
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oral. Health , 03 February 2022
Sec. Oral Infections and Microbes
Volume 3 - 2022 | https://doi.org/10.3389/froh.2022.835902
This article is part of the Research Topic The Role of Epithelial Cell-Microbes Interactions in Oral Health and Disease View all 6 articles
 Maike Paulino da Silva1,2
Maike Paulino da Silva1,2 Viviam de Oliveira Silva2,3
Viviam de Oliveira Silva2,3 Silvana Pasetto2,4
Silvana Pasetto2,4 Ellen Sayuri Ando-Suguimoto1
Ellen Sayuri Ando-Suguimoto1 Dione Kawamoto1
Dione Kawamoto1 Gardênia Márcia Silva Campos Mata1,5
Gardênia Márcia Silva Campos Mata1,5 Ramiro Mendonça Murata6
Ramiro Mendonça Murata6 Marcia Pinto Alves Mayer1
Marcia Pinto Alves Mayer1 Casey Chen2*
Casey Chen2*Aggregatibacter actinomycetemcomitans (Aa) is abundant within the microbial dysbiotic community of some patients with periodontitis. Aa outer membrane protein 29 (OMP29), a member of the OMPA family, mediates the invasion of Aa to gingival epithelial cells (GECs). This study evaluated the effect of OMP29 and its paralogue OMP29par on the response of GECs to Aa. The omp29 or/and omp29par deletion mutants AaΔ29, AaΔ29P, and AaΔ29Δ29P were constructed, and recombinant Aa OMP29His was obtained. Microarray analysis and the evaluation of cxcl-8 gene expression were performed to examine the response of GECs line OBA-09 to Aa and its mutants. The expression of cxcl-8 and its product CXCL-8 was examined in LPS-stimulated OBA-09 cells with Aa OMP29His. Proteomics analysis showed that the deletion of omp29 led to overexpression of both OMP29par and another membrane protein OMP39, the expression of which was further increased in AaΔ29Δ29P. OBA-09 cells challenged with AaΔ29Δ29P exhibited a higher expression of cxcl-8 in comparison to wildtype Aa strain AaD7S or single-deletion mutants AaΔ29 or AaΔ29P. LPS-stimulated OBA-09 cells challenged with Aa OMP29His showed reduced expressions of cxcl-8 and its product CXCL-8. OBA-09 cells challenged with AaΔ29Δ29P in comparison to Aa strain AaD7S resulted in higher expressions of genes involved in apoptosis and inflammatory response such as bcl2, birc3, casp3, c3, ep300, fas, fosb, grb2, il-1α, il-1β, il-6, cxcl-8, nr3c1, prkcq, socs3, and tnfrsf1β and reduced expressions of cd74, crp, faslg, tlr1, and vcam1. The results suggested a novel strategy of Aa, mediated by OMP29 and OMP29par, to evade host immune response by inhibiting CXCL-8 expression and modulating the genes involved in apoptosis and inflammatory response in GECs. Pending further confirmation, the strategy might interfere with the recruitment of neutrophils and dampen the host inflammatory response, leading to a more permissive subgingival niche for bacterial growth.
Aggregatibacter actinomycetemcomitans (Aa) is associated with a rapidly progressing form of periodontitis in young subjects, previously known as localized aggressive periodontitis [1–7]. Recent microbiome data confirmed this association and indicated that Aa is 50 times more abundant in periodontal pockets of Grade C/molar–incisor pattern periodontitis (GC/MIP) patients than in subgingival sites of healthy subjects [8]. Aa can invade non-phagocytic cells, such as gingival epithelial cells, escape from the vacuole, multiply, exit, and spread to adjacent cells [9]. Aa interacts with epithelial cells through adhesins such as OMP100 [10]. In contrast, invasion is mediated by the outer membrane protein 29 (OMP29) via the focal adhesion kinase (FAK) pathway [11]. Furthermore, OMP29 causes apoptosis of gingival epithelial cells (GECs) [12]. The OMP29 and its paralogue can inhibit the classical and mannose-binding lectin complement activation by binding to C4-binding protein [13].
The OMP29 belongs to the OMPA-like family and it is one of the six major OMPs recognized by antibodies in the serum of patients with periodontitis [14]. Furthermore, immunoglobulin G (IgG) titers against OMP29 and Aa serotypes b and c are reduced after successful periodontal treatment in aggressive periodontitis patients [15].
The GECs are the first barrier faced by the subgingival microbial community and play a key role in defense by detecting pathogen-associated molecular patterns (PAMPs) and triggering the immune response to eliminate the infecting pathogen [16]. However, pathogenic bacteria manipulate the host response to promote survival by activating or inhibiting different signaling pathways. Several members of the OMPA-like family were shown to contribute to immune evasion of virulent bacteria through the down-regulation of expression of inflammatory mediators such as CXCL-8 [17, 18]. Other proteins of the OMPA family, such as OMPA from Acinetobacter spp. stimulates the cxcl-8 expression [19] and Aa OMP29 seems to increase CXCL-8 production by human GECs [20]. However, little is known about the role of Aa OMP29 in host response.
Therefore, we aimed to evaluate the role of OMP29 on the immune response of GECs after interaction with Aa. The OMP29 deletion mutants and recombinant OMP29 interacted with GECs and the response was evaluated at gene and protein differential expression levels.
The bacteria strains, plasmids, and primers used in this study are described in Table 1. The Aa D7S-1 wildtype strain [21, 22], designated AaD7S, was used to construct the mutants in omp29 and omp29 paralogue (omp29par) genes. The Aa HK1651 wildtype strain (Genbank Access AY 262734) was used to obtain the OMP29 recombinant protein. Aa strains were grown in a humidified 5% CO2 atmosphere at 37°C (Shel Lab, OR, United States), in modified Trypticase Soy Broth (mTSB) containing 3% trypticase soy broth and 0.6% yeast extract, or on mTSB agar [mTSB with 1.5% agar (Oxoid Ltd, Basingstoke, Hampshire, England)].
