 Amrita Chaudhary1*
Amrita Chaudhary1* Swarnendu Bag1
Swarnendu Bag1 Neeraj Arora2
Neeraj Arora2 Vivek S. Radhakrishnan3Deepak Mishra2
Vivek S. Radhakrishnan3Deepak Mishra2 Geetashree Mukherjee1*
Geetashree Mukherjee1*- 1Department of Histopathology, Tata Medical Center, Kolkata, India
- 2Department of Laboratory Hematology and Molecular Genetics, Tata Medical Center, Kolkata, India
- 3Department of Clinical Hematology, Tata Medical Centre, Kolkata, India
Oral squamous cell carcinoma (OSCC) includes tumors of the lips, tongue, gingivobuccal complex, and floor of the mouth. Prognosis for OSCC is highly heterogeneous, with overall 5-year survival of ~50%, but median survival of just 8–10 months for patients with locoregional recurrence or metastatic disease. A key feature of OSCC is microenvironmental oxygen depletion due to rapid growth of constituent tumor cells, which triggers hypoxia-associated signaling events and metabolic adaptations that influence subsequent tumor progression. Better understanding of leukocyte responses to tissue hypoxia and onco-metabolite expression under low-oxygen conditions will therefore be essential to develop more effective methods of diagnosing and treating patients with OSCC. This review assesses recent literature on metabolic reprogramming, redox homeostasis, and associated signaling pathways that mediate crosstalk of OSCC with immune cells in the hypoxic tumor microenvironment. The likely functional consequences of this metabolic interface between oxygen-starved OSCC and infiltrating leukocytes are also discussed. The hypoxic microenvironment of OSCC modifies redox signaling and alters the metabolic profile of tumor-infiltrating immune cells. Improved understanding of heterotypic interactions between host leukocytes, tumor cells, and hypoxia-induced onco-metabolites will inform the development of novel theranostic strategies for OSCC.
Introduction
Squamous cell carcinoma (SCC) accounts for more than 95% of all cancers affecting the head and neck region, with high rates of associated mortality and morbidity that represent a major public health burden worldwide [1, 2]. In oral squamous cell carcinoma (OSCC), the predominant sites involved include the tongue and the gingivobuccal complex, and mortality rates can be substantially reduced by early detection and prevention strategies [3]. However, despite the development of several high-throughput multimodal diagnostic tools, early stage detection is still problematic; hence, 5-year survival rates in recurrent and metastatic disease remain extremely poor [4]. It remains unclear to what extent radiotherapy or chemotherapy exerts stimulatory or suppressive effects on host leukocyte responses [5]. The immune cell composition, function, and metabolic status are strongly influenced by the tumor microenvironment (TME) [6–8]. The complex dynamics of the OSCC microenvironment alter spatiotemporal distribution and effector functions of infiltrating leukocytes to modify/diminish host defense mechanisms in favor of tumor cell survival [9, 10]. In particular, local oxygen depletion leads to the induction of reactive oxygen species (ROS) that promote cancer cell proliferation and drive autophagic/lysosomal loss of stromal caveolin-1 [an inhibitor of transforming growth factor-β (TGF-β) signaling] in cancer-associated fibroblasts (CAFs), resulting in tumor recurrence and metastasis and affecting patient survival [11–14]. Furthermore, the elevated levels of ROS result in detrimental stabilization of hypoxia-inducible factor (HIF)-1α, which activates pro-angiogenic genes including vascular endothelial growth factor (VEGF) [15–17]. HIF-1α acts as a master regulator of oxygen concentration to stimulate hypoxia-adaptive responses in cells. Immune signaling can be altered through the production of onco-metabolites that may further influence the clinical course of OSCC [18]. Consequently, a better understanding of how hypoxic stress, ROS generation, and onco-metabolites alter immune function in the TME is now a priority issue for the OSCC research community. The present article therefore reviews current knowledge of how redox factors alter leukocyte metabolism to promote the immune suppressive microenvironment of hypoxic OSCC.
Hypoxia and Redox Balance in the Oral Squamous Cell Carcinoma Microenvironment
Hypoxic TME alters local ROS generation and metabolic profile of both constituent tumor cells and infiltrating leukocytes [19–21]. Glycolysis is a metabolic process carried out in cell cytoplasm to generate two ATPs and pyruvates; this pyruvates serves as a fuel for tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) under aerobic conditions [22, 23]. However, under anaerobic conditions, pyruvate is reduced to lactate, and this lactate is secreted into extracellular matrices [24]. The metabolic features of cancer cells are very heterogeneous where OXPHOS and aerobic glycolytic activities are impaired [25].
Intriguingly, cancer cells specifically express pyruvate kinase M (PKM)-2 that oxidizes and generates reduced nicotinamide adenine dinucleotide phosphate (NADPH) to maintain redox buffering; besides, this PKM-2 gene trans-activates HIF-1α target genes, leading to a significant shift in metabolic activity and cancer cell signaling [26, 27]. In solid tumors like OSCC, increased production of ROS, cytokines, and CAFs stimulates the production of pro-angiogenic factors in an attempt to promote neovascularization and enhance survival (Figure 1) [28, 29]. In addition, the cancer cells undergo a metabolic shift from OXPHOS to glycolysis, which produces lactate and increases serum levels of lactate dehydrogenase (LDH), which has been linked with poor survival in patients with OSCC [30]. Tumor cells therefore have the capacity to utilize both OXPHOS and aerobic glycolysis for baseline metabolic activity and rapid energy production via the lactate pathway (Figure 1). In this regard, Otto Warburg proposed that due to mitochondrial defects, the predominant metabolism in cancer cells is aerobic glycolysis rather than OXPHOS. Warburg's historic findings were called Warburg effect [31]. However, the metabolic coupling between OSCC cells and associated stromal cells is mainly determined by growth requirements; these effects are called dual/reverse Warburg effect [32, 33]. A previous NMR-based study suggested that OSCC can contravene the Warburg effect and implicated malonate (a competitive inhibitor of succinate) to induce drastic alterations in the TCA cycle that produce more fatty acid for membrane biogenesis in OSCC [34–36]. Among various glycolytic enzymes, alpha-enolase is crucial to produce phosphoenolpyruvate. As the mortality of OSCC is known to be due to metastasis, enolase in particular seems to play a major role in the malignant transformation of dysplastic epithelium in oral pre-cancer through promoting cell surface receptor enolase [37–40]. In addition to altered glucose metabolism, modified amino acid metabolism also occurs in OSCC. The amplified glutamine catabolism creates glutamine scarcity in hypoxic tumor core and leads to a dramatic histone hypermethylation [41]. In order to better understand the effects of hypoxia on OSCC, we will require new immunological paradigms that consider how dysregulation of crucial metabolic pathways can impact on both tumor growth and host leukocyte responses.
