 Lemchukwu Amaeshi
Lemchukwu Amaeshi Jacqueline N. Poston
Jacqueline N. Poston Mansour Gergi
Mansour Gergi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Hematol. , 26 February 2025
Sec. Blood Cancer
Volume 4 - 2025 | https://doi.org/10.3389/frhem.2025.1499500
Acquired von Willebrand disease (AvWD) is a rare bleeding disorder that results from a structural or functional defect of von Willebrand factor. AvWD is often associated with a variety of underlying diseases, most frequently lymphoproliferative, myeloproliferative, and cardiovascular disorders. In this report, we present a unique case of a patient presenting with AvWD secondary to smoldering myeloma (SMM) but also found to have hereditary telangiectasias (HHT). While AvWD is not part of the defining criteria for a diagnosis of myeloma in patients with SMM, aggressive anti-myeloma treatment in this case led to a complete resolution of the bleeding disorder. Interestingly, since pomalidomide is an effective agent in HHT, a pomalidomide-based treatment was able to manage all 3 of the patient’s hematologic disorders. This case adds to the body of literature supporting the efficacy of aggressive antimyeloma therapy as a definitive treatment in monoclonal gammopathy-associated AvWD and presents an alternative option to traditional supportive therapy, especially in patients with persistent bleeding.
Acquired von Willebrand disease (AvWD) is a rare, acquired bleeding disorder with an estimated prevalence of 0.04 to 0.13% (1); its risk, however, increases with age. AvWD arises from a multitude of causes ranging from mechanical destruction to antibody mediated clearance of von Willebrand factor (vWF) (1). Autoantibody mediated AvWD can be idiopathic, but it is often associated with an underlying hematologic malignancy such as multiple myeloma (MM), chronic lymphocytic leukemia, and monoclonal gammopathy of undetermined significance (MGUS) (2). In these disorders, AvWD can develop via the presence of circulating inhibitory antibodies to VWF. These non-neutralizing autoantibodies form immune complexes with vWF, resulting in clearance from circulation, adsorption of vWF by tumor cells, increased degradation of vWF, and decreased vWF synthesis (3–5).
The treatment of AvWD aims at controlling bleeding episodes, achieving remission, and potentially curing the underlying disease. Several treatment options have been utilized in autoantibody mediated AvWD: desmopressin (DDAVP), vWF/FVIII concentrates, high-dose intravenous immunoglobulins (IVIG), plasmapheresis, anti-fibrinolytic agents, and recombinant activated factor VII (6, 7). While these treatment options can effectively prevent bleeding episodes, they are not curative and often need to be given indefinitely. Treating the underlying disease might be the only potentially curative option when AvWD arises in the setting of a hematologic malignancy. In multiple myeloma, using standard-of-care myeloma-based chemotherapeutic regimens has been shown to cure AvWD (8, 9). However, the approach is less clear when myeloma-directed therapy is not necessarily indicated, such as MGUS and smoldering multiple myeloma (SMM). Data have been limited to case reports, which have suggested improvement and even cure of AvWD with the treatment of MGUS or SMM using a multiagent myeloma-directed therapy (10, 11,12).
Herein, we discuss a unique case of a patient with SMM who presented with recurrent bleeding from AvWD in the setting of previously undiagnosed Hereditary Hemorrhagic Telangiectasia (HHT). Hereditary Hemorrhagic Telangiectasia (HHT) is an autosomal dominant inherited bleeding disorder that affects 1 in 5000-8000 individuals (13). It is characterized by mucocutaneous telangiectasias and arterio-venous malformations (AVM), which can form in the lung, brain, gastrointestinal tract, spine, or liver (14). The earliest clinical manifestation and primary complaint is usually spontaneous, recurrent epistaxis related to nasal mucosal telangiectasias followed by recurrent gastrointestinal (GI) bleeding (15). Interestingly, recent studies have explored the role of an immunomodulatory agent, pomalidomide, initially designed to treat multiple myeloma, as a potential treatment option for epistaxis related to HHT as it alters angiogenesis-related proteins (16). In our case, targeting our patient’s underlying SMM with Daratumumab, Pomalidomide, and Dexamethasone was not just able to cure his AvWD but also to treat the epistaxis related to HHT.