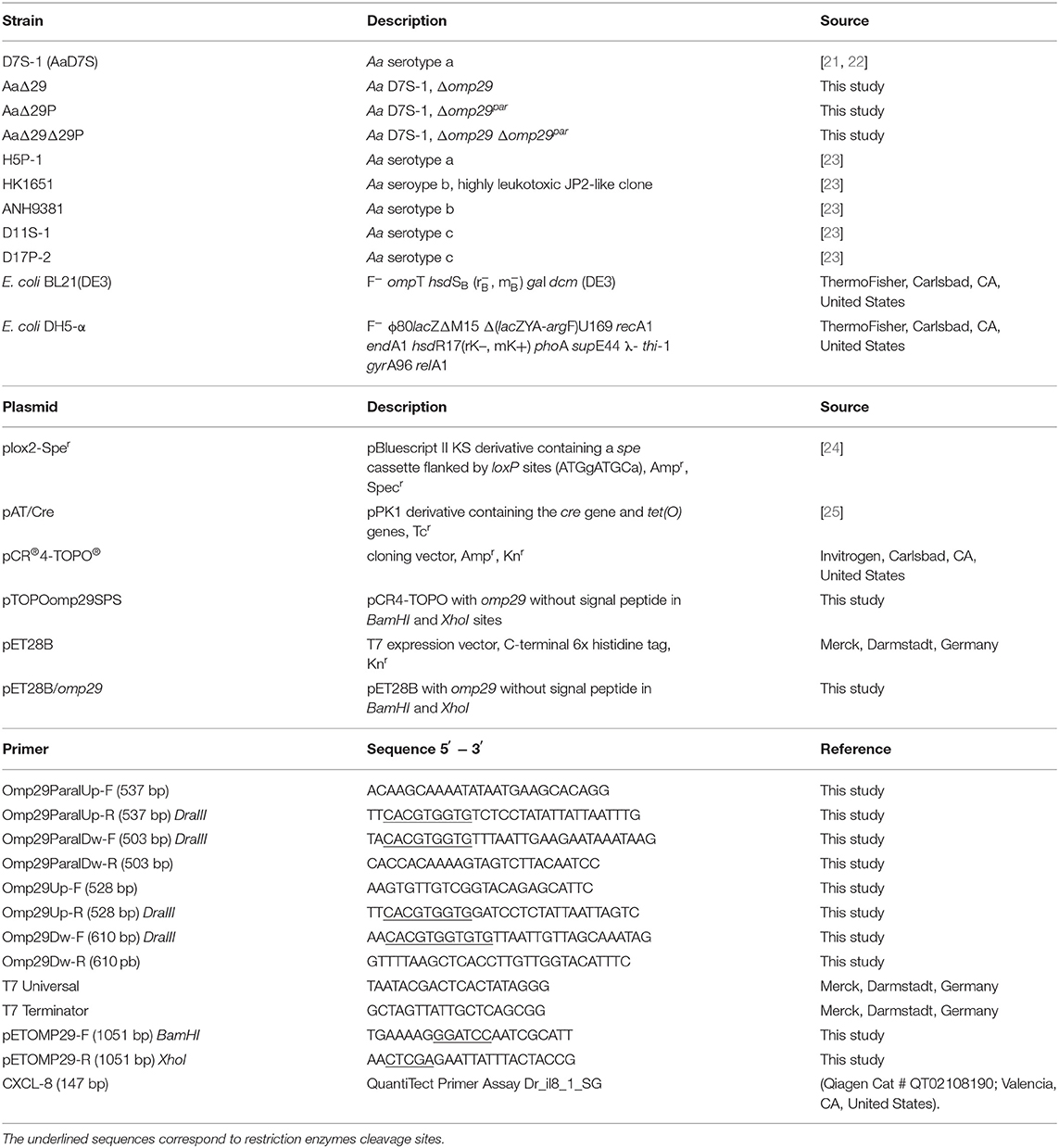
Table 1. Strains, plasmids, and primers used in this study.
The plasmids used in this study were replicated in Escherichia coli strains DH5-α and E. coli BL21(DE3) (Invitrogen, Carlsbad, CA, United States) were cultured by standard methods [26].
Spectinomycin (50 μg/ml, Sigma–Aldrich, St. Louis, MO, United States), tetracycline (4 μg/ml, Sigma–Aldrich), ampicillin (100 μg/ml, Sigma-Aldrich), kanamycin, (50/100 μg/ml, Sigma–Aldrich), and chloramphenicol (50 μg/ml, Sigma–Aldrich) were added to the media when selection of mutants or transformants were needed.
The growth curve was determined in mTSB with the addition of mineral oil to create a microaerophilic condition in 100-well HoneyComb plates (Bioscreen Automation for Microbiology, NJ, United States). The optical density (OD) at 420-580 nm was monitored in a spectrophotometer (Growth Reader Bioscreen C, Oy Growth CurvesAb Ltd., Bioscreen C Type FP-1100-C, FIN-21280 Raisio, Finland) in 1 h intervals for 16 h at 37°C, under agitation. The growth rate was calculated according to the formula: , where N and N0, respectively, correspond to the initial and final OD420−580 at the exponential growth phase and t is the time-course of the growth curve.
Genomic DNA of Aa D7S-1 and Aa HK1651 was obtained by using QIAamp DNA mini kit (Qiagen, Valencia, CA, United States). Plasmids DNA was obtained by using QIAprep Spin Miniprep kit (Qiagen). E. coli DH5-α and BL21(DE3) strains (Invitrogen, Carlsbad, CA, United States) were transformed with pCR 4-TOPO (Invitrogen) and vector pET28B vectors (pET Expression System 28, Novagen), respectively, by electroporation (MicroPulser, BioRad, Hercules, CA, United States). Restriction enzymes, T4 ligase, and Taq DNA polymerase were purchased from New England BioLabs Inc. (Ipswich, MA, United States). PCR was performed with Taq high-fidelity polymerase (Invitrogen), and its products were purified using QIAquick PCR purification and QIAquick gel extraction kits (Qiagen).
Deletion mutants in omp29, omp29par, and both genes were obtained by using the loxP/Cre system [21, 26]. Briefly, the spectinomycin cassette loxP-Specr-loxP was obtained by restriction digestion of plox2/Specr vector [24] with DraIII. The omp29 or omp29par flanking regions were obtained by amplification with primer Omp29Up-F, Omp29Up-R, Omp29Dw-F, Omp29Dw-R, and Omp29ParalUp-F, Omp29ParalUp-R, Omp29ParalDw-F, and Omp29ParalDw-R, respectively, and digested with DraIII. This product was then ligated to the loxP-Specr-loxP fragment and transformed into the competent AaD7S strain.
Transformants were selected in TSYE agar with spectinomycin. The allelic replacement of the target gene was confirmed by PCR and sequencing. The removal of loxP-Specr-loxP of strains was done by transformation with pAT/Cre vector [25], which encodes Cre recombinase.
The omp29 and omp29par deficient mutants of AaD7S were named AaΔ29 and AaΔ29P. The double deletion mutant (referred to as AaΔ29Δ29P) was constructed by the deletion of omp29par from the AaΔ29 strain following the procedure described above, with the exception that the spectinomycin resistance cassette in omp29par was not removed.
The success of the mutation strategy was confirmed by PCR and sequencing (data not shown) using the following primers: Omp29Up-R, Omp29Dw-F, Omp29ParalUp-R, and Omp29ParalDw-F, which annealed downstream and upstream of omp29 and omp29par, respectively.
The Signal 3.0 Server program was used to predict the sequence that encodes the signal peptide of omp29. Based on this prediction, the pET OMP29 F and R primers with BamHI and XhoI restriction sites were used to amplify omp29 from Aa HK1651 (Genbank accession number AY 262734), constructed previously in pCR4-TOPO vector (designated as pTOPOomp29SPS). After digestion, the omp29 amplicon (without the signal peptide) was ligated to pET28B vector (pET Expression System 28, Novagen) and the resulting pET28B/omp29 vector was transformed into E. coli BL21 (DE3). The recombinant plasmids were confirmed by T7 Universal and Terminator primer sets for sequencing.
The OMP29 was expressed by pET28B/omp29 E. coli BL21 (DE3) following the manufacturer's instructions (New England Biolabs, Ipswich, MA, United States). A single colony of pET28B/omp29 E. coli BL21 (DE3) was inoculated into LB broth with kanamycin (50 μg/ml) (Sigma–Aldrich, St. Louis, MO, United States) and incubated under agitation at 120 rpm for 16 h at 37°C. Cells were transferred to fresh LB broths, and incubated under agitation at 250 rpm at 37°C until O.D. 600 nm ≈ 1 was reached. Expression of Aa OMP29His was induced by adding 1 mM of isopropyl β-D-thiogalactoside (IPTG, Sigma–Aldrich, St. Louis, MO, United States) at 37°C and the recombinant protein was observed after 2 h in culture precipitate. Cells were then lysed [27] and Aa OMP29His in cells lysate supernatant was purified by affinity chromatography using nickel chelating resin (Ni-NDA, Invitrogen).
