 Somedeb Ball1
Somedeb Ball1 Najla H. Al Ali2
Najla H. Al Ali2 Akriti G. Jain3
Akriti G. Jain3 Luis E. Aguirre4
Luis E. Aguirre4 Seongseok Yun2Onyee Chan2
Seongseok Yun2Onyee Chan2 Zhuoer Xie2
Zhuoer Xie2 David A. Sallman2
David A. Sallman2 Jeffrey Lancet2Eric Padron2Rami S. Komrokji2Andrew T. Kuykendall2*
Jeffrey Lancet2Eric Padron2Rami S. Komrokji2Andrew T. Kuykendall2*- 1Division of Hematology and Medical Oncology, Vanderbilt Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN, United States
- 2Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, United States
- 3Taussig Cancer Institute, Cleveland Clinic Foundation, Cleveland, OH, United States
- 4Department of Medical Oncology, Adult Leukemia Program, Dana-Farber Cancer Institute, Boston, MA, United States
Background: Disease related anemia in myelofibrosis (MF) is common and prognostically detrimental. Anemia in MF poses a therapeutic challenge as it contributes to poor quality of life and often interferes with JAK inhibitor therapy. Still, the causes for anemia in MF are varied raising the question as to whether all patients with MF-related anemia should be viewed through the same prognostic lens.
Methods: In this retrospective study, we analyzed clinical and genomic data of patients with MF-related anemia using an institutional MF database. Anemia was defined as the requirement of red blood cell transfusions or a hemoglobin level of <10 g/dL at presentation. Multivariable analysis performed using Cox regression formed the basis of a proposed prognostic scoring system for patients with anemic MF.
Results: Among 739 patients with MF, 365 (49.5%) were anemic at presentation. Anemic patients were older, had lower platelet count, lower serum albumin, and higher ferritin level than non-anemic patients. The presence of a JAK2 mutation was less common, whereas mutations in U2AF1 and EZH2 were enriched in the anemic cohort. Blast phase transformation was more common in anemic patients. After a median follow up of 34.5 months, median overall survival (OS) was significantly shorter in anemic vs. non-anemic MF (30.2 vs. 73.9 months; p<0.01). Leukocytosis, thrombocytopenia, low serum albumin, and the presence of a mutation involving SRSF2 or TP53 were independent predictors of inferior OS in anemic MF on multivariable analysis. A proposed prognostic model including these factors stratified anemic MF cohort into low, intermediate, and high-risk categories, with median OS of 69, 37.7, and 11.6 months, respectively (p <0.01).
Conclusions: Our study highlights the heterogeneity of patients with MF and anemia and identifies key prognostic correlates in this subgroup. Our proposed model may help guide therapeutic decision-making in this high-risk cohort.
Introduction
Myelofibrosis (MF) is a chronic myeloid malignancy that can occur either de-novo (primary MF) or after a prior diagnosis of polycythemia vera or essential thrombocythemia (1). Patients with MF have heterogeneous clinical features and variable disease course (1, 2). Almost all patients with MF develop anemia during the course of their disease (3). The causes of anemia in MF vary amongst patients. Contributing factors include an impaired bone marrow niche, ineffective hematopoiesis due to somatically acquired gene mutations, intramedullary hemolysis, organomegaly, chronic inflammation, and an inappropriate erythropoietin response to anemia (4, 5). Across numerous prognostic scoring systems, disease-related anemia consistently demonstrates a detrimental prognostic impact in patients with MF (2, 6, 7).
On the other hand, anemia can also result after treatment with a Janus kinase (JAK) inhibitor such as ruxolitinib. Treatment-induced anemia often leads to suboptimal dosing and is a common reason for therapy discontinuation (8, 9). Studies have shown that treatment-related anemia does not impact clinical response to ruxolitinib (10); however, this data comes from clinical trial populations in which dose modification and treatment discontinuation due to anemia occur less frequently. Often, it can be hard to distinguish disease-related anemia from treatment-related anemia given the frequency of the former and ubiquitous nature of the latter. The recently proposed RR6 model showed that red blood cell transfusion dependence (whether driven by disease or treatment) within first 6 months of ruxolitinib treatment is predictive of worse OS (11). More recently approved JAK inhibitors like momelotinib and pacritinib have shown beneficial effect on MF-related anemia and may provide an alternative for some patients (12–14). Still, questions remain. Should anemia in MF be viewed as uniformly detrimental regardless of the underlying cause? Which patients would benefit from a change in therapy? Answering these questions requires a more nuanced understanding of what factors drive poor outcomes in anemic MF.
