 Raymond Pasman
Raymond Pasman Bastiaan P. Krom
Bastiaan P. Krom Sebastian A. J. Zaat3
Sebastian A. J. Zaat3 Stanley Brul
Stanley Brul
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oral. Health , 07 April 2022
Sec. Oral Infections and Microbes
Volume 3 - 2022 | https://doi.org/10.3389/froh.2022.851786
This article is part of the Research Topic Insights in Oral Infections and Microbes: 2021 View all 9 articles
Candida albicans and Staphylococcus aureus account for most invasive fungal and bacterial bloodstream infections (BSIs), respectively. However, the initial point of invasion responsible for S. aureus BSIs is often unclear. Recently, C. albicans has been proposed to mediate S. aureus invasion of immunocompromised hosts during co-colonization of oral mucosal surfaces. The status of the oral immune system crucially contributes to this process in two distinct ways: firstly, by allowing invasive C. albicans growth during dysfunction of extra-epithelial immunity, and secondly following invasion by some remaining function of intra-epithelial immunity. Immunocompromised individuals at risk of developing invasive oral C. albicans infections could, therefore, also be at risk of contracting concordant S. aureus BSIs. Considering the crucial contribution of both oral immune function and dysfunction, the aim of this review is to provide an overview of relevant aspects of intra and extra-epithelial oral immunity and discuss predominant immune deficiencies expected to facilitate C. albicans induced S. aureus BSIs.
Various niches of the healthy human body are readily colonized by commensal fungi and bacteria. However, numerous of these microbes are opportunistic pathogens, i.e. they become pathogenic when the environment allows it. One major opportunistic pathogen colonizing the oral cavity, gastro- intestinal tract, and vagina of most healthy individuals is Candida albicans [1]. C. albicans is a polymorphic fungus able to grow as relatively harmless yeast and pseudohyphal cells, as well as harmful invasive hyphae [2]. Immunocompromised individuals suffering from suppressed extra epithelial oral immunity are prone to develop oropharyngeal candidiasis (OPC), a local infection of oral mucosa characterized by epithelium invading hyphae. If intra-epithelial immune responses are unable to prevent further growth, invading cells can disseminate and result in life threatening blood stream infections (candidemia) [3, 4]. Candidemia is linked to severe morbidity and mortality, with the latter reaching up to 71% depending on patient age and/or underlying conditions [5]. Approximately one in five candidemia cases is known to be polymicrobial [6, 7], with Staphylococcus aureus, a leading cause of bloodstream infections (BSIs) [7, 8], as one of the most common co-isolated bacterial species [6]. Interestingly, a significant number of patients suffering from staphylococcal BSIs have no reported porte d'entrée [9]. Recent findings suggest that C. albicans invasion of the oral epithelium creates such a porte d'entrée and, thereby, facilitates S. aureus BSIs [10–13]. This process was first hypothesized to be facilitated by hyphae adhering S. aureus moving along with the growing hyphae in a hitchhiking like manner [14]. However, recent research has shown S. aureus to remain situated at the initial point of adhesion during hyphal growth, rendering the co-invasion hypothesis up for debate [11]. Even though the specific mechanisms driving co-invasion and dissemination remain to be determined, it is apparent that it majorly depends on hyphal invasion (both mechanically and with aid of the secreted cytotoxic peptide candidalysin), and the binding of S. aureus to the hyphal agglutinin like sequence 1 and 3 (Als1 and Als3) proteins [11–14]. Importantly, several new lines of evidence also point to a crucial role of the oral immune system in this process [10, 13]. Whereas low level immune suppression is crucial for OPC development and S. aureus co-colonization in murine models, severe immune suppression significantly reduces S. aureus dissemination [13, 15]. Thus far, this reduction has been attributed to a significant reduction in local phagocyte numbers [13]. When present, phagocytes are actively recruited by C. albicans hyphae, but are unable to engulf them and internalize hyphae bound S. aureus instead [11]. S. aureus is notorious for circumventing phagocytic killing and could, thus, utilize phagocytes as a trojan horse while it is transported to draining cervical lymph nodes, facilitating further dissemination to the bloodstream [11, 16]. Thereby, oral immune dysfunction could induce OPC facilitated S. aureus BSIs without instigating candidemia and also account for monomicrobial S. aureus BSIs. Considering the crucial contribution of immune dysfunction, immunocompromised individuals might not only be at increased risk of developing OPC but S. aureus BSIs as well. Due to the fact that immunosuppression affects approximately one in every 16 individuals and is increasing with time, its effect on OPC induced S. aureus BSIs can be more prominent than anticipated [17]. In light of importance of the oral immune system in this process, the aim of this review is to provide a detailed overview of both extra-epithelial and intra-epithelial interactions between the oral immune system and C. albicans and S. aureus. Furthermore, predominant immunosuppressive disorders linked to these interactions and their relation to increased risk of OPC induced S. aureus dissemination will be discussed.
Extra-epithelial oral immunity encompasses immune factors present/secreted in saliva and gingival crevicular fluid (GCF). These immune factors include antimicrobial proteins and antimicrobial peptides (AMPs), oral polymorphonuclear leukocytes and factors of the complement system. Extra-epithelial oral immunity is continuously active to control commensal colonization and prevent pathogenic (over)growth (e.g. of C. albicans). Below all relevant aspects of extra-epithelial oral immunity will be discussed in relation to their general role in the oral cavity as well as their role in C. albicans and S. aureus immunity. Possible mechanisms of both organisms to evade extra-epithelial oral immunity will be covered as well.
Inside the oral cavity cells of the epithelium and salivary glands continuously produce and secrete antimicrobial proteins and AMPs into saliva and GCF (Figure 1) [18–22]. Predominant oral antimicrobial proteins include lysozyme, lactoferrin, and lactoperoxidase and reduce microbial growth by breaking down peptidoglycan residues, sequestering iron, and oxidating various microbial substrates, respectively. Oral AMPs include α-defensins, β-defensins, LL-37, and histatins and likely exert their antimicrobial efficacy through insertion into cell membranes, lethally destabilizing the membrane [23]. In addition to their direct antimicrobial effect, AMPs serve as chemoattractants for immature dendritic cells, neutrophils, monocytes, and various T-cells and induce the secretion of pro-inflammatory cytokines and chemokines [24, 25]. While low levels of AMPs are constitutively expressed and secreted they can be strongly upregulated following the activation of pattern recognition receptors (PRRs) by specific microbial pathogen-associated molecular patterns (PAMPs) [26–29].
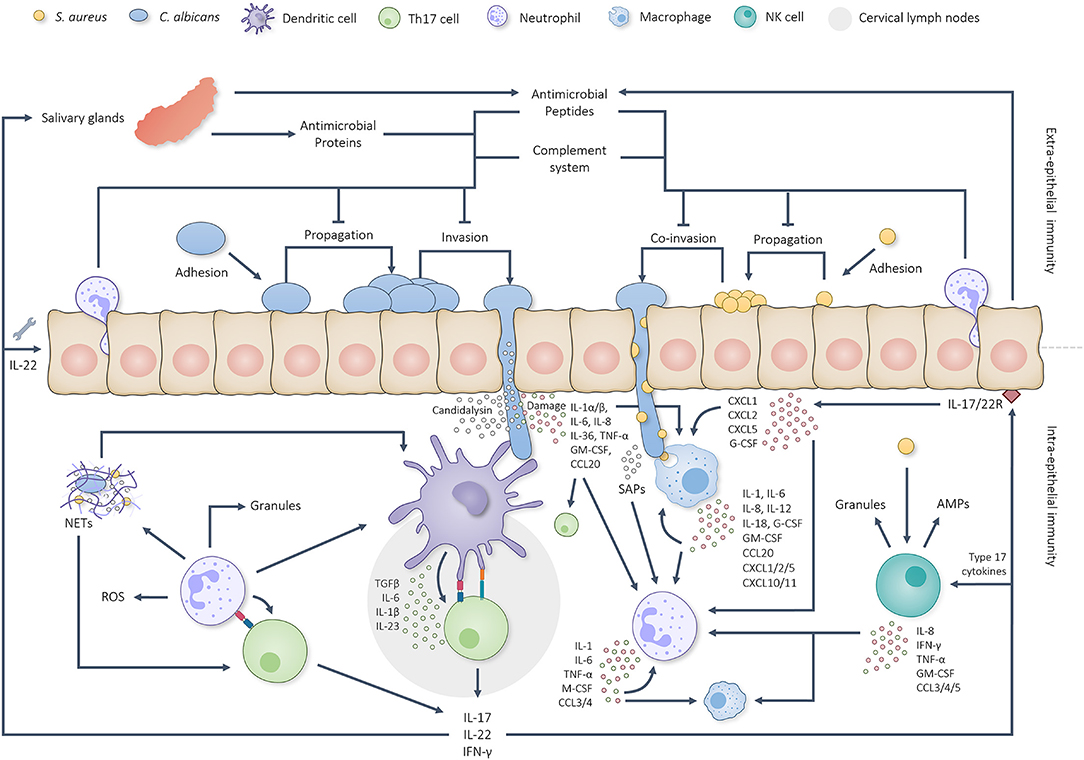
Figure 1. Graphical overview of the intricate interplay between the oral immune system and C. albicans/S. aureus infections. C. albicans first adheres to the oral epithelium, starts propagating and initiates hyphal growth. Extra-epithelial antimicrobial proteins, AMPs, complement factors and neutrophils limit pathogenic overgrowth and tissue invasion. Hyphal invasion, induced tissue damage and candidalysin induce a cascade of intra-epithelial immune reactions. Dendritic cells are able to take up and present pathogenic antigens to naïve T cells in the cervical lymph nodes which, together with IL-1, IL-6 and IL-23, stimulate Th17 differentiation. Simultaneously, oral EC produce IL-1α, IL-1β and IL-36 to activate other type 17 cells. Together, the activated type 17 cells start producing IL-17, IL-22 and IFN-γ. IL-17 and IL-22 sequentially trigger the corresponding receptors on oral ECs and stimulate the production/secretion of both AMPs (also by salivary glands) and chemokines plus aid in the repair of damaged barrier areas. Secreted chemokines attract more neutrophils and macrophages to the site of infection and stimulate their activation. Neutrophils and macrophages phagocytose and break down C. albicans yeast cells, hyphal fragments and S. aureus cells besides which they produce various cytokines and chemokines to further stimulate phagocyte attraction and activation. Phagocyte attraction and activation is also stimulated by candidal and staphylococcal activated NK cells. Neutrophils respond to activation by phagocytosing and killing the threat when possible, producing NETs, secreting granular components and utilizing ROS. Additionally, neutrophils have been found able to prime/activate T cells and APCs besides which they are able to produce extra cytokines and chemokines to stimulate Th17 differentiation and further neutrophil attraction/activation. This positive feedback loop continues until the microbial threat has been eliminated.
Oral epithelial cells (ECs) and cells of the innate immune system utilize PRRs such as Toll-like receptors (TLR), Nucleotide-Binding Oligomerization Domain Receptors (NODs), Protease-Activated Receptors (PARs), C-type lectin receptors (CLR) and RIG-1-like receptors (RLR) to distinguish C. albicans yeast from hyphal PAMPs and adapt their immune response accordingly [30]. Regarding human oral ECs all TLRs have been found present with TLR5 and 7, however, lacking on the surface of buccal epithelial cells [31]. Of these TLRs, TLR2, 4 and 6 have been found to directly affect the mucosal defense against C. albicans through binding of cell wall mannans (TLR2/4/6) and chitin (TLR4) [27, 31]. Additionally, the ephrin type-A receptor 2 (EphA2), has been identified as a non-traditional epithelial PRR able to recognize C. albicans β-glucans [32]. The distinction between yeast and hyphae is mainly accomplished through binding of EC receptor tyrosine kinases (RTKs) such as the human epidermal growth factor receptor 2 (Her2) and EGF, to the hyphal Als3 and Ssa1 proteins [33]. Recently, EGFR has also been found to constitutively associate with the EphA2 receptor to form a physical complex that, in response to Als3, sustains EphA2 activation [34]. Whereas the cell wall composition of C. albicans yeast cells provokes only a weak activation of AMP release, cell wall components of hyphae trigger a more strong and sustained secretion [29, 30]. In response, C. albicans utilizes three mechanisms to reduce AMP efficacy: (1) secretion of proteins able to bind and/or degrade AMPs, (2) extrusion of internalized AMPs via efflux transporters, and (3) downregulation of cellular stress response pathways resulting in cell wall adaptations and both reduced ROS production and ATP efflux crucial for AMP killing [35]. Besides protecting C. albicans cells, secreted AMP inhibiting/degrading proteins are known to provide protection to concurrently present bacteria such as S. aureus [36]. Additional to AMP induction, PAMPs of C. albicans hyphae and produced candidalysin activate the epidermal growth factor receptor (EGFR) and ephrin type-A receptor 2 (EphA2) of oral ECs and, thereby, induce the release of pro-inflammatory cytokines and chemokines which attract neutrophils to the site of infection [37, 38].
Concerning S. aureus, when reaching sufficient biomass and virulence, various PAMPs are able to activate PRRs and promote AMP secretion as well [39–43]. Even though specific research on oral EC recognition of S. aureus is still lacking, general recognition of S. aureus occurs via PRRs, including TLRs (especially TLR2), NOD-2 and TNF-α receptor 1 (TNFR1) which bind staphylococcal specific PAMPs such as cell surface protein A, peptidoglycan, LTA, phenol-soluble modulins (PSMs) and hemolysins [44]. Numerous strains of S. aureus are, however, able to utilize sensor/regulator systems to sense AMPs and adapt their transcriptomic profile accordingly [45]. These transcriptomic shifts can result in the insertion of positively charged D-alanine and L-lysine into the highly negatively charged cell wall lipoteichoic acid and phosphatidylglycerol, respectively, both reducing the affinity of cationic AMPs [45–48]. AMP efficacy can also be directly suppressed by S. aureus via the secretion of the α-defensins inhibiting staphylokinase and LL-37 degrading aureolysin [46, 47]. Whereas C. albicans is known to induce oral epithelial cytokine production, S. aureus has not yet been studied in this context. However, S. aureus is known to induce the secretion of pro-inflammatory cytokines and chemokines in nasal, corneal, and vaginal ECs, rendering it likely that oral ECs could act similarly [49].
A final class of proteins able to prevent extra epithelial oral microbial pathogenesis is immunoglobulins [50]. A key mechanism in prevention of pathogenesis is immune exclusion, primarily facilitated by secretory immunoglobulin A (SIgA), the major immunoglobulin in saliva [51]. Immune exclusion encompasses antibody coating of microbes which limits epithelial contact and invasion of pathogens and antigens [51]. However, whereas SIgA is indeed able to reduce the adherence of C. albicans to human epithelial cells [52], other factors present in saliva, such as secretory component (SC), actually promote adhesion beyond the inhibitory effects of SIgA, rendering immune exclusion of C. albicans unlikely [52, 53]. Even though studies regarding the effects of S. aureus and SIgA are limited, SIgA has been found to abundantly bind to the surface of S. aureus but, however, binding is reduced during biofilm growth [54]. When SIgA is immobilized onto polyvinyl microtiter plates, adhesion of S. aureus is not promoted, suggesting SIgA reduces S. aureus adhesion to at least solid surfaces [55]. Since C. albicans can be considered a solid surface for colonization of S. aureus SIgA could impact this interaction. It should be noted that the effect of this induced immune deficiency on SIgA has not yet been reported [10, 11, 13]. Therefore, the exact role of SIgA in the immune exclusion of S. aureus and the possibility of adhesion promoting salivary proteins and C. albicans remains to be determined.
