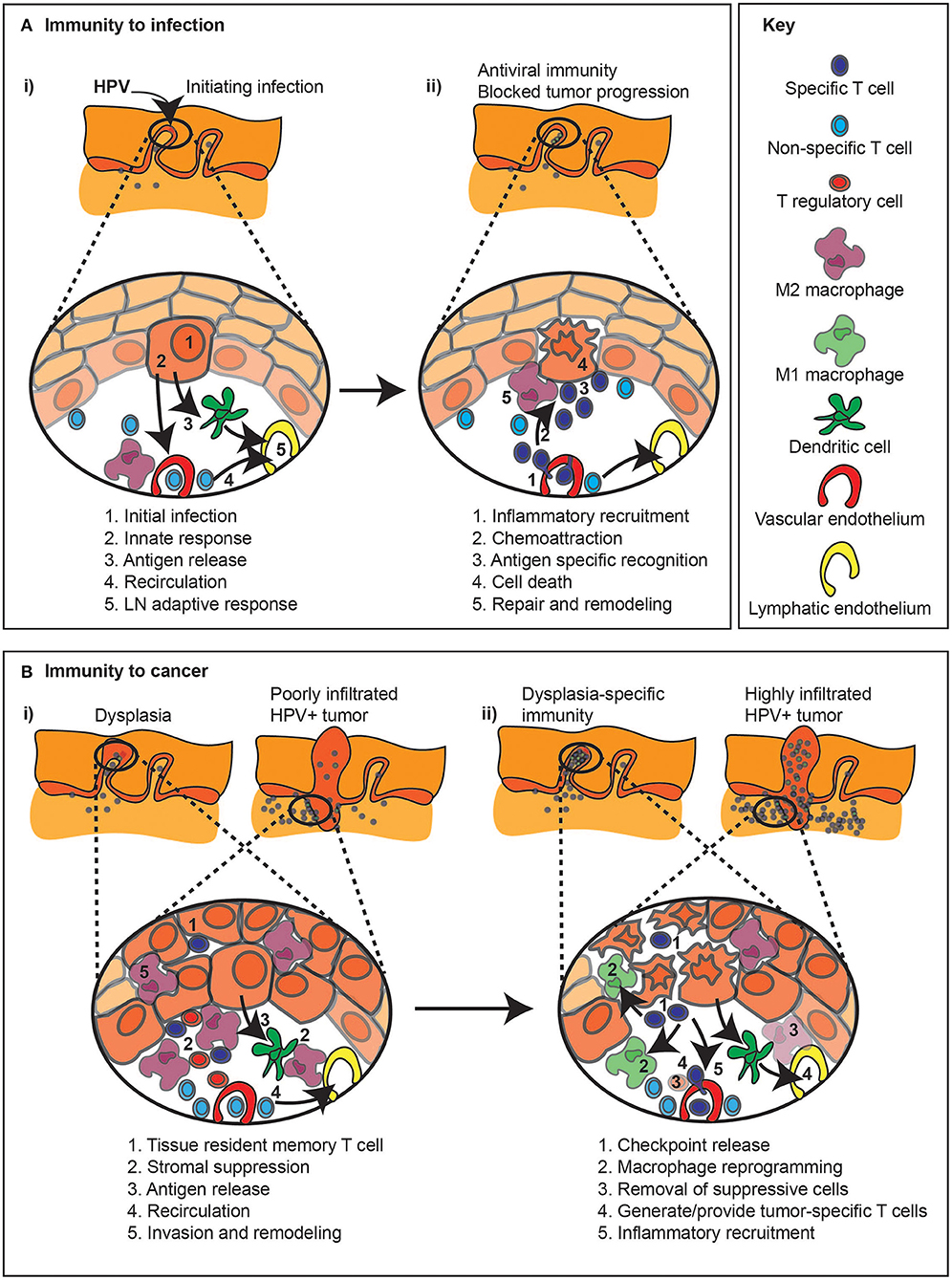
Figure 2. Immune regulation during infection and related immunotherapy effects on pre-existing anti-tumor immune responses. (A) (i) The response to an infection, in this example HPV infection of basal epithelial cells (1), can result in a series of events that result in immunity. Innate sensing of infection (2) can lead to locoregional inflammation (2), as well as antiviral responses in the infected cell. Inflammation combined with antigen release can permit DC migration to lymph nodes (3), so while non-specific cells continue to recirculate through the tissue (4), new immune responses can be initiated in the draining lymph node (5). (ii) Following expansion of antiviral T cells in the draining lymph node, these cells can be preferentially recruited to the site of infection via the impact of inflammation on the vasculature (1). Chemokines released by infected cells can recruit T cells through the stroma to the infection site (2). Cognate recognition of infected cells (3) can lead to their death (4). Dying cells are actively cleared, which can initiate repair and shut down inflammation in the infection site (5). (B) (i) The immune response to cancer, either in early dysplasia or in a malignant tumor, can result in infiltration of tissue resident memory T cells (1), as well as stromal infiltration of T cells, though local immune suppression (2) results in tumor growth dominating over tumor destruction. Ongoing cancer cell death, due to hypoxia or limited growth factor availability can lead to antigen release (3), though stromal suppression can limit the quality of antigen presentation. T cells can continue to recirculate through tumors (4), though they may be restricted to the stroma and poorly interact with cancer cells, and stromal cells can support further invasion of the cancer into surrounding tissues (5). (ii) Immunotherapy with checkpoint inhibitors (1) may derepress local T cells or recirculating T cells to permit destruction of cancer cells. Inflammatory cytokines released by activated T cells can repolarize macrophages in the tumor to limit invasion or immune suppression (2). Alternatively, immunotherapies can be selected to deplete or block the inhibitory effect of suppressive macrophages or Treg (3). Where pre-existing immunity is limited, vaccine or adoptive transfer approaches may provide tumor-specific T cells (4), which can be targeted to the tumor environment with locoregional immunotherapies that generate inflammation in the tumor environment (5).
Evidence for pre-existing immunity determining therapeutic responses
In pre-clinical models, the development of tumor-specific T cells following tumor implantation is dependent on a fully functional immune system [reviewed in Medler et al. [8]]. A failure in any part of that system can result in abnormal immunity. For example, mice deficient in cross-presenting DC via a Batf3 knockout fail to develop functional T cell infiltration in tumors [68]. DC are key for tumors to develop an immune infiltrate even before the tumors become detectable in mice. The degree to which tumors engender T cell immunity early in their development can directly impact their ability to grow in immune competent mice [8, 68]. We discussed this impact for progressing pre-malignant tumor models above. For transplantable tumor models, the injection of cancer cells into mice acts as an initial tumor-specific vaccination event resulting in CD8 T cell immunity which is closely followed by T regulatory cell suppression [69–73]. Thus, even in models that do not progress through pre-malignancy, early immune activation dictates the eventual immune environment of the tumor. This directly impacts treatment responses. For example, we previously demonstrated that the response to combination radiation therapy and checkpoint inhibitor was dependent on the presence of a pre-existing immune responses developed at tumor implantation [74]. When implantation immunity was blocked, these therapies were no longer effective. Therefore, while the goal of combination immunotherapy with radiation therapy has been assumed to result in antigen and endogenous adjuvant release resulting in de novo immune responses to tumors, in our studies the treatment expands existing responses essential for improved outcomes [74]. When considering checkpoint inhibitor immunotherapy this is logical, since checkpoints such as PD-1 and CTLA4 are expressed on antigen experienced T cells. It is reasonable that these therapies remove suppression of an existing population of currently suppressed cells, therefore are dependent on pre-existing immune responses. This is consistent with data in pre-clinical models, where tumor control by combination checkpoint immunotherapy was shown to be dependent on T cells already residing with the tumor, and is lost if immune responses at tumor implantation are blocked [75]. Checkpoint inhibition may not act on all suppressed cells in the tumor, since some may not easily be restored to full function. The phenomenon of tumor-specific T cell exhaustion, akin to that observed in chronic antiviral responses [76], renders T cells unresponsive to formerly effective stimuli [77]. T cell exhaustion has been demonstrated early in tumor progression [78], with exhaustion irreversible by the time solid tumors are detected [78]. As discussed above these T cell populations are very relevant to HNSCC [46] and recent data suggests that terminally exhausted cells in the tumor cannot be restored to full function by checkpoint inhibition. Instead, therapy allows a pre-existing progenitor exhausted population to participate in tumor control [64, 79]. These data suggest that immunotherapy recruits additional immune cells, present in the tumor or recirculating but currently unable to impact tumor growth, to participate in immune control of tumors.
Therapeutic interventions to bring new responses to the tumor
Of course, antagonist anti-CTLA4 and agonists such as anti-OX40 can act as vaccine adjuvants expanding de novo T cell responses following antigen challenge [80–82]. Thus, aside from their effect on existing T cells, there is the potential to bring new cells into the immune response at the tumor. Given the long exposure of patients to tumor-associated antigen, it is unclear whether there exist naïve T cells specific for tumor-associated antigens that have managed to avoid meeting this antigen in their recirculation through lymph nodes. However, it is reasonable that T cells with TCR affinities that are too low for them to functionally participate in endogenous anti-tumor immunity are a potential new source of anti-tumor immunity. For these cells to participate, they need the threshold for activation to be manipulated in their favor (Figure 3). To trigger full activation of CD8 T cells, both the amount of presented antigen and the affinity of the TCR for the antigen-MHC combination matter [83, 84]. This interaction is helped by innate adjuvants, which can increase antigen processing and presentation and upregulate costimulatory molecules such as CD80 and CD86 [85–89] (Figure 3). In addition, successful antigen presentation to CD4 T cells can help CD8 T cell responses, both through cytokines and DC licensing [90–92]. The limiting effect of low TCR affinity for tumor-associated antigen has been demonstrated [93] and results in T cell dysfunction. Importantly, both anti-PD1 and anti-CTLA4 overcome suppressed TCR signaling through effects on phosphatases such as Shp2 and PP2A [94–97] and availability of costimulatory signals [98, 99] altering the signaling threshold of T cells. In pre-clinical models, tumor control with antigens that were suboptimal at engaging T cells is improved on treatment with anti-PD1 and anti-CTLA4 [93]. TGFb acts in part to alter the signaling threshold of T cells, and TGFb inhibition allows T cells to fully activate with normally suboptimal targets [14, 100]. Similarly, novel regulators such as PTPN22 are emerging that can be targeted to improve TCR activation and enhance tumor immune responses [101]. These data mean that even without generating truly de novo immune responses to tumor-associated antigens, our most popular cancer immunotherapies serve to bring suboptimal T cells into the anti-tumor immune response. These data indicate the importance of regulating the activation of pre-existing T cells and improving the response of sub-optimal T cells that were formerly unable to functionally participate. In addition to exploiting pre-existing immunity to alter immune responses to growing tumors, we can restore the function of failed responses to expand tumor immune control. As we will discuss, this may in part account for the beneficial effect of autologous/expanded TIL therapy, and/or repeated infusion of expanded TIL-derived cell products [102].