A 68-year-old male was admitted for severe symptomatic iron deficiency anemia (Hb:8.6 g/dl; Ferritin: 7 ng/ml) due to persistent gastrointestinal (GI) bleeding. A few months prior to this hospitalization, he noticed rectal bleeding. Upper and lower GI endoscopies showed no possible etiology for the bleed. Despite multiple evaluations, he continued to have recurrent bleeding, which led to severe iron deficiency anemia. Additionally, he reported a history of epistaxis since he was ten years old; these episodes resolved spontaneously in early adulthood but recurred a few years before this presentation. He underwent multiple evaluations, including laryngoscopies and nasal exams performed by the otorhinolaryngology team, which did not reveal any arteriovenous malformations (AVM) or mucocutaneous telangiectasias. He denied a family history of bleeding and had three prior surgeries, one of which was a major spinal fusion, which had no complicating bleeding diathesis. On further laboratory testing, he had a normal PTT and PT; however, a factor VIII assay was low at 49%, and VWF antigen and activity were 42% and 22%, respectively; this was accompanied by the absence of high-molecular-weight VWF multimers. His blood group is O negative. These results were confirmed on repeat testing. This seemed to be consistent with AvWD due to the recent history of GI bleeding and the lack of family history of bleeding or bleeding with previous hemostatic challenges. A serum protein electrophoresis (SPEP) and serum free light chain levels were ordered to assess whether an underlying plasma cell disorder was present, and it showed an IgG Kappa monoclonal spike at 0.8 g/dl. His involved-to-uninvolved serum-free light chain ratio was 6.98g/dl. A bone marrow biopsy revealed 40% plasma cell involvement. Further evaluation using cytogenetics and fluorescent in situ hybridization identified a CCND1/IGH fusion, which typically corresponds to a t(11;14) chromosomal abnormality. The patient’s anemia was thought to be primarily related to his iron deficiency, as it was corrected on prior occasions with intravenous iron, and he had no hypercalcemia or renal dysfunction. A staging PET scan did not show any evidence of myeloma-related bone disease, but incidentally showed an AVM in the lung (see image in Supplementary Materials). With these findings, a diagnosis of AvWD associated with SMM was made due to the absence of myeloma-related organ disease. His International Myeloma Working Group risk stratification score was 5, which indicates a 2-year risk of disease progression of 26%. However, the presence of the AVM and the lifelong history of epistaxis were concerning for HHT; therefore, a genetic test was done to explore this further. Intravenous immunoglobulin (IVIG) was started immediately as a potential therapeutic option to control the gastrointestinal bleeding. Within a few weeks of starting IVIG, there was an exponential increase in the levels of VWF antigen and its activity, from 42% at baseline to 131%, and from 22% to 95%, respectively. Additionally, Factor VIII levels rose by over 150%, from 49% at baseline to 136% (Table 1, Figure 1), effectively resolving the patient’s GI bleeding. However, he continued to have intermittent epistaxis despite normalized VWF activity and antigen while on prophylactic IVIG 1 g/kg every three weeks. Genetic testing revealed a pathologic variant in the ENG gene: c.89G>A (p.Cys30Tyr), consistent with a diagnosis of HHT.