Balb/c mice were immunized subcutaneously with 50 μg of purified Aa OMP29His with adjuvant aluminum hydroxide (1:1) in a volume of 200 μl. Booster injections were performed after 7 days, 3, 4, 6, and 7 weeks of the initial challenge. The titers of anti-Aa OMP29His antibodies were determined by immunoenzymatic assay (ELISA). Statistical analysis was performed in GraphPad Prism version 6.0 (GraphPad Software). This assay was approved by the Ethics and Research Committee on Animals of the Institute of Biomedical Sciences of the University of São Paulo under the protocol number 007/11.
The bacterial outer membrane extracts (OMEs) of AaD7S and its mutants AaΔ29, AaΔ29P, and AaΔ29Δ29P were obtained as previously described [28], and outer membrane proteins (OMPs) were resolved by standard SDS-PAGE (10% acrylamide) [29]. Purified Aa OMP29His was also detected after SDS-PAGE using the Bolt Bis-Tris Plus gel (Life Technologies, Carlsbad, CA, United States) and the Bolt Mini Gel Tank (Life Technologies). The gels were stained with Colloidal blue (Life Technologies). OMP29 protein has the approximate size of 29 kDa, however, appears as a 34 kDa band when denatured [14].
After SDS-PAGE, the OMPs corresponding bands of AaD7S and its mutants AaΔ29, AaΔ29P, and AaΔ29Δ29P were excised, eluted with 1% acetic acid/H2O, and analyzed in the mass spectrometer (LTQ Orbitrap Hybrid Mass Spectrometers), and raw data were processed using Proteome Discoverer software (Thermo Scientific). Twelve samples were analyzed. Individual proteins were identified and ranked by protein scores (calculated using SEQUEST algorithm on Proteome Discoverer), which is the sum of all peptide Xcorr values above the specified score threshold. The score threshold is calculated as follows: 0.8 + peptide charge × peptide relevance factor where peptide relevance factor is a parameter with a default value of 0.4. The peptide spectrum matches were used as a surrogate for the relative amounts of individual proteins in each sample.
Following electrophoresis, Aa OMP29His was transferred to a nitrocellulose membrane using an iBlot 2 Dry Blotting System kit (Life Technologies). The membrane was blocked with 5% skimmed milk in TBS-T (20 mM Tris-HCl pH 7.6, 0.8% NaCl and 0.1% Tween 20). Anti-OMP29His polyclonal serum obtained in Balb/c mice and serum of aggressive periodontitis patients colonized by Aa [15] were used as primary antibodies at 1:2,000 dilution in separate membranes. Secondary antibodies consisted of goat anti-mice IgG peroxidase conjugated (Amershan Bioscience, Amershan, Little Chalfont, United Kingdom) or goat anti-human IgG peroxidase conjugated (ThermoFisher, Carlsbad, CA, United States) diluted to 1:10,000. Aa OMP29His bands were detected by using the SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher, Carlsbad, CA, United States).
OBA-09 cells [30], immortalized GEC lineage, were grown at 37°C in humidified 5% CO2 atmosphere in collagen type I pre-coated bottles (BD Bioscience, San Jose, CA, United States) containing complete Keratinocyte Serum-Free Medium (KSFM) (ThermoFisher, Carlsbad, CA, United States) supplemented with insulin, epidermal growth factor, and fibroblast growth factor (complete KSFM) according to the manufacturer's recommendation (ThermoFisher, Carlsbad, CA, United States). After confluent growth, OBA-09 cells were scraped, centrifuged at 593 × g for 10 min at 4°C, resuspended in complete KSFM, added to wells of plates at 1 × 105 cell/ml, and incubated for 16 h.
The interaction assay was performed as previously described [11]. Log phase cultures of AaD7S and its mutants AaΔ29, AaΔ29P, and AaΔ29Δ29P were adjusted to 1 × 108 cell/ml in a complete KSFM and added to OBA-09 cells at a multiplicity of infection of 1:1,000 (OBA-09:bacteria).
After 4 h of interaction, total RNA was isolated using the RNeasy Mini Kit (Qiagen; Valencia, CA, United States) and converted into cDNA using QuantiTect Reverse Transcription Kit (Qiagen).
Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction (qRT-PCR) analysis was carried out to evaluate cxcl-8 relative expression. The reaction was performed using the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Rockford, United States) using the QuantiTect SYBR Green PCR and QuantiTect Primer Assay Cat # QT02108190 (Qiagen; Valencia, CA, United States).
The gene expression of inflammatory response was analyzed by Microarray using the Prime PCR Pathway Plate/Acute Inflammation Response H96 system (Bio-Rad; Hercules, CA, United States), according to the manufacturer's instructions and performed in CFX96 Touch Real-Time PCR Detection Systems (Bio-Rad).
Both Microarray and qRT-PCR data resulted in CT values for each sample. Gene dosages were obtained using the 2−ΔΔCT Livak method [31, 32], considering glyceraldehyde-3-phosphate dehydrogenase (gapdh) as a reference gene and co-culture OBA-09 cell with AaD7S strain as the control. Statistical analysis was performed in GraphPad Prism version 6.0 (GraphPad Software).
The confluent monolayers of OBA-09 cell, as previously described, were added to 24 well/plates (ThermoFisher, Carlsbad, CA, United States) and challenged with E. coli serotype O111:B4 LPS (500 ng/ml; Sigma–Aldrich). Then, aliquots of 1 μg/ml and 10 μg/ml of Aa OMP29His were added to each well. After 4 h of interaction, levels of CXCL-8 in free cell supernatant were established by Bio-Plex Pro Human Chemokine assay kit (Bio-Rad, United States), following the manufacturer's instructions. Those levels were determined by comparison to the standard curve (mean of fluorescence intensity vs. pg/ml), using Bio-Plex software manager 4.0. The results were shown in percentage with CXCL-8 concentration (pg/ml) of the 4 h cultured with OBA-09 being 100% and statistical analysis was performed in GraphPad Prism version 6.0 (GraphPad Software).
Multiple sequence alignment was performed with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) using the default setting. The amino acid sequences of Aa OMP29 and OMP29par of Aa strains HK1651, ANH9381, D7S-1, H5P-1, D11S-1, and D17P-2 (Table 1) were identified and downloaded from our annotated database at http://expression.washington.edu/genetable/script/gene_table_viewer [23].
The sequence alignments of OMP29 and OMP29par of representative strains of serotype a, b, and c are provided in Supplementary Figure 1. The OMP29 is 346 aa long, and the OMP29par is 356 aa long. Each orthologous protein demonstrates 99% amino acid sequence identity among strains. The OMP29 and OMP29par also share approximately 75% amino acid sequence identify.
The recombinant protein Aa OMP29His was obtained, purified, and confirmed by its reaction with mice anti-Aa Omp29His IgG polyclonal and by human aggressive periodontitis sera in a Western-blot assay (Supplementary Figure 2).