In the current study, we aimed to identify clinical and molecular factors that significantly influence outcomes in patients with MF and anemia. From this, we propose distinct prognostic clusters to aid clinicians in better evaluating disease risk, thereby informing critical treatment decisions.
Materials and methods
Study population and datapoints
In this single-center retrospective cohort study, we included all adult patients who presented to Moffitt Cancer Center between 2001 and 2021 with a diagnosis of primary MF per World Health Organization (WHO) 2016 classification criteria, or post-polycythemia vera or post-essential thrombocythemia MF per International Working Group (IWG) criteria (15, 16). The study protocol was approved by the institutional review board at the University of South Florida. Clinical and genomic data on eligible patients were collected from a well-annotated institutional MF database.
Clinical and genomic variables were collected at time of first presentation to our center. Constitutional symptoms were largely recorded by treating physicians as part of routine clinical care. Anemia was defined as the requirement of red blood cell transfusions and/or a hemoglobin level of <10 g/dL at presentation. Patients who started ruxolitinib ≤ 6 months prior to presentation were excluded from this analysis to better isolate disease-related rather than JAK inhibitor-driven anemia.
Cytogenetic findings were used to categorize patients into ‘favorable’, ‘unfavorable’, and ‘very high risk’ categories (17). Data on relevant somatic mutations was obtained as part of routine clinical care using an in-house targeted DNA sequencing panel for myeloid malignancies. Although this panel has evolved over the years, it has always had coverage for JAK2, MPL, CALR, TET2, DNMT3A, ASXL1, SRSF2, U2AF1, EZH2, IDH1, IDH2, KRAS, NRAS, RUNX1, and TP53. A minimum variant allele frequency of 5% was used to call single nucleotide variants, and a cutoff of 10% was used for insertions or deletions. Pathogenicity of variants was determined by an internally relied on variant classification system, based on joint consensus recommendations of the Association for Molecular Pathology, American Society of Clinical Oncology, and the College of American Pathologists, in conjunction with review of COSMIC and cBioPortal databases as needed.
Statistical analysis
Categorical variables were summarized in the form of numbers and percentages. Standard descriptive statistics were used for the continuous variables. Categorical variables were compared using the Fisher’s exact test or Chi-squared test, as applicable. Comparisons of continuous variables were performed with the Wilcoxon rank sum test or Kruskal–Wallis test, as appropriate. OS was estimated using the Kaplan-Meier method with survival estimates being compared using log rank test. OS was calculated from the time of presentation. A univariate Cox regression analysis identified variables that correlated with OS. For serum albumin, median level in study cohort was used as cut-off for regression analysis. All variables with a p value of <0.10 were included in the subsequent multivariate analyses. Due to missing genomic data in a significant proportion of patients, two multivariable analyses were performed. The first was performed on the entire cohort and only included clinical variables. The second was only performed on patients with available genomic information. From these findings, a novel prognostic scoring system was proposed, incorporating the independent predictors for OS on multivariate analyses with scores assigned based on magnitude of hazard ratios (HR). All statistical analyses were conducted in IBM SPSS statistics (version 29).
Results
Clinical and genomic characteristics
Among total 737 patients with MF, 365 (49.5%) patients were anemic at presentation, including 205 patients who required transfusions. The median age at presentation was 68.5 (range, 18.9-91.4) years. Median time from MF diagnosis to presentation was 2.8 months. Clinical and genomic characteristics of study population are shown in Table 1. Patients with anemic MF were significantly older (median age, 69.8 vs. 67.2 years; p <0.01). Anemic patients were more likely to be diagnosed with primary MF (72% vs. 65%; p = 0.04). Patients with anemic MF had a lower median WBC (6.9 x 109/L vs. 11 x 109/L; p <0.01) and platelet count (146 x 109/L vs. 279 x 109/L; p <0.01) than non-anemic patients. Serum erythropoietin and ferritin levels were significantly higher in anemic MF (median, 91 vs. 18 mIU/ml, and 567 vs. 136 microgram/L, respectively; p <0.01). Median serum albumin was 4.1 gm/dL in anemic patients vs. 4.4 gm/dL in others (p <0.01). Patients with anemic MF had higher frequency of advanced (MF 2 or 3) marrow fibrosis.