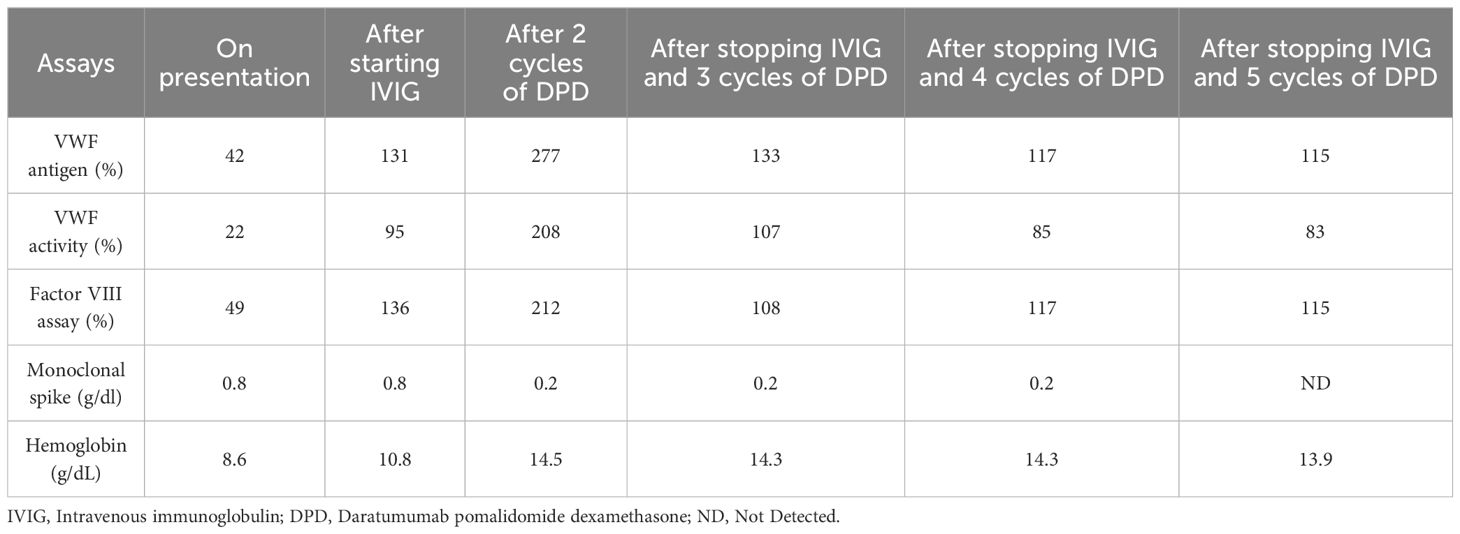
Table 1. Evolution of coagulation and myeloma parameters with treatment.
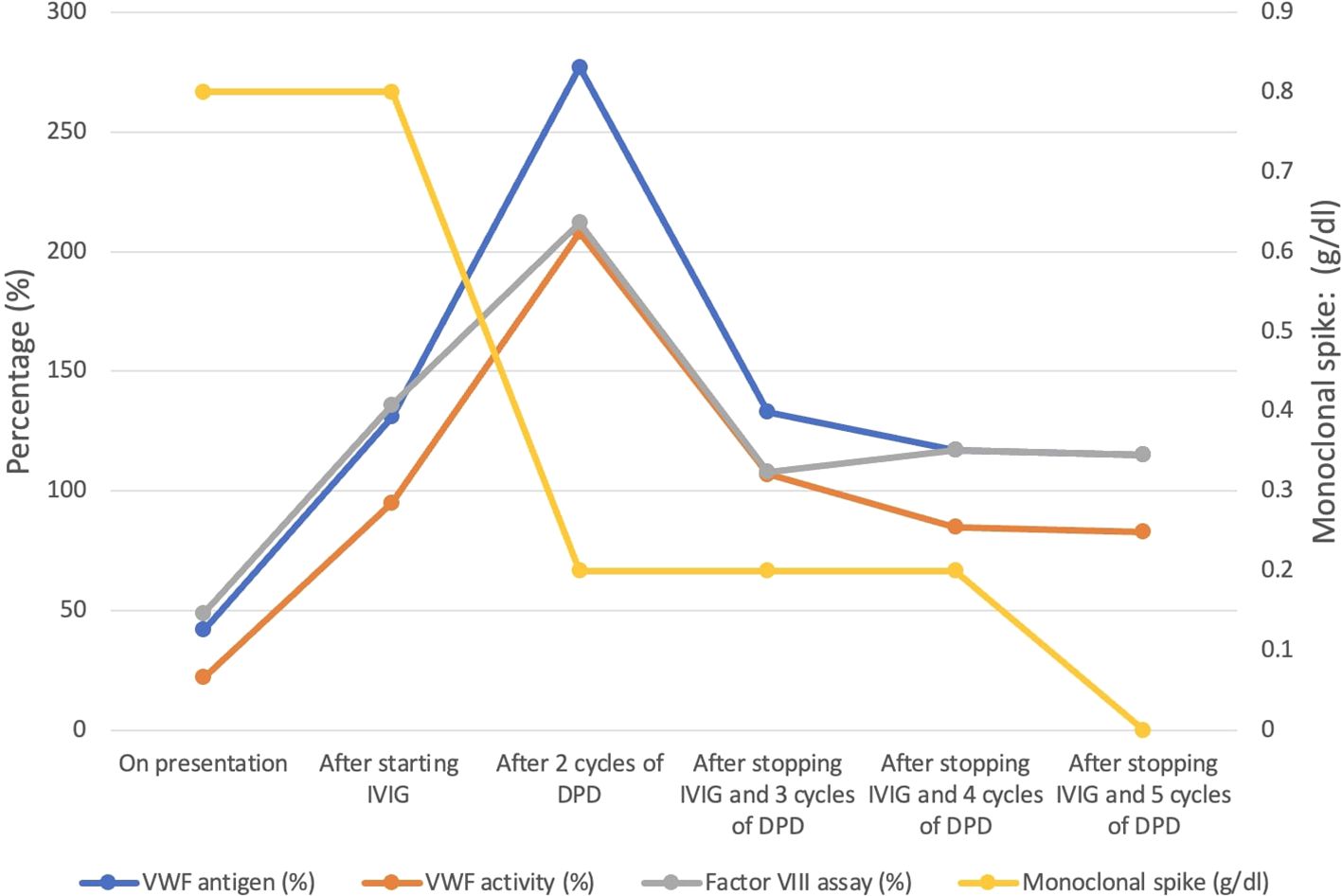
Figure 1. Evolution of coagulation and myeloma parameters with treatment.
Although IVIG was preventing spontaneous GI bleeding from AvWD, the patient continued to have epistaxis, thought to be due to his underlying HHT. Given the limited data on pomalidomide mitigating HHT related epistaxis, combining pomalidomide with a multiagent anti-myeloma regimen was considered to concurrently treat his HHT related epistaxis and his SMM, potentially curing his AvWD while also reducing the frequency of his epistaxis. He was started on the combination of daratumumab, pomalidomide, and dexamethasone (DPD). The patient initially remained on IVIG; however, after two cycles of treatment with DPD, his IgG kappa monoclonal spike decreased from 0.8 g/dl to 0.2 g/dl, and after five cycles, it became undetectable. He did not experience recurrent epistaxis or any other bleeding. Subsequently, IVIG was discontinued. Although there was a slight decrease in the levels of VWF antigen, VWF activity, and Factor VIII assay after stopping IVIG, they remained within normal limits (Table 1). The patient tolerated the treatment very well, with only grade 1 fatigue reported as a side effect. He was monitored closely thereafter, and he continued to have normal VWF levels with no further bleeding episodes. The patient continues to respond to the treatment. Lenalidomide as maintenance therapy in SMM has been shown to delay its progression to multiple myeloma (17). Given that pomalidomide belongs to the same class as lenalidomide and has a mechanism