Aa mutants in omp29, omp29par and in both genes were constructed and characterized. All strains presented similar growth rates in mTSB medium (Supplementary Figure 3). Analysis of OME revealed differences in the protein profile between the wildtype and the omp29 and omp29par deletion mutants (Figure 1). To better assess the profile, the distinct bands were excised from the gel and evaluated by mass spectrophotometry. The identity and the relative amounts of individual proteins differentially expressed were determined (Supplementary Table 1).
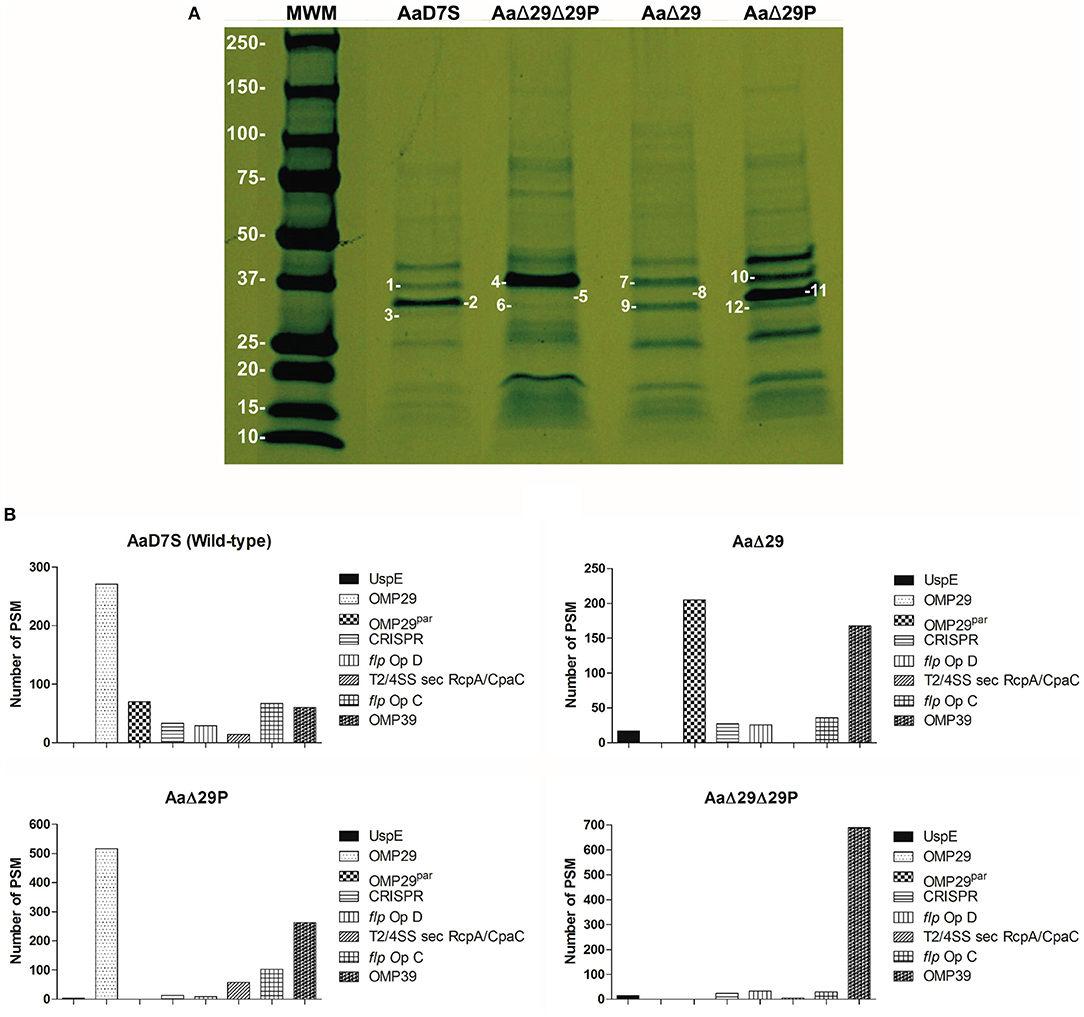
Figure 1. Outer membrane extracts profile of Aa D7S-1 strains. (A) Outer membrane protein profiles of Aa grown until mid-log phase were determined by SDS-PAGE analysis: MWM: Molecular Weight Marker—Precision Plus Protein Standard, Bio-Rad, in kDa. Gel slices numbered 1-12 showed the following OMPs: AaD7S, 1-OMP39, 2-OMP29; 3-OMP29par; AaΔ29Δ29P, 4-OMP39, 5-OMP29 absent; 6-OMP29par absent; AaΔ29, 7-OMP39, 8-OMP29 absent; 9-OMP29par; AaΔ29P, 10-OMP39, 11-OMP29; 12-OMP29par absent). Photograph of a representative gel of at least two independent assays. (B) Proteomic analysis of detected peptides or peptide spectrum matches (PSM) for the 8 major proteins: (Universal stress protein UspE, OMP29, OMP29par, CRISP-associated protein (CRISP), flp operon protein D (flp Op D), type II/IV secretion system secretin RcpA/CpaC (T2/4SS sec RcpA/CpaC), and flp operon protein (flp Op C) and OMP39) after combining gel #1-3, gel #4-6, gel #7-9, and gel #10-12, to determine whether some proteins were differentially expressed in AaD7S and its mutants AaΔ29Δ29P, AaΔ29, and AaΔ29P, respectively. AaD7S, Aa D7S-1 wildtype strain; AaΔ29, Aa D7S-1 omp29 mutant strain; AaΔ29P, Aa D7S-1 omp29par mutant strain; AaΔ29Δ29P, Aa D7S-1 omp29 and omp29par mutant strain.
Proteomic analysis indicated that OMP29 and OMP29par were simultaneously expressed by the wildtype strain AaD7S, whereas the target proteins were absent in AaΔ29, AaΔ29P, and AaΔ29Δ29P. The deletion of omp29 led to overexpression of at least 3 times of both OMP29par and OMP39. The deletion of OMP29par resulted in a 2 times increase in OMP29 levels and 4 times increase in OMP39 levels (Figure 1). Furthermore, the double deletion in strain AaΔ29Δ29P resulted in the overexpression of OMP39 (11 times increase compared to the AaD7S). The relative expression levels of other proteins also changed in the mutants, although not as pronounced as seen for OMP39. The physiological significance of these changes is unknown but may represent a compensatory mechanism.
CXCL-8 plays a significant role in the pathogenesis of periodontitis. In this study, we aimed to confirm the effect of OMP29 and OMP29par on the inhibition of cxcl-8 transcription. The effect of the 4 h interaction of Aa wildtype and its mutants with GECs on cxcl-8 transcription was evaluated by RT-qPCR. The AaΔ29Δ29P mutant promoted a significant increase in cxcl-8 transcripts levels in GECs compared to the wildtype. However, the deletion of omp29 or omp29par did not result in cxcl-8 regulation, suggesting functional redundancy of OMP29 and OMP29par in the regulation of cxcl-8 expression (Figure 2A).