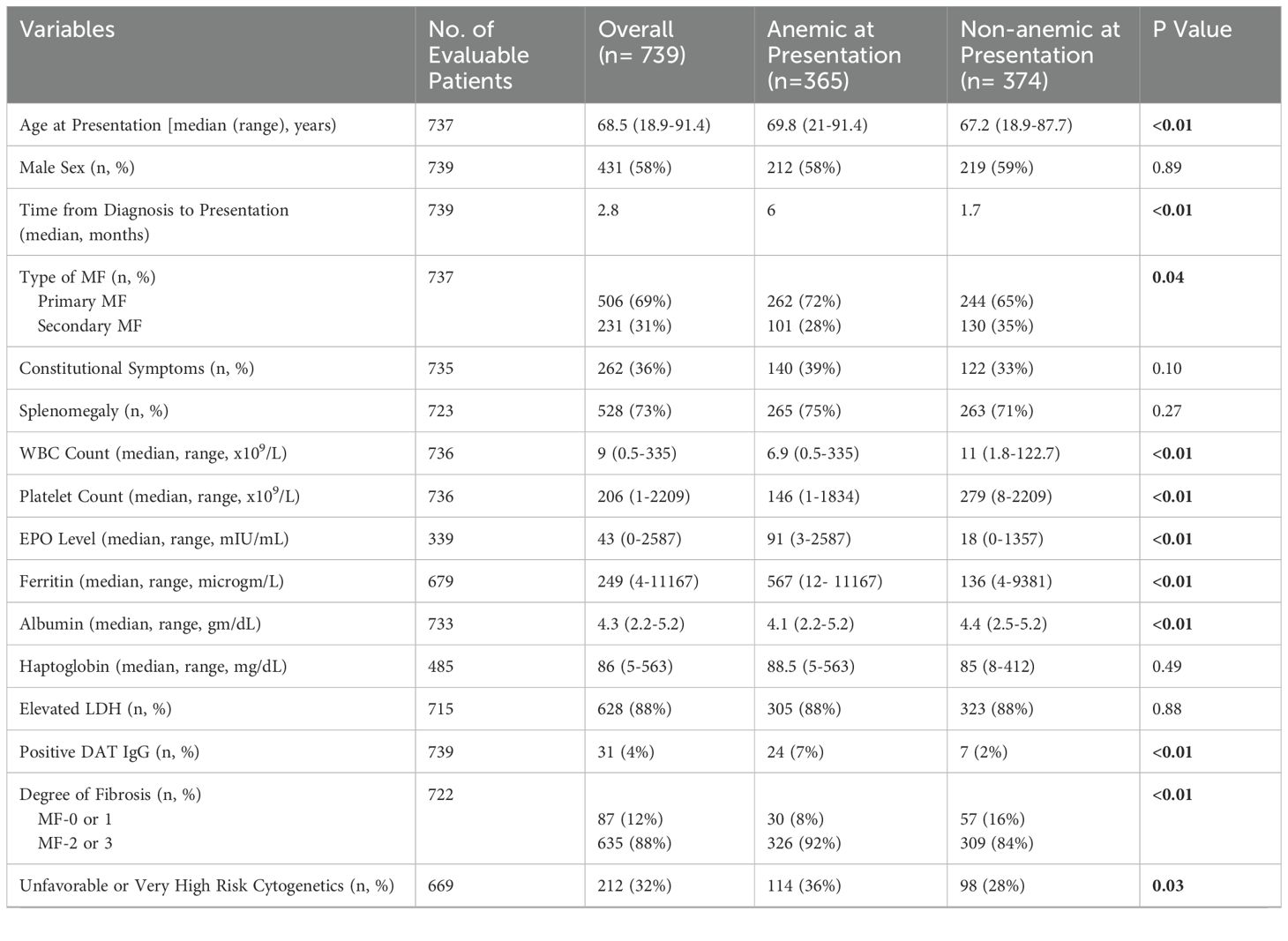
Table 1. Clinical and genomic characteristics of study population.