of action similar to lenalidomide, along with its efficacy in HHT, we continued him on pomalidomide. However, we stopped daratumumab to reduce the treatment burden and the number of visits to the clinic or infusion center. Although his AVM was repaired through endovascular embolization, we plan to conduct surveillance annual PET CT scans. On the single agent Pomalidomide, the patient continues to have an excellent response to treatment without recurrence of his von Willebrand disease or epistaxis. We intend to treat him for 24 months.
This is a case of a patient with an undiagnosed inherited bleeding disorder whose bleeding phenotype increased after developing AvWD from SMM. Careful clinical history and observing persistent epistaxis after normalizing his VWF levels with IVIG led to a diagnosis of HHT. By adding pomalidomide, which is effective for HHT and SMM, to his myeloma treatment regimen, the patient’s bleeding resolved from the triple effect of treating AvWD, SMM, and HHT: ‘hitting three birds with one stone.’
Since the initial report of AvWD in a patient with systemic lupus erythematosus over 50 years ago, most of our understanding of this disease in terms of clinical and laboratory features, as well as treatment approaches, has largely been based on case reports, case series, and retrospective studies. This is because the disease is rare and underreported, and most cases are asymptomatic (10, 18, 19). Lymphoproliferative disorders are the most common cause of AvWD. In these disorders, sequestration of high VWF multimers after binding to monoclonal antibodies is the recognized pathophysiologic mechanism, similar to inherited type 2A and type 3 vWD (20, 21).