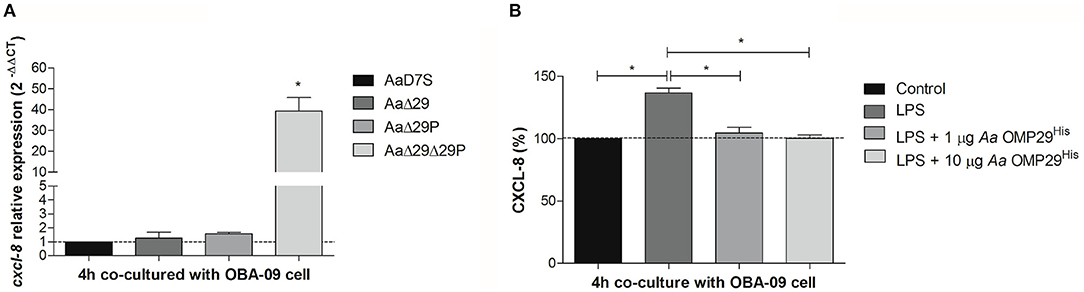
Figure 2. Chemokine CXCL-8 expression. (A) cxcl-8 relative transcription. Aa wildtype strain (AaD7S) and its mutant strains (AaΔ29, Aa D7S-1 omp29 mutant strain; AaΔ29P, Aa D7S-1 omp29par mutant strain; AaΔ29Δ29P, Aa D7S-1 omp29 and omp29par mutant strain) were grown in mTSB until mid-exponential phase and then co-cultured with confluent monolayers of OBA-09 cells for 4 h in complete KSF medium. After the RNA extraction and cDNA synthesis, qRT-PCR was performed and relative mRNA levels of cxcl-8 were evaluated using gapdh as reference. OBA-09 cell was co-cultured with AaD7S as a control (dotted line). *One-way ANOVA followed by Dunnett's post-test (significant, p < 0.05). (B) CXCL-8 production. OBA-09 cells were grown in KSF medium and challenged with E. coli serotype O111:B4 LPS followed by treatment with 1 μg/ml and 10 μg/ml Aa OMP29His for 4 h. CXCL-8 levels in cell supernatants were evaluated by ELISA, and data were reported as the percentages of CXCL levels in relation to non-inoculated cells (control) (dotted line). LPS, lipopolysaccharide; Aa OMP29His, OMP29 recombinant protein of Aa HK1651. *One-way ANOVA followed by Tukey's post-test (significant, p < 0.05).
To determine whether the effect on cxcl-8 expression was due to the compensatory expression of other proteins such as OMP39 in the mutants, we investigated the interaction between OBA-09 cell and Aa OMP29His (Figure 2B). E. coli LPS was used to trigger CXCL-8 expression in OBA-09 cells [33, 34], and cells were challenged with 1 or 10 μg/ml of Aa OMP29His. After 4 h of interaction, the expression of CXCL-8 was determined at the protein level. The analyses indicated that both concentrations of Aa OMP29His could inhibit the transcription of cxcl-8, resulting in reduced CXCL-8 levels in OBA-9 cells supernatants compared to control cells (Figure 2B).
After observing the effect of OMP29 on CXCL-8 expression, we explored the gene expression profile of host acute inflammatory response to AaD7S and its mutants. The results showed modulatory effects (at least two-fold) of 21 genes (Table 2; for gene function, see Supplementary Table 2). Sixteen genes had increased (bcl2, birc3, casp3, c3, ep300, fas, fosb, grb2, il-1[ENTX]X003B1[/ENTX], il-1β, il-6, cxcl-8, nr3c1, prkcq, socs3, and tnfrsf1β) and five had genes decreased (cd74, crp, faslg, tlr1, and vcam1) expression in OBA-09 cells co-cultured with AaΔ29Δ29P strain compared to AaD7S. The data suggested a potential role of OMP29 and its paralogue in the host response of GECs. Nevertheless, these findings suggested a possible influence of OMP29 and its paralogue in anti-apoptotic (e.g., bcl2, birc3, grb2, prkcq) and pro-apoptotic (e.g., casp3, c3, ep300, fosb, nr3c1, crp, and socs3) pathways. Furthermore, the interaction with the strain AaΔ29Δ29P down-regulated transcription of cd74, faslg, tlr1, and vcam1 and up-regulated fas and tnfrsf1β. The faslg and fas also participate in pro-apoptotic pathways. Finally, interaction with the strain AaΔ29Δ29P upregulated transcription of il-1[ENTX]X003B1[/ENTX], il-1β, il-6, cxcl-8, and tnfrsf1β, encoding cytokines and chemokines involved in the inflammatory response. Notably, the effect promoted by the absence of OMP29 or OMP29par was not as intense as observed for the double mutant, possibly due to the compensation mechanism induced by their absence (Table 2). Alternatively, the effects described above may be due to yet to be characterized pleiotropic effects of the deletion of omp29 and omp29par.
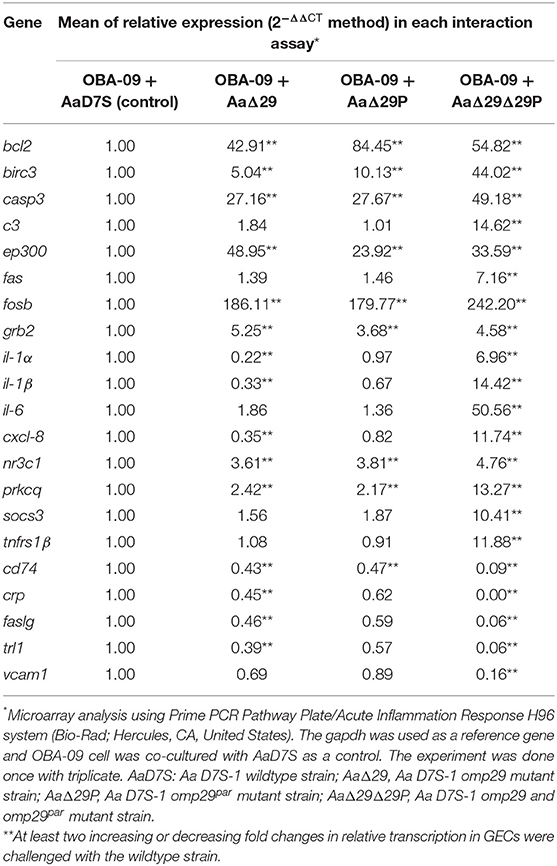
Table 2. Inflammatory response of OBA-09 cell after 4 h of interaction with Aa strains.
To improve our understanding of the mechanisms involved in periodontitis associated with Aa, we aimed to evaluate the effect of Aa OMP29 on the interaction with gingival epithelial cells. Outer membrane protein 29 and the other five outer membrane proteins of Aa (OMP100, OMP64, OMP39, OMP16, and OMP18) were recognized by antibodies in the serum of patients with periodontitis of high rate of progression in young subjects [9]. OMP29 is an OMPA-like protein and its paralogous OMP29par was identified in our study and a in recently published article [13].
We have noticed increased and compensatory expressions of other OMPs associated with the deletion of omp29 and omp29par. For example, the deletion of omp29 led to an over-expression of OMP29par, and the deletion of omp29 or omp29par led to an over-expression of OMP39. We have also noted the changes in the transcription profiles of GECs challenged with the defective mutants.
The more robust alterations in mRNA levels were observed for the double mutant than when GECs were challenged with the single mutant. This is likely due to the functional redundancy and compensatory expression of OMP29 or OMP29par in the corresponding single-deletion mutants, which may exert similar effects to GECs as the wildtype AaD7S.