Oral polymorphonuclear leukocytes (PMNs), mainly neutrophils, constantly migrate into the oral cavity and patrol mucosal surfaces to detect and respond to present microorganisms [56, 57]. When PAMPs are detected, neutrophils phagocytose the pathogen (when possible) and release neutrophil extracellular traps (NETs), reactive oxygen species (ROS), cytokines, chemokines, and granules containing, amongst others, the serine proteases elastase, cathepsin G and proteinase 3 (Figures 1, 2) [27, 58–60].
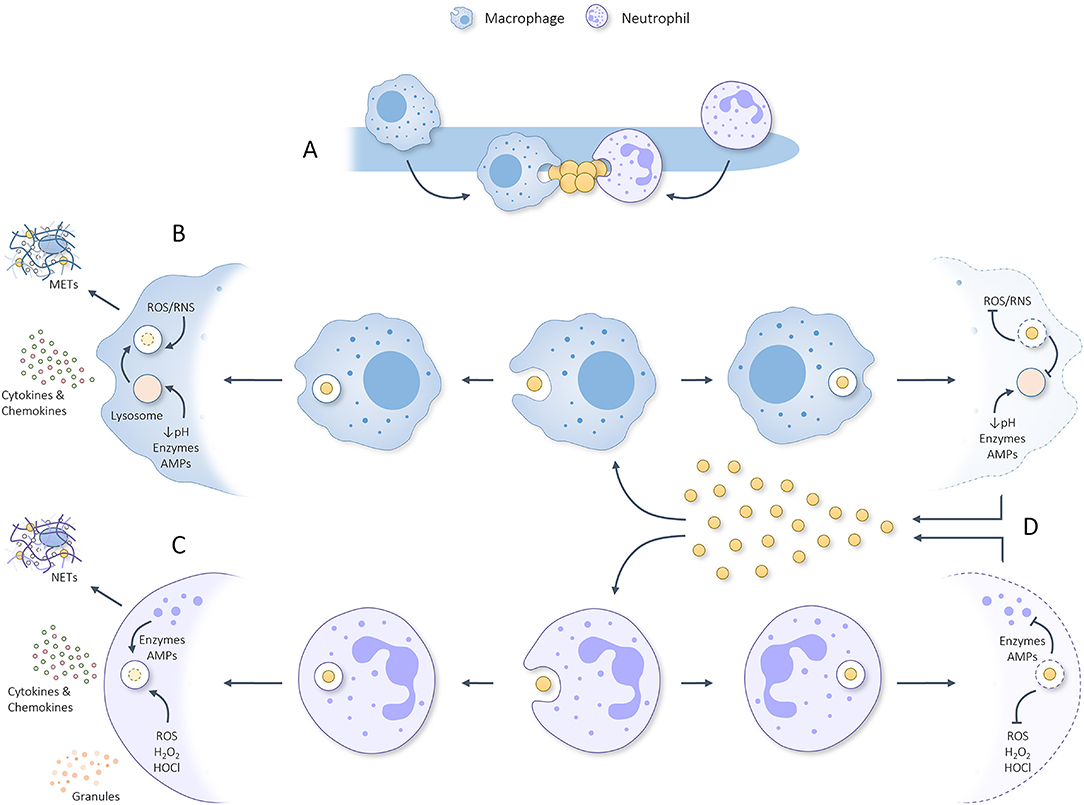
Figure 2. Graphical summary regarding (A) the attraction of macrophages and neutrophils toward C. albicans hyphae which sequentially are scavenged for parts/attached microbes that can be phagocytosed. (B) Once phagocytosed by macrophages, the phagosome containing S. aureus will be fused with a lysosome to form a phagolysosome that utilizes ROS, RNS, acidity, degrading enzymes and AMPs to eliminate the phagocytosed threat. In response to phagocytosing S. aureus, macrophages can produce METs and secrete pro-inflammatory cytokines/chemokines which help attract/activate T cells, NK cells, dendritic cells and neutrophils. (C) Once phagocytosed by neutrophils, granules start fusing with the phagosome during a process deemed degranulation and utilize various kinds of ROS, degrading enzymes and AMPs to eliminate the phagocytosed threat. In response to phagocytosis/activation, neutrophils can produce NETs and secrete granules plus pro-inflammatory cytokines/chemokines. (D) S. aureus has developed numerous ways to inhibit phagocytic killing by macrophages and neutrophils rendering it able to survive the harsh phagosomal/phagolysosomal environment, propagate, and kill the concerning phagocyte. Released staphylococcal cells can, thereafter, be phagocytosed to repeat the process. This misemployment of phagocytes could aid S. aureus dissemination to various other body sites and initiate lethal infection.
Neutrophils are attracted to the site of infection by locally secreted cytokines and chemokines where they also directly respond to candidal cell wall components via both their PRRs (TLR2, TLR4, TLR9, Dectin-1, Dectin-2, Dectin-3, DC-SIGN, and MINCLE) and secreted C. albicans Sap proteins [27, 61]. While phagocytosis of C. albicans yeast cells and smaller hyphae is still feasible, phagocytosis of larger hyphae present during candidiasis is not possible [62–64]. C. albicans cells that are phagocytosed can resist neutrophil killing to a certain extent but are, nevertheless, effectively blocked in their survival, growth, and escape [65, 66]. Regarding hyphae that cannot be phagocytosed, NETs majorly contributes to candidal killing. Evidently, C. albicans hyphae are more prone to trigger NET release compared to yeast cells [67]. NETs are networks of extracellular fibers primarily made up of neutrophil DNA and aid in hyphal killing by trapping C. albicans cells and binding/concentrating present antimicrobial factors [67, 68]. Oral PMNs release 13 times more NET material compared to circulating PMNs, rendering NETs particularly important in oral immunity [69]. Additional to phagocytosis and NETs, neutrophils are generally able to damage/kill fungal cells by releasing ROS which damage fungal and bacterial DNA, RNA and proteins [70]. However, neutrophilic killing mechanisms described above have been mainly studied using non-oral PMNs and planktonic cultures. C. albicans biofilms actually reduce the release of NETs and ROS, inhibiting the main antifungal effectors of neutrophils [71, 72]. Besides aiding in C. albicans killing, neutrophils also act as immune stimulators by releasing serine proteases such as elastase or cathepsin G [73]. These proteases act as immune stimulators by: (1) promoting pro-inflammatory cytokine and chemokine production [59, 74–76], (2) promoting secretion and aggregation of thrombocytes [77], (3) activating natural killer (NK) cells, T-cells and B lymphocytes, (4) increasing NK cell cytotoxicity and cytokine production [78], and (5) activating various chemoattractants [78–80].
In contrast to C. albicans hyphae, S. aureus cells can be readily phagocytosed by neutrophils. Following phagocytosis, phagosomes containing S. aureus utilize PMN-derived oxygen-dependent factors such as O2−, hydrogen peroxide (H2O2), hypochlorous acid (HOCl) and other ROS to kill the internalized threat (Figure 2) [81, 82]. Oral PMNs have been shown to be functionally capable of producing and utilizing ROS (both intracellular and extracellular) for microbial killing [57]. Additionally, neutrophils utilize PMN-derived oxygen–independent factors, including, elastase, proteinase-3, azurocidin, cathepsins, lysozyme, and various AMPs which are introduced into the phagosome by various granules during a process known as degranulation (Figure 2) [83–85]. Using various degranulation markers oral PMNs have been shown to have upregulated degranulation and increased exocytosis of granular content compared to peripheral PMNs [57]. The state of degranulation in oral PMNs has been suggested to be adjusted in accordance to the number of present oral PMNs relative to the bacterial load with the expression of degranulation markers increasing most during periodontitis [57]. Together, the oxygen-dependent and independent factors elicit highly effective antibacterial effects. Even though the studies mentioned above mainly focus on general PMN responses, similar responses are likely to occur inside the oral cavity. Interestingly, even though oral PMNs are more prone to adhere and internalize bacteria compared to circulating PMNs, they show a reduced capacity for E. coli and, therefore, possibly S. aureus as well [69]. The reduced killing efficacy of PMNs is limited further by the ability of S. aureus to inhibit phagocytosis and phagosomal killing [46, 47]. Importantly, S. aureus is able to prevent oxidative phagosomal killing by; (1) utilizing two superoxide dismutases to convert superoxide into hydrogen peroxide and molecular oxygen, (2) degrading hydrogen peroxide to water and oxygen, (3) producing the antioxidant carotenoid staphyloxanthin, (4) repairing oxidative damaged proteins, and (5) by directly decreasing ROS production [46, 47]. Moreover, AMPs utilized during non-oxidative killing can be impaired by S. aureus, as discussed earlier, through cell surface alterations and secretion of AMP inhibiting/degrading proteins. By inhibiting neutrophilic oxidative and non-oxidative killing mechanisms S. aureus cells can survive inside the phagosome, propagate, and produce cytolytic toxins which induce neutrophil osmotic lysis and necrosis [46, 47]. Subsequentially, viable S. aureus cells are released into the surrounding environment following which the process of phagocytosis, survival, staphylococcal propagation and neutrophil death can be repeated (Figure 2). If this process occurs outside the original invasion zone, S. aureus cells could be able to utilize this mechanism as means for dissemination. Additional to phagocytosis, neutrophils secrete NETs, ROS, pro-inflammatory cytokines and chemokines in response to S. aureus [46, 47, 86]. However, S. aureus is a potent neutralizer of NETs by using nuclease and adenosine synthase to convert them to deoxyadenosine [46, 87, 88]. Staphylococcal NET neutralization could provide C. albicans with protection against this anti-hyphal immune response. Besides phagocytosis and NET circumvention, S. aureus is able to inhibit neutrophil chemoattraction, PRR activation, extravasation, calcium mobilization, and actin polymerization [47]. Nevertheless, during co-infection with C. albicans, phagocytes are still actively attracted to the hyphae regardless of S. aureus, suggesting the chemotactic inhibition of S. aureus to either be suppressed during pathogenic co-culture or overruled by hyphal chemoattraction [11]. Finally, neutrophil secreted serine proteases elastase, proteinase 3, and cathepsin G are also inhibited by S. aureus [46, 47]. Therefore, S. aureus is indeed recognized and phagocytosed by neutrophils but has the ability to circumvent phagocytosis, survive within the phagolysosomes, and kill the neutrophil. Moreover S. aureus is able to reduce extracellular neutrophil responses that also affect C. albicans, granting both organisms protection during co-infection.
The complement system is one of the first extra-epithelial antimicrobial immune responses and is actively present in human saliva and GCF [89–92]. Following mucosal damage or gingival inflammation, the complement system initiates fast and efficiently via a combination of the classical, alternative and lectin pathways plus indirectly via the coagulation and fibrinolysis systems [89]. Regarding C. albicans and S. aureus immunity the complement factors B, C3, C4, C5-C9, and mannan binding lectin (MBL) are considered the main effectors [89, 93, 94]. C5-C9 are known to form the membrane attack complex (MAC) which is characterized as the direct antifungal response of the complement system by forming pores in the cell surface [89]. Even though studies regarding complement responses to C. albicans and S. aureus have not yet been performed in relation to the oral cavity, the oral response is expected to act similarly.
However, even though MACs are considered antifungal, they are not able to effectively lyse candidal cells, indicating the complement system to play a more modulatory role in C. albicans immunity [89]. Immune modulation by the complement system is facilitated through opsonization of C. albicans cells with C3 and Mannose-binding lectin (MBL) which promote phagocytosis by local phagocytes when possible [95, 96]. Furthermore, MBL is able to stimulate ROS production by nearby neutrophils which are readily attracted to the site of infection via complement factor C5a, a cleavage product of C5 [97, 98]. However, C. albicans is able to evade the complement system by masking itself for complement activation, cleaving/blocking complement proteins and recruiting complement regulators [28, 89]. While the antifungal mechanisms governed by the complement system remain modulatory, its direct antibacterial effects are more prominent.
Regarding S. aureus, MAC formation is able to directly damage and kill the bacterium by successfully puncturing the bacterial cell surface [90]. The complement system is also capable to reduce staphylococcal/EC adherence [99]. However, this is likely overcome during pathogenic candidal growth since the presence of C. albicans is known to significantly enhance the presence of S. aureus on the tongue of immunocompromised mice [10, 11, 13]. Similar to C. albicans, S. aureus has developed various mechanisms to avoid complement mediated killing, including the inhibition of MAC formation [100], inhibition of the classical and lectin pathways [101], and the utilization of various extracellular proteases which efficiently degrade crucial complement factors [102]. Since both organisms possess mechanisms to inhibit the complement system, their inhibitory potency likely adds up during co-infection.
To summarize, antimicrobial proteins, AMPs, neutrophils, and complement factors present in saliva and GCF are able to balance microbial growth to prevent overgrowth and pathogenesis (Figure 1). Thereby, saliva and GCF play key roles in maintaining homeostasis in the oral cavity. However, numerous evasion strategies allow C. albicans and S. aureus to reduce antimicrobial efficacy and result in disease. Loss of immune fitness obviously increases the incidence of infections originating from the oral cavity. When pathogenic hyphal growth of C. albicans is not limited, hyphae will grow invasively into the oral epithelium and trigger intra-epithelial immunity. During invasive hyphal growth S. aureus will translocate into the oral tissue and contribute to the immune activation as well.
Once invaded, C. albicans and S. aureus stimulate intra-epithelial immunity, including the type 17 response, further attraction/activation of both neutrophils and macrophages (assisted by Th1 activation), the NK response, and B cell response (Figure 1). Moreover, intra-epithelial immunity promotes extra-epithelial immunity by stimulating the production/secretion of AMPs by salivary glands and epithelial cells.