MF patients with anemia more frequently harbored unfavorable or very high-risk cytogenetics than patients without disease-driven anemia (36% vs. 28%; p =0.03). Mutations in JAK2 were less common in anemic than non-anemic subgroup (57% vs. 65%; p =0.02), whereas mutations involving U2AF1 (20% vs. 3%; p <0.01) and EZH2 (11% vs. 6) were more prevalent in patients with anemic MF.
Treatment and outcomes
In our study population, the treatment modalities across all lines of therapy included erythropoiesis stimulating agents (ESA) (32%), hydroxyurea (34%), thalidomide or lenalidomide (24%), ruxolitinib (38%), androgen (8%), and interferon (5%). Use of ESA, thalidomide or lenalidomide, and androgen was more common, and treatment with hydroxyurea and ruxolitinib was less common in the anemic MF subgroup (Table 1). Overall, 14% of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT), with this intervention being more commonly used in anemic patients (17% vs. 11%; p = 0.03).
Rate of blast phase transformation was significantly higher in patients with anemic MF than those without anemia (14% vs. 7%; p <0.01). After a median follow up duration of 34.5 months, median OS was 45.4 (95% confidence interval [CI]: 39.9-50.8) months for the entire study population. Anemia at presentation was associated with significantly worse OS in patients with MF (30.2 [95% CI: 24-36.4] vs. 73.9 [95% CI: 54.6-93.3] months; p <0.01).
Prognostic factors for overall survival
In the univariate analysis (Table 2), variables associated with an inferior OS in the anemic MF cohort (n=365) included age > 65 years (HR 2.07; p <0.01), constitutional symptoms (HR 1.59; p <0.01), WBC count >25 x 109/L (HR 3.23; p <0.01), platelet count <100 x 109/L (HR 1.57; p <0.01), peripheral blasts ≥ 1% (HR 1.66; p <0.01), serum albumin <4.1 gm/dL (HR 1.86; p <0.01), and the presence of an ASXL1 (HR 1.23; p <0.01), EZH2 (HR 1.22; p <0.01), SRSF2 (HR 1.21; p =0.01), or TP53 (HR 1.22; p <0.01) mutation.
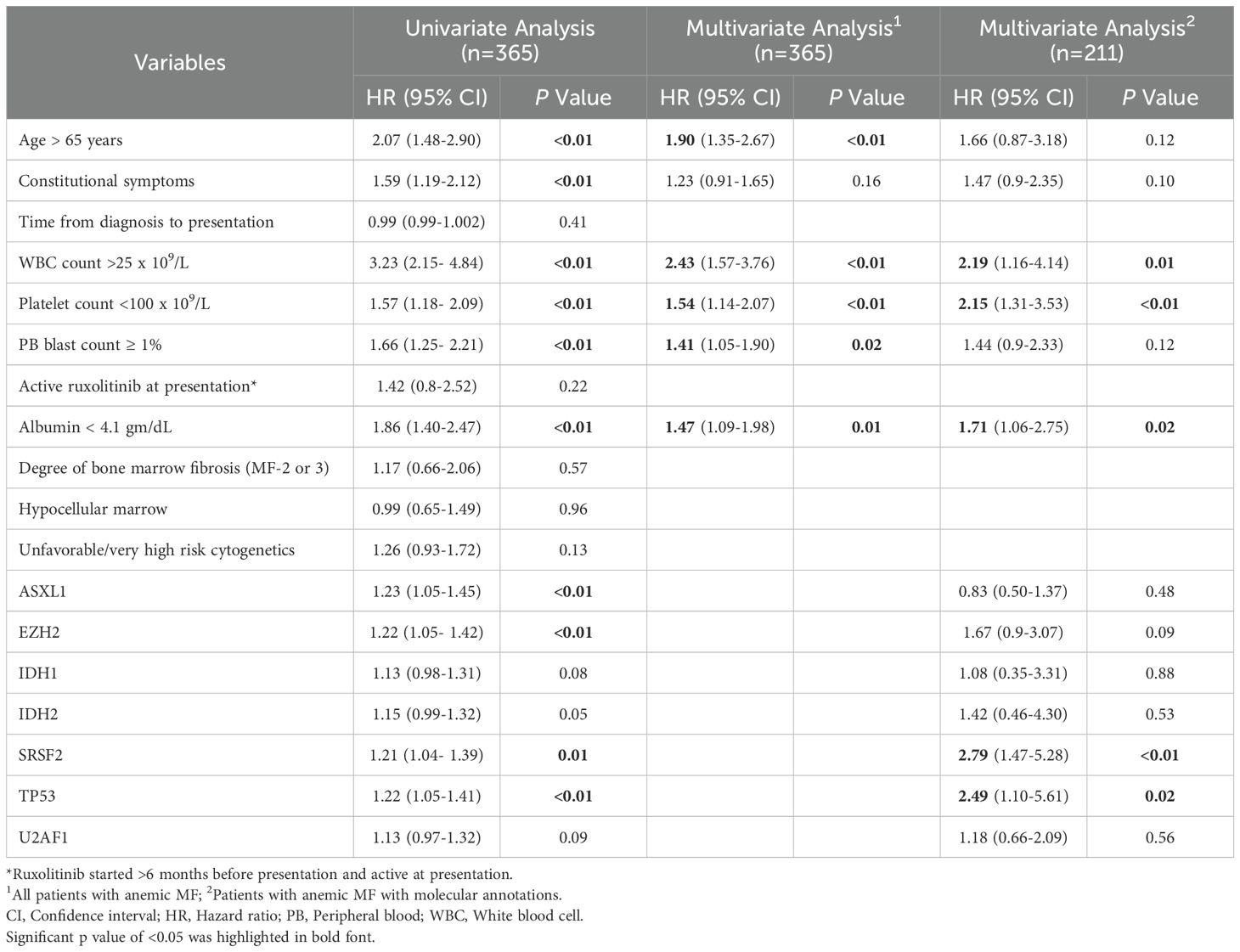