The study examined the role of OMP29 in the expression of cxcl-8 in GECs. We focused on CXCL-8 since this chemokine is associated with inflammatory tissue destruction in periodontitis and periodontal tissue homeostasis. The expression of cxcl-8 was higher in OBA-09 cells challenged with AaΔ29Δ29P than with the wildtype AaD7S. To rule out the effects associated with the compensatory expressions of other proteins such as OMP39, we tested the recombinant Aa OMP29His and demonstrated its ability to suppress cxcl-8 transcription and CXCL-8 production in LPS-stimulated OBA-9 cells. The results suggested that OMP29 and OMP29par were involved in the inhibition of cxcl-8. Our results were in disagreement with another study showing the induction of CXCL-8 by OMP29 in gingival epithelial cells [20]. Previous studies extracted OMP29 directly from Aa outer membrane, which may contain contaminants that affect the outcomes of the experiments [11–14, 20, 35].
Previous data reported that Aa OMP29 promotes apoptosis in GECs via TGF-βR/smad2 pathway by binding fibronectin (Fn) to ease Fn/integrinβ1/FAK signaling-dependent TGF-β release from the extracellular matrix [12]. The primary mechanism of defense of the host cell against intracellular bacteria is the induction of apoptosis. However, obligate intracellular pathogens such as Chlamydia [36] may have anti-apoptotic mechanisms to promote their survival in the intracellular environment. This study showed that the expression of several genes involved in apoptotic pathways was altered in OBA-09 cells when co-cultured with the double mutant strain AaΔ29Δ29P, suggesting that OMP29 or OMP29par may regulate apoptosis of GECs, influencing the intracellular survival of Aa. However, both pro-apoptotic genes (casp3, c3, ep300, fosb, nr3c1, crp, and socs3) and anti-apoptotic (bcl2, birc3, grb2, and prkcq) were affected and the net effect to apoptosis is not apparent. The interaction with the AaΔ29Δ29P strain down-regulated transcription of cellular receptors and adhesion, highlighting the faslg and fas which also participate in pro-apoptotic pathways. Finally, AaΔ29Δ29P upregulated the transcription of il-1a, il-1β, il-6, cxcl-8, and tnfrsf1β, encoding cytokines and chemokines involved in the inflammatory response. The data suggested a potential role of OMP29 and its paralogue to the host response of GECs. Alternatively, the effect may be due to changes in virulence expression of Aa after the deletion of omp29 and omp29par. Further investigation is needed to understand the influence of OMP29 and OMP29par on the host response of GECs to Aa. This study did not address the net impact of OMP29 and OMP29par on GECs. We will explore the role and the mechanisms of these proteins in the apoptosis of GECs in future studies.
The study is limited by its in vitro assessment of the virulence determinants of the serotype a AaD7S. The innate immune response of epithelial cells may differ when challenged with other Aa serotypes with different virulence potentials [37–39]. In addition, we evaluated only the response of epithelial cells monolayers to the bacterial challenges and not the response of the oral mucosa with multiple immune cells. Moreover, given the plethora of microorganisms at the oral epithelium, the overall responses are triggered by the whole community and not by a single species [40].
Aa is known to exhibit strain-to-strain variation in virulence. The results of this study did not exclude the possibility that different Aa strains may exhibit distinct effects on GECs. However, the effects of OMP29 and OMP29par in different Aa strains on GECs are likely similar. First, OMP29 and OMP29par are highly conserved among genetically distanced Aa strains (Supplementary Figure 1). Second, the recombinant OMP29 in our study was derived from a serotype b strain HK1651, and exhibited similar inhibitory properties demonstrated in serotype a strain AaD7S that expressed this protein. Finally, a preliminary analysis of the upstream regions of omp29 and omp29par did not reveal any apparent structural variations that may lead to differential gene expression (data not shown). Additional studies are needed to resolve this question.
It is worth noting the clinical implication of the inhibition of CXCL-8 by OMP29 of Aa. CXCL-8 is a potent chemoattractant for polymorphonuclear cells into the infected tissue [41]. A balanced proportion of neutrophils in the periodontal tissues is essential for homeostasis, and both too high or too low numbers of neutrophils contribute to periodontitis [40]. Thus, the inhibitory activity in GECs response by OMP29 may break this balance, decreasing the numbers of neutrophils at the infected sites, allowing a more permissive niche for bacteria replication. This transient inhibition could delay the recruitment of neutrophils, and therefore facilitate the initial colonization of this pathogen and other microorganisms [42]. Other pathogens such as Porphyromonas gingivalis inhibit the production of CXCL-8 in periodontal tissues by secreting a serine phosphatase (SerB) [43]. OMP29 may be an additional tool of Aa to alter the microenvironment of subgingival biofilm, allowing bacterial survival and tissue destruction. Also, OMP29 may act in synergy with Aa secretin HofQ, which uptakes CXCL-8 [44], limiting CXCL-8 availability in periodontal tissues. These data are in accordance with a study in humans that reported that Aa-negative gingival crevicular fluid (GCF) contained higher concentrations of CXCL-8 than sites harboring Aa [45].
In conclusion, our study suggested that OMP29 and its paralogue of Aa may suppress the expression of CXCL-8, modulate the host responses in epithelial cells, and permit initial colonization of Aa.
The original contributions presented in the study are included in the article/Supplementary Files, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by Ethics and Research Committee on Animals of the Institute of Biomedical Sciences of the University of São Paulo.
CC: supervision, investigation, conceptualization and design of the study, formal analysis, writing original draft, and review and editing. MM: supervision, investigation, conceptualization and design of the study, writing review, and editing. RM: design of the study and writing original draft. GM: formal/statistical analysis, writing original draft, and review and editing. DK and VS: investigation, formal analysis, and writing original draft. EA-S: investigation, formal analysis and writing original draft, and review and editing. SP: investigation and writing original draft. MS: investigation, formal analysis, design of the study, writing original draft, and review and editing. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by FAPESP (Grants 2011/18683-1, 2012/05911-9, and 2015/18273-9), CAPES PNPD (Grant 88887.466596/2019-00), and NIH (Grant R01 DE012212).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The OBA-9 cells used in this study were kindly given by Dr. Shinya Murakami (Osaka University, Japan).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/froh.2022.835902/full#supplementary-material
Supplementary Figure 1. Multiple Sequence Aligment of OMP29 and OMP29par among Aa strains of different serotypes. The multiple sequence alignments of OMP29 (A) and OMP29par (B) of representative Aa strains of serotype a, b and c (HK1651, ANH9381, D7S-1, H5P-1, D11S-1, and D17P-2) was performed with Clustal Omega using the default setting. Each protein demonstrates 99% amino acid sequence identify among strains. These two homologous proteins also share approximately 75% amino acid sequence identify.
Supplementary Figure 2. SDS-PAGE, ELISA and Immunoblot of Aa OMP29His. (A) 10% polyacrylamide gel of SDS-PAGE with aliquot of Aa OMP29His (OMP29 recombinant protein of A. actinomycetemcomitans HK1651). E. coli BL21 (DE3) transformed with pET28B/omp29 vector was cultured by standard methods for 2 h with 1 mM IPTG for the purpose of protein expression induction. MWM: Molecular Weight Marker - Precision Plus Protein Standard, Bio-Rad, in kDa. The arrow indicates a band corresponding to Aa OMP29His heated with approximately 38.44 kDa (predicted by the website www.bioinformatics.org). (B) ELISA Optical Density (O.D.) 490nm data at 63 days after primary immunization and booster injections at 7 days, 3, 4, 6 and 7 weeks, using different serum dilutions (1:500, 1:1,000, 1:2,000 and 1:5,000) from immunized mice against Aa OMP29His and non-immunized controls. *Two-way ANOVA followed by post-test of Tukey, p < 0.0001. (C) 10% polyacrylamide gel of SDS-PAGE with aliquot of Aa OMP29His. (D) Western Blot: (D1) Detection of OMP29His using mice polyclonal anti-Aa OMP29His serum. (D2) Detection of Aa OMP29His using serum from aggressive periodontitis patient.