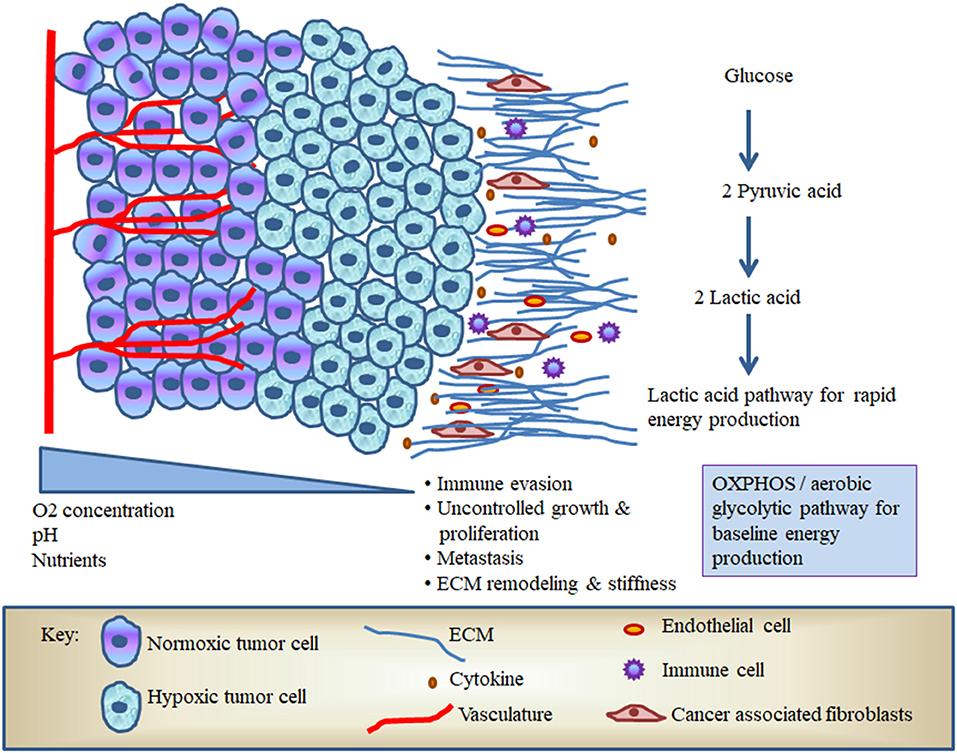
Figure 1. The hypoxic microenvironment of oral squamous cell carcinoma (OSCC).
Mitochondrial Homeostasis and Immune Dysfunction in Oral Squamous Cell Carcinoma
In the hypoxic/acidic TME, reduced OXPHOS and electron transport chain (ETC) activity in local immune cells lead to altered mitochondrial membrane potential and impaired generation of ATP [42, 43]. The immune system not only kills cancerous cells but also modifies the TME in three phases—elimination, equilibrium, and escape [44]. The growing and transformed cells can be eradicated by immune response in the elimination phase; however, immune selection and reorganization create an immune resistant environment, namely, the equilibrium phase [45]. Consequently, immune surveillance escapes to kill tumor cells, and tumor cells grow in an uncontrolled manner [46]. In the last phase of cancer immuno-editing (i.e., “escape” phase), cancer cells produce large amounts of “pro-tolerogenic” kynurenine catalyzed by indoleamine 2,3-dioxygenase (IDO) processing of tryptophan [47]. Tryptophan catabolites have affinity to bind aryl hydrocarbon receptors (AhRs) of mitochondria, which persuade mitochondrial dysfunction in T cells and natural killer (NK) cells [48, 49]. Thus, under conditions of acute tryptophan depletion, central mitochondrial metabolic processes and synthesis of NADPH are disrupted such that infiltrating immune cells will undergo apoptosis rather than eradicating the tumor [50].
Heterotypic Immune Modulation in Hypoxic Oral Squamous Cell Carcinoma
Hypoxic OSCC reprograms cellular metabolism in order to modify the repertoire of infiltrating immune cells toward a more tumor-permissive profile [51]. For example, macrophages located within hypoxic tumors tend to polarize toward an “anti-inflammatory” M2 phenotype, whereas cytotoxic T lymphocytes shift from glycolysis to OXPHOS-based metabolism (Figure 2) [52, 53]. Since essentially all OSCC tumors are subject to hypoxia upon reaching a certain mass, it is important to study how innate and adaptive immune cells alter their metabolism under these conditions in order to fully understand their influence on disease progression.
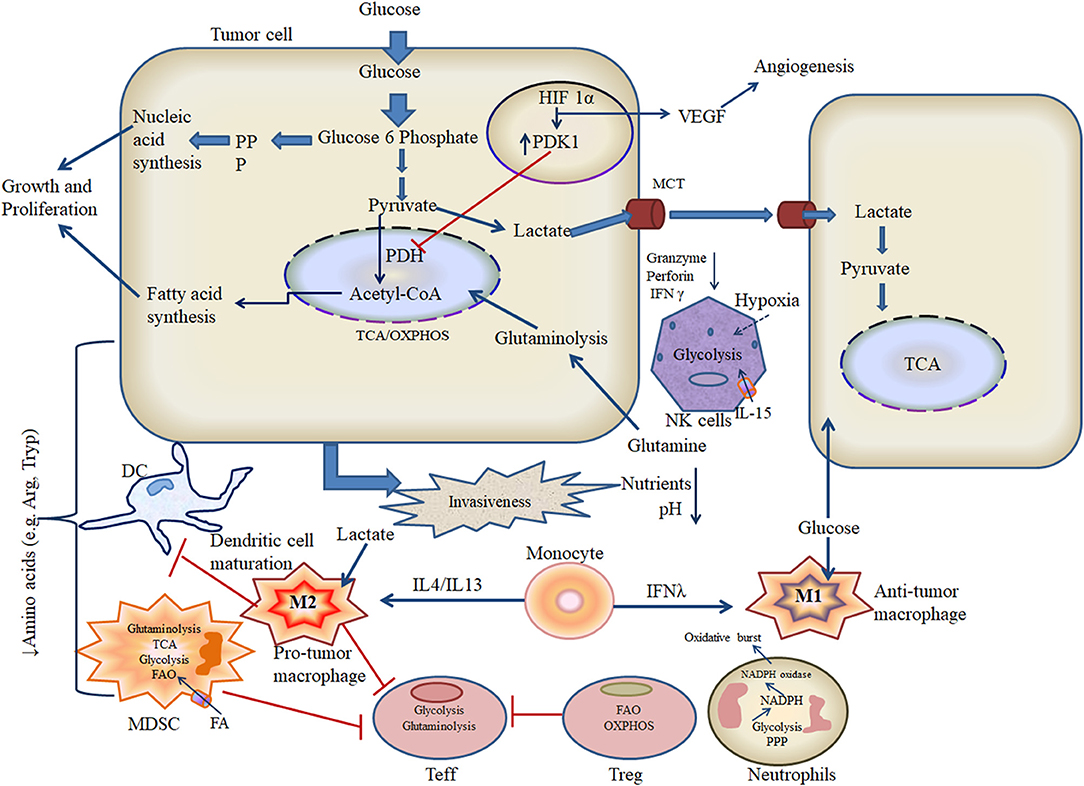
Figure 2. Metabolic crosstalk of tumor and immune cells in the hypoxic microenvironment.