Symptomatic AvWD presents as 1) mucocutaneous bleeding, as seen in our patient, and 2) excessive bleeding following trauma or a surgical procedure. Therefore, treatment goals are to control acute bleeding episodes, prevent bleeding in high-risk situations, and ultimately treat the underlying pathology (2, 7, 19). Established therapies for AvWD include desmopressin (DDAVP), vWF/FVIII concentrates, antifibrinolytic agents, high-dose intravenous immunoglobulins, and recombinant activated factor VII (rFVIIa) (6, 7). We used IVIG to control the bleeding episodes in our patient. Through anti-idiotypic antibodies and modulation of B-cell and T-cell activities, IVIG can neutralize the effect of circulating autoantibodies produced by the abnormal plasma cells, which drive immunologic clearance of VWF in AvWD (22, 23). In a report of 10 patients with AvWD due to MGUS, IVIG produced more sustained levels of VWF compared to DDAVP or VWF concentrates (24). However, the benefits of IVIG are limited to IgG MGUS.
Although IVIG is effective in controlling acute bleeding episodes and preventing excessive intra/post-operative bleeding, the effects are temporary, and repeated dosing may be necessary. In some cases of significant bleeding episodes, a combination of IVIG and VWF/FVIII concentrates may be required. Although IVIG neutralizes the circulating autoantibodies, it has little effect on the plasma cells producing autoantibodies (12). Myeloma-based therapies, through their direct toxic effect on the plasma cell clones producing these antibodies, can theoretically lead to more lasting remission of bleeding episodes. While such therapies are not routinely used to address AvWD without symptomatic myeloma, there have been documented cases of long-term remission of AvWD with myeloma-based therapies refractory to traditional AvWD treatments (11, 12). In a case report by Micahel Iarossi et al., Daratumumab, Lenalidomide, and Dexamethasone successfully treated AvWD in a patient with SMM (10). Similar to these cases, our patient had SMM and clinically significant AvWD, requiring IVIG; however, our patient also had HHT, which made our case unique. IVIG was insufficient to control the bleeding episodes related to HHT, so we instituted a myeloma-based regimen. Our decision to use a multi-drug combination regimen -pomalidomide, dexamethasone, and daratumumab - stems from reports showing durable responses in SMM associated AvWD with treatment intensification regimens (11) and the effectiveness of pomalidomide in HHT. Pomalidomide is a third-generation immunomodulatory drug (IMiD) with significant activity in multiple myeloma (MM) and has been shown to yield positive results in patients with MM who are refractory to lenalidomide (25, 26). They work by directly suppressing or killing clonal plasma cells and activating T-cells and natural killer cells, which aid in killing malignant plasma cell clones (26). In addition, pomalidomide has been shown to be efficacious in HHT, likely by impacting angiogenesis by interfering with VEGF (27, 28). In a recent randomized, double-blind, placebo-controlled trial – PATH-HHT trial - pomalidomide reduced epistaxis in patients with HHT compared to placebo (29). Thalidomide, a first-generation IMiD used for treating multiple myeloma, is more readily available and affordable than pomalidomide and is equally effective in HHT, especially in resource-limited settings (30). However, its significant toxicity profile, including painful peripheral neuropathy, renders it a less favorable option. Interestingly, lenalidomide, which is widely available and less toxic than thalidomide, has not been studied in HHT. Clinical trials evaluating lenalidomide in HHT could provide valuable insights into its efficacy and safety, thus expanding therapeutic options for the HHT patient population.
Our case, rare and unique, provided the golden opportunity where the use of a myeloma-based regimen with pomalidomide as the backbone would effectively treat both bleeding disorders – AvWD and HHT - and prevent the progression of SMM to multiple myeloma, which occurs at 10% annually (31). This case adds to the existing body of literature demonstrating that treating asymptomatic plasma cell disorders, such as MGUS and SMM, is effective in curing and preventing further bleeding with AvWD. Although myeloma treatment regimens can be toxic, the burden associated with severe iron deficiency anemia, recurrent bleeding, and poor quality of life may justify this toxicity.
Patient Perspective: ‘I want to share with you how the treatment has affected my life. A year ago, I had been experiencing unexplained bleeding for over two years from my nose and rectal cavity. I had no idea what was happening to my health, and I was discouraged. Before melanoma, Von Willebrand Disease, and HHT affected my life, I was very active outdoors, enjoying photography, fly fishing, raptor birding in wildlife areas, and long bike rides. All these activities required a great deal of energy, which I lost.’ The treatment made a positive change in my life with no further bleeding, and I am starting to regain my energy. I want to thank everyone for my health care and the treatment.’