Supplementary Figure 3. Growth curve of A. actinomycetemcomitans strains. Bacteria grown overnight were diluted in mTSB medium and grown for 16h to obtain the growth curve and the outer membrane extracts. Growth curve: cell density at O.D.420−580 were determined hourly. Each point represents the mean values of two independent cultures. AaD7S: A. actinomycetemcomitans D7S-1 wild-type strain; AaΔ29: A. actinomycetemcomitans D7S-1 omp29 mutant strain; AaΔ29P: A. actinomycetemcomitans D7S-1 omp29par mutant strain; AaΔ29Δ29P: A. actinomycetemcomitans D7S-1 omp29 and omp29par mutant strain; mTSB: media not inoculated was used as a control. The growth rates for each strain in the exponential phase were 1.2 h−1, 1.04 h−1, 1.03 h−1, 1.02 h−1 and 0.92 h−1 for AaD7S, AaΔ29, AaΔ29P and AaΔ29Δ29P, respectively.
Supplementary Table 1. Identification and the amounts of number of peptide spectrum matches (PSM) of proteins with high scoring peptides from gel slices number 1–12, according to Figure 1A.
Supplementary Table 2. Function of the statistically relevant genes analyzed by the microarray assay.
1. Slots J, Listgarten MA. Bacteroides gingivalis, Bacteroides intermedius and Actinobacillus actinomycetemcomitans in human periodontal diseases. J Clin Periodontol. (1988) 15:85–93. doi: 10.1111/j.1600-051X.1988.tb00999.x
2. Fine DH, Patil AG, Loos BG. Classification and diagnosis of aggressive periodontitis. J Clin Periodontol. (2018) 45:S95–S111. doi: 10.1111/jcpe.12942
3. Timmerman MF, Van der Weijden GA, Abbas F, Arief EM, Armand S, Winkel EG, et al. Untreated periodontal disease in Indonesian adolescents. longitudinal clinical data and prospective clinical and microbiological risk assessment. J Clin Periodontol. (2000) 27:932–42. doi: 10.1034/j.1600-051x.2000.027012932.x
4. Van der Velden U, Abbas F, Armand S, Loos BG, Timmerman MF, Van der Weijden GA, et al. Java project on periodontal diseases. the natural development of periodontitis: risk factors, risk predictors and risk determinants. J Clin Periodontol. (2006) 33:540–8. doi: 10.1111/j.1600-051X.2006.00953.x
5. Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, et al. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol. (2007) 45:3859–69. doi: 10.1128/JCM.00653-07
6. Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet. (2008) 371:237–42. doi: 10.1016/S0140-6736(08)60135-X
7. Hoglund Aberg C, Kwamin F, Claesson R, Dahlen G, Johansson A, Haubek D. Progression of attachment loss is strongly associated with presence of the JP2 genotype of Aggregatibacter actinomycetemcomitans: a prospective cohort study of a young adolescent population. J Clin Periodontol. (2014) 41:232–41. doi: 10.1111/jcpe.12209
8. Amado P, Kawamoto D, Albuquerque-Souza E, Franco DC, Saraiva L, Casarin R., et al. Oral and fecal microbiome in molar-incisor pattern periodontitis. Front Cell Infect Microbiol. (2020) 10:583761. doi: 10.3389/fcimb.2020.583761
9. Meyer DH, Mintz KP, Fives-Taylor PM. Models of invasion of enteric and periodontal pathogens into epithelial cells: a comparative analysis. Crit Ver Oral Biol Med. (1997) 8:389–409. doi: 10.1177/10454411970080040301
10. Asakawa R, Komatsuzawa H, Kawai T, Yamada S, Goncalves RB, Izumi S, et al. Outer membrane protein 100, a versatile virulence factor of Actinobacillus actinomycetemcomitans. Mol Microbiol. (2003) 50:1125–39. doi: 10.1046/j.1365-2958.2003.03748.x
11. Kajiya M, Komatsuzawa H, Papantonakis A, Seki M, Makihira S, Ouhara K, et al. Aggregatibacter actinomycetemcomitans Omp29 is associated with bacterial entry to gingival epithelial cells by F-actin rearrangement. PloS ONE. (2011) 6:e18287. doi: 10.1371/journal.pone.0018287
12. Yoshimoto T, Fujita T, Kajiya M, Ouhara K, Matsuda S, Komatsuzawa H, et al. Aggregatibacter actinomycetemcomitans outer membrane protein 29 (Omp29) induces TGF-β-regulated apoptosis signal in human gingival epithelial cells via fibronectin/integrinβ1/FAK cascade. Cell. Microbiol. (2016) 18:1723–38. doi: 10.1111/cmi.12607
13. Lindholm M, Min Aung K, Nyunt Wai S, Oscarsson J. Role of OmpA1 and OmpA2 in Aggregatibacter actinomycetemcomitans and Aggregatibacter aphrophilus serum resistance. J Oral Microbiol. (2018) 11:1536192. doi: 10.1080/20002297.2018.1536192
14. Komatsuzawa H, Asakawa R, Kawai T, Ochiai K, Fujiwara T, Taubman MA, et al. Identification of six major outer membrane proteins from Actinobacillus actinomycetemcomitans. Gene. (2002) 288:195–201. doi: 10.1016/S0378-1119(02)00500-0
15. Rebeis ES, Albuquerque-Souza E, Paulino da Silva M, Giudicissi M, Mayer M, Saraiva L. Effect of periodontal treatment on Aggregatibacter actinomycetemcomitans colonization and serum IgG levels against A. actinomycetemcomitans serotypes and Omp29 of aggressive periodontitis patients. Oral Dis. (2019) 25:569–79. doi: 10.1111/odi.13010
16. Groeger S, Meyle J. Oral mucosal epithelial cells. Front Immunol. (2019) 10:208. doi: 10.3389/fimmu.2019.00208
17. March C, Moranta D, Regueiro V, Llobet E, Tomás A, Garmendia J, et al. Klebsiella pneumoniae outer membrane protein A is required to prevent the activation of airway epithelial cells. J Biol Chem. (2011) 286:9956–9967. doi: 10.1074/jbc.M110.181008
18. Selvaraj SK, Prasadarao NV. Escherichia coli K1 inhibits proinflammatory cytokine induction in monocytes by preventing NF-kappaB activation. J Leukoc Biol. (2005) 78:544–54. doi: 10.1189/jlb.0904516
19. Ofori-Darko E, Zavros Y, Rieder G, Tarlé SA, Van Antwerp M, Merchant JL. An OmpA-like protein from Acinetobacter spp. stimulates gastrin and interleukin-8 promoters. Infect Immun. (2000) 68:3657–66. doi: 10.1128/IAI.68.6.3657-3666.2000
20. Kishimoto A, Fujita T, Shiba H, Komatsuzawa H, Takeda K, Kajiya M, et al. Irsogladine maleate abolishes the increase in interleukin-8 levels caused by outer membrane protein 29 from Aggregatibacter (Actinobacillus) actinomycetemcomitans through the ERK pathway in human gingival epithelial cells. J Periodontal Res. (2008) 43:508–13. doi: 10.1111/j.1600-0765.2007.01059.x
21. Wang Y, Goodman SD, Redfield RJ, Chen C. Natural transformation and DNA uptake signal sequences in Actinobacillus actinomycetemcomitans. J Bacteriol. (2002) 184:3442–9. doi: 10.1128/JB.184.13.3442-3449.2002
22. Chen C, Kittichotirat W, Chen W, Downey JS, Si Y, Bumgarner R. Genome sequence of naturally competent Aggregatibacter actinomycetemcomitans serotype a strain D7S-1. J Bacteriol. (2010) 192:2643–4. doi: 10.1128/JB.00157-10
23. Kittichotirat W, Bumgarner RE, Asikainen S, Chen C. Identification of the pangenome and its components in 14 distinct aggregatibacter actinomycetemcomitans strains by comparative genomic analysis. PloS ONE. (2011) 6:e22420. doi: 10.1371/journal.pone.0022420
24. Cheng YA, Jee J, Hsu G, Huang Y, Chen C, Lin CP. A markerless protocol for genetic analysis of Aggregatibacter actinomycetemcomitans. J Formos Med Assoc. (2014) 113:114–23. doi: 10.1016/j.jfma.2012.05.005
25. Fujise O, Wang Y, Chen W, Chen C. Adherence of Aggregatibacter actinomycetemcomitans via serotype-specific polysaccharide antigens in lipopolysaccharides. Oral Microbiol Immunol. (2008) 23:226–33. doi: 10.1111/j.1399-302X.2007.00416.x
26. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. ed. New York, NY: Cold Spring Harbor Laboratory Press (1989).