Hypoxia “Edits” Immune Signaling
The OSCC microenvironment has two forms of immune responses: innate and adaptive. Innate immune responses are non-specific and instant against pathogens, allergens, and non-self proteins. Phagocytes [myeloid-derived suppressor cells (MDSCs), neutrophils, monocytes, and macrophages] and NK cells are the main cells of innate immunity. Phagocytes engulf the foreign particles and digest through lysosomal enzymes, whereas NK cells kill the foreign bodies using altered major histocompatibility complex I (MHC class I) proteins, perforin, and granzyme-mediated apoptosis [54]. Dendritic cells (DCs) serve as a crucial link between innate and adaptive immune responses under physiological conditions. DCs process antigens and present them to T lymphocytes via MHC class I or II [55]. Adaptive immunity is composed of mainly T and B lymphocytes; B cells are professional antigen-presenting cells (APCs) that can activate T cells in tertiary lymphoid structures, allowing coordination of B and T cell responses in OSCC [56, 57]. In head and neck squamous cell carcinoma (HNSCC), regulatory T cells (Treg) situated in the center of the tumor mass have been reported to be more strongly immunosuppressive than circulating Treg [58]). Treg can impede T effector (Teff) cell function to reduce antitumor activity and contribute to poor prognosis in multiple types of cancer. Activated CD8+ Teff are dominant antitumor cells that secrete granzymes, perforin, and pro-inflammatory cytokines, such as tumor necrosis factor (TNF) and interferon (IFN)-γ, whereas CD4+ T cells can either inhibit or promote tumor cell activity via the specific activities listed here [59, 60].
The macrophage subtypes M1 and M2 are activated in response to microbial and cancer-derived stimuli, respectively [61]. M1 polarization of macrophages is induced by T helper type 1 (TH1) cytokines such as IFN-γ and signaling through signal transducer and activator of transcription 1 (STAT1), whereas M2 polarization is promoted by T helper type 2 (TH2) cytokines such as IL-4 and IL-13 that trigger the STAT6 pathway [62]. Functionally, M1 macrophages produce pro-inflammatory cytokines, ROS and reactive nitrogen species (RNS), while mediating antigen presentation via MHC class II molecules. M1 macrophages also actively phagocytose pathogens and are considered to suppress tumor development [63]. In contrast, M2 macrophages are activated by cytokines including IL-4/IL-13, IL-10, TGF-β, and glucocorticoids that promote secretion of anti-inflammatory mediators. Further, M2 macrophages inhibit the lytic activity of CD8+ cytotoxic T cells [55]. Despite their opposing roles, both M1 and M2 macrophages can coexist within the same tumor.
Tumor cells can secrete IL-10, colony-stimulating factor (CSF)-1, and various chemokines [C-C motif chemokine ligand (CCL)-2, CCL-18, CCL-17, and C-X-C motif chemokine ligand (CXCL)-4] that appear to favor M2 polarization [64]. In addition to cytokine expression, hypoxic tumors can further direct macrophage phenotypes and responses via release of exosomes loaded with soluble factors and suppressive micro RNA [65]. In human HNSCC, the acidic TME has been reported to promote HIF-1α activation and tumor-associated macrophage (TAM) expression of M2-specific markers CSF1R and CD163, as well as driving concomitant production of arginase and VEGF [66, 67]. NK cells have the capacity to kill tumor cells and activate antitumor T cell responses by secreting IFN-λ and cytotoxic molecules such as granzyme and perforin, but these activities can be severely restricted by the concomitant presence of MDSCs [68, 69]. MDSCs are known for their immune suppressive activity for both innate and adaptive immunity. The two subtypes of MDSCs are monocytic (M-MDSCs) and polymorphonuclear (PMN-MDSCs), which have variable capacities to inhibit the function of activated CD8+ T cells [70, 71]. MDSCs are thought to suppress T cell responses by expressing a range of inhibitory factors including arginase, inducible nitric oxide synthase (iNOS), TGF-β, IL-10, cyclooxygenase (COX)-2, and IDO [72]. HIF-1α appears to play a key role in this immunosuppressive process by driving the expression of cytokines that promote MDSC infiltration of the tumor mass [73]. In addition, granulocyte MDSCs (G-MDSCs) are classified as T cell-suppressive neutrophils because of similar morphology and cell surface markers as mature neutrophils [74, 75]. Neutrophils migrate toward and infiltrate tumors under the influence of potent chemokines such as IL-8, after which these cells appear to enhance tumor proliferation and are correlated with poor survival in solid cancers [76, 77]. Several clinical observations indicate that neutrophil activity is further modulated under the influence of the TME to assist cancer development [78]. However, tumor-associated neutrophils (TANs) may exert a dual role, since these cells appear capable of promoting either CD8+ T cell activity or tumor progression, depending on the prevailing level of TGF-β within the TME [79].
Likewise, with the polarization of TAMs, TANs exhibit two polarization phenotypes, i.e., N1 (antitumor neutrophils) and N2 (pro-tumor neutrophils), where TGF-β signaling plays a vital role [80]. Obstruction of TGF-β signaling or type I IFNs activates N1 phenotype with accretion of TNF-α and type 1 IFN, whereas augmentation in TGF-β signaling leads to N2 phenotype with high levels of neutrophil elastase (NE) and arginase in oral cancers [81]. Immature DCs are activated by pathogen-associated molecular patterns such as toll-like receptor (TLR) ligands, thereafter migrating to lymphoid organs and presenting antigen to T cells in the context of MHC [82]. In this process, phosphoinositide 3-kinase (PI3K)/Akt signaling pathway regulates the metabolic switch through inhibiting AMP-activated protein kinase and promotes glycolysis [83]. While adenosine signaling limits DC activation, ATP detection by P2YR and P2XR promotes DC migration and IL-1 secretion, respectively [84]. Hypoxic TME not only alters the innate immune signaling but also modifies adaptive immune signaling. Likewise, under hypoxic conditions, B cell caspase signaling is activated and kinase complex mammalian target of rapamycin complex 1 (mTORC1) pathway is reduced, leading to cell death via apoptosis [85]. B cells also secrete IL-10 under hypoxic stress; however, in-depth molecular pathway is not well-characterized [86]. The role of HIF-1α transcription factor in tumor-infiltrating T cells remains unclear [87]. However, in cancer cells, HIF-1α interaction with hypoxia response element (HRE) in the programmed cell death ligand 1 (PD-L1) promoter can trigger rapid expression of this immune checkpoint molecule, which is also capable of signaling more effectively in the lactate-rich TME [59, 88]. PD-L1 ligation of programmed cell death protein 1 (PD-1) on Teff can inhibit T cell receptor (TCR) signaling and attenuates the PI3K/Akt and Ras/mitogen activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathways to restrict antitumor responses [89, 90]. Accordingly, antibodies targeting the PD-1: PD-L1 axis and other immune checkpoints have the ability to restore glucose levels in the hypoxic TME and have proven highly effective in the treatment of OSCC [91–93]. In particular, abnormal metabolic processes within cancer cells can generate neo-antigens that are presented by MHC class I molecules on the cell surface of antigen-presenting cells for recognition by CD8+ cytotoxic T cells [94].