The type 17 response is mainly governed by antigen presenting cells, such as dendritic cells (DCs) and Langerhans cells (specialized subset of DCs), which patrol the soft oral tissues for foreign antigens [103]. Oral mucosa, except for sublingual mucosa, are relatively non-absorptive, indicating the need for antigens to penetrate the epithelium in order to induce downstream immune responses [104]. When DCs encounter fungal and/or bacterial antigens they are activated and start sampling the antigen and present it on the cell surface using major histocompatibility complex (MHC) class II molecules [105]. Following antigen uptake, DCs also start to mature and travel to the draining lymph nodes where naïve T-cells are able to bind DCs and their presented antigens. Following maturation, DCs produce different cytokines according to the PRR and molecular activation. Fungal and bacterial activated DCs produce, amongst others, IL-6 and TGFβ which, together with MHCII antigen binding and the supporting production of IL-1β and IL-23, drive naïve T-cells into Th17 differentiation (Figure 1) [105]. The main goal of differentiated Th17 cells is to migrate to infected tissues and support local immune responses by producing IL-17, IL-22 and IFN-γ. Additional IL-17 is produced by local cells of the innate immune system, including γδ T cells, mucosa-associated invariant T (MAIT) cells, NK T cells, innate lymphoid cells (ILCs), T cell receptor αβ+ cells (TCR αβ+), natural Th17 cells (nTh17) and Foxp3+ T regulatory cell (Treg)–like cells which, together with Th17 cells, are deemed the type 17 response [106, 107]. Even though cells of the type 17 response only constitute a small fraction of the oral cell population (i.e. 1% of the gingival CD4 T cell population) they are indispensable for intra-epithelial immunity [27, 103]. Additional to intra-epithelial immunity, the type 17 response stimulates extra-epithelial oral immunity through IL-17 induced production/secretion of AMPs and chemokines [27, 103]. Th17 produced IL-22 plays an intricate role in epithelial repair by stimulating survival and proliferation of the basal oral epithelium to replenish damaged IL-17 receptor-expressing ECs [108]. Moreover, IL-22 is responsible for preventing accumulation of apoptotic inflammatory cells and preservation of genome integrity [108]. In summary, even though the type 17 response does not directly kill fungi or bacteria, it does crucially stimulates other aspects of oral antifungal/bacterial immunity.
C. albicans is able to activate the type 17 response via hyphal PAMPs and through candidalysin. Candidalysin activates the type 17 response by activating oral ECs, as discussed earlier, into releasing IL-1α/β which stimulate the proliferation of innate Th17 lymphocytes. Moreover, candidalysin synergistically acts with IL-17 to promote more oral EC activation and, thus, creating a positive feedback loop of candidalysin induced type 17 activation [109]. However, C. albicans has developed mechanisms to sense IL-17 and induce autophagy, resulting in efficient recycling of nutrients and cellular components, clearance of aggregates and improved resistance to environmental stresses such as antifungal drugs [110]. The binding of IL-17 also increases candidal adhesion and filamentous growth resulting in the formation of biofilms which are known to be more resistant to antifungal immune responses and treatments [110, 111].
Whereas C. albicans is able to use the type 17 response to induce autophagy for emergency survival, S. aureus is able to reduce T-cell numbers by producing various toxins that directly kill local T-cells [112]. Moreover, staphylococcal cell wall components and produced phenol soluble modulins have been found to suppress T cell proliferation and differentiation [112, 113]. S. aureus is also able to stimulate the expansion of T-cell suppressive immune cells such as granulocytic and monocytic myeloid-derived suppressor cells [112, 113]. Together, IL-17 triggered biofilm formation of C. albicans could grant S. aureus protection while generic T cell inhibition of S. aureus further reduces type 17 response efficacy during co-infection.
In contrast to neutrophils, macrophages are not present outside of the oral epithelium and, therefore, only contribute to intra-epithelial immunity. Oral macrophages react to invaded microbial threats by phagocytosing and killing them (Figures 1, 2). Similar to neutrophils, macrophages cannot phagocytose larger intact hyphae but scavenge hyphal cells for associated microbes that can be phagocytosed [11, 62–64].
The attraction to and recognition of C. albicans cells by macrophages is stimulated by fungal cell wall glycosylation [63, 64] and involves both PRRs (TLR2, TLR4, TLR9, Dectin-1, Dectin-2, Dectin-3, DC-SIGN, and MINCLE) and recognition of secreted C. albicans Sap proteins [27, 61]. Following C. albicans recognition and/or phagocytosis, macrophages secrete pro-inflammatory cytokines and chemokines [27]. Together with the cytokines and chemokines secreted by oral ECs, these molecules help attract and activate more macrophages and neutrophils [27]. Macrophages can contribute to C. albicans killing through phagocytosis and producing macrophage extracellular traps (METs) [114]. Following phagocytosis C. albicans is, however, able to alter its transcriptional profile to survive nutrient-depleted and acidic phagosomes [115]. Furthermore, C. albicans induces cell wall remodeling to initiate an intraphagocytic yeast to hyphal transition able to induce pyroptosis, an inflammatory mediated lytic programmed cell death, as well as rupturing of the macrophage membrane [115]. METs contribute to the antifungal response similar to NETs and are likely released before C. albicans is able to induce macrophage pyroptosis [116]. As discussed for extra-epithelial immunity, neutrophils prevent overgrowth of C. albicans yeast cells by phagocytosis, prevent/limit hyphal growth by releasing NETs and ROS, and stimulate further immune activation by secreting serine proteases.
In line with their general response, macrophages play a crucial role in the immune response against S. aureus infections (Figure 2) [49]. Macrophages recognize S. aureus through a combination of receptors, including scavenger receptors, complement receptors and Fc receptors [117]. S. aureus activated macrophages produce various cytokines and chemokines which attract T cells, NK cells, DCs, more macrophages, and neutrophils to the site of infection (Figures 1, 2) [118]. When activated, macrophages engulf staphylococcal cells into a phagosome which, unlike in neutrophils, fuses with a lysosome to form a bactericidal phagolysosome (Figure 2) [117]. Following S. aureus interaction, macrophages can either provoke a pro-inflammatory or an anti-inflammatory response. The pro-inflammatory response entails active phagocytosis, the production of intracellular ROS, nitric oxide (NO), pro-inflammatory cytokines, AMPs (including hepcidin and calprotectin), and enzymes, as well as acidification of the phagolysosome, nutrient restriction, and autophagy [49]. Similar to the antifungal response, macrophages target extracellular bacteria using macrophage extracellular traps (METs) [114]. In response to recurrent S. aureus infections macrophages are able to generate a longer term memory by increasing their pro-inflammatory responsiveness [119]. Importantly, recurrent S. aureus infections also induce a Th1 memory response which results in additional IFN-γ production and macrophage activation [120–122]. In contrast, S. aureus infections involving mechanisms such as biofilm growth induce an anti-inflammatory macrophage response, actually impairing phagocytosis [123]. Additionally, S. aureus is able to avoid macrophage killing by preventing phagolysosome acidification [124], restricting ROS/RNS production [125], and preventing autophagy by blocking autophagic flux [126]. Using these mechanisms, S. aureus is able to survive inside macrophages, proliferate and ultimately kill the phagocyte, resulting in the release of viable S. aureus cells which are again internalized by other macrophages to repeat the process (Figure 2) [124, 127, 128]. When co-phagocytosis of C. albicans and S. aureus occurs, the survival mechanisms utilized by both organisms could enhance intracellular survival and macrophage killing.
Even though the intra-epithelial neutrophil response operates identically to the extra-epithelial response discussed earlier, there are some additions. Namely, intra-epithelial neutrophils are able to interact with, activate and act as antigen presenting cells (APCs) [129]. Moreover, produced NETs activate APCs and directly prime T cells, highlighting the importance of neutrophils in both antifungal immunity and immune regulation [129]. Due to the fact that the intra-epithelial macrophage and neutrophil responses are stimulated by all aspects of C. albicans/S. aureus oral immunity (Figures 1, 2) they can be considered the main effectors.
Besides the type 17, macrophage, and neutrophil response, NK cells also contribute to intra-epithelial C. albicans/S. aureus immunity. Whereas NK cells constitute approximately 5–20% of the peripheral blood mononuclear cell population, they comprise a substantial part of the gingival lymphoid cell population and elicit both direct and indirect antifungal responses by phagocytosis and releasing cytotoxic granular content [130, 131].
Direct antifungal responses mainly involve phagocytosis and the release of granular contents, including the apoptosis such as granzyme B and the pore-forming molecules granulysin and perforin [130, 132, 133]. Due to the fact that depletion of NK cells in immunocompetent mice does not increase C. albicans susceptibility, NK cells are expected to play a more immunomodulatory role in C. albicans immunity [133]. Immune modulation by NK cells is achieved through production of inflammatory mediators able to attract and activate macrophages and neutrophils [130, 132, 133]. NK cell cytokine production is also induced by activated DCs and contributes to sustaining neutrophil antifungal response [134]. While most NK cell studies have mainly been focussed on systemic fungal infections, similar NK cell functions are likely to contribute to mucosal C. albicans immunity [27].
Regarding antibacterial responses, NK cells elicit both direct and indirect effects [135]. Direct antibacterial effects are facilitated by inducing microbial apoptosis through releasing granular components and various AMPs [135, 136]. Indirect NK cell responses again mainly include the production of inflammatory mediators. NK cells, while mainly activated by type 17 cytokines, are also able to be activated directly by S. aureus and by staphylococcal products, likely contributing to the control of staphylococcal infections prior to T cell activation [137–140]. Interestingly, even though depletion of NK cells does not influence C. albicans susceptibility, it does increase the susceptibility to staphylococcal infections [139]. This is partly attributable to an NK cell induced promotion of S. aureus phagocytosis by macrophages [139]. Research regarding evasion strategies of C. albicans and S. aureus against the NK cell response is, unfortunately, still lacking.
Finally, the B cell response also contributes to intra-epithelial oral immunity, albeit playing a minor role in C. albicans/S. aureus immunity [141–144].
While patients suffering from agammaglobulinemia hardly or do not produce immunoglobulins they do show a normal functioning antifungal immune response [144]. Nevertheless, patients undergoing B cell depletion therapy exhibit an increased susceptibility to fungal infections attributable to reduced Th1/Th17 responses, whereas bacterial infection rates remain relatively normal, indicating B cells to contribute to C. albicans immunity in an immunoglobulin independent manner [141, 142].
In contrast to antifungal immunity, B-cell-deficient mice do not show a difference in bacterial infection related mortality and clearance, indicating B cells to not significantly contribute to S. aureus immunity [143]. Nevertheless, vaccination of mice with a sublethal doses of live S. aureus does provoke the production of specific antibodies against a wide variety of staphylococcal antigens [145]. Correspondingly, patients suffering from S. aureus skin and soft tissue infections, prosthetic joint infections, and pediatric hematogenous osteomyelitis establish a repository of memory B cells which are able to produce antibodies against a variety of S. aureus exotoxins [146, 147]. However, immunoglobulins acting directly on S. aureus have not yet been identified, which is likely attributable to the fact that B cells are not able to directly bind S. aureus cells [148].
Altogether, the above mentioned extra-epithelial immune responses inhibit propagation of invaded C. albicans and S. aureus while, additionally, stimulating the repair of damaged tissue. Even though C. albicans and S. aureus have developed numerous mechanisms to evade these immune responses, the oral immune system of healthy individuals is capable of preventing and/or limiting their pathogenicity to maintain homeostasis. However, in immunocompromised hosts this no longer holds true, allowing for pathogenic growth of C. albicans and with it possibly the initiation of S. aureus BSIs.
To date, in vitro and murine model studies regarding oral polymicrobial C. albicans/S. aureus infections have established that; (1) immune suppression is essential for the development of OPC, (2) OPC increases the presence of S. aureus on the tongue surface, (3) invasive OPC facilitates infiltration of S. aureus into the underlying substratum via, amongst others, the production of candidalysin, (4) severe immune suppression significantly reduces dissemination of S. aureus [10–13, 15]. Therefore, the role of the oral immune system appears to be paradoxical: whereas low levels of suppression allow for OPC development and S. aureus dissemination, severe suppression significantly reduces the dissemination potency of S. aureus, even though OPC development is more severe. The immune system in immunocompromised individuals could, thereby, first allow invasive candidal growth due to lack of activity while remaining immune activity sequentially aids in S. aureus dissemination. However, while immune deficiencies, either congenital (primary) or acquired (secondary), are known predisposing factors for OPC and S. aureus BSIs separately, they have not yet been described with regard to OPC induced staphylococcal BSIs [149–152]. Therefore, predominant immune deficiencies separately known to increase the risk for OPC as well as S. aureus BSIs will be discussed.
The vast majority of immune deficiencies related mucocutaneous candidiasis incidences are attributable to diabetes mellitus, cancer, human immunodeficiency virus (HIV) infection, and prescribed corticoids [152–157]. S. aureus BSIs have been associated with similar immune deficiencies, fortifying the notion that immune deficiency induced OPC could concordantly increase the risk of S. aureus BSI development [149–151, 158]. Interestingly, once instigated, S. aureus BSIs do not show a higher mortality rate among immunocompromised patients, indicating immune deficiencies to only contribute to BSI onset but not further lethality [159, 160]. Due to the fact that diabetes mellitus, HIV, cancer, and use of prescribed corticoids comprise the majority of immune deficiencies linked to both C. albicans and S. aureus infections separately, their effects on the oral immune response to both organisms will be discussed. Additional immune deficiencies, affecting aspects of the oral C. albicans and S. aureus immune response, are listed in Table 1.
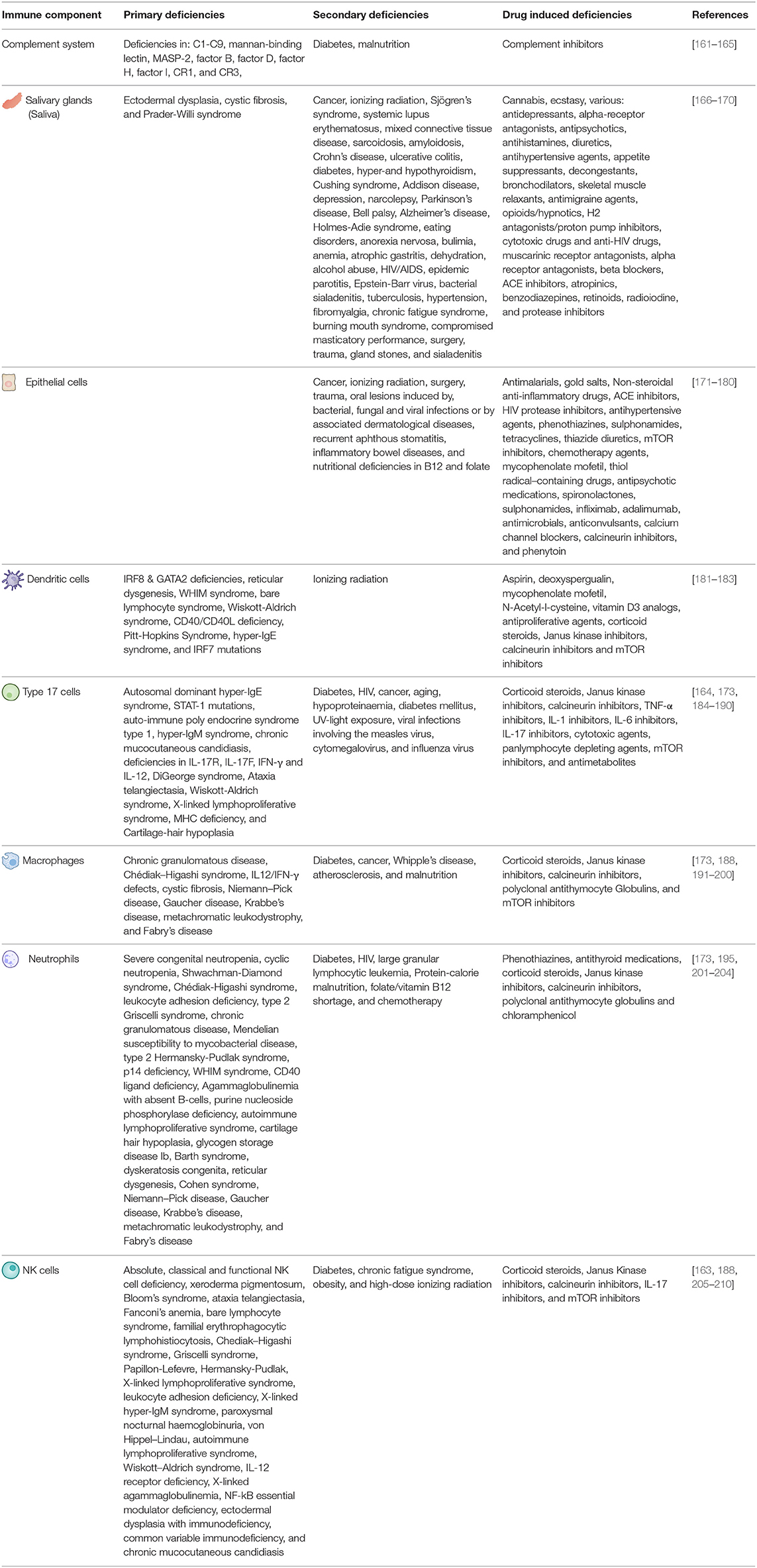
Table 1. Primary, secondary, and drug induced immune deficiencies able to affect the various aspects or oral C. albicans and S. aureus immunity.