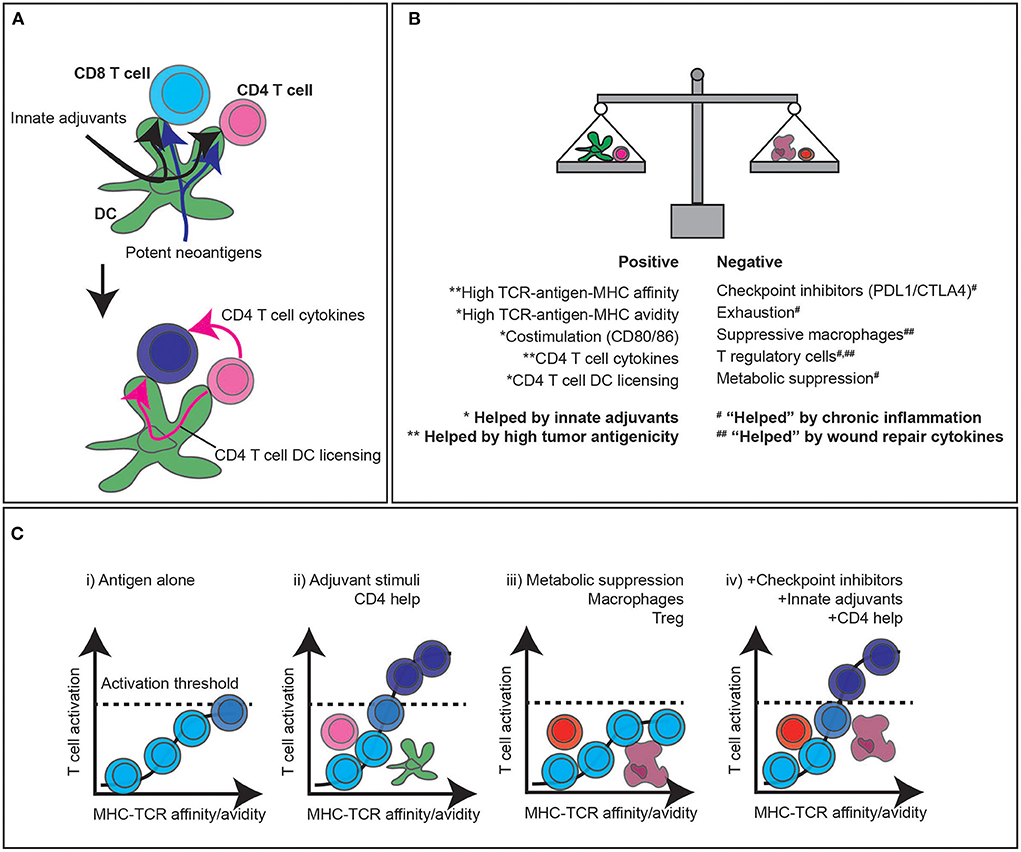
Figure 3. CD4 T cell help, adjuvant signals, and immunotherapy to enhance CD8 T cell responses to tumors. (A) The classic three cell model of CD8 T cell activation relies on antigens being presented to both CD4 and CD8 T cells by dendritic cells. Innate adjuvants present in the infection improve antigen processing and presentation, as well as upregulate costimulatory molecules. Highly antigenic targets increase the likelihood of high affinity antigens being available for both CD8 targeting and CD4 T cell help. (B) The balance of positive and negative stimuli dictate whether T cells can contribute to anti-tumor responses. Broadly, highly antigenic tumors with endogenous adjuvant support can improve the likelihood of T cell responses, which are countered by a range of negative regulatory features of the tumor environment. (C) High affinity antigen-specific T cell responses have the potential to stimulate T cells independently, but the likelihood of passing the critical activation thresholds is increased by CD4 help and adjuvant signals to cross-presenting DC and directly presenting cancer cells. A range of suppressive factors in the tumor can decrease the ability of T cells to pass the critical activation threshold to control tumors, and current immunotherapies can overcome these limitations to overcome suppression and bring new T cells into the response that were formerly unable to cross the activation threshold to participate in tumor control.
Adoptive T cell therapies to overcome suboptimal pre-existing immunity
Adoptive TIL-based T cell therapy platforms in HNSCC
The initial success of autologous TIL therapy in melanoma relied on access to rIL-2 in quantities necessary for clinical use [103–105] and was then enhanced further by incorporation of preparative non-myeloablative (NMA) conditioning [106–109]. The mechanisms of this therapy appear to rely on optimizing transferred T cell expansion in vivo to generate diverse T cell phenotypes that can overcome ongoing T cell dysfunction in the patient [110, 111]. This established a standard three-step therapeutic sequence for conventional TIL therapy, consisting of: (i) conditioning; (ii) cell infusion; and (iii) cytokine support. Despite these successes in melanoma, successful experiences with TIL therapy have been limited in epithelial malignancies, including HNSCC. For example, in 2015 through an institutional research protocol, we identified a 21-year-old patient with treatment-refractory recurrent OSCC, for whom ex vivo cultured TIL showed a high degree of reactivity and specificity by in vitro assay to his autologous primary tumor cell line, but not to HLA-matched, allogeneic tumor cell line controls. Under an FDA single-patient IND, autologous TIL were expanded under cGMP conditions and he was treated with standard cyclophosphamide/fludarabine NMA conditioning followed 1 week later by cell infusion and high dose IL-2 support, but unfortunately, he did not respond to therapy [112]. In late 2018, Iovance released preliminary results from a phase II trial of conventional TIL therapy, including cyclophosphamide/fludarabine NMA conditioning and high dose IL-2 support (LN-145) in 2nd line or later relapsed and metastatic HNSCC (NCT03083873) [113]. Patients had a median of 3 prior therapies. Responses were observed in 4 of 17 patients (anatomic subsite and HPV-status not reported). A follow-up Iovance phase II trial of LN-145 plus a concurrent single dose of pembrolizumab, in immunotherapy-naïve relapsed and metastatic (r/m) HNSCC, (NCT03645928), reported initial results in late 2021 with confirmed partial or complete (PR and CR, respectively), in 5 of 18 patients, and unconfirmed clinical response in an additional two patients, for overall response rate of 39% [114]. Patients had a median of one prior line of therapy. Again, anatomic subsites and HPV-status were not reported, so it is unknown if responses included OSCC. It is plausible that responses were primarily in HPV-associated cases [115–118].
TCR-Transduced adoptive T cell therapy in HNSCC
Promising results from an NCI group, first reported in late 2018, used a gene-modified TCR-transduced adoptive T cell therapy (TCR-T) targeting HPV16 E7 protein [119–122] viral tumor-associated antigen (HLA-A*02:01 or A*02:06 restricted) for T cell mediated eradication of HPV16-associated tumors (NCT02858310) [123]. Complete regression of most tumors was observed with 6 of 12 treated patients. The majority of these patients were refractory to prior PD-1 blockade including all the HNSCC patients (ORR 50%, all PR/no CR) [124]. Among the four HNSCC patients, two responses were observed, including one patient who had been previously treated on the Iovance trial of LN-145 conventional TIL therapy at our center. Intra-patient tissue genomic analysis representing dichotomous tumor responses, revealed known resistance mechanisms involving defects in antigen presentation and interferon response pathways consistent with immune editing.
In 2020, Adaptimmune reported the ADP-A2M4 single-arm, phase II pilot trial of MAGE-A4 targeted, HLA-A*02:01 restricted TCR-T therapy in combination with pembrolizumab for first-line recurrent/metastatic, HPV-agnostic HNSCC. This trial has since closed due to slow accrual of MAGE-A4 expressing & HLA-matched eligible participants (NCT03132922) [125]. In 2021, Immatics reported preliminary results from the IMA203 phase I dose-escalation trial of MAGE-A4/A8 (PRAME) targeted, HLA-A*02:01 restricted TCR-T therapy for treatment-refractory advanced solid tumors (NCT03686124) [126] at the SITC annual meeting. Of 16 participants, three HNSCC patients were treated, but none with durable response more than 12 weeks (subsite and HPV-status not specified, but OSCC eligible). At this time, both companies are moving forward with next generation TCR-T products incorporating a CD8a coreceptor and affinity optimized TCR [127, 128], OSCC patients may be eligible in future trials given prior interest in HNSCC for MAGE-A4 targeting.
Chimeric antigen receptor T cell therapy in HNSCC
Systemic delivery of CAR-T targeting HNSCC surface antigens, to date been shown to be unsafe due to the unacceptable risk of on-target/off-tumor toxicities [129, 130]. CAR-T may be inferior to TCR-T in the long-term, due to more rapid exhaustion, despite more potent initial effector and killing function [131–133]. However, CAR-T intratumoral delivery in OSCC may hold promise as a means of TME remodeling and immune priming. A phase 1 dose-escalation, single center trial of intratumorally delivered pan-ErbB CAR-T in HNSCC (NCT01818323) [134] was reported at AACR in 2017 [135] and at ASCO in 2018 [136], using T1E28ζ, a CAR containing a promiscuous ErbB ligand (which engages 8/9 ErbB homo/heterodimers) coupled to a CD28 + CD3ζ endodomain and 4αβ, an IL-4-responsive chimeric cytokine receptor which enables IL-4-driven selective CAR T-cell enrichment/expansion during manufacture [137]. CAR T-cell dose was escalated from 1 × 10e7 to 1 × 10e9 T-cells administered as a single treatment, by multifocal intra-tumoral injection without lymphodepletion and no dose limiting toxicities were observed. Three observations in this study point to tumor remodeling effects: (i) tumor growth slowed to stable disease in 10 of 16 patients, despite rapid disease progression on study entry; (ii) CAR-T cells remained undetectable in the circulation throughout; and (iii) a rapid complete response was observed in a patient who subsequently received aPD-1 + T-VEC, since durable for >3 years.