Hypoxia Modifies the Resting Metabolic Status of Immune Cells
The resting metabolic status and associated effector functions of local immune cells play vital roles in determining the nature of host antitumor responses. In particular, glucose transport regulates pyruvate flow into the TCA cycle and is essentially “rate-limiting” for host immunity, since leukocytes typically require rapid energy generation in order to achieve full activation. The hypoxic TME is a key driver of M2 polarization in infiltrating macrophages likely via the expression of specific cytokine signals that activate nuclear factor (NF)-κB, although the underlying mechanism has yet to be precisely defined in OSCC [95, 96]. Like other myeloid lineage cells, macrophage mitochondria can generate both superoxide and NO, which react to form the powerful oxidant peroxynitrite, which is highly toxic to cancer cells [97, 98]. While some investigators have reported that mitochondrial ROS (mROS) stimulate macrophage expression of pro-inflammatory cytokines, other researchers have instead observed the induction of an anti-inflammatory phenotype; hence, further study is required to fully understand these events [99–102]. The glycolytic reprogramming of TAM is regulated by oxygen sensors including prolyl-hydroxylases (PHDs) and is accompanied by proton pumping and acidification of M2 macrophages that subsequently impair antitumor responses [103]. Similarly, while resting NK cells typically utilize OXPHOS, exposure to high doses of tissue damage-associated cytokine IL-15 stimulates conversion to glycolytic activity [104]. For tumor-infiltrating neutrophils, the principal metabolic pathways employed are aerobic glycolysis and the pentose phosphate pathway (PPP), which support chemotaxis and microbicidal activities, respectively (Figure 2) [105]. Metabolic shift toward PPP is also required for formation of neutrophil extracellular traps, which envelope and attach to the circulating cancer cells and expedite metastasis to distant sites [106]. Tumor-associated MDSCs predominantly utilize fatty acid (FA)-β oxidation (FAO) and thus display high rates of oxygen consumption (Figure 2) [107]. In the hypoxic TME, MDSCs display potent immunosuppressive activity, which depends on the endoplasmic reticulum (ER) stress response transcription factor CCAAT-enhancer-binding protein homologous protein (CHOP) [108]. DCs are the critical components of the immune system against cancer as they have robust antigen-presenting ability to educate T cells [109, 110]. Upon microbial stimulation, DCs typically shift from OXPHOS to glycolysis; however, in TME, DCs promote immune suppression through galectin-1 [111]. In this regard, how metabolic profiles influence DC function and tumor progression in vivo is not yet well-defined [56, 112]. Activated B cells can secrete antibodies that can bind and induce tumor cell killing, but these processes can be strongly influenced by mitochondrial generation of ROS and heme synthesis. While the presence of CD20+ B cells within the TME indicates a good prognosis in lung cancer, gastric cancer, and melanoma, the role played by B cells in HNSCC has yet to be fully investigated. Variable hypoxia across the developing OSCC tumor is thought to alter tissue distribution of local B cells, which generate immune complexes and produce cytokines that then modify myeloid cell function to assist tumor progression [113]. B cells utilize glycolytic metabolism during early development in the bone marrow. Later survival, maturation, and functional activity of B cells are instead regulated by HIF-1α and depend on glucose transporters and phosphofructokinase. The oncogenic Myc expression in B cells hindered the oxidation of acetyl-CoA in TCA cycle as it persuades lactate dehydrogenase for conversion of pyruvate (glycolysis intermediate) to lactate [114]. Naive T cells employ OXPHOS and fatty acid metabolism before shifting glycolysis in order to support Teff functions [112]. Memory T cells also depend on OXPHOS for energy generation in the resting state, whereas Treg favor fatty acid oxidation. It is noteworthy that while glycolytic metabolism predominates among Teff and Treg, both populations have been observed to maintain OXPHOS within the TME (Figure 2), which may contribute to detrimental cancer-associated inflammation, further mutation, and eventual metastasis [115, 116]. Metabolic alterations in the T cell pool may also impede the differentiation of Teff while increasing the generation of Treg and “exhausted” populations, thus further supporting cancer evasion of host immunity [50].
Hypoxic Oral Squamous Cell Carcinoma Produces Immunosuppressive Onco-Metabolites
The metabolic products of cancer cells (onco-metabolites) are intimately linked with the control of the immune cell function [117]. For example, the microenvironment of OSCC is characterized by hypoxia, low pH, and elevated lactate levels, which disturb ETC operation and leads to deposition of citrate and succinate [21]. Citrate is converted into acetyl-CoA and utilized in several biosynthetic pathways. Oxidation of succinate produces ROS and promotes HIF-1α activation. Hypoxia-generated lactate also drives macrophage differentiation toward an M2 phenotype [118]. TAMs are unable to utilize extracellular arginine due to rapid enzymatic breakdown by arginase and must instead use extracellular glutamine to produce this “semi-essential” amino acid [119]. TANs also produce high levels of arginase to disrupt TCR signaling. Increased lactate concentration in the hypoxic TME favors decreased NK cell expression of granzyme/perforin and NKp46, leading to reduced anticancer cytolytic activity (Figure 2) [120]. The nuclear factor of activated T cells (NFAT) has also been implicated in downregulation of NK cell activity via an increase in cancer-associated lactate dehydrogenase expression [121]. Other hypoxia-induced onco-metabolites such as adenosine and lactic acid have previously been reported to impair DC activation (Figure 2) [122]. The tumor-associated dendritic cells (TADCs) stimulate arginase, which then depletes arginine in the extracellular matrix (ECM) and arginine scarcity impairs CD8+ T cell responses [123]. Some stable onco-metabolites (like kynurenine and quinolinate) along with specific cytokine milieu promote AhR signaling in non-functional Treg, Foxp3+-induced Treg (iTreg), and TH17 cells. These signaling pathways further de-differentiate Treg, iTreg, and TH17 cells into functional iTreg and endorse immune tolerance [124, 125]. IDO contributes to the tolerogenic ability of DCs to inhibit Teff functions and promotes Treg activity [126, 127]. The by-products (highly reactive aldehyde) of anomalous lipid peroxidation (triggered by ROS) create ER stress on TADCs and lower antitumor responses [128]. In addition, acylcarnitine and 2-hydroxyglutarate (2-HG) have been identified as a prominent onco-metabolite in HNSCC [129]. This 2-HG skews TH17 polarization and alters Treg metabolism by promoting the OXPHOS and destabilizing HIF-1α [130, 131].