The global burden of diabetes is increasing continuously and is currently responsible for over 1.5 million deaths per year, ranking it within the top 10 causes of death [211, 212]. Individuals suffering from diabetes are more susceptible to C. albicans and S. aureus infections due to both a reduced salivary flow and impaired immune responses, including suppression of cytokine production, defects in phagocytosis, dysfunction of immune cells and a decreased complement response [213–215]. Evidently, peripheral blood mononuclear cell (PBMCs) isolated from diabetic patients have a lowered cytokine production and take on a more anti-inflammatory phenotype [216–218]. However, macrophages do increase their phagocytosis rate in response to C. albicans or S. aureus during hyperglycaemia [216, 217]. Therefore, even though macrophages increase bacterial phagocytosis, they have a reduced intracellular killing efficacy and lowered immune stimulatory capacity [218]. Besides macrophages, neutrophils of diabetic patients are impaired in their attraction, migration, ROS production, NET formation, and degranulation [213]. Hyperglycaemia is also known to decrease the efficacy of NK cells and the complement system by inhibiting their degranulation capacity and C3/C4 pathway activation, respectively [213, 218]. Interestingly, under hyperglycaemic conditions S. aureus is able to deplete serum C3 levels, reducing complement-mediated killing as well [219]. Altogether, hyperglycaemia allows for invasive C. albicans and S. aureus growth due to a reduced salivary flow and impaired extra-epithelial neutrophil and complement responses. Once invaded, the reduced intra-epithelial type 17, macrophage, neutrophil, and NK cell responses could allow for sequential phagocytic uptake, survival and dissemination of S. aureus. Interestingly, the risk of developing diabetes is significantly higher amongst individuals suffering from HIV infections [220].
Approximately 37.7 million individuals live with HIV/AIDS worldwide while HIV-related diseases are responsible for about 680,000 deaths each year [221]. HIV especially burdens low-income countries, ranking it within the top 10 of causes of death [222]. One of the main causes of HIV induced mortality is a secondary infection which develops due to a progressive decline of CD4 T cells [223]. The predominantly affected CD4 T cell class during HIV infections is Th17, resulting in both cellular depletion and dysfunction [224–226]. Four mechanisms are known to inflict HIV induced T cell decline; (1) direct viral killing of infected cells, (2) induced apoptosis of infected cells, (3) killing of infected cells by CD8 T cells, and (4) defects in T cell regeneration together with destruction of CD4 T cell progenitors within the thymus [223]. The decline in CD4 T cells will eventually reach a critical point at which the immune system is no longer capable of preventing opportunistic pathogenic infections of, for instance, C. albicans and S. aureus. This is well reflected by the fact that oral candidiasis induced lesions are commonly observed during HIV infections and are significantly associated with a low CD4 T cell count [227–231]. Correspondingly, HIV patients are at an increased risk of developing S. aureus infections and BSIs which, besides T cell depletion, are also linked to reduced neutrophil counts (neutropenia) [232–235]. Neutropenia is frequently observed among HIV patients and is strongly associated with reduced CD4 T cell numbers and an increased viral load [236]. Importantly, neutrophils still present during HIV infections are known to actually contribute to T cell dysfunction by suppressing T cell responses [237]. Besides T cells, macrophages and dendritic cells can also be infected with HIV. However, macrophages are known to resist HIV infection much better than CD4 T cells besides which they are less sensitive to cytotoxic killing of the virus, also producing and releasing relatively low amounts of viral particles when killing does occur [238]. Taken together, the reduction in extra-epithelial neutrophils most likely allows for invasive OPC development which is able to progress due to intra-epithelial CD4 T cell depletion, also reducing extra-epithelial AMP responses and barrier repair. Following the development of invasive OPC, S. aureus will co-invade and likely utilize the reduced intra-epithelial neutrophil count to disseminate. Interestingly, the immunocompromised state of HIV patients also significantly increases their risk of developing various types of cancer linked to C. albicans and S. aureus infections [239, 240].
Cancer is currently responsible for 10 million deaths per year, ranking it as one of the leading causes of mortality [241]. Besides its direct lethal effects, cancer and its treatments also increase the risk for infection [242, 243]. The increased risk of infection observed among cancer patients depends on various risk factors such as the type of cancer (i.e. lymphoma or acute leukemia versus a solid tumor), reduced cellular functioning as a result of cytotoxic or immunosuppressive therapies, usage of indwelling devices (e.g. catheters), the severity/duration of induced neutropenia, surgery, and gastrointestinal mucositis due to chemotherapy [243]. Due to the vast variation in cancer types and treatments it is not possible to summarize their effects on the aspects of oral immune responses relevant to C. albicans and S. aureus infections. Nevertheless, studies concerning human cancers and murine cancer models and have shown various relevant cancer induced peripheral immune perturbations which have been extensively reviewed elsewhere [244]. It must be noted that not every perturbation is yet known to occur in every cancer type. Known perturbation related cancer types and mouse models are summarized in detail by Hiam-Galvez et al. [244]. In brief, cancer is able to induce: (1) expansion of immature immunosuppressive neutrophils and monocytes due to aberrant haematopoiesis, (2) a decrease in dendritic cell subsets and dendritic cell maturation, (3) lymphopenia, (4) reduction of CD4 T cell function, (5) an increase in regulatory T and B cells able to dampen immune responses, and (6) a suppressed NK phenotype through a decreased number of activating receptors and cytotoxic potential as well as increased number of inhibitory receptor numbers [244]. Interestingly, surgical resection of the tumor can reverse various of these peripheral immune perturbations in multiple murine breast and colon cancer models, suggesting the observed immune effects to indeed be induced by the tumor itself [245]. Wheter this also holds true for humans and other cancer types remains to be determined. Besides the cancer itself, treatments can also affect peripheral immune responses but remain highly dependent on the type of treatment and cancer [244]. Treatment induced perturbations, include neutropenia, lymphodepletion, myeloid cell expansion, reduced APC function and T cell responses, and increased numbers of immunosuppressive polymorphonucleocytes and monocytes [244, 246, 247]. Additionally, various chemotherapies and radiation therapy affecting the oral cavity can inflict damage to the epithelial lining of the oral cavity and the salivary glands, allowing oral infections to instigate more easily [171, 172, 247–249]. Moreover, high dosage radiation therapies covering the oral cavity can eliminate small oral mucosal blood vessels and/or result in severe mucositis with ulceration which both induce necrosis of oral soft tissues [243]. Radiation may also damage the serous cells of salivary glands, leading to xerostomia (dry mouth) which, in relation to neck and head radiation, is significantly linked to an increased risks of C. albicans and S. aureus infections [248, 249].
Altogether, no general claim can be made concerning cancer and treatment induced peripheral immune perturbations. However, it remains clear that induced immunosuppression, damage the oral epithelial barrier, and induced xerostomia can allow for OPC development and more easy invasion of C. albicans and S. aureus. Interestingly, corticoids, the remaining risk factor for C. albicans and S. aureus infections, are commonly administered to cancer patients for their anti-cancer and anti-swelling effects [250].
Corticoids, while not considered as direct immunocompromised conditions, are commonly prescribed immunosuppressants and are utilized to induce immunosuppression in murine co-infection models involving C. albicans and S. aureus [12–14, 251]. Whereas the positive feedback loop of the C. albicans/S. aureus immune response triggers pro-inflammatory effects (Figure 1), endogenous glucocorticoids play an essential role in both limiting and resolving inflammation to prevent adverse effects [252]. Corticoids are derived from the glucocorticoid family of steroid hormones and are, therefore, able to dampen immune responses in inflammatory disorders such as allergies, asthma, and autoimmune diseases, besides which they are beneficial for other disease such as cancer [173, 223, 250]. Importantly, corticoids are also generally used in murine models to induce an immunocompromised state. The immunosuppressive effects of corticoids are mainly facilitated through binding intracellular glucocorticoid receptors expressed by the majority of human cell types [223, 253]. Due to the fact that multiple cell types express these receptors, corticoids induce multiple anti-inflammatory effects. It is generally accepted that corticoids reduce the level and function of CD4 T cells besides which they reduce pro-inflammatory responses of macrophages and monocytes, resulting in a reduction of: (1) pro-inflammatory cytokine production, (2) chemotaxis, (3) migration, (4) phagocytosis, (5) oxidative burst and free radical generation, and (6) nitric oxide secretion [223, 253–256]. However, various studies regarding corticoid treatment and neutrophils have also reported upregulation of pro-inflammatory responses besides which there have been contradictory studies regarding the inhibition/stimulation of corticoid induced neutrophil apoptosis, highlighting the complexity of the corticoid response [256]. This complexity is further emphasized by Th17 glucocorticoid sensitivity. While Th17 cells were first considered insensitive to corticoids, Th17 cells in patients suffering from psoriasis and related disorders are corticoid sensitive, indicating corticoid sensitivity to also depend on the immunopathology [257]. Nonetheless, the anti-inflammatory effects of corticoids on lymphocytes, neutrophils, monocytes/macrophages, and other immune effector cells are known to increase the susceptibility to invasive candidiasis [254]. Additionally, prognosis of S. aureus bacteraemia is negatively affected by the use of corticoid steroids and other immunosuppressives [258–261]. Even though the relation between C. albicans/S. aureus infections and corticoid use is apparent, the mechanism driving this relation remains unclear.
To summarize, the most common immune deficiencies linked to both C. albicans infections and S. aureus BSIs include diabetes mellitus, HIV, cancer, and prescribed corticoids. Diabetes induces immune deficiency through xerostomia and reduced extra-epithelial neutrophil and complement responses as well as impaired intra-epithelial type 17, macrophage and neutrophil responses. The reduction of extra-epithelial neutrophils during HIV infections likely allows for OPC development which is able to progress due to reduced intra-epithelial Th17 responses. S. aureus could co-invade during OPC progression and utilize the reduction in neutrophil count to disseminate. Regarding cancer, immune deficiencies arise from various aspects of the disease and its treatment with induced neutropenia as the main effector. Finally, use of corticoids induces various immune deficiencies, including reduced type 17, neutrophil and macrophage responses. Each of these immune deficiencies are able to facilitate OPC development and could, thereby, account for S. aureus BSIs.
A strict balance between pathogenic C. albicans growth and oral immune activation normally inhibits hyphal invasion and ensures oral homeostasis. When oral immunity is compromised OPC is able to develop and invade oral mucosal tissue. In murine models, when S. aureus is additionally present, it is able to co-invade with C. albicans and utilize phagocytes present in the tissue to disseminate to draining lymph nodes. Whether this holds true for human co-infections remains to be determined. Considering that: (1) C. albicans and S. aureus are a common member of the human oral microbiome [262], (2) approximately one in five cases of candidemia are polymicrobial [6], (3) C. albicans instigated S. aureus BSI, in theory, do not have to concur with candidemia, and (4) a significant number of patients suffering from staphylococcal BSIs have no reported porte d'entrée [9], human S. aureus BSIs could be facilitated by OPC. Immunocompromised individuals, additional to developing OPC, could, therefore, also be at risk of contracting concordant S. aureus BSIs. Considering the crucial contribution of the oral immune system in this process, the aim of this review was to provide a detailed overview concerning all relevant aspects of oral C. albicans and S. aureus immunity (Figure 1) and to discuss the predominant immune deficiencies able to allow for OPC induced S. aureus BSIs (Table 1). Based on current literature it is evident that when the oral epithelium is not broken due to physical intervention, loss of extra-epithelial oral immune fitness during immune deficiencies such as diabetes mellitus, HIV, cancer, and the use of corticoid steroids can facilitate invasive growth of C. albicans and result in breakage of the oral epithelial barrier. Through this porte d'entrée, oral bacteria associated with C. albicans, such as S. aureus, are granted access to the underlaying substratum. Sequentially, phagocytes are actively attracted to the site of invasion and start scavenging C. albicans hyphae while phagocytosing attached staphylococcal cells. S. aureus cells able to survive phagocytic killing can proliferate and kill the phagocyte, releasing more bacteria in the surrounding to repeat the process (Figure 2). In contrast to OPC development, this staphylococcal dissemination process is severely hampered by excessive immune suppression. Ultimately, S. aureus is either granted direct access to the bloodstream or by surviving inside phagocytes during migration. Once disseminated, S. aureus (either accompanied by C. albicans or not) can initiate lethal infections throughout the body. Due to the fact that various immune dysfunctions are able to instigate OPC, through loss of oral immune fitness, they are likely able to also function as facilitator of S. aureus BSIs and should, therefore, be considered a potent risk factor for OPC induced S. aureus dissemination. Besides S. aureus, C. albicans is able to interact with other oral BSI inducing bacteria such as Staphylococcus epidermidis [263], Enterococcus faecalis [264], and Streptococcus gordonii [265, 266], the first, second, and fourth most common co-isolated bacteria during C. albicans BSIs, respectively [6]. Therefore, the described mechanism of oral C. albicans and immune system facilitated bacterial invasion and dissemination during immunosuppression could be a general BSI inducing mechanism for bacteria able to directly interact with C. albicans.