Additional modification of T cell therapy platforms
With the advent of widely available tumor sequencing, tumor neoantigen redirected T cell therapies [138] (such as TCR-T) hold enormous promise for truly tumor-unique targeting without off-tumor/on-target concerns (Figure 4A). These have already demonstrated initial, though limited, success in multiple epithelial malignancies [139–143] including cholangiocarcinoma [102], colon [144, 145], and breast cancer [146, 147]. Similarly, we recently demonstrated the potential of KRAS-specific TCR-T for pancreatic cancer [148]. By extension, HRAS mutations have been identified as a conserved neoantigen potential target for redirected T cell therapy in OSCC [149]. Numerous synthetic biology approaches are being brought to bear in order to enhance cell-intrinsic characteristics and cell product manufacturing [142, 150] including: selection of optimal T cell subpopulations for transduction (including non-viral transduction); manipulation of cell culture to promote favorable phenotype and improve yield during the manufacturing process; transduction of druggable growth receptors to introduce control of ex vivo and in vivo proliferative kinetics; TCR affinity and signaling enhancements, membrane-anchored cytokines to promote in vivo persistence; conversion switches to couple an inhibitory cell surface receptor with a stimulatory cytosolic tail, and thus armor the cells for trafficking, survival and effector function on entry to a hostile TME. These cell-intrinsic enhancements undoubtedly will increase therapeutic efficacy. Nevertheless, the foremost challenge to TCR-based cell therapy approaches remains ineffective antigen presentation without which T cell-dependent immunotherapy, whether through immune checkpoint inhibitor or adoptive cell therapy, cannot exert an effect. MHC class I down-regulation and alterations in antigen processing and presentation are prevalent in HNSCC (selective HLA loss of 37% in primary lesions; total HLA loss of 15% in primary lesions and 40% in metastatic lesions) [151] and are well-described [152–158]. Several groups have demonstrated that MHC class I down-regulation portends a poor prognosis in OSCC specifically [2, 159] and in HNSCC more broadly [160–162] and represents a mechanism for immune escape and acquired resistance to immunotherapy, generally [163]. NK cell-based therapies, which can detect and eliminate tumor cells via MHC-independent mechanisms, have been proposed to overcome this challenge in HNSCC. NK-92 cells [164] engineered to express endoplasmic reticulum-retained IL-2 [165] and a second generation CAR targeting PD-L1 [166] (PD-L1 t-haNK) were shown by the NIDCD group to eradicate MOC1 tumors in vitro in a PD-L1-dependent fashion [167]. In an OSCC model system of adoptive T cell therapy immune escape, using UM-SCC-1 and UM-SCC-47 oral cancer cell lines with varying admixtures of HLA-expressing and HLA-nul tumor cells, the addition of PD-L1 t-haNK in vitro was able to salvage response and prevent clonal outgrowth of escape variant tumor cells [167]. The NIDCD group further demonstrated synergy of combinatorial PD-L1 t-haNK, PD-1 blockade, and IL-15 superagonist in the MOC1 syngeneic mouse oral cancer model laying the groundwork for a recently activated phase II trial (n = 55) of this triplet combination in 2nd line recurrent/metastatic HNSCC (NCT04847466). A potential advantage of CAR-NK cell therapy over CAR-T therapy is a very reduced incidence of graft vs. host disease due to limited longevity. Tracking studies indicate that PD-L1 t-haNK cells persist in tumors for up to 72 h, suggesting that to ensure the maintenance of cells in situ, weekly or more frequent dosing may be necessary.
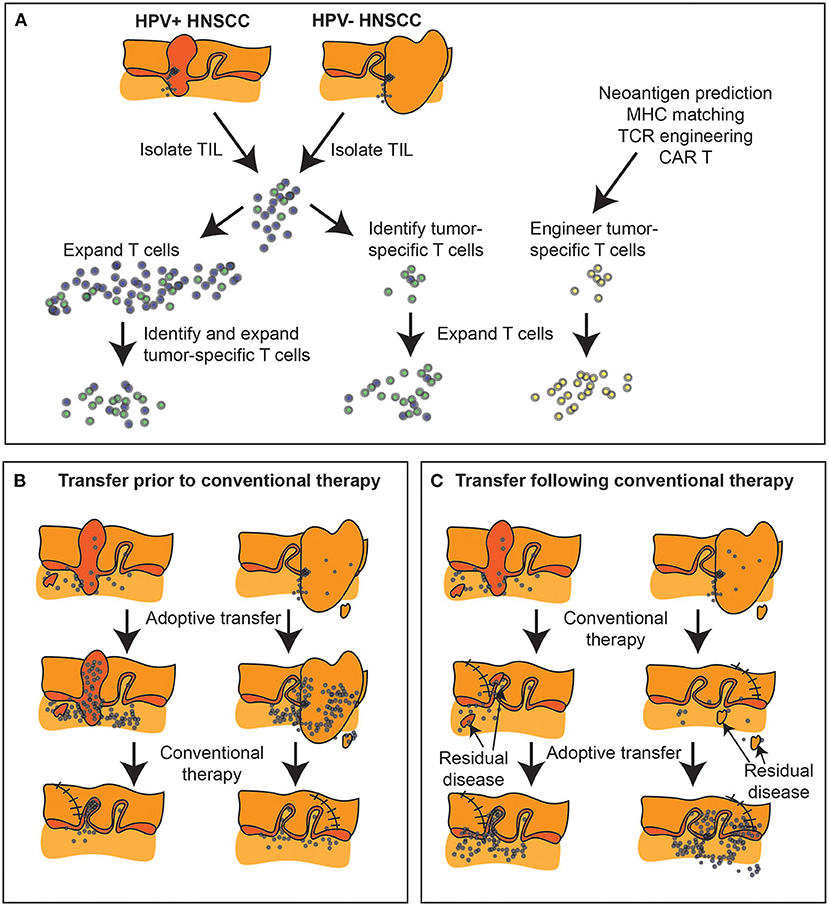
Figure 4. Expanding tumor-specific T cell immunity via adoptive transfer. (A) Biopsies and surgical specimen can be a source of tumor infiltrating lymphocytes (TIL), which can be expanded ex vivo and tumor-specific cells isolated either by response, or by initial phenotype. Alternatively, tumor-specific T cells can be engineered if both antigen and MHC match known TCR specificities, or using chimeric antigen receptors (CAR). (B) Adoptive transfer can occur prior to conventional therapy to alter the tumor immune environment prior to further treatment, or (C) can follow conventional therapy to target residual disease. At present, with surgical samples the dominant source of TIL, adoptive transfer is generally delivered following completion of conventional multimodality treatment or in the metastatic setting.
Targeting the tumor immune environment to enhance adoptive transfer
Myeloid cells represent a potential impediment to effective adoptive cell therapy approaches in OSCC [168]. In HNSCC patients an expanded population of CD34+ myeloid cells that can suppress T cells has long been identified [169, 170], which match the consensus definition of myeloid-derived suppressor cells [171]. Using murine oral cancer (MOC) C57BL/6 syngeneic tumor models, PI3Kδ/γ inhibition or anti-Ly6G depletion inhibits the impact of the expanded myeloid populations and enhances responses to PD-L1 mAb in T-cell–inflamed MOC1, but not in non-T-cell–inflamed MOC2 tumors [172]. Similarly, using the same models anti-Ly6G depletion was shown to improve tumor control following anti-CTLA-4 treatment in T-cell inflamed MOC1 tumors, but again not in non-T-cell inflamed MOC2 tumors [173]. Recent studies demonstrate that SX-682, an oral CXCR1/2 inhibitor, disrupts MDSC trafficking, and improves tumor control by anti-PD1 therapy in an oral cancer model [174], as previously shown in other tumor models [175, 176]. These data demonstrate that where T cells are abundant but suppressed, myeloid targeting may optimize T cell control of tumors, though in poorly infiltrated tumors there may be limited benefit. However, this blockade of CXCR1/2 also enhances tumor infiltration, activation, and therapeutic efficacy of adoptively transferred murine NK cells [177], suggesting that cellular therapies can be improved by therapeutic modulation of the tumor environment. In addition, cellular therapies may be able to directly deplete these expanded myeloid cells, since the PD-L1 targeting CAR-NK (PD-L1 t-haNK) was able to preferentially lyse MDSC in the PBMC fraction of peripheral blood cells [166]. Thus, in addition to targeting PD-L1-expressing cancer cells [166], the on-target off-tumor lysis of normal cells may modify the tumor immune environment.