Immune-Tumor Metabolic Switch Under Hypoxia
Tumor hypoxia is characterized by local tissue acidification and nutrient depletion, thereby creating metabolic competition and generating active biomolecules that influence cancer cell interactions with host leukocytes. Competition for key metabolites along with cholesterol esterification, release of adenosine, and expression of prostaglandin E2 inhibits effector T cells [132]. Hypoxic OSCC modifies pro- and antitumoral γδ T cell populations via exosomes [11]. OSCC can also express a range of different TLRs, with high levels of TLR-2 and TLR-4 correlating with tumor progression and chemoresistance, respectively. HIF-1 can deregulate TLR3 and TLR4 in OSCC cell lines under hypoxia stress, leading to potent effects on tumor cell survival, proliferation, and metastatic potential [133].
Concluding Remarks and Future Directions
Hypoxia-related metabolic stress inhibits the activity of host immune cells to support oncogenic transformation and inflammation in OSCC. The hypoxia signaling in immune cells not only alters the glycolysis but also modifies other metabolic pathways like amino acid, FAO, PPP, and TCA, resulting in onco-metabolite production. Further, these onco-metabolites disturb the redox balance, mitochondrial function, and ATP production through aerobic glycolysis in OSCC.
In future studies, it will be important to elucidate the correlation between spatial distributions of immune cells in hypoxic/non-hypoxic OSCC. Novel and advanced therapeutic approaches like interfering with HIF-1α signaling in immune cells through antisense or small interfering RNA, modulating the metabolic status of immune cells through gene editing technology [clusters of regularly interspaced short palindromic repeats–caspase 9 (CRISPR-Cas9)], and designing new smart oxygen-sensitive chimeric antigen receptor (CAR) T cell may provide new insight to overcome the challenges associated with hypoxic OSCC in the future.
Author Contributions
AC conceptualized, conceived, and wrote the manuscript. SB conceptualized and organized the review. NA guided and revised the manuscript. VR revised and approved the manuscript. DM guided, revised and approved the manuscript. GM guided to write, organized and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AC and SB acknowledged the Department of Biotechnology, Government of India, and New Delhi for providing research fellowship as Scientist C under the project: Systems Medicine Cluster (SyMeC, Project Reference: No./BT/Med-II/NIBMG/SyMeC/2014/Vol II).
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo MM, et al. Cancer incidence and mortality worldwide, sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:359–86. doi: 10.1002/ijc.29210
2. Gupta B, Johnson NW, and Kumar N. Global epidemiology of head and neck cancers: a continuing challenge. Oncology. (2016) 91:13–23. doi: 10.1159/000446117
3. Larsen SR, Johansen J, Sørensen JA, and Krogdahl A. The prognostic significance of histological features in oral squamous cell carcinoma. J Oral Path Med. (2009) 38:657–62. doi: 10.1111/j.1600-0714.2009.00797.x
4. Rogers SN, Brown JS, Woolgar JA, Lowe D, Magennis P, Shaw RJ, et al. Survival following primary surgery for oral cancer. Oral Oncol. (2009) 45:201–11. doi: 10.1016/j.oraloncology.2008.05.008
5. Carvalho HDA, and Villar RC. Radiotherapy and immune response, the systemic effects of a local treatment. Clinics. (2018) 73:e557s. doi: 10.6061/clinics/2018/e557s
6. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature Med. (2015) 21:938. doi: 10.1038/nm.3909
7. Lei Y, Xie Y, Tan YS, Prince ME, Moyer JS, Nör J, et al. Telltale tumor infiltrating lymphocytes (TIL) in oral, head and neck cancer. Oral Oncol. (2016) 61:159–65. doi: 10.1016/j.oraloncology.2016.08.003
8. Kong H, and Chandel NS. Regulation of redox balance in cancer and T cells. J Biol Chem. (2018) 293:7499–507. doi: 10.1074/jbc.TM117.000257
9. Chakraborty P, Karmakar T, Arora N, and Mukherjee G. Immune and genomic signatures in oral (head and neck) cancer. Heliyon. (2018) 4:e00880. doi: 10.1016/j.heliyon.2018.e00880
10. Purohit V, Simeone DM, and Lyssiotis CA. Metabolic regulation of redox balance in cancer. Cancers. (2019) 11:955. doi: 10.3390/cancers11070955
11. Fu P, Chen F, Pan Q, Zhao X, Zhao C, Cho WCS, et al. The different functions and clinical significances of caveolin-1 in human adenocarcinoma and squamous cell carcinoma. OncoTargets Ther. (2017) 10:819. doi: 10.2147/OTT.S123912
12. Bag S, Conjeti S, Das RK, Pal M, Anura A, Paul RR, et al. Computational analysis of p63+ nuclei distribution pattern by graph theoretic approach in an oral pre-cancer (sub-mucous fibrosis). J Pathol Inform. (2013) 4:35. doi: 10.4103/2153-3539.124006
13. Chen X, Song M, Zhang B, and Zhang Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longevity. (2016) 2016:1580967. doi: 10.1155/2016/1580967
14. Liou GY, and Storz P. Reactive oxygen species in cancer. Free Radic Res. (2010) 44:479–96. doi: 10.3109/10715761003667554
15. Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. (2004) 24:9695–704. doi: 10.1128/MCB.24.22.9695-9704.2004
16. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy, the bright side of the moon. Exp Mol Med. (2020) 52:1–12. doi: 10.1038/s12276-020-0384-2
17. Sobhakumari A, Love-Homan L, Fletcher EV, Martin SM, Parsons AD, Spitz K, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE. (2012) 7:e0048175. doi: 10.1371/journal.pone.0048175
18. Li Y, Patel SP, Roszik J, and Qin Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment, new approaches for combinational immunotherapy. Front Immunol. (2018) 9:1591. doi: 10.3389/fimmu.2018.01591
19. Eckert AW, Wickenhauser C, Salins PC, Kappler M, Bukur J, and Seliger B. Clinical relevance of the tumor microenvironment and immune escape of oral squamous cell carcinoma. J Transl Med. (2016) 14:85. doi: 10.1186/s12967-016-0828-6
20. Peltanova B, Raudenska M, and Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma, a systematic review. Mol Cancer. (2019) 18:63. doi: 10.1186/s12943-019-0983-5
21. Epstein T, Gatenby RA, and Brown JS.The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE. (2017) 12:e0185085. doi: 10.1371/journal.pone.0185085