Even though candidemia has been found to coincide with bacterial BSIs in approximately 20% of the cases [6, 7], C. albicans-induced dissemination of bacteria has not yet been directly observed in humans. In contrast, studies using murine and in vitro models have provided strong evidence to support the risk of C. albicans induced S. aureus invasion and dissemination during immunosuppression [10–13, 15]. Moreover, while the overall structure of the immune system of mice and humans is similar, discrepancies do exist and have been extensively reviewed elsewhere [267]. Importantly, numerous in vitro studies used to investigate immune responses to C. albicans and S. aureus. While this response is often described as a general response, it could differ from in vivo oral immune responses. As OPC-facilitated invasion of S. aureus represents a new porte d'entrée with potentially life-threatening consequences, future research, unraveling the role of the oral immune system in, and the mechanism of, invasion and dissemination of bacteria such as S. aureus in humans are important next steps in development of life-saving preventive strategies.
RP wrote the original draft. BK, SZ, and SB reviewed and edited the manuscript and conceptualized the review. SB acquired funding. All authors contributed to the article and approved the submitted version.
The work of RP in the laboratory of SB, in collaboration with the labs of BK and SZ, was funded by strategic investments of the University of Amsterdam in the framework of the Research Priority areas Host-Microbiome Interactions and Personal Microbiome Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflictof interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nobile CJ, Johnson AD. Candida albicans biofilms and human disease. Annu Rev Microbiol. (2015) 69:71–92. doi: 10.1146/annurev-micro-091014-104330
2. Staniszewska M, Bondaryk M, Siennicka K, Kurzatkowski W. Ultrastructure of Candida albicans pleomorphic forms: phase-contrast microscopy, scanning and transmission electron microscopy. Polish J Microbiol. (2012) 61:129–35. doi: 10.33073/pjm-2012-016
3. Sims CR, Ostrosky-Zeichner L, Rex JH. Invasive candidiasis in immunocompromised hospitalized patients. Arch Med Res. (2005) 36:660–71. doi: 10.1016/j.arcmed.2005.05.015
4. Patil S, Rao RS, Majumdar B, Anil S. Clinical appearance of oral candida infection and therapeutic strategies. Front Microbiol. (2015) 6. doi: 10.3389/fmicb.2015.01391
5. McCarty TP, White CM, Pappas PG. Candidemia and invasive candidiasis. Infect Dis Clin North Am. (2021) 35:389–413. doi: 10.1016/j.idc.2021.03.007
6. Klotz SA, Chasin BS, Powell B, Gaur NK, Lipke PN. Polymicrobial bloodstream infections involving Candida species: analysis of patients and review of the literature. Diagn Microbiol Infect Dis. (2007) 59:401–6. doi: 10.1016/j.diagmicrobio.2007.07.001
7. Pien BC, Sundaram P, Raoof N, Costa SF, Mirrett S, Woods CW, et al. The clinical and prognostic importance of positive blood cultures in adults. Am J Med. (2010) 123:819–28. doi: 10.1016/j.amjmed.2010.03.021
8. Diekema DJ, Hsueh P-R, Mendes RE, Pfaller MA, Rolston K V, Sader HS, et al. The microbiology of bloodstream infection: 20-year trends from the SENTRY antimicrobial surveillance program. Antimic Agent Chemot. (2019) 63. doi: 10.1128/AAC.00355-19
9. del Rio A, Cervera C, Moreno A, Moreillon P, Miró JM. Patients at risk of complications of Staphylococcus aureus bloodstream infection. Clin Infect Dis. (2009) 48:S246–53. doi: 10.1086/598187
10. Schlecht LM, Peters BM, Krom BP, Freiberg JA, Hänsch GM, Filler SG, et al. Systemic Staphylococcus aureus infection mediated by Candida albicans hyphal invasion of mucosal tissue. Microbiology. (2015) 161:168–81. doi: 10.1099/mic.0.083485-0
11. Allison DL, Scheres N, Willems HME, Bode CS, Krom BP, Shirtliff ME. The host immune system facilitates disseminated Staphylococcus aureus disease due to phagocytic attraction to Candida albicans during coinfection: a case of bait and switch. Infect Immun. (2019) 87. doi: 10.1128/IAI.00137-19
12. Kong EF, Kucharíková S, Van Dijck P, Peters BM, Shirtliff ME, Jabra-Rizk MA. Clinical implications of oral candidiasis: host tissue damage and disseminated bacterial disease. Infect Immun. (2015) 83:604–13. doi: 10.1128/IAI.02843-14
13. Van Dyck K, Viela F, Mathelié-Guinlet M, Demuyser L, Hauben E, Jabra-Rizk MA, et al. Adhesion of Staphylococcus aureus to Candida albicans during co-infection promotes bacterial dissemination through the host immune response. Front Cell Infect Microbiol. (2021) 10. doi: 10.3389/fcimb.2020.624839
14. Schlecht LM, Peters BM, Krom BP, Freiberg JA, Hänsch GM, Filler SG, et al. Systemic Staphylococcus aureus infection mediated by Candida albicans hyphal invasion of mucosal tissue. Microbiology. (2015) 161:168–81. doi: 10.1099/mic.0.08345-0
15. Solis N V, Filler SG. Mouse model of oropharyngeal candidiasis. Nat Protoc. (2012) 7:637–42. doi: 10.1038/nprot.2012.011
16. Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol. (2011) 9:215–22. doi: 10.1038/nrmicro2508
17. Patel M, Chen J, Kim S, Garg S, Flannery B, Haddadin Z, et al. Analysis of marketscan data for immunosuppressive conditions and hospitalizations for acute respiratory illness, United States. Emerg Infect Dis. (2020) 26:1720–30. doi: 10.3201/eid2608.191493
18. Kumar B, Kashyap N, Avinash A, Chevvuri R, Sagar MK, Shrikant K. The composition, function and role of saliva in maintaining oral health: a review. Int J Contemp Dent Med. (2017) 2017. doi: 10.15713/ins.ijcdmr.121
19. Feller L, Altini M, Khammissa RAG, Chandran R, Bouckaert M, Lemmer J. Oral mucosal immunity. Oral Surg Oral Med Oral Pathol Oral Radiol. (2013) 116:576–83. doi: 10.1016/j.oooo.2013.07.01324119522
20. Devine DA. Antimicrobial peptides in defence of the oral and respiratory tracts. Mol Immunol. (2003) 40:431–43. doi: 10.1016/S0161-5890(03)00162-7
21. Alalwani M, Kharma M, Aws G. Profound study for functions of antimicrobial peptides in prevention of oral disease. Br J Med Med Res. (2016) 14:1–10. doi: 10.9734/BJMMR/2016/24528
22. Wertz PW, de Szalay S. Innate antimicrobial defense of skin and oral mucosa. Antibiotics. (2020) 9:159. doi: 10.3390/antibiotics9040159
23. Khurshid Z, Naseem M, Sheikh Z, Najeeb S, Shahab S, Zafar MS. Oral antimicrobial peptides: types and role in the oral cavity. Saudi Pharm J. (2016) 24:515–24. doi: 10.1016/j.jsps.2015.02.015
24. Chen X, Takai T, Xie Y, Niyonsaba F, Okumura K, Ogawa H. Human antimicrobial peptide LL-37 modulates proinflammatory responses induced by cytokine milieus and double-stranded RNA in human keratinocytes. Biochem Biophys Res Commun. (2013) 433:532–7. doi: 10.1016/j.bbrc.2013.03.024
25. Yang D, Chen Q, Chertov O, Oppenheim JJ. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J Leukoc Biol. (2000) 68:9–14. doi: 10.1189/jlb.68.1.9
26. Jang J-H, Shin HW, Lee JM, Lee H-W, Kim E-C, Park SH. An overview of pathogen recognition receptors for innate immunity in dental pulp. Mediators Inflamm. (2015) 2015:1–12. doi: 10.1155/2015/794143
27. Zhou Y, Cheng L, Lei YL, Ren B, Zhou X. The interactions between Candida albicans and mucosal immunity. Front Microbiol. (2021) 12. doi: 10.3389/fmicb.2021.652725
29. Nikou S-A, Kichik N, Brown R, Ponde N, Ho J, Naglik J, et al. Candida albicans interactions with mucosal surfaces during health and disease. Pathogens. (2019) 8:53. doi: 10.3390/pathogens8020053
30. Mukaremera L, Lee KK, Mora-Montes HM, Gow NAR. Candida albicans yeast, pseudohyphal, and hyphal morphogenesis differentially affects immune recognition. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00629
31. Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, et al. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J Clin Invest. (2007) 117:3664–72. doi: 10.1172/JCI28115
32. Swidergall M, Solis N V, Lionakis MS, Filler SG. EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat Microbiol. (2018) 3:53–61. doi: 10.1038/s41564-017-0059-5
33. Pavlova A, Sharafutdinov I. Recognition of Candida albicans and role of innate type 17 immunity in oral candidiasis. Microorganisms. (2020) 8:1340. doi: 10.3390/microorganisms8091340
34. Swidergall M, Solis N V, Phan QT, Lazarus MD, Wang Z, Filler SG. Activation of the epidermal growth factor receptor initiates innate immune responses during oropharyngeal candidiasis. BioRxiv. (2018) 491076. doi: 10.1101/491076
35. Swidergall M, Ernst JF. Interplay between Candida albicans and the Antimicrobial Peptide Armory. Eukaryot Cell. (2014) 13:950–7. doi: 10.1128/EC.00093-14
36. Swidergall M, Ernst AM, Ernst JF. Candida albicans mucin Msb2 is a broad-range protectant against antimicrobial peptides. Antimicrob Agents Chemother. (2013) 57:3917–22. doi: 10.1128/AAC.00862-13
37. Ho J, Yang X, Nikou S-A, Kichik N, Donkin A, Ponde NO, et al. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun. (2019) 10:2297. doi: 10.1038/s41467-019-09915-2
38. Swidergall M, Solis N V, Millet N, Huang MY, Lin J, Phan QT, et al. Activation of EphA2-EGFR signaling in oral epithelial cells by Candida albicans virulence factors. PLoS Pathog. (2021) 17:e1009221. doi: 10.1371/journal.ppat.1009221
39. Kurokawa K, Lee H, Roh K-B, Asanuma M, Kim YS, Nakayama H, et al. The triacylated ATP binding cluster transporter substrate-binding lipoprotein of staphylococcus aureus functions as a native ligand for toll-like receptor 2. J Biol Chem. (2009) 284:8406–11. doi: 10.1074/jbc.M809618200
40. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. (2003) 278:8869–72. doi: 10.1074/jbc.C200651200
41. Hruz P, Zinkernagel AS, Jenikova G, Botwin GJ, Hugot J-P, Karin M, et al. NOD2 contributes to cutaneous defense against Staphylococcus aureus through -toxin-dependent innate immune activation. Proc Natl Acad Sci. (2009) 106:12873–8. doi: 10.1073/pnas.0904958106
42. Perfetto B, Donnarumma G, Criscuolo D, Paoletti I, Grimaldi E, Tufano MA, et al. Bacterial components induce cytokine and intercellular adhesion molecules-1 and activate transcription factors in dermal fibroblasts. Res Microbiol. (2003) 154:337–44. doi: 10.1016/S0923-2508(03)00084-6
43. Tufano MA, Cipollaro de. l'Ero G, Ianniello R, Galdiero M, Galdiero F. Protein A and other surface components of Staphylococcus aureus stimulate production of IL-1 alpha, IL-4, IL-6, TNF and IFN-gamma. Eur Cytokine Netw. (1991) 2:361–6.
44. Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. (2005) 18:521–40. doi: 10.1128/CMR.18.3.521-540.2005
45. Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol Microbiol. (2007) 66:1136–47. doi: 10.1111/j.1365-2958.2007.05986.x
46. McGuinness W, Kobayashi S, DeLeo F. Evasion of neutrophil killing by Staphylococcus aureus. Pathogens. (2016) 5:32. doi: 10.3390/pathogens5010032
47. Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. Epic Immune Battles of History: Neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol. (2017) 7. doi: 10.3389/fcimb.2017.00286
48. Joo H-S, Otto M. Mechanisms of resistance to antimicrobial peptides in staphylococci. Biochim Biophys Acta - Biomembr. (2015) 1848:3055–61. doi: 10.1016/j.bbamem.2015.02.009
49. Pidwill GR, Gibson JF, Cole J, Renshaw SA, Foster SJ. The role of macrophages in Staphylococcus aureus infection. Front Immunol. (2021) 11. doi: 10.3389/fimmu.2020.620339
50. Brandtzaeg P. Secretory immunity with special reference to the oral cavity. J Oral Microbiol. (2013) 5:20401. doi: 10.3402/jom.v5i0.20401
51. Brandtzaeg P. mucosal immunity: induction, dissemination, and effector functions. Scand J Immunol. (2009) 70:505–15. doi: 10.1111/j.1365-3083.2009.02319.x
52. Holmes AR, Bandara BMK, Cannon RD. Saliva promotes candida albicans adherence to human epithelial cells. J Dent Res. (2002) 81:28–32. doi: 10.1177/002203450208100107
53. van der Wielen P, Holmes A, Cannon R. Secretory component mediates Candida albicans binding to epithelial cells. Oral Dis. (2016) 22:69–74. doi: 10.1111/odi.12397
54. Heo S-M, Choi K-S, Kazim LA, Reddy MS, Haase EM, Scannapieco FA, et al. Host defense proteins derived from human saliva bind to Staphylococcus aureus. Infect Immun. (2013) 81:1364–73. doi: 10.1128/IAI.00825-12
55. Biesbrock AR, Reddy MS, Levine MJ. Interaction of a salivary mucin-secretory immunoglobulin A complex with mucosal pathogens. Infect Immun. (1991) 59:3492–7. doi: 10.1128/iai.59.10.3492-3497.1991
56. Domnich M, Riedesel J, Pylaeva E, Kürten CHL, Buer J, Lang S, et al. Oral neutrophils: underestimated players in oral cancer. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.565683
57. Rijkschroeff P, Loos BG, Nicu EA. Oral polymorphonuclear neutrophil contributes to oral health. Curr Oral Heal Reports. (2018) 5:211–20. doi: 10.1007/s40496-018-0199-6
58. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
59. Pham CTN. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. (2006) 6:541–50. doi: 10.1038/nri1841
60. Niemiec MJ, Grumaz C, Ermert D, Desel C, Shankar M, Lopes JP, et al. Dual transcriptome of the immediate neutrophil and Candida albicans interplay. BMC Genomics. (2017) 18:696. doi: 10.1186/s12864-017-4097-4
61. Gabrielli E, Sabbatini S, Roselletti E, Kasper L, Perito S, Hube B, et al. In vivo induction of neutrophil chemotaxis by secretory aspartyl proteinases of Candida albicans. Virulence. (2016) 7:819–25. doi: 10.1080/21505594.2016.1184385
62. Rudkin FM, Bain JM, Walls C, Lewis LE, Gow NAR, Erwig LP. Altered dynamics of candida albicans phagocytosis by macrophages and PMNs when both phagocyte subsets are present. MBio. (2013) 4. doi: 10.1128/mBio.00810-13
63. McKenzie CGJ, Koser U, Lewis LE, Bain JM, Mora-Montes HM, Barker RN, et al. Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect Immun. (2010) 78:1650–8. doi: 10.1128/IAI.00001-10
64. Lewis LE, Bain JM, Lowes C, Gillespie C, Rudkin FM, Gow NAR, et al. Stage specific assessment of candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLoS Pathog. (2012) 8:e1002578. doi: 10.1371/journal.ppat.1002578
65. Miramón P, Dunker C, Windecker H, Bohovych IM, Brown AJP, Kurzai O, et al. Cellular responses of candida albicans to phagocytosis and the extracellular activities of neutrophils are critical to counteract carbohydrate starvation, oxidative and nitrosative stress. PLoS ONE. (2012) 7:e52850. doi: 10.1371/journal.pone.0052850
66. Ermert D, Niemiec MJ, Röhm M, Glenthøj A, Borregaard N, Urban CF. Candida albicans escapes from mouse neutrophils. J Leukoc Biol. (2013) 94:223–36. doi: 10.1189/jlb.0213063
67. Urban CF, Nett JE. Neutrophil extracellular traps in fungal infection. Semin Cell Dev Biol. (2019) 89:47–57. doi: 10.1016/j.semcdb.2018.03.020
68. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PLoS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
69. Moonen CGJ, Hirschfeld J, Cheng L, Chapple ILC, Loos BG, Nicu EA. Oral neutrophils characterized: chemotactic, phagocytic, and neutrophil extracellular trap (NET) formation properties. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.00635
70. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
71. Xie Z, Thompson A, Sobue T, Kashleva H, Xu H, Vasilakos J, et al. Candida albicans biofilms do not trigger reactive oxygen species and evade neutrophil killing. J Infect Dis. (2012) 206:1936–45. doi: 10.1093/infdis/jis607
72. Kernien JF, Johnson CJ, Bayless ML, Chovanec JF, Nett JE. Neutrophils from patients with invasive candidiasis are inhibited by candida albicans biofilms. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.587956
73. Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. (2000) 12:201–10. doi: 10.1016/S1074-7613(00)80173-9
74. Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem. (2011) 286:24638–48. doi: 10.1074/jbc.M110.201988
75. Brignone C, Munoz O, Batoz M, Rouquette-Jazdanian A, Cousin J. Proteases produced by activated neutrophils release soluble CD23 fragments endowed with proinflammatory effects. FASEB J. (2001) 15:2027–9. doi: 10.1096/fj.00-0773fje
76. Thomas MP, Whangbo J, McCrossan G, Deutsch AJ, Martinod K, Walch M, et al. Leukocyte protease binding to nucleic acids promotes nuclear localization and cleavage of nucleic acid binding proteins. J Immunol. (2014) 192:5390–7. doi: 10.4049/jimmunol.1303296
77. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. (2000) 275:6819–23. doi: 10.1074/jbc.275.10.6819
78. Gao S. Cathepsin G and its role in inflammation and autoimmune diseases. Arch Rheumatol. (2018) 33:498–504. doi: 10.5606/ArchRheumatol.2018.6595
79. Sørensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, et al. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. (2001) 97:3951–9. doi: 10.1182/blood.V97.12.3951
80. Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al. Ll-37, the neutrophil granule–and epithelial cell–derived cathelicidin, utilizes formyl peptide receptor–like 1 (Fprl1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. (2000) 192:1069–74. doi: 10.1084/jem.192.7.1069
81. Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal. (2013) 18:642–60. doi: 10.1089/ars.2012.4827
82. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. (2014) 15:602–11. doi: 10.1038/ni.2921
83. Borregaard N, Sørensen OE, Theilgaard-Mönch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. (2007) 28:340–5. doi: 10.1016/j.it.2007.06.002
84. Hirsch JG, Cohn ZA. Degranulation of polymorphonuclear leucocytes following phagocytosis of microorganisms. J Exp Med. (1960) 112:1005–14. doi: 10.1084/jem.112.6.1005
85. Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics. (2005) 4:1503–21. doi: 10.1074/mcp.M500143-MCP200
86. Ledo C, Gonzalez CD, Garofalo A, Sabbione F, Keitelman IA, Giai C, et al. Protein A modulates neutrophil and keratinocyte signaling and survival in response to Staphylococcus aureus. Front Immunol. (2021) 11. doi: 10.3389/fimmu.2020.524180
87. Speziale P, Pietrocola G. Staphylococcus aureus induces neutrophil extracellular traps (NETs) and neutralizes their bactericidal potential. Comput Struct Biotechnol J. (2021) 19:3451–7. doi: 10.1016/j.csbj.2021.06.012
88. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus aureus Degrades Neutrophil Extracellular Traps to Promote Immune Cell Death. Science. (2013) 342:863–6. doi: 10.1126/science.1242255
89. Harpf V, Rambach G, Würzner R, Lass-Flörl C, Speth C. Candida and complement: new aspects in an old battle. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.01471
90. Heesterbeek DAC, Angelier ML, Harrison RA, Rooijakkers SHM. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J Innate Immun. (2018) 10:455–64. doi: 10.1159/000491439
91. Andoh A, Fujiyama Y, Kimura T, Uchihara H, Sakumoto H, Okabe H, et al. Molecular characterization of complement components (C3, C4, and factor B) in human saliva. J Clin Immunol. (1997) 17:404–7. doi: 10.1023/A:1027320425291
92. Courts FJ, Boackle RJ, Fudenberg HH, Silverman MS. Detection of functional complement components in gingival crevicular fluid from humans with periodontal disease. J Dent Res. (1977) 56:327–31. doi: 10.1177/00220345770560032001
93. Eichenberger EM, Dagher M, Ruffin F, Park L, Hersh L, Sivapalasingam S, et al. Complement levels in patients with bloodstream infection due to Staphylococcus aureus or Gram-negative bacteria. Eur J Clin Microbiol Infect Dis. (2020) 39:2121–31. doi: 10.1007/s10096-020-03955-z
94. Hallett AF, Cooper R. Complement activation in Staphylococcus aureus bacteraemia. Clin Exp Immunol. (1980) 40:306–11.
95. Zipfel PF, Skerka C. Complement, Candida, and cytokines: The role of C5a in host response to fungi. Eur J Immunol. (2012) 42:822–5. doi: 10.1002/eji.201242466
96. Densen P. Human Complement Deficiency States and Infection. Complement Heal. Dis., Dordrecht: Springer Netherlands. (1993). p. 173–97. doi: 10.1007/978-94-011-2214-6_6
97. Li D, Dong B, Tong Z, Wang Q, Liu W, Wang Y, et al. MBL-Mediated opsonophagocytosis of Candida albicans by human neutrophils is coupled with intracellular dectin-1-triggered ROS production. PLoS ONE. (2012) 7:e50589. doi: 10.1371/journal.pone.0050589
98. Hünniger K, Bieber K, Martin R, Lehnert T, Figge MT, Löffler J, et al. A second stimulus required for enhanced antifungal activity of human neutrophils in blood is provided by anaphylatoxin C5a. J Immunol. (2015) 194:1199–210. doi: 10.4049/jimmunol.1401845
99. Cunnion KM, Frank MM. Complement activation influences staphylococcus aureus adherence to endothelial cells. Infect Immun. (2003) 71:1321–7. doi: 10.1128/IAI.71.3.1321-1327.2003
100. Jang KO, Lee YW, Kim H, Chung DK. Complement inactivation strategy of staphylococcus aureus using decay-accelerating factor and the response of infected HaCaT cells. Int J Mol Sci. (2021) 22:4015. doi: 10.3390/ijms22084015
101. Woehl JL, Stapels DAC, Garcia BL, Ramyar KX, Keightley A, Ruyken M, et al. The extracellular adherence protein from staphylococcus aureus inhibits the classical and lectin pathways of complement by blocking formation of the C3 proconvertase. J Immunol. (2014) 193:6161–71. doi: 10.4049/jimmunol.1401600
102. Jusko M, Potempa J, Kantyka T, Bielecka E, Miller HK, Kalinska M, et al. Staphylococcal proteases aid in evasion of the human complement system. J Innate Immun. (2014) 6:31–46. doi: 10.1159/000351458
103. Gaffen SL, Moutsopoulos NM. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci Immunol. (2020) 5:eaau4594. doi: 10.1126/sciimmunol.aau4594
104. Hovav A-H. Dendritic cells of the oral mucosa. Mucosal Immunol. (2014) 7:27–37. doi: 10.1038/mi.2013.42
105. Hilligan KL, Ronchese F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell Mol Immunol. (2020) 17:587–99. doi: 10.1038/s41423-020-0465-0
106. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
107. Sobkowiak MJ, Davanian H, Heymann R, Gibbs A, Emgård J, Dias J, et al. Tissue-resident MAIT cell populations in human oral mucosa exhibit an activated profile and produce IL-17. Eur J Immunol. (2019) 49:133–43. doi: 10.1002/eji.201847759
108. Aggor FEY, Break TJ, Trevejo-Nuñez G, Whibley N, Coleman BM, Bailey RD, et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17–mediated immunity to oral mucosal candidiasis. Sci Immunol. (2020) 5:eaba0570. doi: 10.1126/sciimmunol.aba0570
109. Verma AH, Richardson JP, Zhou C, Coleman BM, Moyes DL, Ho J, et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol. (2017) 2. doi: 10.1126/sciimmunol.aam8834
110. Zelante T, Iannitti RG, De Luca A, Arroyo J, Blanco N, Servillo G, et al. Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat Commun. (2012) 3:683. doi: 10.1038/ncomms1685
111. Yu Q, Jia C, Dong Y, Zhang B, Xiao C, Chen Y, et al. Candida albicans autophagy, no longer a bystander: Its role in tolerance to ER stress-related antifungal drugs. Fungal Genet Biol. (2015) 81:238–49. doi: 10.1016/j.fgb.2015.02.008
112. Teymournejad O, Montgomery CP. Evasion of immunological memory by S. aureus infection: implications for vaccine design. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.633672
113. Schreiner J, Kretschmer D, Klenk J, Otto M, Bühring H-J, Stevanovic S, et al. Staphylococcus aureus phenol-soluble modulin peptides modulate dendritic cell functions and increase in vitro priming of regulatory T cells. J Immunol. (2013) 190:3417–26. doi: 10.4049/jimmunol.1202563
114. Aulik NA, Hellenbrand KM, Czuprynski CJ. Mannheimia haemolytica and its leukotoxin cause macrophage extracellular trap formation by bovine macrophages. Infect Immun. (2012) 80:1923–33. doi: 10.1128/IAI.06120-11
115. Muñoz JF, Delorey T, Ford CB Li BY, Thompson DA, Rao RP, et al. Coordinated host-pathogen transcriptional dynamics revealed using sorted subpopulations and single macrophages infected with Candida albicans. Nat Commun. (2019) 10:1607. doi: 10.1038/s41467-019-09599-8
116. Loureiro A, Pais C, Sampaio P. Relevance of macrophage extracellular traps in C. albicans killing. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.02767
117. Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol. (2008) 9:639–49. doi: 10.1038/nrm2447
118. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. (2014) 5. doi: 10.3389/fimmu.2014.00491
119. Chan LC, Rossetti M, Miller LS, Filler SG, Johnson CW, Lee HK, et al. Protective immunity in recurrent Staphylococcus aureus infection reflects localized immune signatures and macrophage-conferred memory. Proc Natl Acad Sci. (2018) 115:E11111–9. doi: 10.1073/pnas.1808353115
120. Brown AF, Murphy AG, Lalor SJ, Leech JM, O'Keeffe KM, Mac Aogáin M, et al. Memory Th1 cells are protective in invasive Staphylococcus aureus infection. PLoS Pathog. (2015) 11:e1005226. doi: 10.1371/journal.ppat.1005226
121. Kolata JB, Kühbandner I, Link C, Normann N, Vu CH, Steil L, et al. The fall of a dogma? Unexpected high T-cell memory response to staphylococcus aureus in humans. J Infect Dis. (2015) 212:830–8. doi: 10.1093/infdis/jiv128
122. Bröker B, Mrochen D, Péton V. The T Cell Response to Staphylococcus aureus. Pathogens. (2016) 5:31. doi: 10.3390/pathogens5010031
123. Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy. (2011) 66:396–403. doi: 10.1111/j.1398-9995.2010.02498.x
124. Jubrail J, Morris P, Bewley MA, Stoneham S, Johnston SA, Foster SJ, et al. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol. (2016) 18:80–96. doi: 10.1111/cmi.12485
125. Grayczyk JP, Alonzo F. Staphylococcus aureus lipoic acid synthesis limits macrophage reactive oxygen and nitrogen species production to promote survival during infection. Infect Immun. (2019) 87. doi: 10.1128/IAI.00344-19
126. O'Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of autophagy in phagocytes facilitates staphylococcus aureus bloodstream infection. Infect Immun. (2015) 83:3445–57. doi: 10.1128/IAI.00358-15
127. Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, et al. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun. (2000) 68:5385–92. doi: 10.1128/IAI.68.9.5385-5392.2000
128. Flannagan RS, Heit B, Heinrichs DE. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol. (2016) 18:514–35. doi: 10.1111/cmi.12527
129. Li Y, Wang W, Yang F, Xu Y, Feng C, Zhao Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun Signal. (2019) 17:147. doi: 10.1186/s12964-019-0471-y
130. Schmidt S, Zimmermann S-Y, Tramsen L, Koehl U, Lehrnbecher T. Natural killer cells and antifungal host response. Clin Vaccine Immunol. (2013) 20:452–8. doi: 10.1128/CVI.00606-12
131. Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. (2016) 9:1163–72. doi: 10.1038/mi.2015.136
132. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.01869
133. Voigt J, Hünniger K, Bouzani M, Jacobsen ID, Barz D, Hube B, et al. Human natural killer cells acting as phagocytes against candida albicans and mounting an inflammatory response that modulates neutrophil antifungal activity. J Infect Dis. (2014) 209:616–26. doi: 10.1093/infdis/jit574
134. Whitney PG, Bär E, Osorio F, Rogers NC, Schraml BU, Deddouche S, et al. Correction: syk signaling in dendritic cells orchestrates innate resistance to systemic fungal infection. PLoS Pathog. (2018) 14:e1007366. doi: 10.1371/journal.ppat.1007366
135. Schmidt S, Ullrich E, Bochennek K, Zimmermann S-Y, Lehrnbecher T. Role of natural killer cells in antibacterial immunity. Expert Rev Hematol. (2016) 9:1119–27. doi: 10.1080/17474086.2016.1254546
136. Minns D, Smith KJ, Alessandrini V, Hardisty G, Melrose L, Jackson-Jones L, et al. The neutrophil antimicrobial peptide cathelicidin promotes Th17 differentiation. Nat Commun. (2021) 12:1285. doi: 10.1038/s41467-021-21533-5
137. Haller D, Serrant P, Granato D, Schiffrin EJ, Blum S. Activation of human NK cells by staphylococci and lactobacilli requires cell contact-dependent costimulation by autologous monocytes. Clin Vaccine Immunol. (2002) 9:649–57. doi: 10.1128/CDLI.9.3.649-657.2002
138. D'Orazio JA, Burke GW, Stein-Streilein J. Staphylococcal enterotoxin B activates purified NK cells to secrete IFN-gamma but requires T lymphocytes to augment NK cytotoxicity. J Immunol. (1995) 154:1014–23.