Table 2. Results of univariate and multivariate Cox-regression analysis in anemic myelofibrosis cohort.
Two separate multivariate analyses were performed. In the first multivariate analysis, we used data from entire anemic MF cohort (n=365) and included all clinical and laboratory variables with borderline significance (p <0.10) on univariate analysis. Age >65 years (HR 1.90; p <0.01), WBC count >25 x 109/L (HR 2.43; p <0.01), platelet count <100 x 109/L (HR 1.54; p <0.01), peripheral blasts ≥ 1% (HR 1.41; p =0.02), and serum albumin <4.1 gm/dL (HR 1.47; p =0.01) were the independent prognostic factors for OS.
The second multivariate analysis included only the anemic MF cases with available baseline molecular information (n=211). In this analysis, clinical, laboratory and genomic variables were included. WBC count >25 x 109/L (HR 2.19, 95% CI: 1.16-4.14; p =0.01), platelet count <100 x 109/L (HR 2.15, 95% CI: 1.31-3.53; p <0.01), serum albumin <4.1 gm/dL (HR 1.71, 95% CI: 1.06-2.75; p =0.02), SRSF2 mutation (HR 2.79, 95% CI: 1.47-5.28; p <0.01), and TP53 mutation (HR 2.49, 95% CI: 1.10-5.61; p =0.02) were the independent predictors of OS in patients with MF and anemia (Table 2, Figure 1).

Figure 1. Multivariate analysis forest plot for overall survival in patients with anemic myelofibrosis. Alb, Albumin; CI, Confidence interva; k, Thousand; WBC, White blood cell.
Development of prognostic scoring system
Based upon these findings, we developed a prognostic scoring system comprised of the five significant variables in the multivariate analysis on molecularly annotated cohort of anemic MF. This score included serum albumin <4.1 gm/dL (1 point), WBC count >25 x 109/L (1.5 points), platelet count <100 x 109/L (1.5 points), TP53 mutation (1.5 points), and SRSF2 mutation (2 points). Patients in the low risk (no risk factor; aggregate score 0), intermediate risk (1 risk factor; aggregate score 1 to 2), and high risk (>1 risk factor; aggregate score >2) categories had distinct median OS estimates of 69, 37.7, and 11.6 months, respectively (p <0.01) (Figure 2).