22. Zheng JIE. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation. Oncol Lett. (2012) 4:1151–7. doi: 10.3892/ol.2012.928
23. Ashton TM, McKenna WG, Kunz-Schughart LA, and Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. (2018) 24:2482–90. doi: 10.1158/1078-0432.CCR-17-3070
24. Romero-Garcia S, Moreno-Altamirano MMB, Prado-Garcia H, and Sánchez-García FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. (2016) 7:52. doi: 10.3389/fimmu.2016.00052
25. Jose C, Bellance N, and Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma? Biochim. Biophys. Acta. (2011) 1807:552–61. doi: 10.1016/j.bbabio.2010.10.012
26. McGarry T, Biniecka M, Veale DJ, and Fearon U. Hypoxia, oxidative stress and inflammation. Free Rad Biol Med. (2018) 125:15–24. doi: 10.1016/j.freeradbiomed.2018.03.042
27. Hamanaka RB, and Chandel NS.Warburg effect and redox balance. Science. (2011) 334:1219–20. doi: 10.1126/science.1215637
28. Al Tameemi W, Dale TP, Al-Jumaily RMK, and Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol. (2019) 7:4. doi: 10.3389/fcell.2019.00004
29. Johnson S, De Costa AM, and Young M. Effect of the premalignant and tumor microenvironment on immune cell cytokine production in head and neck cancer. Cancers (Basel). (2014) 6:756–70. doi: 10.3390/cancers6020756
30. Wei J, Wu J, Xu W, Nie H, Zhou R, and Wang R. Salvanic acid B inhibits glycolysis in oral squamous cell carcinoma via targeting PI3K/AKT/HIF-1α signaling pathway. Cell Death Dis. (2018) 9:599. doi: 10.1038/s41419-018-0623-9
31. Ngo H, Tortorella SM, Ververis K, and Karagiannis TC. The Warburg effect, molecular aspects and therapeutic possibilities. Mol Biol Rep. (2015) 42:825–34. doi: 10.1007/s11033-014-3764-7
32. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget. (2017) 8:57813. doi: 10.18632/oncotarget.18175
33. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic coupling and the Reverse Warburg Effect in cancer, Implications for novel biomarker and anticancer agent development. Semin Oncol. (2017) 44:198–203. doi: 10.1053/j.seminoncol.2017.10.004
34. Bag S, Banerjee DR, Basak A, Das AK, Pal M, Banerjee R, et al. NMR (1H and 13C) based signatures of abnormal choline metabolism in oral squamous cell carcinoma with no prominent Warburg effect. Biochem Biophys Res Commun. (2015) 459:574–8. doi: 10.1016/j.bbrc.2015.02.149
35. Sant'Anna-Silva ACB, Santos GC, Campos SPC, Oliveira Gomes AM, Pérez-Valencia JA, and Rumjanek FD. Metabolic profile of oral squamous carcinoma cell lines relies on a higher demand of lipid metabolism in metastatic cells. Front Oncol. (2018) 8:13. doi: 10.3389/fonc.2018.00013
36. Chen X, and Yu D. Metabolomics study of oral cancers. Metabolomics. (2019) 15:22. doi: 10.1007/s11306-019-1483-8
37. Tsai ST, Chien IH, Shen WH, Kuo YZ, Jin YT, and Wong TY. ENO1, a potential prognostic head and neck cancer marker, promotes transformation partly via chemokine CCL20 induction. Eur J Cancer. (2010) 46:1712–23. doi: 10.1016/j.ejca.2010.03.018
38. Bag S, Dutta D, Chaudhary A, Sing BC, Pal M, Ray AK, et al. Identification of α-enolase as a prognostic and diagnostic precancer biomarker in oral submucous fibrosis. J Clin Pathol. (2018) 71:228–38. doi: 10.1136/jclinpath-2017-204430
39. Bag S, Dutta D, Chaudhary A, Sing BC, Banerjee R, Pal M, et al. NanoLC MALDI MS/MS based quantitative metabolomics reveals the alteration of membrane biogenesis in oral cancer. RSC Adv. (2016) 6:62420–33. doi: 10.1039/C6RA07001A
40. Munir R, Lisec J, Swinnen JV, and Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. (2019) 120:1090–8. doi: 10.1038/s41416-019-0451-4
41. Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nature Cell Biol. (2016) 18:1090–101. doi: 10.1038/ncb3410
42. Chattopadhyay E, and Roy B. Altered mitochondrial signalling and metabolism in cancer. Front Oncol. (2017) 7:43. doi: 10.3389/fonc.2017.00043
43. Chen Y, Zhou Z, and Min W. Mitochondria, oxidative stress and innate immunity. Front Physiol. (2018) 9:1487. doi: 10.3389/fphys.2018.01487
44. Mittal D, Gubin MM, Schreiber RD, and Smyth MJ. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
45. Kim R, Emi M, and Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunol. (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
46. Fridman WH. From cancer immune surveillance to cancer immunoediting: birth of modern immuno-oncology. J Immunol. (2018) 201:825–6. doi: 10.4049/jimmunol.1800827
47. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, Halmos G, and Székvölgyi L. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
48. Anderson G, and Maes M. Interactions of tryptophan and its catabolites with melatonin and the alpha 7 nicotinic receptor in central nervous system and psychiatric disorders: role of the aryl hydrocarbon receptor and direct mitochondria regulation. Internat J Tryptophan Res. (2017) 10:1178646917691738. doi: 10.1177/1178646917691738
49. Wang X, Li S, Liu L, Jian Z, Cui T, Yang Y, et al. Role of the aryl hydrocarbon receptor signaling pathway in promoting mitochondrial biogenesis against oxidative damage in human melanocytes. J Dermatol Sci. (2019) 96:33–41. doi: 10.1016/j.jdermsci.2019.09.001
50. Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
51. Canning M, Guo G, Yu M, Myint C, Groves M, Byrd K, et al. Heterogeneity of the head and neck squamous cell carcinoma immune landscape and its impact on immunotherapy. Front Cell Dev Biol. (2019) 7:52. doi: 10.3389/fcell.2019.00052
52. Meng W, Hao Y, He C, Li L, and Zhu G. Exosome-orchestrated hypoxic tumor microenvironment. Mol Cancer. (2019) 18:57. doi: 10.1186/s12943-019-0982-6
53. Li Y, and Zhu B. Metabolism of cancer cells and immune cells in the tumor microenvironment. Front Immunol. (2018) 9:3080. doi: 10.3389/fimmu.2018.03080
54. Pandya PH, Murray ME, Pollok KE, and Renbarger JL. The immune system in cancer pathogenesis, potential therapeutic approaches. J Immunol Res. (2016) 2016:4273943. doi: 10.1155/2016/4273943
55. Veglia F, and Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. (2017) 45:43–51. doi: 10.1016/j.coi.2017.01.002
56. Cano RLE, and Lopera HDE. Introduction to T and B lymphocytes. In: Anaya JM, Shoenfeld Y, Rojas-Villarraga A, Levy RA, Cervera R, editors. Autoimmunity: From Bench to Bedside [Internet]. Bogota: El Rosario University Press (2013) 77–95.