139. Small C-L, McCormick S, Gill N, Kugathasan K, Santosuosso M, Donaldson N, et al. NK Cells play a critical protective role in host defense against acute extracellular staphylococcus aureus bacterial infection in the lung. J Immunol. (2008) 180:5558–68. doi: 10.4049/jimmunol.180.8.5558
140. Genardi S, Visvabharathy L, Cao L, Morgun E, Cui Y, Qi C, et al. Type II Natural Killer T Cells contribute to protection against systemic methicillin-resistant Staphylococcus aureus Infection. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.610010
141. Li R, Rezk A, Li H, Gommerman JL, Prat A, Bar-Or A. Antibody-independent function of human b cells contributes to antifungal t cell responses. J Immunol. (2017) 198:3245–54. doi: 10.4049/jimmunol.1601572
142. Kamar N, Milioto O, Puissant-Lubrano B, Esposito L, Pierre MC, Mohamed AO, et al. Incidence and predictive factors for infectious disease after rituximab therapy in kidney-transplant patients. Am J Transplant. (2010) 10:89–98. doi: 10.1111/j.1600-6143.2009.02785.x
143. Gjertsson I, Hultgren OH, Stenson M, Holmdahl R, Tarkowski A. Are B lymphocytes of importance in severe Staphylococcus aureus infections? Infect Immun. (2000) 68:2431–4. doi: 10.1128/IAI.68.5.2431-2434.2000
144. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-Linked agammaglobulinemia. Medicine. (2006) 85:193–202. doi: 10.1097/01.md.0000229482.27398.ad
145. Selle M, Hertlein T, Oesterreich B, Klemm T, Kloppot P, Müller E, et al. Global antibody response to Staphylococcus aureus live-cell vaccination. Sci Rep. (2016) 6:24754. doi: 10.1038/srep24754
146. Pelzek AJ, Shopsin B, Radke EE, Tam K, Ueberheide BM, Fenyö D, et al. Human memory B cells targeting staphylococcus aureus exotoxins are prevalent with skin and soft tissue infection. Mbio. (2018) 9. doi: 10.1128/mBio.02125-17
147. Radke EE, Brown SM, Pelzek AJ, Fulmer Y, Hernandez DN, Torres VJ, et al. Hierarchy of human IgG recognition within the Staphylococcus aureus immunome. Sci Rep. (2018) 8:13296. doi: 10.1038/s41598-018-31424-3
148. Nygaard TK, Kobayashi SD, Freedman B, Porter AR, Voyich JM, Otto M, et al. Interaction of staphylococci with human B cells. PLoS ONE. (2016) 11:e0164410. doi: 10.1371/journal.pone.0164410
149. Souli M, Ruffin F, Choi S-H, Park LP, Gao S, Lent NC, et al. Changing characteristics of Staphylococcus aureus bacteremia: results from a 21-year, prospective, longitudinal study. Clin Infect Dis. (2019) 69:1868–77. doi: 10.1093/cid/ciz112
150. Smit J, Søgaard M, Schønheyder HC, Nielsen H, Frøslev T, Thomsen RW. Diabetes and risk of community-acquired Staphylococcus aureus bacteremia: a population-based case–control study. Eur J Endocrinol. (2016) 174:631–9. doi: 10.1530/EJE-16-0023
151. Larsen M, Harboe Z, Ladelund S, Skov R, Gerstoft J, Pedersen C, et al. Major but differential decline in the incidence of Staphylococcus aureus bacteraemia in HIV-infected individuals from 1995 to 2007: a nationwide cohort study. HIV Med. (2011) 13:45–53. doi: 10.1111/j.1468-1293.2011.00937.x
152. Subhashini M. A cross sectional study on clinicomycological aspects of mucocutaneous candidiasis in a tertiary care center. Int J Dermatology Clin Res. (2018) 4:003–7. doi: 10.17352/2455-8605.000026
153. Subhashini M. A Study on clinical patterns of mucocutaneous candidiasis in immunosuppressed patients. Int J Dermatology Clin Res. (2017) 3:032–4. doi: 10.17352/2455-8605.000024
154. Bartholomew GA, Rodu B, Bell DS. Oral candidiasis in patients with diabetes mellitus: a thorough analysis. Diabetes Care. (1987) 10:607–12. doi: 10.2337/diacare.10.5.607
155. Nguyen TD, Nguyen TTH, Tran QV. The incidence of oral candidiasis in patients with diabetes mellitus: a cross-sectional study in southern Vietnam. J Crit Rev. (2020) 7. doi: 10.31838/jcr.07.04.17
156. Ueta E, Osaki T, Yoneda K, Yamamoto T. Prevalence of diabetes mellitus in odontogenic infections and oral candidiasis: an analysis of neutrophil suppression. J Oral Pathol Med. (1993) 22:168–74. doi: 10.1111/j.1600-0714.1993.tb01051.x
157. Lamey P-J, Darwaza A, Fisher BM, Samaranayake LP, Macfarlane TW, Frier BM. Secretor status, candidal carriage and candidal infection in patients with diabetes mellitus. J Oral Pathol Med. (1988) 17:354–7. doi: 10.1111/j.1600-0714.1988.tb01549.x
158. Bello-Chavolla OY, Bahena-Lopez JP, Garciadiego-Fosass P, Volkow P, Garcia-Horton A, Velazquez-Acosta C, et al. Bloodstream infection caused by S. aureus in patients with cancer: a 10-year longitudinal single-center study. Support Care Cancer. (2018) 26:4057–65. doi: 10.1007/s00520-018-4275-1
159. Sasson G, Bai AD, Showler A, Burry L, Steinberg M, Ricciuto DR, et al. Staphylococcus aureus bacteremia in immunosuppressed patients: a multicenter, retrospective cohort study. Eur J Clin Microbiol Infect Dis. (2017) 36:1231–41. doi: 10.1007/s10096-017-2914-y
160. Camp J, Glaubitz L, Filla T, Kaasch AJ, Fuchs F, Scarborough M, et al. Impact of immunosuppressive agents on clinical manifestations and outcome of staphylococcus aureus bloodstream infection: a propensity score–matched analysis in 2 large, prospectively evaluated cohorts. Clin Infect Dis. (2021) 73:1239–47. doi: 10.1093/cid/ciab385
161. Belot A, Cimaz R. Monogenic forms of systemic lupus erythematosus: new insights into SLE pathogenesis. Pediatr Rheumatol. (2012) 10:21. doi: 10.1186/1546-0096-10-21
162. Fernandez J. Overview of Immunodeficiency Disorders. MSD Man Prof Version 2021. Available online at: https://www.msdmanuals.com/en-gb/professional/immunology-allergic-disorders/immunodeficiency-disorders/overview-of-immunodeficiency-disorders (accessed September 9, 2021).
164. McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy, Asthma Clin Immunol. (2018) 14:61. doi: 10.1186/s13223-018-0290-5
165. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of current complement therapeutics. Mol Immunol. (2019) 114:341–52. doi: 10.1016/j.molimm.2019.07.030
166. Ahmadi F, Davoodi P, Dalband M, Hendi SS. Saliva as a mirror of the body health. Avicenna J Dent Res. (2018) 1:41–55.
167. Delli K, Spijkervet FKL, Vissink A. Salivary Gland Diseases: Infections, Sialolithiasis and Mucoceles. (2014). p. 135–48. doi: 10.1159/000358794
168. Lo Russo L, Guida L, Di Masi M, Buccelli C, Giannatempo G, Di Fede O, et al. Adverse drug reactions in the oral cavity. Curr Pharm Des. (2012) 18:5481–96. doi: 10.2174/138161212803307518
169. Lopes JP, Stylianou M, Backman E, Holmberg S, Jass J, Claesson R, et al. Evasion of immune surveillance in low oxygen environments enhances candida albicans virulence. MBio. (2018) 9. doi: 10.1128/mBio.02120-18
170. Tschoppe P, Wolgin M, Pischon N, Kielbassa AM. Etiologic factors of hyposalivation and consequences for oral health. Quintessence Int. (2010) 41:321–33.
171. Koray M, Tosun T. Oral Mucosal Trauma and Injuries. Trauma Dent, IntechOpen. (2019). doi: 10.5772/intechopen.81201
172. Wang S-S, Tang Y-L, Pang X, Zheng M, Tang Y-J, Liang X-H. The maintenance of an oral epithelial barrier. Life Sci. (2019) 227:129–36. doi: 10.1016/j.lfs.2019.04.029
173. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids — new mechanisms for old drugs. N Engl J Med. (2005) 353:1711–23. doi: 10.1056/NEJMra050541
174. Kozlak ST, Walsh SJ, Lalla R V. Reduced dietary intake of vitamin B12 and folate in patients with recurrent aphthous stomatitis. J Oral Pathol Med. (2010) 39:42–3. doi: 10.1111/j.1600-0714.2009.00867.x
175. Lankarani KB. Oral manifestation in inflammatory bowel disease: a review. World J Gastroenterol. (2013) 19:8571. doi: 10.3748/wjg.v19.i46.8571
176. Lawson W, Blitzer A. Inflammatory and neoplastic lesions of the oral cavity. Clin Dermatol. (1987) 5:43–58. doi: 10.1016/0738-081X(87)90007-1
177. Porter S, Gueiros LA, Leão JC, Fedele S. Risk factors and etiopathogenesis of potentially premalignant oral epithelial lesions. Oral Surg Oral Med Oral Pathol Oral Radiol. (2018) 125:603–11. doi: 10.1016/j.oooo.2018.03.008
178. Weusten B. Aphthous ulcers and vitamin B12 deficiency. Neth J Med. (1998) 53:172–5. doi: 10.1016/S0300-2977(98)00096-5
179. Wong T, Yap T, Wiesenfeld D. Common benign and malignant oral mucosal disease. Aust J Gen Pract. (2020) 49:568–73. doi: 10.31128/AJGP-02-20-5250-01
180. Xu X, He J, Xue J, Wang Y, Li K, Zhang K, et al. Oral cavity contains distinct niches with dynamic microbial communities. Environ Microbiol. (2015) 17:699–710. doi: 10.1111/1462-2920.12502
181. Bigley V, Barge D, Collin M. Dendritic cell analysis in primary immunodeficiency. Curr Opin Allergy Clin Immunol. (2016) 16:530–40. doi: 10.1097/ACI.0000000000000322
182. Lagaraine C, Lebranchu Y. Effects of immunosuppressive drugs on dendritic cells and tolerance induction. Transplantation. (2003) 75:37S−42S. doi: 10.1097/01.TP.0000067950.90241.1D
183. Manda K, Glasow A, Paape D, Hildebrandt G. Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Front Oncol. (2012) 2. doi: 10.3389/fonc.2012.00102
184. Chinen J, Shearer WT. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol. (2010) 125:S195–203. doi: 10.1016/j.jaci.2009.08.040
185. McDonald DR. TH17 deficiency in human disease. J Allergy Clin Immunol. (2012) 129:1429–35. doi: 10.1016/j.jaci.2012.03.034
186. Parampalli Yajnanarayana S, Stübig T, Cornez I, Alchalby H, Schönberg K, Rudolph J, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol. (2015) 169:824–33. doi: 10.1111/bjh.13373
187. Verstappen GM, Corneth OBJ, Bootsma H, Kroese FGM. Th17 cells in primary Sjögren's syndrome: Pathogenicity and plasticity. J Autoimmun. (2018) 87:16–25. doi: 10.1016/j.jaut.2017.11.003
188. Wiseman AC. Immunosuppressive Medications. Clin J Am Soc Nephrol. (2016) 11:332–43. doi: 10.2215/CJN.08570814
189. Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T cell dysfunction in cancer immunity and immunotherapy. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01719
190. Yang P, Qian F, Zhang M, Xu A, Wang X, Jiang B, et al. Th17 cell pathogenicity and plasticity in rheumatoid arthritis. J Leukoc Biol. (2019) 106:1233–40. doi: 10.1002/JLB.4RU0619-197R
191. Redmond HP. Impaired macrophage function in severe protein-energy malnutrition. Arch Surg. (1991) 126:192. doi: 10.1001/archsurg.1991.01410260080011
192. Schrijvers DM, De Meyer GRY, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. (2005) 25:1256–61. doi: 10.1161/01.ATV.0000166517.18801.a7
193. De Vries LCS, Duarte JM, De Krijger M, Welting O, Van Hamersveld PHP, Van Leeuwen-Hilbers FWM, et al. A JAK1 selective kinase inhibitor and tofacitinib affect macrophage activation and function. Inflamm Bowel Dis. (2019) 25:647–60. doi: 10.1093/ibd/izy364
194. Desnues B, Ihrig M, Raoult D, Mege J-L. Whipple's disease: a macrophage disease. Clin Vaccine Immunol. (2006) 13:170–8. doi: 10.1128/CVI.13.2.170-178.2006
195. Douglas SD, Musson RA. Phagocytic defects—Monocytes/macrophages. Clin Immunol Immunopathol. (1986) 40:62–8. doi: 10.1016/0090-1229(86)90069-3
196. Ferrer IR, Araki K, Ford ML. Paradoxical aspects of rapamycin immunobiology in transplantation. Am J Transplant. (2011) 11:654–9. doi: 10.1111/j.1600-6143.2011.03473.x
197. Jennings C, Kusler B, Jones PP. Calcineurin inactivation leads to decreased responsiveness to LPS in macrophages and dendritic cells and protects against LPS-induced toxicity in vivo. Innate Immun. (2009) 15:109–20. doi: 10.1177/1753425908100928
198. Lévêque M, Le Trionnaire S, Del Porto P, Martin-Chouly C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J Cyst Fibros. (2017) 16:443–53. doi: 10.1016/j.jcf.2016.10.011
199. Rahman FZ, Hayee B, Chee R, Segal AW, Smith AM. Impaired macrophage function following bacterial stimulation in chronic granulomatous disease. Immunology. (2009) 128:253–9. doi: 10.1111/j.1365-2567.2009.03112.x
200. Ramirez-Alejo N, Santos-Argumedo L. Innate Defects of the IL-12/IFN-γ Axis in Susceptibility to Infections by Mycobacteria and Salmonella. J Interf Cytokine Res. (2014) 34:307–17. doi: 10.1089/jir.2013.0050
201. Frater JL. How I investigate neutropenia. Int J Lab Hematol. (2020) 42:121–32. doi: 10.1111/ijlh.13210
202. Imbert S, Bresler P, Boissonnas A, Gauthier L, Souchet L, Uzunov M, et al. Calcineurin inhibitors impair neutrophil activity against Aspergillus fumigatus in allogeneic hematopoietic stem cell transplant recipients. J Allergy Clin Immunol. (2016) 138:860–8. doi: 10.1016/j.jaci.2016.02.026
203. Mitchell TS, Moots RJ, Wright HL. Janus kinase inhibitors prevent migration of rheumatoid arthritis neutrophils towards interleukin-8, but do not inhibit priming of the respiratory burst or reactive oxygen species production. Clin Exp Immunol. (2017) 189:250–8. doi: 10.1111/cei.12970
204. Rezaei N, Moazzami K, Aghamohammadi A, Klein C. Neutropenia and primary immunodeficiency diseases. Int Rev Immunol. (2009) 28:335–66. doi: 10.1080/08830180902995645
205. Chen J, Liu X, Zeng Z, Li J, Luo Y, Sun W, et al. Immunomodulation of NK cells by ionizing radiation. Front Oncol. (2020) 10. doi: 10.3389/fonc.2020.00874
206. Morteau O, Blundell S, Chakera A, Bennett S, Christou CM, Mason PD, et al. Renal transplant immunosuppression impairs natural killer cell function in vitro and in vivo. PLoS ONE. (2010) 5:e13294. doi: 10.1371/journal.pone.0013294
207. Nocturne G, Pascaud J, Ly B, Tahmasebi F, Mariette XJAK. inhibitors alter NK cell functions and may impair immunosurveillance against lymphomagenesis. Cell Mol Immunol. (2020) 17:552–3. doi: 10.1038/s41423-019-0320-3
208. Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect. (2002) 4:1545–58. doi: 10.1016/S1286-4579(02)00038-2
209. Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol. (2006) 6:399–409. doi: 10.1097/ACI.0b013e3280106b65
210. Vitale C, Chiossone L, Cantoni C, Morreale G, Cottalasso F, Moretti S, et al. The corticosteroid-induced inhibitory effect on NK cell function reflects down-regulation and/or dysfunction of triggering receptors involved in natural cytotoxicity. Eur J Immunol. (2004) 34:3028–38. doi: 10.1002/eji.200425418
211. Khan MAB, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J. Epidemiology of type 2 diabetes – global burden of disease and forecasted trends. J Epidemiol Glob Health. (2019) 10:107. doi: 10.2991/jegh.k.191028.001
212. Loke A. Diabetes 2021:1. Available online at: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed October 18, 2021).
213. Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev. (2020) 16:442–9. doi: 10.2174/1573399815666191024085838
214. Critchley JA, Carey IM, Harris T, DeWilde S, Hosking FJ, Cook DG. Glycemic control and risk of infections among people with type 1 or type 2 diabetes in a large primary care cohort study. Diabetes Care. (2018) 41:2127–35. doi: 10.2337/dc18-0287
215. Malicka B, Kaczmarek U, Skośkiewicz-Malinowska K. Prevalence of Xerostomia and the Salivary Flow Rate in Diabetic Patients. Adv Clin Exp Med. (2014) 23:225–33. doi: 10.17219/acem/37067
216. Janssen AWM, Stienstra R, Jaeger M, van Gool AJ, Joosten LAB, Netea MG, et al. Understanding the increased risk of infections in diabetes: innate and adaptive immune responses in type 1 diabetes. Metabolism. (2021) 121:154795. doi: 10.1016/j.metabol.2021.154795
217. Tan KS, Lee KO, Low KC, Gamage AM, Liu Y, Tan G-YG, et al. Glutathione deficiency in type 2 diabetes impairs cytokine responses and control of intracellular bacteria. J Clin Invest. (2012) 122:2289–300. doi: 10.1172/JCI57817
218. Berrou J, Fougeray S, Venot M, Chardiny V, Gautier J-F, Dulphy N, et al. Natural killer cell function, an important target for infection and tumor protection, is impaired in type 2 diabetes. PLoS ONE. (2013) 8:e62418. doi: 10.1371/journal.pone.0062418
219. Hair PS, Echague CG, Rohn RD, Krishna NK, Nyalwidhe JO, Cunnion KM. Hyperglycemic conditions inhibit C3-mediated immunologic control of Staphylococcus aureus. J Transl Med. (2012) 10:35. doi: 10.1186/1479-5876-10-35
220. Brown TT. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med. (2005) 165:1179. doi: 10.1001/archinte.165.10.1179
221. Doychinov N. HIV/AIDS. World Heal Organ 2021. Available online at: https://www.who.int/data/gho/data/themes/hiv-aids (accessed October 26, 2021).
222. WHO. The top 10 causes of death 2020. Available online at: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed October 26, 2021).
224. Wacleche V, Landay A, Routy J-P, Ancuta P. The Th17 lineage: from barrier surfaces homeostasis to autoimmunity, cancer, and HIV-1 pathogenesis. Viruses. (2017) 9:303. doi: 10.3390/v9100303
225. Bixler SL, Mattapallil JJ. Loss and dysregulation of Th17 cells during HIV infection. Clin Dev Immunol. (2013) 2013:1–9. doi: 10.1155/2013/852418
226. Valverde-Villegas JM, Matte MCC, Medeiros RM de, Chies JAB. New insights about treg and Th17 cells in HIV infection and disease progression. J Immunol Res. (2015) 2015:1–14. doi: 10.1155/2015/647916
227. Nanteza M, Tusiime JB, Kalyango J, Kasangaki A. Association between oral candidiasis and low CD4+ count among HIV positive patients in Hoima Regional Referral Hospital. BMC Oral Health. (2014) 14:143. doi: 10.1186/1472-6831-14-143
228. Bravo IM, Correnti M, Escalona L, Perrone M, Brito A, Tovar V, et al. Prevalence of oral lesions in HIV patients related to CD4 cell count and viral load in a Venezuelan population. Med Oral Patol Oral Cir Bucal. (2006) 11:E33–9.
229. Kerdpon D, Pongsiriwet S, Pangsomboon K, Iamaroon A, Kampoo K, Sretrirutchai S, et al. Oral manifestations of HIV infection in relation to clinical and CD4 immunological status in northern and southern Thai patients. Oral Dis. (2004) 10:138–44. doi: 10.1046/j.1601-0825.2003.00990.x
230. Naidu S, Thakur R, Singh A, Rajbhandary S, Mishra R, Sagtani A. Oral lesions and immune status of HIV infected adults from eastern Nepal. J Clin Exp Dent. (2013) 5:e1–7. doi: 10.4317/jced.50888
231. Bodhade AS, Ganvir SM, Hazarey VK. Oral manifestations of HIV infection and their correlation with CD4 count. J Oral Sci. (2011) 53:203–11. doi: 10.2334/josnusd.53.203
232. Nguyen MH. Nasal carriage of and infection with Staphylococcus aureus in HIV-infected patients. Ann Intern Med. (1999) 130:221. doi: 10.7326/0003-4819-130-3-199902020-00026
233. Senthilkumar A, Kumar S, Sheagren JN. Increased incidence of staphylococcus aureus bacteremia in hospitalized patients with acquired immunodeficiency syndrome. Clin Infect Dis. (2001) 33:1412–6. doi: 10.1086/322656
234. Adesida SA, Abioye OA, Bamiro BS, Amisu KO, Badaru SO, Coker AO. Staphylococcal bacteraemia among human immunodeficiency virus positive patients at a screening center in Lagos, Nigeria. Beni-Suef Univ J Basic Appl Sci. (2017) 6:112–7. doi: 10.1016/j.bjbas.2016.08.006
235. Ferreira D, da Silva G, Cavalcante F, do Carmo F, Fernandes L, Moreira S, et al. Methicillin-resistant Staphylococcus aureus in HIV patients: Risk factors associated with colonization and/or infection and methods for characterization of isolates – a systematic review. Clinics. (2014) 69:770–6. doi: 10.6061/clinics/2014(11)11
236. Shi X, Sims MD, Hanna MM, Xie M, Gulick PG, Zheng Y-H, et al. Neutropenia during HIV infection: adverse consequences and remedies. Int Rev Immunol. (2014) 33:511–36. doi: 10.3109/08830185.2014.893301
237. Bowers NL, Helton ES, Huijbregts RPH, Goepfert PA, Heath SL, Hel Z. Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Pathog. (2014) 10:e1003993. doi: 10.1371/journal.ppat.1003993
238. Koppensteiner H, Brack-Werner R, Schindler M. Macrophages and their relevance in Human Immunodeficiency Virus Type I infection. Retrovirology. (2012) 9:82. doi: 10.1186/1742-4690-9-82
239. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. (2018) 378:1029–41. doi: 10.1056/NEJMra1615896
240. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. (2007) 370:59–67. doi: 10.1016/S0140-6736(07)61050-2
241. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
242. Rolston KVI. Infections in cancer patients with solid tumors: a review. Infect Dis Ther. (2017) 6:69–83. doi: 10.1007/s40121-017-0146-1
243. Niederhuber JE, Armitage JO, Doroshow JH, Kastan MB, Tepper JE. Abeloff's Clinical Oncology. 6th ed., Elsevier. (2020). p. 544–64.
244. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. (2021) 21:345–59. doi: 10.1038/s41568-021-00347-z
245. Allen BM, Hiam KJ, Burnett CE, Venida A, DeBarge R, Tenvooren I, et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med. (2020) 26:1125–34. doi: 10.1038/s41591-020-0892-6
246. Shaked Y. The pro-tumorigenic host response to cancer therapies. Nat Rev Cancer. (2019) 19:667–85. doi: 10.1038/s41568-019-0209-6
247. Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia. Cancer. (2004) 100:228–37. doi: 10.1002/cncr.11882
248. Karbach J, Walter C, Al-Nawas B. Evaluation of saliva flow rates, Candida colonization and susceptibility of Candida strains after head and neck radiation. Clin Oral Investig. (2012) 16:1305–12. doi: 10.1007/s00784-011-0612-1
249. Ligtenberg AJM, Almståhl A. Xerostomia and the Oral Microflora. Dry Mouth, Berlin, Heidelberg: Springer Berlin Heidelberg. (2015). p. 81–101. doi: 10.1007/978-3-642-55154-3_6
250. Lossignol D. A little help from steroids in oncology. J Transl Intern Med. (2016) 4:52–4. doi: 10.1515/jtim-2016-0011
251. Wallace BI, Kenney B, Malani PN, Clauw DJ, Nallamothu BK, Waljee AK. Prevalence of immunosuppressive drug use among commercially insured US adults, 2018-2019. JAMA Netw Open. (2021) 4:e214920. doi: 10.1001/jamanetworkopen.2021.4920
252. Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. (2002) 20:125–63. doi: 10.1146/annurev.immunol.20.082401.104914
253. Quatrini L, Ugolini S. New insights into the cell- and tissue-specificity of glucocorticoid actions. Cell Mol Immunol. (2021) 18:269–78. doi: 10.1038/s41423-020-00526-2
254. Lionakis MS, Kontoyiannis DP. Glucocorticoids and invasive fungal infections. Lancet. (2003) 362:1828–38. doi: 10.1016/S0140-6736(03)14904-5
255. Pasternak Y, Yarden-Bilavsky H, Kodman Y, Zoldan M, Tamary H, Ashkenazi S. Inhaled corticosteroids increase blood neutrophil count by decreasing the expression of neutrophil adhesion molecules Mac-1 and L-selectin. Am J Emerg Med. (2016) 34:1977–81. doi: 10.1016/j.ajem.2016.07.003
256. Ronchetti S, Ricci E, Migliorati G, Gentili M, Riccardi C. How glucocorticoids affect the neutrophil life. Int J Mol Sci. (2018) 19:4090. doi: 10.3390/ijms19124090
257. Banuelos J, Cao Y, Shin SC, Lu NZ. Immunopathology alters Th17 cell glucocorticoid sensitivity. Allergy. (2017) 72:331–41. doi: 10.1111/all.13051
258. Forsblom E, Nurmi A-M, Ruotsalainen E, Järvinen A. Should all adjunctive corticosteroid therapy be avoided in the management of hemodynamically stabile Staphylococcus aureus bacteremia? Eur J Clin Microbiol Infect Dis. (2016) 35:471–9. doi: 10.1007/s10096-015-2563-y
259. Kaech C, Elzi L, Sendi P, Frei R, Laifer G, Bassetti S, et al. Course and outcome of Staphylococcus aureus bacteraemia: a retrospective analysis of 308 episodes in a Swiss tertiary-care centre. Clin Microbiol Infect. (2006) 12:345–52. doi: 10.1111/j.1469-0691.2005.01359.x
260. Hill PC, Birch M, Chambers S, Drinkovic D, Ellis-Pegler RB, Everts R, et al. Prospective study of 424 cases of Staphylococcus aureus bacteraemia: determination of factors affecting incidence and mortality. Intern Med J. (2001) 31:97–103. doi: 10.1111/j.1444-0903.2001.00029.x
261. Choi S-H, Cho SY, Park J-H, Chung J-W. Impact of infectious-disease specialist consultations on outcomes of Staphylococcus aureus bacteremia in a hospital with a low volume of patients with S. aureus bacteremia J Infect. (2011) 62:181–5. doi: 10.1016/j.jinf.2010.12.002
262. McCormack MG, Smith AJ, Akram AN, Jackson M, Robertson D, Edwards G. Staphylococcus aureus and the oral cavity: An overlooked source of carriage and infection? Am J Infect Control. (2015) 43:35–7. doi: 10.1016/j.ajic.2014.09.015
263. Beaussart A, Herman P, El-Kirat-Chatel S, Lipke PN, Kucharíková S, Van Dijck P, et al. Single-cell force spectroscopy of the medically important Staphylococcus epidermidis–Candida albicans interaction. Nanoscale. (2013) 5:10894. doi: 10.1039/c3nr03272h
264. Krishnamoorthy AL, Lemus AA, Solomon AP, Valm AM, Neelakantan P. Interactions between Candida albicans and Enterococcus faecalis in an Organotypic Oral Epithelial Model. Microorganisms. (2020) 8:1771. doi: 10.3390/microorganisms8111771
265. Mosailova N, Truong J, Dietrich T, Ashurst J. Streptococcus gordonii : a rare cause of infective endocarditis. Case Rep Infect Dis. (2019) 2019:1–2. doi: 10.1155/2019/7127848
266. Bamford C V, Nobbs AH, Barbour ME, Lamont RJ, Jenkinson HF. Functional regions of Candida albicans hyphal cell wall protein Als3 that determine interaction with the oral bacterium Streptococcus gordonii. Microbiology. (2015) 161:18–29. doi: 10.1099/mic.0.083378-0
Keywords: bloodstream infection (BSI), Candida albicans, Staphylococcus aureus, oral, immunocompromised
Citation: Pasman R, Krom BP, Zaat SAJ and Brul S (2022) The Role of the Oral Immune System in Oropharyngeal Candidiasis-Facilitated Invasion and Dissemination of Staphylococcus aureus. Front. Oral. Health 3:851786. doi: 10.3389/froh.2022.851786
Received: 10 January 2022; Accepted: 25 February 2022;
Published: 07 April 2022.
Edited by:
Georgios N. Belibasakis, Karolinska Institutet (KI), SwedenReviewed by:
Richard Cannon, University of Otago, New ZealandCopyright © 2022 Pasman, Krom, Zaat and Brul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stanley Brul, cy5icnVsQHV2YS5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.