Immunotherapy combinations for adoptive transfer
Under the conventional three-step therapeutic sequence of TIL therapy (conditioning/cell infusion/cytokine support), it is worthy to note here that the non-myeloablative conditioning regimen of cyclophosphamide/fludarabine (Cy/Flu)—which has modest activity in HNSCC [178, 179]—has been unchanged for decades. This represents an area of opportunity for remodeling of the tumor immune microenvironment in the current context of multiple bioactive immune modulating agents that are newly available or in clinical development. This can be put to the test, either alone or in combination with chemotherapy, as immune induction during the existing preparative window preceding cell infusion. The observation that CD40 signaling in APCs may remodel the TME [180] and that effective T cell therapy results in T cell-directed and CD40-mediated remodeling of the tumor stroma [181], suggests that this approach has potential to alter the contours of the tumor immune environment. In pre-clinical models, CD40-CD40L interactions were necessary to develop pre-existing T cell responses in tumors and therefore essential for the response to immunotherapy and conventional radiation therapy combinations [74]. In pre-clinical models CD40 agonist antibodies can enhance the expansion of adoptively transferred T cells in vivo and boost their antitumor activity [182]. Using the KPC model of intraepithelial pancreatic tumorigenesis, cDC1 dysfunction and apoptosis was shown to be neutralized in vivo by IL-6 blockade, and CD40-agonist restored cDC1 maturation and abundance, resulting in enhanced control of tumor outgrowth [183]. Standard conditioning Cy/Flu lymphodepletion was observed to induce peripheral MDSC expansion in melanoma patients treated with TIL therapy [184], and was associated with reduced TIL persistence and poorer survival. In the B16/pmel-1 model, IL-6 driven differentiation of mobilized hematopoietic progenitor cells following standard Cy/Flu conditioning was shown to mediate ACT failure, and ACT function could be rescued by IL-6 blockade administered the day prior to conditioning Cy/Flu [184]. Future studies will need to demonstrate if the combination of CD40-agonist and IL-6 blockade (e.g., tocilizumab) can synergize toward favorable TME remodeling and immune induction in the preparative context of TCR-T or other ACT approaches.
Together, these data indicate that even where tumors are poorly infiltrated with tumor-specific T cells, such cells can be engineered ex vivo to enhance tumor infiltration. Moreover, while the tumor blocks T cell function, ex vivo engineering can build resistance into the transferred cells. Finally, where antigen presentation is a limitation, exploiting NK cell therapy or the effects of combination with conventional therapies may improve the visibility of tumors to the immune system [185–187]. At present, adoptive T cell therapies are not well-integrated into the conventional treatment paradigm of HNSCC. These are currently experimental therapies for patients who have failed existing treatments or for whom conventional alternatives are not available. As with other immunotherapies, adoptive transfer may be used to change the immune environment of the tumor in conjunction with chemotherapy, radiation therapy, and surgery in HNSCC (Figures 4B,C). As with all therapies, timing may be key, since chemotherapy and radiation therapy can be unforgiving to T cells. Moreover, biomaterial-based intraoperative T cell or immunotherapy delivery may be an efficient means to regulate the immune environment for subsequent control of logoregional disease [188, 189]. The fact that HNSCC is a disease conventionally treated with multimodality therapy makes it essential that novel immunotherapies are appropriately integrated into current treatment paradigms to convert poor responding patients to cures.
Conclusions
Almost every category of immunotherapy has proven effective in controlling HNSCC tumors in pre-clinical models. These include innate adjuvants, T cell checkpoint inhibitors, T cell costimulators, adoptively transferred T cells, myeloid regulatory targets, vaccines, and a wide range of alternative immune focused therapies. Despite this, there are only a handful of approved immunotherapies for HNSCC in the clinic. It is normal to see promising pre-clinical therapies fail in clinical trials, but it is always valuable to understand the biology behind the wide difference between successes in pre-clinical models and clinical trials.
Firstly, there is a significant impact of pre-clinical model selection. Some tumor models are more responsive to treatment than others. In part this is explained by immunogenicity [8], where some tumors are so close to the precipice of cure that almost any intervention can result in success. In our experience the CT26 colorectal carcinoma is one such model that is responsive to almost all T cell targeted therapies that we test in combination with radiation therapy. It could be argued that it would be more accurate to report the response of 8 different tumor models, rather than eight genetically identical mice each given the same genetically identical cancer cells. There may well be patients who are also highly responsive and so may be well-matched to therapies that succeed in CT26 tumors, but the diversity in tumor genetics, patient genetics, and patient immune status across a clinical trial are vastly greater than the differences between any two pre-clinical models. The limitation in pre-clinical models should be understood and embraced, since they are just models. However, clinical translation of any agent that has only cured one or two highly responsive pre-clinical models does not seem reasonable. We would argue that translation requires a higher bar. Yet, the clinical development of anti-CTLA4 and anti-PD-1 blocking antibodies for cancer was based on single agent treatment of highly responsive CT26 and MC38 tumors [190]. As single agents these fail in most melanoma models [191–194] and most immunotherapies fail in mice with authentic spontaneous genetically engineered tumors of various origin in the original host [195, 196], yet ipilimumab and nivolumab are each effective therapies for melanoma patients [197, 198]. Clearly, we cannot over-rely on the authenticity of pre-clinical models. Secondly, through experimental design we can increase the likelihood of cures in any given pre-clinical model. For example, mice treated with immunotherapies within the first few days of tumor implantation can be cured by many agents, but these therapies fail if the tumor is first allowed to establish [74]. In part this is due to the vaccine effect of tumor implantation as discussed above [74], but also the fact that mature suppressive environments have not yet developed [9]. At the point that single agent immunotherapies start to fail, there may be a further therapeutic window where combination therapies can be effective. Therefore, to test combination therapies we must select models and timings where single agents fail, otherwise we cannot show a need or benefit for the combination. Model selection and experimental design are a normal part of pre-clinical testing, can dramatically impact the success of any given treatment, and should be carefully interpreted to help understand whether they will be relevant to actual clinical scenarios in patients [199].
Due to the complexity and impracticality of testing multiple agents in patients to evaluate their ability to change the tumor immune environment from a negative to a positive state, we may need to turn to ex vivo patient-derived modeling. Simple tumor explant models can provide a rapid readout of patient-specific responses to immunotherapy agents [189–202]. In these systems, the short ex vivo response of small fragments of fresh tumor tissue can have predictive power for patient responses to the same therapy [202]. Tools that help us predict how a patient's tumor might respond to a therapy can help us select the optimum treatment for an individual. Repeated sampling may allow us to determine whether their immune environment has shifted into one that predicts better OS with conventional therapy, and therefore could allow us to gradually shift a predicted unresponsive patient into a predicted responsive patient. In this setting window of opportunity trials can provide valuable information on patient specific responses to diverse interventions with the potential to shift their immune status [203, 204]. We would propose that treatments would focus on generating a tumor specific CD4 and CD8 T cell population in the tumor, and may rely on therapies that can bring suboptimal T cells into the tumor to become functional anti-tumor effectors. In this, way we can begin to find responses for HNSCC patients who are currently poorly served by current treatments.
As we have discussed, pre-malignancy represents the first opportunity for intervention to alter the course of disease (Table 1) and this has a clear relevance to OSCC. Preventing progression in high-risk patients will have a major impact by avoiding malignancy; however, we propose that even in patients that progress, it is possible that intervention can alter the immune environment and thus impact the response to conventional therapies. It seems clear that a wide range of current interventions are highly dependent on the pre-existing immune status of the patient. As we have discussed, analysis of the tumor immune environment of patients with advanced cancers has identified T cell populations that recognize tumor-associated antigens and are linked to improved outcomes in patients. Providing these cells represent a logical target for intervention in patients that lack such cells (Table 1), and innovative adoptive transfer approaches are in development for HNSCC. However, the mere presence of T cell infiltrates in tumors is generally insufficient. HNSCC patients with high T cell infiltrates are only cured following treatment, and it is reasonable that providing T cells will be a good first step for poor prognosis patients before proceeding to conventional therapy. While the ideal goal would be an immune panacea, the history of HNSCC therapy has been and remains multimodal therapy. Integrating immunotherapy into HNSCC treatment is a practical reality for the foreseeable future.
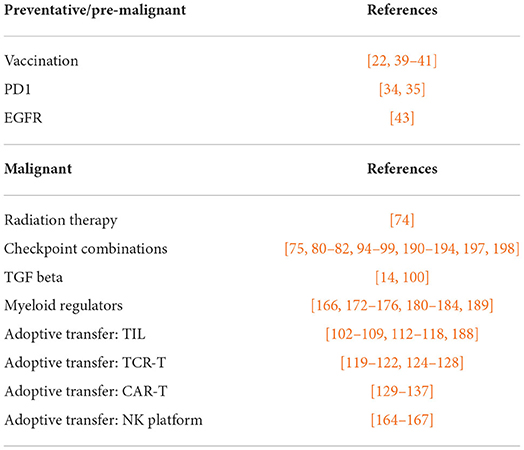
Table 1. Immunotherapy interventions discussed.
Author contributions
TD, MG, RL, and SS were jointly responsible for the preparation, writing, and editing of the manuscript and figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NCI R01CA182311, NCI R01CA244142, and by the Providence Foundation.