57. Lechner A, Schlößer HA, Thelen M, Wennhold K, Rothschild SI, Gilles R, et al. Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. Oncoimmunol. (2019) 8:1535293. doi: 10.1080/2162402X.2018.1535293
58. Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. (2013) 109:2629–35. doi: 10.1038/bjc.2013.645
59. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
60. Siska PJ, and Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. (2015) 36:257–64. doi: 10.1016/j.it.2015.02.007
61. Henze AT, and Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Inv. (2016) 126:3672–9. doi: 10.1172/JCI84427
62. Binnemars-Postma K, Bansal R, Storm G, and Prakash J. Targeting the Stat6 pathway in tumor-associated macrophages reduces tumor growth and metastatic niche formation in breast cancer. FASEB J. (2018) 32:969–78. doi: 10.1096/fj.201700629R
63. Genard G, Lucas S, and Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies, radiotherapy versus chemo-and immunotherapies. Front Immunol. (2017) 8:828. doi: 10.3389/fimmu.2017.00828
64. Sica A, and Mantovani A. Macrophage plasticity and polarization, in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
65. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, and Low JK. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. (2019) 38:5158–73. doi: 10.1038/s41388-019-0782-x
66. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, and Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
67. Ohashi T, Aoki M, Tomita H, Akazawa T, Sato K, Kuze B, et al. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. (2017) 108:1128–34. doi: 10.1111/cas.13244
68. Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, et al.The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front Immunol. (2013) 4:490. doi: 10.3389/fimmu.2013.00490
69. Krasnova Y, Putz EM, Smyth MJ, and Souza-Fonseca-Guimaraes F. Bench to bedside: NK cells and control of metastasis. Clin Immunol. (2017) 177:50–9. doi: 10.1016/j.clim.2015.10.001
70. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
71. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
72. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nature Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
73. Parker KH, Beury DW, and Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
74. Aarts CE, and Kuijpers TW. Neutrophils as myeloid-derived suppressor cells. European J Clin Invest. (2018) 48:e12989. doi: 10.1111/eci.12989
75. Zilio S, and Serafini P. Neutrophils and granulocytic MDSC: the janus god of cancer immunotherapy. Vaccines. (2016) 4:31. doi: 10.3390/vaccines4030031
76. Egners A, Erdem M, and Cramer T.The response of macrophages and neutrophils to hypoxia in the context of cancer and other inflammatory diseases. Mediat Inflamm. (2016) 2016:2053646. doi: 10.1155/2016/2053646
77. Manfroi B, Moreaux J, Righini C, Ghiringhelli F, Sturm N, and Huard B.Tumor-associated neutrophils correlate with poor prognosis in diffuse large B-cell lymphoma patients. Blood Cancer J. (2018) 8:66. doi: 10.1038/s41408-018-0099-y
78. Shen M, Hu P, Donskov F, Wang G, Liu Q, and Du J. Tumor-associated neutrophils as a new prognostic factor in cancer, a systematic review and meta-analysis. PLoS ONE. (2014) 9:e98259. doi: 10.1371/journal.pone.0098259
79. Grecian R, Whyte MK, and Walmsley SR. The role of neutrophils in cancer. Br Med Bull. (2018) 128:5–14. doi: 10.1093/bmb/ldy029
80. Dumitru CA, Fechner MK, Hoffmann TK, Lang S, and Brandau S. A novel p38-MAPK signaling axis modulates neutrophil biology in head and neck cancer. J Leukoc Biol. (2012) 91:591–8. doi: 10.1189/jlb.0411193
81. Jablonska E, Puzewska W, Grabowska Z, Jablonski J, and Talarek L. VEGF, IL-18 and NO production by neutrophils and their serum levels in patients with oral cavity cancer. Cytokine. (2005) 30:93–9. doi: 10.1016/j.cyto.2004.12.004
82. Metelmann HR, Hyckel P, and Podmelle F. Oral cancer treatment and immune targets–A role for dendritic cells? J CranioMaxill Surg. (2012) 40:103–4. doi: 10.1016/j.jcms.2011.03.009
83. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
84. Pearce EJ, and Everts B. Dendritic cell metabolism. Nat Rev Immunol. (2015) 15:18–29. doi: 10.1038/nri3771
85. Gough NR. Hypoxia limits IgG-producing B cells. Sci Signal. (2016) 9:ec209. doi: 10.1126/scisignal.aaj1881
86. Meng X, Grötsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun. (2018) 9:1–17. doi: 10.1038/s41467-017-02683-x
87. Gerriets VA, and Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. (2012) 33:168–73. doi: 10.1016/j.it.2012.01.010
88. Wang Y, Wang H, Yao H, Li C, Fang JY, and Xu J. Regulation of PD-L1, emerging routes for targeting tumor immune evasion. Front Pharmacol. (2018) 9:536. doi: 10.3389/fphar.2018.00536
89. Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, and Baldwin AS. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinase α (IKKα). J Biol Chem. (2014) 289:25227–40. doi: 10.1074/jbc.M114.554881
90. Kortum RL, Rouquette-Jazdanian AK, and Samelson LE. Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. (2013) 34:259–68. doi: 10.1016/j.it.2013.02.004
91. Kim J. Regulation of immune cell functions by metabolic reprogramming. J Immunol Res. (2018) 2018:8605471. doi: 10.1155/2018/8605471
92. Maruse Y, Kawano S, Jinno T, Matsubara R, Goto Y, Kaneko N, et al. Significant association of increased PD-L1 and PD-1 expression with nodal metastasis and a poor prognosis in oral squamous cell carcinoma. Int J Oral Max Surg. (2018) 47:836–45. doi: 10.1016/j.ijom.2018.01.004
93. Mei Z, Huang J, Qiao B, and Lam AKY. Immune checkpoint pathways in immunotherapy for head and neck squamous cell carcinoma. Int J Oral Sci. (2020) 12:1–9. doi: 10.1038/s41368-020-0084-8
94. Wei X, Chen F, Xin K, Wang Q, Yu L, Liu B, et al. Cancer-testis antigen peptide vaccine for cancer immunotherapy: progress and prospects. Trans Oncol. (2019) 12:733–8. doi: 10.1016/j.tranon.2019.02.008
95. Formentini L, Santacatterina F, Núñez de Arenas C, Stamatakis K, López-Martínez D, Logan A, et al. Mitochondrial ROS production protects the intestine from inflammation through functional M2 macrophage polarization. Cell Rep. (2017) 19:1202–13. doi: 10.1016/j.celrep.2017.04.036
96. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. (2014) 5:5350–68. doi: 10.18632/oncotarget.2110
97. Fraszczak J, Trad M, Janikashvili N, Cathelin D, Lakomy D, Granci V, et al. Peroxynitrite-dependent killing of cancer cells and presentation of released tumor antigens by activated dendritic cells. J Immunol. (2010) 184:1876–84. doi: 10.4049/jimmunol.0900831