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
LA: Writing – original draft, Writing – review & editing. MG: Conceptualization, Writing – original draft, Writing – review & editing. JP: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2025.1499500/full#supplementary-material
1. Kumar S, Pruthi RK, Nichols WL. Acquired von Willebrand disease. Mayo Clin Proc. (2002) 77:181–7. doi: 10.4065/77.2.181
2. Federici A, Budde U, Castaman G, Rand J, Tiede A. Current diagnostic and therapeutic approaches to patients with acquired von willebrand syndrome: A 2013 update. Semin Thromb Hemost. (2013) 39:191–201. doi: 10.1055/s-0033-1334867
3. Richard C, Cuadrado MA, Prieto M, Batlle J, López Fernández MF, Rodriguez Salazar ML, et al. Acquired von Willebrand disease in multiple myeloma secondary to absorption of von Willebrand factor by plasma cells. Am J Hematol. (1990) 35:114–7. doi: 10.1002/ajh.2830350210
4. van Genderen PJ, Vink T, Michiels JJ, van ‘t Veer MB, Sixma JJ, van Vliet HH. Acquired von Willebrand disease caused by an autoantibody selectively inhibiting the binding of von Willebrand factor to collagen. Blood. (1994) 84:3378–84.
5. Mohri H, Hisanaga S, Mishima A, Fujimoto S, Uezono S, Okubo T. Autoantibody inhibits binding of von Willebrand factor to glycoprotein Ib and collagen in multiple myeloma: recognition sites present on the A1 loop and A3 domains of von Willebrand factor. Blood Coagul Fibrinolysis. (1998) 9:91–7. doi: 10.1097/00001721-199801000-00012
6. Federici AB. Therapeutic approaches to acquired von Willebrand syndrome. Expert Opin Investig Drugs. (2000) 9:347–54. doi: 10.1517/13543784.9.2.347
7. Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood. (2011) 117:6777–85. doi: 10.1182/blood-2010-11-297580
8. Katagiri S, Akahane D, Amano K, Ohyashiki K. Long-term remission of acquired von Willebrand syndrome associated with multiple myeloma using bortezomib and dexamethasone therapy. Haemophilia. (2016) 22:e557–e559. doi: 10.1111/hae.13077
9. Ouadii A, Pauline C, Sophie V, Antoine H, Aurore P, Teresa B. Management of a young patient with high-risk multiple myeloma complicated by acquired von willebrand syndrome: A diagnostic and therapeutic emergency. Plasmatology. (2024) 18. doi: 10.1177/26348535231222994
10. Iarossi M, Vekemans MCM, Weynants N, Hermans C. Acquired von willebrand syndrome associated with a smoldering multiple myeloma, successfully treated by daratumumab, lenalidomide, and dexamethasone. Acta Haematol. (2024) 147:587–91. doi: 10.1159/000536650
11. Jeryczynski G, Agis H, Eichinger-Hasenauer S, Krauth MT. Successful treatment of acquired von Willebrand syndrome associated with monoclonal gammopathy. Wien Klin Wochenschr. (2022) 134:478–82. doi: 10.1007/s00508-022-02012-3
12. Abou-Ismail MY, Rodgers GM, Bray PF, Lim MY. Acquired von Willebrand syndrome in monoclonal gammopathy – A scoping review on hemostatic management. Res Pract Thromb Haemost. (2021) 5:356–65. doi: 10.1002/rth2.12481
13. Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist’s perspective. Haematologica. (2018) 103:1433–43. doi: 10.3324/haematol.2018.193003
14. Grigg C, Anderson D, Earnshaw J. Diagnosis and treatment of hereditary hemorrhagic telangiectasia. Ochsner J. (2017) 17:157–61.
15. Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. (1989) 32:291–7. doi: 10.1002/ajmg.1320320302
16. Al-Samkari H, Kasthuri RS, Iyer VN, Pishko AM, Decker JE, Weiss CR, et al. PATH-HHT, a double-blind, randomized, placebo-controlled trial in hereditary hemorrhagic telangiectasia demonstrates that pomalidomide reduces epistaxis and improves quality of life. Blood. (2023) 142:LBA–3-LBA-3. doi: 10.1182/blood-2023-191983
17. Mateos MV, Hernández MT, Giraldo P, de la Rubia J, de Arriba F, López Corral L, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. New Engl J Med. (2013) 369:438–47. doi: 10.1056/NEJMOA1300439
18. Simone JV, Cornet JA, Abildgaard CF. Acquired von Willebrand’s syndrome in systemic lupus erythematosus. Blood. (1968) 31:806–12. doi: 10.1182/blood.V31.6.806.806
19. Franchini M, Mannucci PM. Acquired von Willebrand syndrome: focused for hematologists. Haematologica. (2020) 105:2032–7. doi: 10.3324/haematol.2020.255117
20. Michiels JJ, Budde U, van der Planken M, van Vliet HH, Schroyens W, Berneman Z. Acquired von Willebrand syndromes: clinical features, aetiology, pathophysiology, classification and management. Best Pract Res Clin Haematol. (2001) 14:401–36. doi: 10.1053/beha.2001.0141
21. Federici AB, Rand JH, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. (2000) 84:345–9. Erratum in: Thromb Haemost 2000 Oct;84(4):739.
22. Boros P, Gondolesi G, Bromberg JS. High dose intravenous immunoglobulin treatment: Mechanisms of action. Liver Transplantation. (2005) 11:1469–80. doi: 10.1002/lt.20594
23. Dicke C, Schneppenheim S, Holstein K, Spath B, Bokemeyer C, Dittmer R, et al. Distinct mechanisms account for acquired von Willebrand syndrome in plasma cell dyscrasias. Ann Hematol. (2016) 95:945–57. doi: 10.1007/s00277-016-2650-x
24. Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood. (1998) 92:2707–11. doi: 10.1182/blood.V92.8.2707
25. McCurdy AR, Lacy MQ. Pomalidomide and its clinical potential for relapsed or refractory multiple myeloma: an update for the hematologist. Ther Adv Hematol. (2013) 4:211–6. doi: 10.1177/2040620713480155
26. Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. (2010) 24:22. doi: 10.1038/LEU.2009.236
27. Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. (2010) 16:420–8. doi: 10.1038/nm.2131
28. Swaidani S, Kundu S, Samour M, Silver B, Parambil J, Thomas S, et al. Pomalidomide reduces bleeding and alters expression of angiogenesis-related proteins in patients with hereditary hemorrhagic telangiectasia. Blood. (2019) 134:5761–1. doi: 10.1182/blood-2019-127344
29. Zhang E, Virk ZM, Rodriguez-Lopez J, Al-Samkari H. Hereditary hemorrhagic telangiectasia may be the most morbid inherited bleeding disorder in women. Blood Adv. (2024) 8:3166–72. doi: 10.1182/bloodadvances.2023011961
30. Harrison L, Kundra A, Jervis P. The use of thalidomide therapy for refractory epistaxis in hereditary haemorrhagic telangiectasia: systematic review. J Laryngol Otol. (2018) 132:866–71. doi: 10.1017/S0022215118001536
31. Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. (2010) 24:1121. doi: 10.1038/LEU.2010.60
Keywords: acquired von Willebrand disease, smoldering multiple myeloma, hereditary hemorrhagic telangiectasias, pomalidomide, daratumumab, IVIg, case report
Citation: Amaeshi L, Poston JN and Gergi M (2025) Acquired von Willebrand disease associated with smoldering multiple myeloma and hereditary hemorrhagic telangiectasia successfully treated with daratumumab, pomalidomide, and dexamethasone: hitting three birds with one stone? – case report. Front. Hematol. 4:1499500. doi: 10.3389/frhem.2025.1499500
Received: 20 September 2024; Accepted: 04 February 2025;
Published: 26 February 2025.
Edited by:
Attaya Suvannasankha, Indiana University, United StatesReviewed by:
Matilde Scaldaferri, AOU Città della Salute e della Scienza di Torino, ItalyCopyright © 2025 Amaeshi, Poston and Gergi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lemchukwu Amaeshi, bGFtYWVzaGlAbW9udGVmaW9yZS5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.