Conflict of interest
RL receives research funding from Clinigen, Incute, Celldex, and Bristol Myers Squibb. MG receives research funding from Bristol Myers Squibb and VIR Biotechnology. SS receives research funding from IMV Inc. and consulting fees from Stanford Burham Prebys Medical Discover Institute. Funders had no role in the preparation of the manuscript.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. (2020) 6:92. doi: 10.1038/s41572-020-00224-3
2. Feng Z, Bethmann D, Kappler M, Ballesteros-Merino C, Eckert A, Bell RB, et al. Multiparametric immune profiling in HPV- oral squamous cell cancer. JCI Insight. (2017) 2:e93652. doi: 10.1172/jci.insight.93652
3. Mukherjee G, Bag S, Chakraborty P, Dey D, Roy S, Jain P, et al. Density of CD3+ and CD8+ cells in gingivo-buccal oral squamous cell carcinoma is associated with lymph node metastases and survival. PLoS ONE. (2020) 15:e0242058. doi: 10.1371/journal.pone.0242058
4. Diao P, Jiang Y, Li Y, Wu X, Li J, Zhou C, et al. Immune landscape and subtypes in primary resectable oral squamous cell carcinoma: prognostic significance and predictive of therapeutic response. J Immunother Cancer. (2021) 9:e002434. doi: 10.1136/jitc-2021-002434
5. Danilova L, Anagnostou V, Caushi JX, Sidhom JW, Guo H, Chan HY, et al. The mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay: a sensitive platform for monitoring antitumor immunity. Cancer Immunol Res. (2018) 6:888–99. doi: 10.1158/2326-6066.CIR-18-0129
6. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. (2018) 9:2724. doi: 10.1038/s41467-018-05072-0
7. Blair TC, Alice AF, Zebertavage L, Crittenden MR, Gough MJ. The dynamic entropy of tumor immune infiltrates: the impact of recirculation, antigen-specific interactions, and retention on T cells in tumors. Front Oncol. (2021) 11:653625. doi: 10.3389/fonc.2021.653625
8. Medler TR, Blair TC, Crittenden MR, Gough MJ. Defining immunogenic and radioimmunogenic tumors. Front Oncol. (2021) 11:667075. doi: 10.3389/fonc.2021.667075
9. Tormoen GW, Crittenden MR, Gough MJ. Role of the immunosuppressive microenvironment in immunotherapy. Adv Radiat Oncol. (2018) 3:520–6. doi: 10.1016/j.adro.2018.08.018
10. Obradovic A, Graves D, Korrer M, Wang Y, Roy S, Naveed A, et al. Immunostimulatory cancer-associated fibroblast subpopulations can predict immunotherapy response in head and neck cancer. Clin Cancer Res. (2022) 28:2094–109. doi: 10.1158/1078-0432.CCR-21-3570
11. Gunderson AJ, Yamazaki T, McCarty K, Phillips M, Alice A, Bambina S, et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PLoS ONE. (2019) 14:e0211117. doi: 10.1371/journal.pone.0211117
12. Plzak J, Boucek J, Bandurova V, Kolar M, Hradilova M, Szabo P, et al. The head and neck squamous cell carcinoma microenvironment as a potential target for cancer therapy. Cancers. (2019) 11:440. doi: 10.3390/cancers11040440
13. Young KH, Gough MJ, Crittenden M. Tumor immune remodeling by TGFbeta inhibition improves the efficacy of radiation therapy. Oncoimmunology. (2015) 4:e955696. doi: 10.4161/21624011.2014.955696
14. Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat Commun. (2020) 11:1749. doi: 10.1038/s41467-020-15404-8
15. Zhou L, Zeng Z, Egloff AM, Zhang F, Guo F, Campbell KM, et al. Checkpoint blockade-induced CD8+ T cell differentiation in head and neck cancer responders. J Immunother Cancer. (2022) 10:e004034. doi: 10.1136/jitc-2021-004034
16. Gough MJ, Killeen N, Weinberg AD. Targeting macrophages in the tumour environment to enhance the efficacy of alphaOX40 therapy. Immunology. (2012) 136:437–47. doi: 10.1111/j.1365-2567.2012.03600.x
17. Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. (2008) 68:5206–15. doi: 10.1158/0008-5472.CAN-07-6484
18. Jagadeeshan S, Prasad M, Ortiz-Cuaran S, Gregoire V, Saintigny P, Elkabets M. Adaptive responses to monotherapy in head and neck cancer: interventions for rationale-based therapeutic combinations. Trends Cancer. (2019) 5:365–90. doi: 10.1016/j.trecan.2019.04.004
19. Duhen R, Fesneau O, Samson KA, Frye AK, Beymer M, Rajamanickam V, et al. PD-1 and ICOS co-expression identifies tumor-reactive CD4 T cells in human solid tumors. J Clin Invest. (2022) 132:e15821. doi: 10.1172/JCI156821
20. Oliva M, Spreafico A, Taberna M, Alemany L, Coburn B, Mesia R, et al. Immune biomarkers of response to immune-checkpoint inhibitors in head and neck squamous cell carcinoma. Ann Oncol. (2019) 30:57–67. doi: 10.1093/annonc/mdy507
21. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
22. Chaturvedi AK, Graubard BI, Broutian T, Pickard RKL, Tong ZY, Xiao W, et al. Effect of prophylactic human papillomavirus (HPV) vaccination on oral HPV infections among young adults in the United States. J Clin Oncol. (2018) 36:262–7. doi: 10.1200/JCO.2017.75.0141
23. Agalliu I, Gapstur S, Chen Z, Wang T, Anderson RL, Teras L, et al. Associations of oral alpha-, beta-, and gamma-human papillomavirus types with risk of incident head and neck cancer. JAMA Oncol. (2016) 2:599–606. doi: 10.1001/jamaoncol.2015.5504
24. Conway C, Graham JL, Chengot P, Daly C, Chalkley R, Ross L, et al. Elucidating drivers of oral epithelial dysplasia formation and malignant transformation to cancer using RNAseq. Oncotarget. (2015) 6:40186–201. doi: 10.18632/oncotarget.5529
25. Banerjee AG, Bhattacharyya I, Vishwanatha JK. Identification of genes and molecular pathways involved in the progression of premalignant oral epithelia. Mol Cancer Ther. (2005) 4:865–75. doi: 10.1158/1535-7163.MCT-05-0033
26. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. (2015) 517:576–82. doi: 10.1038/nature14129
27. Foy JP, Bertolus C, Ortiz-Cuaran S, Albaret MA, Williams WN, Lang W, et al. Immunological and classical subtypes of oral premalignant lesions. Oncoimmunology. (2018) 7:e1496880. doi: 10.1080/2162402X.2018.1496880
28. Gannot G, Gannot I, Vered H, Buchner A, Keisari Y. Increase in immune cell infiltration with progression of oral epithelium from hyperkeratosis to dysplasia and carcinoma. Br J Cancer. (2002) 86:1444–8. doi: 10.1038/sj.bjc.6600282
29. Foy JP, Tortereau A, Caulin C, Le Texier V, Lavergne E, Thomas E, et al. The dynamics of gene expression changes in a mouse model of oral tumorigenesis may help refine prevention and treatment strategies in patients with oral cancer. Oncotarget. (2016) 7:35932–45. doi: 10.18632/oncotarget.8321
30. Hanna GJ, Villa A, Mistry N, Jia Y, Quinn CT, Turner MM, et al. Comprehensive immunoprofiling of high-risk oral proliferative and localized leukoplakia. Cancer Res Commun. (2021) 1:30–40. doi: 10.1158/2767-9764.CRC-21-0060
31. Koike K, Dehari H, Ogi K, Shimizu S, Nishiyama K, Sonoda T, et al. Prognostic value of FoxP3 and CTLA-4 expression in patients with oral squamous cell carcinoma. PLoS ONE. (2020) 15:e0237465. doi: 10.1371/journal.pone.0237465
32. Seminerio I, Descamps G, Dupont S, de Marrez L, Laigle JA, Lechien JR, et al. Infiltration of FoxP3+ regulatory t cells is a strong and independent prognostic factor in head and neck squamous cell carcinoma. Cancers. (2019) 11:227. doi: 10.3390/cancers11020227
33. Chao JL, Korzinkin M, Zhavoronkov A, Ozerov IV, Walker MT, Higgins K, et al. Effector T cell responses unleashed by regulatory T cell ablation exacerbate oral squamous cell carcinoma. Cell Rep Med. (2021) 2:100399. doi: 10.1016/j.xcrm.2021.100399
34. Chen Y, Li Q, Li X, Ma D, Fang J, Luo L, et al. Blockade of PD-1 effectively inhibits in vivo malignant transformation of oral mucosa. Oncoimmunology. (2018) 7:e1388484. doi: 10.1080/2162402X.2017.1388484
35. Shi Y, Xie TX, Leach DG, Wang B, Young S, Osman AA, et al. Local Anti-PD-1 delivery prevents progression of premalignant lesions in a 4NQO-oral carcinogenesis mouse model. Cancer Prev Res. (2021) 14:767–78. doi: 10.1158/1940-6207.CAPR-20-0607
36. Bouaoud J, Foy JP, Tortereau A, Michon L, Lavergne V, Gadot N, et al. Early changes in the immune microenvironment of oral potentially malignant disorders reveal an unexpected association of M2 macrophages with oral cancer free survival. Oncoimmunology. (2021) 10:1944554. doi: 10.1080/2162402X.2021.1944554
37. Kouketsu A, Sato I, Oikawa M, Shimizu Y, Saito H, Tashiro K, et al. Regulatory T cells and M2-polarized tumour-associated macrophages are associated with the oncogenesis and progression of oral squamous cell carcinoma. Int J Oral Maxillofac Surg. (2019) 48:1279–88. doi: 10.1016/j.ijom.2019.04.004
38. Pellicioli ACA, Bingle L, Farthing P, Lopes MA, Martins MD, Vargas PA. Immunosurveillance profile of oral squamous cell carcinoma and oral epithelial dysplasia through dendritic and T-cell analysis. J Oral Pathol Med. (2017) 46:928–33. doi: 10.1111/jop.12597
39. Nielsen KJ, Jakobsen KK, Jensen JS, Gronhoj C, Von Buchwald C. The effect of prophylactic HPV vaccines on oral and oropharyngeal HPV infection-a systematic review. Viruses. (2021) 13:1339. doi: 10.3390/v13071339
40. Wang C, Dickie J, Sutavani RV, Pointer C, Thomas GJ, Savelyeva N. Targeting head and neck cancer by vaccination. Front Immunol. (2018) 9:830. doi: 10.3389/fimmu.2018.00830
41. De Costa AM, Justis DN, Schuyler CA, Young MR. Administration of a vaccine composed of dendritic cells pulsed with premalignant oral lesion lysate to mice bearing carcinogen-induced premalignant oral lesions stimulates a protective immune response. Int Immunopharmacol. (2012) 13:322–30. doi: 10.1016/j.intimp.2012.05.004
42. Taoudi Benchekroun M, Saintigny P, Thomas SM, El-Naggar AK, Papadimitrakopoulou V, Ren H, et al. Epidermal growth factor receptor expression and gene copy number in the risk of oral cancer. Cancer Prev Res. (2010) 3:800–9. doi: 10.1158/1940-6207.CAPR-09-0163
43. Shin DM, Zhang H, Saba NF, Chen AY, Nannapaneni S, Amin AR, et al. Chemoprevention of head and neck cancer by simultaneous blocking of epidermal growth factor receptor and cyclooxygenase-2 signaling pathways: preclinical and clinical studies. Clin Cancer Res. (2013) 19:1244–56. doi: 10.1158/1078-0432.CCR-12-3149
44. van den Bulk J, Verdegaal EME, Ruano D, Ijsselsteijn ME, Visser M, van der Breggen R, et al. Neoantigen-specific immunity in low mutation burden colorectal cancers of the consensus molecular subtype 4. Genome Med. (2019) 11:87. doi: 10.1186/s13073-019-0697-8
45. Canale FP, Ramello MC, Nunez N, Araujo Furlan CL, Bossio SN, Gorosito Serran M, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8(+) T cells. Cancer Res. (2018) 78:115–28. doi: 10.1158/0008-5472.CAN-16-2684
46. Eberhardt CS, Kissick HT, Patel MR, Cardenas MA, Prokhnevska N, Obeng RC, et al. Functional HPV-specific PD-1(+) stem-like CD8 T cells in head and neck cancer. Nature. (2021) 597:279–84. doi: 10.1038/s41586-021-03862-z
47. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol. (2016) 16:102–11. doi: 10.1038/nri.2015.10
48. Ward MJ, Thirdborough SM, Mellows T, Riley C, Harris S, Suchak K, et al. Tumour-infiltrating lymphocytes predict for outcome in HPV-positive oropharyngeal cancer. Br J Cancer. (2014) 110:489–500. doi: 10.1038/bjc.2013.639
49. Matlung SE, Wilhelmina van Kempen PM, Bovenschen N, van Baarle D, Willems SM. Differences in T-cell infiltrates and survival between HPV+ and HPV- oropharyngeal squamous cell carcinoma. Future Sci OA. (2016) 2:FSO88. doi: 10.4155/fso.15.88
50. Montler R, Bell RB, Thalhofer C, Leidner R, Feng Z, Fox BA, et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin Transl Immunol. (2016) 5:e70. doi: 10.1038/cti.2016.16
51. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. (2016) 45:1135–47. doi: 10.1016/j.immuni.2016.10.021
52. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. (2016) 45:1122–34. doi: 10.1016/j.immuni.2016.10.032
53. Ben Khelil M, Godet Y, Abdeljaoued S, Borg C, Adotevi O, Loyon R. Harnessing antitumor CD4(+) T cells for cancer immunotherapy. Cancers. (2022) 14:260. doi: 10.3390/cancers14010260
54. Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, et al. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity. (2020) 52:183–99.e9. doi: 10.1016/j.immuni.2019.11.014
55. Kortekaas KE, Santegoets SJ, Sturm G, Ehsan I, van Egmond SL, Finotello F, et al. CD39 identifies the CD4(+) tumor-specific T-cell population in human cancer. Cancer Immunol Res. (2020) 8:1311–21. doi: 10.1158/2326-6066.CIR-20-0270
56. Russell S, Angell T, Lechner M, Liebertz D, Correa A, Sinha U, et al. Immune cell infiltration patterns and survival in head and neck squamous cell carcinoma. Head Neck Oncol. (2013) 5:24.