98. Griffiths HR, Gao D, and Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. (2017) 12:50–7. doi: 10.1016/j.redox.2017.01.023
99. Laskin DL, Sunil VR, Gardner CR, and Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Ann Review Pharmacol Toxicol. (2011) 51:267–88. doi: 10.1146/annurev.pharmtox.010909.105812
100. Li X, Fang P, Mai J, Choi ET, Wang H, and Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. (2013) 6:19. doi: 10.1186/1756-8722-6-19
101. Mittal M, Siddiqui MR, Tran K, Reddy SP, and Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Sign. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
102. Naik E, and Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. (2011) 208:417–20. doi: 10.1084/jem.20110367
103. Benmoussa K, Garaude J, and Acín-Pérez R. How mitochondrial metabolism contributes to macrophage phenotype and functions. J Mol Biol. (2018) 430:3906–21. doi: 10.1016/j.jmb.2018.07.003
104. Yongsheng Li, Wan YY, and Zhu B. Immune cell metabolism in tumor microenvironment. Adv Exp Med Biol. (2017) 1011:163–96. doi: 10.1007/978-94-024-1170-6_5
105. Azevedo EP, Rochael NC, Guimarães-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril-and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem. (2015) 290:22174–83. doi: 10.1074/jbc.M115.640094
106. Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int J Cancer. (2017) 140:2321–30. doi: 10.1002/ijc.30635
107. Al-Khami AA, Rodriguez PC, and Ochoa AC. Metabolic reprogramming of myeloid-derived suppressor cells (MDSC) in cancer. Oncoimmunol. (2016) 5:e1200771. doi: 10.1080/2162402X.2016.1200771
108. Cubillos-Ruiz JR, Bettigole SE, and Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. (2017) 168:692–706. doi: 10.1016/j.cell.2016.12.004
109. Gowda VMA, and Smitha T. The dendritic cell tool for oral cancer treatment. J Oral Maxill Pathol. (2019) 23:326. doi: 10.4103/jomfp.JOMFP_325_19
110. Ostrand-Rosenberg S, Sinha P, Beury DW, and Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Sem Cancer Biol. (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
111. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nature Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
112. Mu CY, Huang JA, Chen Y, Chen C, and Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Nature Rev Immunol. (2015) 15:18–29. doi: 10.1007/s12032-010-9515-2
113. Krzywinska E, and Stockmann C. Hypoxia, metabolism and immune cell function. Biomed. (2018) 6:56. doi: 10.3390/biomedicines6020056
114. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. (2000). Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 275:21797–800. doi: 10.1074/jbc.C000023200
115. Konjar S, and Veldhoen M. Dynamic metabolic state of tissue resident CD8 T cells. Front Immunol. (2019) 10:1683. doi: 10.3389/fimmu.2019.01683
116. Jung J, Zeng H, and Horng T. Metabolism as a guiding force for immunity. Nature Cell Biol. (2019) 21:85–93. doi: 10.1038/s41556-018-0217-x
117. Palazon A, Goldrath AW, Nizet V, and Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
118. Saravia J, Raynor JL, Chapman NM, Lim SA, and Chi H. Signaling networks in immunometabolism. Cell Res. (2020) 30:1–15. doi: 10.1038/s41422-020-0301-1
119. Newsholme P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr. (2001) 131:2515–22. doi: 10.1093/jn/131.9.2515S
120. Husain Z, Huang Y, Seth P, and Sukhatme VP. Tumor-derived lactate modifies antitumor immune response, effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
121. Chambers AM, Lupo KB, and Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol. (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
122. Wculek SK, Khouili SC, Priego E, Heras-Murillo I, and Sancho D. Metabolic control of dendritic cell functions, digesting information. Front Immunol. (2019) 10:775. doi: 10.3389/fimmu.2019.00775
123. Tran Janco JM, Lamichhane P, Karyampudi L, and Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. (2015) 194:2985–91. doi: 10.4049/jimmunol.1403134
124. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell–like and Foxp3+ regulatory T cells. Nat Immunol. (2010) 11:846. doi: 10.1038/ni.1915
125. Hill M, Tanguy-Royer S, Royer P, Chauveau C, Asghar K, Tesson L, et al. IDO expands human CD4+ CD25high regulatory T cells by promoting maturation of LPS-treated dendritic cells. Eur J Immunol. (2007) 37:3054–62. doi: 10.1002/eji.200636704
126. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
127. Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2, 3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. (2007) 67:792–801. doi: 10.1158/0008-5472.CAN-06-2925
128. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
129. Mukherjee PK, Funchain P, Retuerto M, Jurevic RJ, Fowler N, Burkey B, et al. Metabolomic analysis identifies differentially produced oral metabolites, including the oncometabolite 2-hydroxyglutarate, in patients with head and neck squamous cell carcinoma. BBA Clin. (2017) 7:8–15. doi: 10.1016/j.bbacli.2016.12.001
130. Böttcher M, Renner K, Berger R, Mentz K, Thomas S, Cardenas-Conejo ZE, et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunol. (2018) 7:e1445454. doi: 10.1080/2162402X.2018.1445454
131. Lyssiotis CA, and Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. (2017) 27:863–75. doi: 10.1016/j.tcb.2017.06.003
132. Li L, Cao B, Liang X, Lu S, Luo H, Wang Z, et al. Microenvironmental oxygen pressure orchestrates an anti-and pro-tumoral γδ T cell equilibrium via tumor-derived exosomes. Oncogene. (2019) 38:2830–43. doi: 10.1038/s41388-018-0627-z
Keywords: hypoxia, immune cells, metabolism, microenvironment, oral cancer
Citation: Chaudhary A, Bag S, Arora N, Radhakrishnan VS, Mishra D and Mukherjee G (2020) Hypoxic Transformation of Immune Cell Metabolism Within the Microenvironment of Oral Cancers. Front. Oral. Health 1:585710. doi: 10.3389/froh.2020.585710
Received: 21 July 2020; Accepted: 13 November 2020;
Published: 16 December 2020.
Edited by:
Daniel Lambert, The University of Sheffield, United KingdomReviewed by:
Samapika Routray, All India Institute of Medical Sciences Bhubaneswar, IndiaCraig Murdoch, The University of Sheffield, United Kingdom
Copyright © 2020 Chaudhary, Bag, Arora, Radhakrishnan, Mishra and Mukherjee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geetashree Mukherjee, Z2VldGFzaHJlZS5tdWtoZXJqZWVAdG1ja29sa2F0YS5jb20=; Amrita Chaudhary, Y2hhdWRoYXJ5YnQ4QGdtYWlsLmNvbQ==