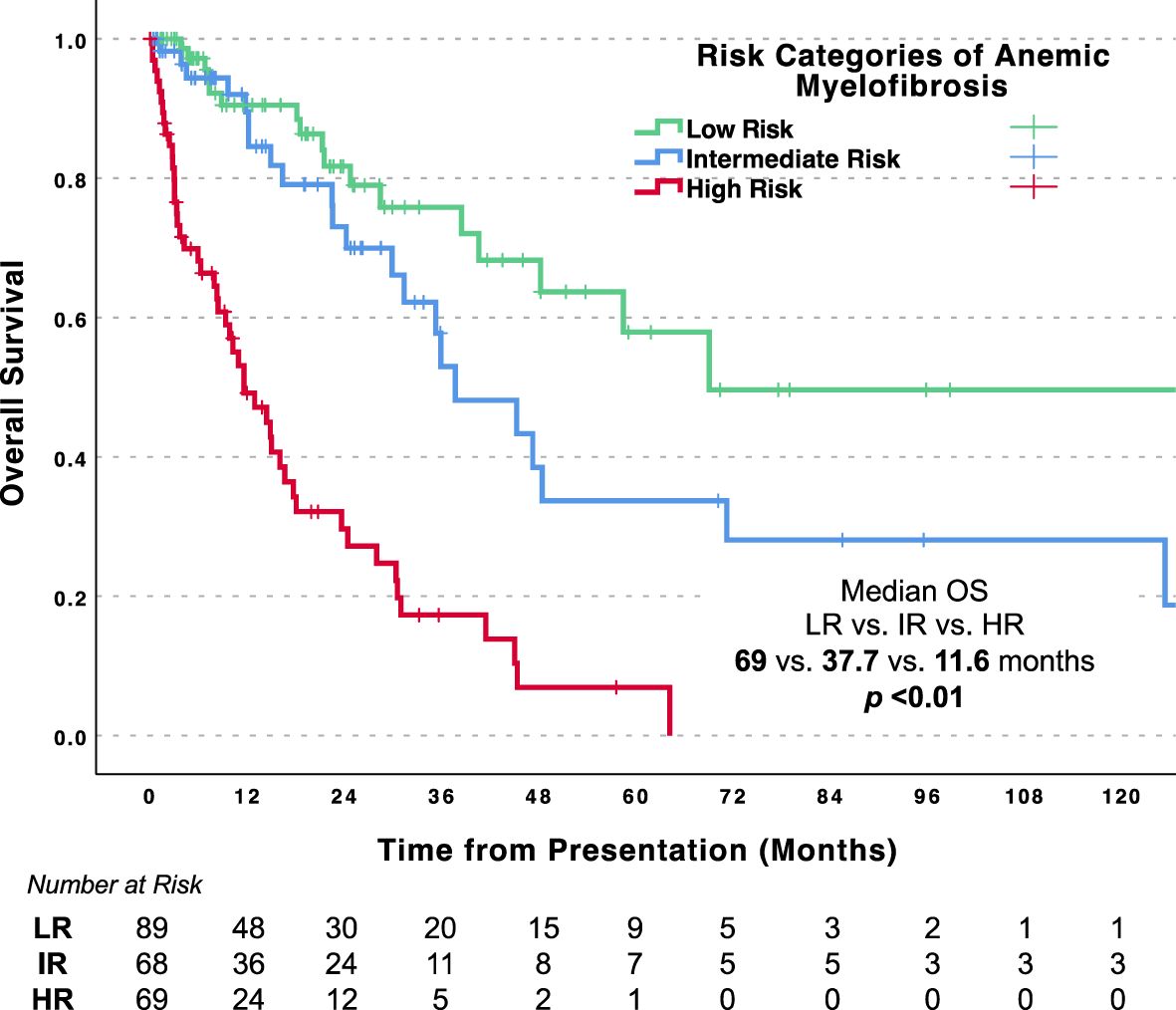
Figure 2. Overall survival for patients with anemic myelofibrosis by risk categories. LR, Low risk; IR, Intermediate risk; HR, High risk.
Discussion
In this single center analysis, we validated several key characteristics of anemic MF which have been previously shown. Patients with disease-related anemia at presentation were significantly older and had higher proportion of primary MF compared to non-anemic cohort (3, 17, 18). In addition, patients with MF and anemia had lower WBC and platelet counts, lower serum albumin levels, higher ferritin and were more likely to harbor high-risk genetic features (2, 3, 6). In line with these findings, blast phase transformation was more common and overall survival was worse in this subgroup (10).