57. Wood O, Woo J, Seumois G, Savelyeva N, McCann KJ, Singh D, et al. Gene expression analysis of TIL rich HPV-driven head and neck tumors reveals a distinct B-cell signature when compared to HPV independent tumors. Oncotarget. (2016) 7:56781–97. doi: 10.18632/oncotarget.10788
58. Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. (2021) 184:6101–18.e13. doi: 10.1016/j.cell.2021.11.007
59. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. (2014) 35:571–80. doi: 10.1016/j.it.2014.09.006
60. Gunderson AJ, Rajamanickam V, Bui C, Bernard B, Pucilowska J, Ballesteros-Merino C, et al. Germinal center reactions in tertiary lymphoid structures associate with neoantigen burden, humoral immunity and long-term survivorship in pancreatic cancer. Oncoimmunology. (2021) 10:1900635. doi: 10.1080/2162402X.2021.1900635
61. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA. (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
62. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. (2012) 122:899–910. doi: 10.1172/JCI45817
63. Egelston CA, Avalos C, Tu TY, Rosario A, Wang R, Solomon S, et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight. (2019) 4:e130000. doi: 10.1172/jci.insight.130000
64. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. (2019) 50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021
65. Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. (2015) 350:568–71. doi: 10.1126/science.aab3291
66. Senba M, Mori N. Mechanisms of virus immune evasion lead to development from chronic inflammation to cancer formation associated with human papillomavirus infection. Oncol Rev. (2012) 6:e17. doi: 10.4081/oncol.2012.e17
67. Steinbach A, Riemer AB. Immune evasion mechanisms of human papillomavirus: an update. Int J Cancer. (2018) 142:224–9. doi: 10.1002/ijc.31027
68. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. (2008) 322:1097–100. doi: 10.1126/science.1164206
69. Bursuker I, North RJ. Immunological consequences of tumor excision: from active immunity to immunological memory. Int J Cancer. (1986) 37:275–81. doi: 10.1002/ijc.2910370216
70. North RJ, Dye ES. Ly 1+2- suppressor T cells down-regulate the generation of Ly 1-2+ effector T cells during progressive growth of the P815 mastocytoma. Immunology. (1985) 54:47–56.
71. Mills CD, North RJ. Ly-1+2- suppressor T cells inhibit the expression of passively transferred antitumor immunity by suppressing the generation of cytolytic T cells. Transplantation. (1985) 39:202–8. doi: 10.1097/00007890-198502000-00018
72. Bursuker I, North RJ. Suppression of generation of concomitant antitumor immunity by passively transferred suppressor T cells from tumor-bearing donors. Cancer Immunol Immunother. (1985) 19:215–8. doi: 10.1007/BF00199229
73. North RJ, Bursuker I. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2- suppressor T cells down-regulate the generation of Ly-1-2+ effector T cells. J Exp Med. (1984) 159:1295–311. doi: 10.1084/jem.159.5.1295
74. Crittenden MR, Zebertavage L, Kramer G, Bambina S, Friedman D, Troesch V, et al. Tumor cure by radiation therapy and checkpoint inhibitors depends on pre-existing immunity. Sci Rep. (2018) 8:7012. doi: 10.1038/s41598-018-25482-w
75. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. (2014) 2:3. doi: 10.1186/2051-1426-2-3
76. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
77. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol. (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
78. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. (2016) 45:389–401. doi: 10.1016/j.immuni.2016.07.011
79. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
80. Bell RB, Leidner RS, Crittenden MR, Curti BD, Feng Z, Montler R, et al. OX40 signaling in head and neck squamous cell carcinoma: overcoming immunosuppression in the tumor microenvironment. Oral Oncol. (2016) 52:1–10. doi: 10.1016/j.oraloncology.2015.11.009
81. Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti-OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. Eur J Immunol. (2007) 37:157–66. doi: 10.1002/eji.200636428
82. Kuhns MS, Epshteyn V, Sobel RA, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates the size, reactivity, and function of a primed pool of CD4+ T cells. Proc Natl Acad Sci USA. (2000) 97:12711–6. doi: 10.1073/pnas.220423597
83. Campillo-Davo D, Flumens D, Lion E. The quest for the best: how TCR affinity, avidity, and functional avidity affect TCR-engineered T-cell antitumor responses. Cells. (2020) 9:1720. doi: 10.3390/cells9071720
84. Gilfillan CB, Hebeisen M, Rufer N, Speiser DE. Constant regulation for stable CD8 T-cell functional avidity and its possible implications for cancer immunotherapy. Eur J Immunol. (2021) 51:1348–60. doi: 10.1002/eji.202049016
85. Miconnet I, Koenig S, Speiser D, Krieg A, Guillaume P, Cerottini JC, et al. CpG are efficient adjuvants for specific CTL induction against tumor antigen-derived peptide. J Immunol. (2002) 168:1212–8. doi: 10.4049/jimmunol.168.3.1212
86. Sun S, Kishimoto H, Sprent J. DNA as an adjuvant: capacity of insect DNA and synthetic oligodeoxynucleotides to augment T cell responses to specific antigen. J Exp Med. (1998) 187:1145–50. doi: 10.1084/jem.187.7.1145
87. Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med. (1997) 186:1623–31. doi: 10.1084/jem.186.10.1623
88. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. (2008) 8:279–89. doi: 10.1038/nri2215
89. Matzinger P. An innate sense of danger. Semin Immunol. (1998) 10:399–415. doi: 10.1006/smim.1998.0143
90. Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. (1998) 393:480–3. doi: 10.1038/31002
91. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. (1998) 393:474–8. doi: 10.1038/30989
92. Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. (1998) 393:478–80. doi: 10.1038/30996
93. Westcott PMK, Sacks NJ, Schenkel JM, Ely ZA, Smith O, Hauck H, et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat Cancer. (2021) 2:1071–85. doi: 10.1038/s43018-021-00247-z
94. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv. (2020) 6:eaay4458. doi: 10.1126/sciadv.aay4458
95. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
96. Schneider H, Rudd CE. Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun. (2000) 269:279–83. doi: 10.1006/bbrc.2000.2234
97. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity. (2000) 13:313–22. doi: 10.1016/S1074-7613(00)00031-5
98. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
99. Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science. (2019) 364:558–66. doi: 10.1126/science.aav7062
100. Arumugam V, Bluemn T, Wesley E, Schmidt AM, Kambayashi T, Malarkannan S, et al. TCR signaling intensity controls CD8+ T cell responsiveness to TGF-beta. J Leukoc Biol. (2015) 98:703–12. doi: 10.1189/jlb.2HIMA1214-578R
101. Ho WJ, Croessmann S, Lin J, Phyo ZH, Charmsaz S, Danilova L, et al. Systemic inhibition of PTPN22 augments anticancer immunity. J Clin Invest. (2021) 131:e146950. doi: 10.1172/JCI146950
102. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
103. Donohue JH, Rosenstein M, Chang AE, Lotze MT, Robb RJ, Rosenberg SA. The systemic administration of purified interleukin 2 enhances the ability of sensitized murine lymphocytes to cure a disseminated syngeneic lymphoma. J Immunol. (1984) 132:2123–8.
104. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. (1986) 233:1318–21. doi: 10.1126/science.3489291
105. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
106. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
107. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
108. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
109. Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. (2010) 33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc
110. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. (2005) 202:907–12. doi: 10.1084/jem.20050732
111. Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev. (2014) 257:264–76. doi: 10.1111/imr.12135
112. Leidner R, Curti BD, Payne RM, Urba W, Bahjat KS, Koguchi Y, et al. Adoptive TIL in HPV-negative oral scca. J Immunother Cancer. (2015) 3:P26. doi: 10.1186/2051-1426-3-S2-P26
113. Leidner RS, Sukari A, Chung CH, Ohr J, Haigentz M, Cohen EEW, et al. A phase 2, multicenter study to evaluate the efficacy and safety of autologous tumor infiltrating lymphocytes (LN-145) for the treatment of patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck (HNSCC). J Clin Oncol. (2018) 36:TPS6096. doi: 10.1200/JCO.2018.36.15_suppl.TPS6096
114. O'Malley D, Lee S, Psyrri A, Sukari A, Thomas S, Wenham R, et al. 492 Phase 2 efficacy and safety of autologous tumor-infiltrating lymphocyte (TIL) cell therapy in combination with pembrolizumab in immune checkpoint inhibitor-naïve patients with advanced cancers. J Immunother Cancer. (2021) 9:A523–4. doi: 10.1136/jitc-2021-SITC2021.492
115. Riemer AB, Keskin DB, Zhang G, Handley M, Anderson KS, Brusic V, et al. A conserved E7-derived cytotoxic T lymphocyte epitope expressed on human papillomavirus 16-transformed HLA-A2+ epithelial cancers. J Biol Chem. (2010) 285:29608–22. doi: 10.1074/jbc.M110.126722
116. Ramos CA, Narala N, Vyas GM, Leen AM, Gerdemann U, Sturgis EM, et al. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes for adoptive immunotherapy of HPV-associated malignancies. J Immunother. (2013) 36:66–76. doi: 10.1097/CJI.0b013e318279652e
117. Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. (2017) 356:200–5. doi: 10.1126/science.aak9510
118. Mercier-Letondal P, Marton C, Deschamps M, Ferrand C, Vauchy C, Chenut C, et al. Isolation and characterization of an HLA-DRB1*04-restricted HPV16-E7 T cell receptor for cancer immunotherapy. Hum Gene Ther. (2018) 29:1202–12. doi: 10.1089/hum.2018.091
119. Jin J, Gkitsas N, Fellowes VS, Ren J, Feldman SA, Hinrichs CS, et al. Enhanced clinical-scale manufacturing of TCR transduced T-cells using closed culture system modules. J Transl Med. (2018) 16:13. doi: 10.1186/s12967-018-1384-z
120. Jin BY, Campbell TE, Draper LM, Stevanovic S, Weissbrich B, Yu Z, et al. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI insight. (2018) 3:e99488. doi: 10.1172/jci.insight.99488
121. Helman SR, Stevanovic S, Campbell TE, Kwong MLM, Doran SL, Faquin WC, et al. Human papillomavirus T-cell cross-reactivity in cervical cancer: implications for immunotherapy clinical trial design. JAMA Netw Open. (2018) 1:e180706. doi: 10.1001/jamanetworkopen.2018.0706
122. Norberg SM, Nagarsheth N, Doran S, Kanakry JA, Adhikary S, Schweitzer C, et al. Regression of epithelial cancers following T cell receptor gene therapy targeting human papillomavirus-16 E7. Blood. (2018) 132:492. doi: 10.1182/blood-2018-99-117017
123. Doran SL, Stevanovic S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, et al. T-Cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J Clin Oncol. (2019) 37:2759–68. doi: 10.1200/JCO.18.02424
124. Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. (2021) 27:419–25. doi: 10.1038/s41591-020-01225-1
125. Cohen EE, Dunn L, Neupane P, Gibson M, Leidner R, Savvides P, et al. 976TiP SPEARHEAD-2 trial design: A phase II pilot trial of ADP-A2M4 in combination with pembrolizumab in patients with recurrent or metastatic head and neck cancer. Ann Oncol. (2020) 31:S685. doi: 10.1016/j.annonc.2020.08.1091
126. Wermke M, Tsimberidou A-M, Mohamed A, Mayer-Mokler A, Satelli A, Reinhardt C, et al. 959 Safety and anti-tumor activity of TCR-engineered autologous, PRAME-directed T cells across multiple advanced solid cancers at low doses – clinical update on the ACTengine® IMA203 trial. J Immunother Cancer. (2021) 9:A1009. doi: 10.1136/jitc-2021-SITC2021.959
127. Wu F, Hong J, Du N, Wang Y, Chen J, He Y, et al. Long-Term outcomes of neoadjuvant chemotherapy in locally advanced gastric cancer/esophagogastric junction cancer: a systematic review and meta-analysis. Anticancer Agents Med Chem. (2022) 22:143–51. doi: 10.2174/1871520621666210315091932
128. Bajwa G, Lanz I, Cardenas M, Brenner MK, Arber C. Transgenic CD8alphabeta co-receptor rescues endogenous TCR function in TCR-transgenic virus-specific T cells. J Immunother Cancer. (2020) 8:e001487. doi: 10.1136/jitc-2020-001487
129. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
130. Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. (2013) 31:999–1008. doi: 10.1038/nbt.2725
131. Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. (2018) 36:215–9. doi: 10.1038/nbt.4090
132. Hinrichs CS. Molecular pathways: breaking the epithelial cancer barrier for chimeric antigen receptor and T-cell receptor gene therapy. Clin Cancer Res. (2016) 22:1559–64. doi: 10.1158/1078-0432.CCR-15-1294
133. Wachsmann TLA, Wouters AK, Remst DFG, Hagedoorn RS, Meeuwsen MH, van Diest E, et al. Comparing CAR and TCR engineered T cell performance as a function of tumor cell exposure. Oncoimmunology. (2022) 11:2033528. doi: 10.1080/2162402X.2022.2033528
134. van Schalkwyk MC, Papa SE, Jeannon JP, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev. (2013) 24:134–42. doi: 10.1089/humc.2013.144
135. Papa S, Adami A, Metoudi M, Achkova D, Schalkwyk Mv, Pereira AP, et al. Abstract CT118: T4 immunotherapy of head and neck squamous cell carcinoma using pan-ErbB targeted CAR T-cells. Cancer Res. (2017) 77:CT118. doi: 10.1158/1538-7445.AM2017-CT118
136. Papa S, Adami A, Metoudi M, Achkova D, van Schalkwyk M, Parente Pereira A, et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). J Clin Oncol. (2018) 36:3046. doi: 10.1200/JCO.2018.36.15_suppl.3046
137. Larcombe-Young D, Papa S, Maher J. PanErbB-targeted CAR T-cell immunotherapy of head and neck cancer. Expert Opin Biol Ther. (2020) 20:965–70. doi: 10.1080/14712598.2020.1786531
138. Kessels HW, Wolkers MC, van den Boom MD, van der Valk MA, Schumacher TN. Immunotherapy through TCR gene transfer. Nat Immunol. (2001) 2:957–61. doi: 10.1038/ni1001-957
139. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
140. Schumacher TN, Scheper W, Kvistborg P. Cancer neoantigens. Annu Rev Immunol. (2019) 37:173–200. doi: 10.1146/annurev-immunol-042617-053402
141. Tran E, Robbins PF, Rosenberg SA. 'Final common pathway' of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. (2017) 18:255–62. doi: 10.1038/ni.3682
142. June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. (2015) 7:280ps7. doi: 10.1126/scitranslmed.aaa3643
143. Bethune MT, Joglekar AV. Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr Opin Biotechnol. (2017) 48:142–52. doi: 10.1016/j.copbio.2017.03.024
144. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. (2015) 350:1387–90. doi: 10.1126/science.aad1253
145. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
146. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–730. doi: 10.1038/s41591-018-0040-8
147. Zacharakis N, Huq LM, Seitter SJ, Kim SP, Gartner JJ, Sindiri S, et al. Breast cancers are immunogenic: immunologic analyses and a phase ii pilot clinical trial using mutation-reactive autologous lymphocytes. J Clin Oncol. (2022) 40:1741–54. doi: 10.1200/JCO.21.02170
148. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med. (2022) 386:2112–9. doi: 10.1056/NEJMoa2119662
149. Le X, Marcelo K, Coleman N, Hopkins J, Balsara B, Leoni M, et al. 893P Clinico-genetic profiling of HRAS mutant head and neck squamous cell carcinoma (HNSCC). Ann Oncol. (2021) 32:S799–800. doi: 10.1016/j.annonc.2021.08.1303
150. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. (2017) 545:423–31. doi: 10.1038/nature22395
151. Ferris RL, Hunt JL, Ferrone S. Human leukocyte antigen (HLA) class I defects in head and neck cancer: molecular mechanisms and clinical significance. Immunol Res. (2005) 33:113–33. doi: 10.1385/IR:33:2:113
152. Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin Cancer Res. (2006) 12:3890–5. doi: 10.1158/1078-0432.CCR-05-2750
153. Leibowitz MS, Andrade Filho PA, Ferrone S, Ferris RL. Deficiency of activated STAT1 in head and neck cancer cells mediates TAP1-dependent escape from cytotoxic T lymphocytes. Cancer Immunol Immunother. (2011) 60:525–35. doi: 10.1007/s00262-010-0961-7
154. Lopez-Albaitero A, Nayak JV, Ogino T, Machandia A, Gooding W, DeLeo AB, et al. Role of antigen-processing machinery in the in vitro resistance of squamous cell carcinoma of the head and neck cells to recognition by CTL. J Immunol. (2006) 176:3402–9. doi: 10.4049/jimmunol.176.6.3402
155. Matsui M, Ikeda M, Akatsuka T. High expression of HLA-A2 on an oral squamous cell carcinoma with down-regulated transporter for antigen presentation. Biochem Biophys Res Commun. (2001) 280:1008–14. doi: 10.1006/bbrc.2000.4234
156. Meissner M, Whiteside TL, van Kuik-Romein P, Valesky EM, van den Elsen :PJ, Kaufmann R, et al. Loss of interferon-gamma inducibility of the MHC class II antigen processing pathway in head and neck cancer: evidence for post-transcriptional as well as epigenetic regulation. Br J Dermatol. (2008) 158:930–40. doi: 10.1111/j.1365-2133.2008.08465.x
157. Grandis JR, Falkner DM, Melhem MF, Gooding WE, Drenning SD, Morel PA. Human leukocyte antigen class I allelic and haplotype loss in squamous cell carcinoma of the head and neck: clinical and immunogenetic consequences. Clin Cancer Res. (2000) 6:2794–802. Available online at: https://aacrjournals.org/clincancerres/article/6/7/2794/288296/Human-Leukocyte-Antigen-Class-I-Allelic-and?searchresult=1
158. Hammerman PS, Hayes DN, Grandis JR. Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov. (2015) 5:239–44. doi: 10.1158/2159-8290.CD-14-1205
159. Bandoh N, Ogino T, Katayama A, Takahara M, Katada A, Hayashi T, et al. HLA class I antigen and transporter associated with antigen processing downregulation in metastatic lesions of head and neck squamous cell carcinoma as a marker of poor prognosis. Oncol Rep. (2010) 23:933–9. doi: 10.3892/or_00000717
160. Meissner M, Reichert TE, Kunkel M, Gooding W, Whiteside TL, Ferrone S, et al. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin Cancer Res. (2005) 11:2552–60. doi: 10.1158/1078-0432.CCR-04-2146
161. Ogino T, Bandoh N, Hayashi T, Miyokawa N, Harabuchi Y, Ferrone S. Association of tapasin and HLA class I antigen down-regulation in primary maxillary sinus squamous cell carcinoma lesions with reduced survival of patients. Clin Cancer Res. (2003) 9:4043–51. Available online at: https://aacrjournals.org/clincancerres/article/9/11/4043/202632/Association-of-Tapasin-and-HLA-Class-I-Antigen?searchresult=1
162. Ogino T, Shigyo H, Ishii H, Katayama A, Miyokawa N, Harabuchi Y, et al. HLA class I antigen down-regulation in primary laryngeal squamous cell carcinoma lesions as a poor prognostic marker. Cancer Res. (2006) 66:9281–9. doi: 10.1158/0008-5472.CAN-06-0488
163. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
164. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. (1994) 8:652–8.
165. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM II, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
166. Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J Immunother Cancer. (2020) 8:e000450. doi: 10.1136/jitc-2019-000450
167. Lee MY, Robbins Y, Sievers C, Friedman J, Abdul Sater H, Clavijo PE, et al. Chimeric antigen receptor engineered NK cellular immunotherapy overcomes the selection of T-cell escape variant cancer cells. J Immunother Cancer. (2021) 9:e002128. doi: 10.1136/jitc-2020-002128
168. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol. (2016) 58:59–70. doi: 10.1016/j.oraloncology.2016.05.002
169. Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young MR. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res. (1995) 1:95–103.
170. Young MR, Wright MA, Lozano Y, Prechel MM, Benefield J, Leonetti JP, et al. Increased recurrence and metastasis in patients whose primary head and neck squamous cell carcinomas secreted granulocyte-macrophage colony-stimulating factor and contained CD34+ natural suppressor cells. Int J Cancer. (1997) 74:69–74. doi: 10.1002/(SICI)1097-0215(19970220)74:1<69::AID-IJC12>3.0.CO;2-D
171. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425; author reply 426. doi: 10.1158/0008-5472.CAN-06-3037
172. Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res. (2017) 77:2607–19. doi: 10.1158/0008-5472.CAN-16-2534
173. Clavijo PE, Moore EC, Chen J, Davis RJ, Friedman J, Kim Y, et al. Resistance to CTLA-4 checkpoint inhibition reversed through selective elimination of granulocytic myeloid cells. Oncotarget. (2017) 8:55804–20. doi: 10.18632/oncotarget.18437
174. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI insight. (2019) 4:e126853. doi: 10.1172/jci.insight.126853
175. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. (2017) 543:728–32. doi: 10.1038/nature21676
176. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
177. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res. (2020) 26:1420–31. doi: 10.1158/1078-0432.CCR-19-2625
178. Huber MH, Lippman SM, Benner SE, Shirinian M, Dimery IW, Dunnington JS, et al. A phase II study of ifosfamide in recurrent squamous cell carcinoma of the head and neck. Am J Clin Oncol. (1996) 19:379–82. doi: 10.1097/00000421-199608000-00012
179. Sandler A, Saxman S, Bandealy M, Heilman D, Monaco F, McClean J, et al. Ifosfamide in the treatment of advanced or recurrent squamous cell carcinoma of the head and neck: a phase II hoosier oncology group trial. Am J Clin Oncol. (1998) 21:195–7. doi: 10.1097/00000421-199804000-00021
180. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. (2011) 331:1612–6. doi: 10.1126/science.1198443
181. Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AHR, Ugel S, et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell. (2016) 30:377–90. doi: 10.1016/j.ccell.2016.08.004
182. Liu C, Lewis CM, Lou Y, Xu C, Peng W, Yang Y, et al. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J Immunother. (2012) 35:276–82. doi: 10.1097/CJI.0b013e31824e7f43
183. Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med. (2020) 217:e20190673. doi: 10.1084/jem.20190673
184. Innamarato P, Kodumudi K, Asby S, Schachner B, Hall M, Mackay A, et al. Reactive myelopoiesis triggered by lymphodepleting chemotherapy limits the efficacy of adoptive T cell therapy. Mol Ther. (2020) 28:2252–70. doi: 10.1016/j.ymthe.2020.06.025
185. Zebertavage LK, Alice A, Crittenden MR, Gough MJ. Transcriptional upregulation of NLRC5 by radiation drives STING- and interferon-independent MHC-I expression on cancer cells and T cell cytotoxicity. Sci Rep. (2020) 10:7376. doi: 10.1038/s41598-020-64408-3
186. Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. (2018) 9:3868. doi: 10.1038/s41467-018-06300-3
187. Chapuis AG, Afanasiev OK, Iyer JG, Paulson KG, Parvathaneni U, Hwang JH, et al. Regression of metastatic merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-I. Cancer Immunol Res. (2014) 2:27–36. doi: 10.1158/2326-6066.CIR-13-0087
188. Sharon S, Baird JR, Bambina S, Kramer G, Blair TC, Jensen SM, et al. A platform for locoregional T-cell immunotherapy to control HNSCC recurrence following tumor resection. Oncotarget. (2021) 12:1201–13. doi: 10.18632/oncotarget.27982
189. Baird JR, Bell RB, Troesch V, Friedman D, Bambina S, Kramer G, et al. Evaluation of explant responses to STING ligands: personalized immunosurgical therapy for head and neck squamous cell carcinoma. Cancer Res. (2018) 78:6308–19. doi: 10.1158/0008-5472.CAN-18-1652
190. Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, et al. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS ONE. (2016) 11:e0161779. doi: 10.1371/journal.pone.0161779
191. van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. (1999) 190:355–66. doi: 10.1084/jem.190.3.355
192. Li B, VanRoey M, Wang C, Chen TH, Korman A, Jooss K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor–secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res. (2009) 15:1623–34. doi: 10.1158/1078-0432.CCR-08-1825
193. Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol Res. (2016) 4:845–57. doi: 10.1158/2326-6066.CIR-16-0060
194. Lelliott EJ, Cullinane C, Martin CA, Walker R, Ramsbottom KM, Souza-Fonseca-Guimaraes F, et al. A novel immunogenic mouse model of melanoma for the preclinical assessment of combination targeted and immune-based therapy. Sci Rep. (2019) 9:1225. doi: 10.1038/s41598-018-37883-y
195. McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci USA. (2016) 113:E6409–17. doi: 10.1073/pnas.1613601113
196. Wisdom AJ, Mowery YM, Hong CS, Himes JE, Nabet BY, Qin X, et al. Single cell analysis reveals distinct immune landscapes in transplant and primary sarcomas that determine response or resistance to immunotherapy. Nat Commun. (2020) 11:6410. doi: 10.1038/s41467-020-19917-0
197. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
198. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
199. Gough MJ, Sharon S, Crittenden MR, Young KH. Using preclinical data to design combination clinical trials of radiation therapy and immunotherapy. Semin Radiat Oncol. (2020) 30:158–72. doi: 10.1016/j.semradonc.2019.12.002
200. Sharon S, Duhen T, Bambina S, Baird J, Leidner R, Bell B, et al. Explant modeling of the immune environment of head and neck cancer. Front Oncol. (2021) 11:611365. doi: 10.3389/fonc.2021.611365
201. Powley IR, Patel M, Miles G, Pringle H, Howells L, Thomas A, et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br J Cancer. (2020) 122:735–44. doi: 10.1038/s41416-019-0672-6
202. Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat Med. (2021) 27:1250–61. doi: 10.1038/s41591-021-01398-3
203. Aroldi F, Lord SR. Window of opportunity clinical trial designs to study cancer metabolism. Br J Cancer. (2020) 122:45–51. doi: 10.1038/s41416-019-0621-4
Keywords: OSCC (oral squamous cell carcinoma), HNSCC (head and neck squamous cell carcinoma), CD8, CD4, pre-malignancies, TIL (tumor infiltrating lymphocytes), immunotherapy
Citation: Duhen T, Gough MJ, Leidner RS and Stanton SE (2022) Development and therapeutic manipulation of the head and neck cancer tumor environment to improve clinical outcomes. Front. Oral. Health 3:902160. doi: 10.3389/froh.2022.902160
Received: 22 March 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Sabrina Wurzba, McGill University, CanadaReviewed by:
Wei-fa Yang, The University of Hong Kong, Hong Kong SAR, ChinaJason Y. K. Chan, The Chinese University of Hong Kong, China
Moshe Elkabets, Ben-Gurion University of the Negev, Israel
Copyright © 2022 Duhen, Gough, Leidner and Stanton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Gough, TWljaGFlbC5nb3VnaEBwcm92aWRlbmNlLm9yZw==