27. Ando ES, De-Gennaro LA, Faveri M, Feres M, DiRienzo JM, Mayer MP. Immune response to cytolethal distending toxin of Aggregatibacter actinomycetemcomitans in periodontitis patients. J Periodontal Res. (2010) 45:471–80. doi: 10.1111/j.1600-0765.2009.01260.x
28. Mintz KP. Identification of an extracellular matrix protein adhesin, EmaA, which mediates the adhesion of Actinobacillus actinomycetemcomitans to collagen. Microbiology (Reading). (2004) 150:2677–88. doi: 10.1099/mic.0.27110-0
29. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. (1970) 227:680–5. doi: 10.1038/227680a0
30. Kusumoto Y, Hirano H, Saitoh K, Yamada S, Takedachi M, Nozaki T, et al. Human gingival epithelial cells produce chemotactic factors interleukin-8 and monocyte chemoattractant protein-1 after stimulation with Porphyromonas gingivalis via toll-like receptor 2. J Periodontol. (2004) 75:370–9. doi: 10.1902/jop.2004.75.3.370
31. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta C(T)) Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
32. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. (2008) 3:1101–8. doi: 10.1038/nprot.2008.73
33. Takahashi N, Kobayashi M, Takaki T, Takano K, Miyata M, Okamatsu Y., et al. Actinobacillus actinomycetemcomitans lipopolysaccharide stimulates collagen phagocytosis by human gingival fibroblasts. Oral Microbiol Immunol. (2008) 23:259–64. doi: 10.1111/j.1399-302X.2007.00414.x
34. Wei C, Tan CK, Xiaoping H, Junqiang J. Acanthoic acid inhibits LPS-induced inflammatory response in human gingival fibroblasts. Inflammation. (2015) 38:896–901. doi: 10.1007/s10753-014-0051-7
35. Wilson ME. IgG antibody response of localized juvenile periodontitis patients to the 29 kilodalton outer membrane protein of Actinobacillus actinomycetemcomitans. J Periodontol. (1991) 62:211–8. doi: 10.1902/jop.1991.62.3.211
36. Messinger JE, Nelton E, Feeney C, Gondek DC. Chlamydia infection across host species boundaries promotes distinct sets of transcribed anti-apoptotic factors. Front Cell Infect Microbiol. (2015) 5:96. doi: 10.3389/fcimb.2015.00096
37. Asikainen S, Lai CH, Alaluusua S, Slots J. Distribution of Actinobacillus actinomycetemcomitans serotypes in periodontal health and disease. Oral Microbiol Immunol. (1991) 6:115–118. doi: 10.1111/j.1399-302X.1991.tb00462.x
38. Kawamoto D, Ando ES, Longo PL, Nunes AC, Wikström M, Mayer MP. Genetic diversity and toxic activity of Aggregatibacter actinomycetemcomitans isolates. Oral Microbiol Immunol. (2009) 24:493–501. doi: 10.1111/j.1399-302X.2009.00547.x
39. Dogan B, Chen J, Çiftlikli SY, Huang J, Kadir T, Alniak AK, Chen C. Occurrence and serotype distribution of Aggregatibacter actinomycetemcomitans in subjects without periodontitis in Turkey. Arch Oral Biol. (2016) 61:125–9. doi: 10.1016/j.archoralbio.2015.10.023
40. Moutsopoulos NM, Konkel JE. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol. (2018) 39:276–87. doi: 10.1016/j.it.2017.08.005
41. Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. (2002) 72:847–55. doi: 10.1189/jlb.72.5.847
42. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. (2011) 10:497–506. doi: 10.1016/j.chom.2011.10.006
43. Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Ver Microbiol. (2010) 8:481–90. doi: 10.1038/nrmicro2337
44. Ahlstrand T, Torittu A, Elovaara H, Välimaa H, Pöllänen MT, Kasvandik S, et al. Interactions between the Aggregatibacter actinomycetemcomitans secretin HofQ and host cytokines indicate a link between natural competence and interleukin-8 uptake. Virulence. (2018) 9:1205–23. doi: 10.1080/21505594.2018.1499378
45. Gonçalves PF, Klepac-Ceraj V, Huang H, Paster BJ, Aukhil I, Wallet SM, et al. Correlation of Aggregatibacter actinomycetemcomitans detection with clinical/immunoinflammatory profile of localized aggressive periodontitis using a 16S rRNA microarray method: a cross-sectional study. PloS ONE. (2013) 8:e85066. doi: 10.1371/journal.pone.0085066
Keywords: Aggregatibacter actinomycetemcomitans, outer membrane protein 29, outer membrane protein 29 paralogue, aggressive periodontitis, gingival epithelial cells, inflammatory response, virulence factors
Citation: Silva MP, Silva VO, Pasetto S, Ando-Suguimoto ES, Kawamoto D, Mata GMSC, Murata RM, Mayer MPA and Chen C (2022) Aggregatibacter actinomycetemcomitans Outer Membrane Proteins 29 and 29 Paralogue Induce Evasion of Immune Response. Front. Oral. Health 3:835902. doi: 10.3389/froh.2022.835902
Received: 15 December 2021; Accepted: 03 January 2022;
Published: 03 February 2022.
Edited by:
Georgios N. Belibasakis, Karolinska Institutet (KI), SwedenReviewed by:
Anders Johansson, Umeå University, SwedenCopyright © 2022 Silva, Silva, Pasetto, Ando-Suguimoto, Kawamoto, Mata, Murata, Mayer and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Casey Chen, Y2NjaGVuQHVzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.