Beyond this, we assessed whether all patients with MF and anemia should be viewed as high-risk. We identified clinical and genetic variables that are specifically prognostic in this anemic subgroup. Interestingly, we found that patients with MF and anemia have a unique set of variables that hold prognostic significance. Thrombocytopenia, leukocytosis, low serum albumin, and the presence of an SRSF2 or TP53 mutation hold prognostic significance in this cohort and MF at large (6); however, variables such as age, peripheral blast percentage, presence of constitutional symptoms, and presence of ASXL1 or U2AF1 mutations did not demonstrate prognostic significance (2, 3, 6). Several studies have demonstrated the value of serum albumin in cachexia and overall prognosis in patients with cancers including MF (18, 19). We previously showed that serum albumin level could be prognostic in patients with MF and disease-related thrombocytopenia (20), and can serve as a dynamic marker for outcomes in patients with MF treated with ruxolitinib (21). Findings of our current study help to hone in on the variables that drive outcomes in patients with an anemic phenotype.
We leveraged these findings to build a prognostic system that could stratify patients by risk. Interestingly, we found a low-risk subgroup that lacked leukocytosis, thrombocytopenia, low albumin, and high-risk mutations (SRSF2 and TP53), which comprised more than a third of the anemic cohort. This subgroup ultimately demonstrated an expected survival that closely resembled that of non-anemic patients. Alternatively, patients who harbored at least two risk factors for high-risk disease exhibited a median OS of less than one year. These findings highlight the importance of appropriate risk stratification for clinical management in patients with anemic MF, where traditional MF prognostic models may be of limited value (2, 6). Clinicians can use this model to identify patients with anemic MF (intermediate and high risk), who could potentially benefit from immediate evaluation for allo-HSCT.
Our study is strengthened by the use of a large molecularly annotated cohort of patients with MF with long follow-up and well documented outcomes. However, this single center retrospective study has some clear limitations. Differentiating between disease-related and treatment-induced anemia in MF can be challenging. We tried to limit the influence of ruxolitinib-induced anemia by excluding those who had been on ruxolitinib for less than 6 months at the time of baseline assessment as treatment-induced anemia is most common in this window (9). Still, we likely included some patients whose anemia was driven, at least in part, by treatment. Additionally, this analysis was observational, and we were not able to evaluate the responses to specific therapies in patients with anemic MF or assess how these therapies may impact outcome. Since we did not routinely use structured symptom assessment form, the data on constitutional symptoms was largely dependent on the subjective assessment by treating physicians.
In conclusion, our study highlights the unique clinico-molecular prognostic correlates for MF with disease-related anemia and proposes a novel prognostic model for effective risk stratification in these patients. Anemic MF often defines a high-risk disease state; however, an evolving treatment landscape that includes less myelosuppressive JAK inhibitors with ACVR1 inhibitor activity (13, 14), improved feasibility of allo-HSCT, and promising agents in development offers new hope (22). We hope that our findings will support individualized, risk-adapted treatment decisions for patients with anemic MF.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by University of South Florida Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
SB: Data curation, Formal Analysis, Project administration, Writing – original draft, Writing – review & editing. NA: Data curation, Writing – review & editing. AJ: Data curation, Writing – review & editing. LA: Data curation, Writing – review & editing. SY: Writing – review & editing. OC: Writing – review & editing. ZX: Writing – review & editing. DS: Writing – review & editing. JL: Writing – review & editing. EP: Writing – review & editing. RK: Writing – review & editing. AK: Conceptualization, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
SB, NA, LA, SY, ZX: No conflict of interest to declare; AGJ: Rigel- speaker’s bureau; OC: Jazz- research funding, BMS- consultancy, honoraria and membership on an entity's board of directors or advisory committees, Syndax- membership on an entity's board of directors or advisory committees, AbbVie- honoraria and research funding, Aptitude Health- honoraria; DS: served on the advisory board or panel for Agios, Avencell, BlueBird Bio, BMS, Dark Blue, Jasper Therapeutics, Kite, Magenta Therapeutics, NKARTA, Novartis, Rigel Shattuck Labs, Servier, Syndax, Syros; consultancy with AbbVie, Gilead, Molecular Partners AG, Takeda; JL: consultancy- Dedham Group, Jasper Therapeutics, Astellas, Dava Oncology, Novartis, Agios/Servio, Boxer Capital, Jazz, BerGenBio, Millenium Pharma/Takeda, ElevateBio Management, Daiichi Sankyo, AbbVie, Servier; research funding- Syntrix Pharmaceuticals, Celgene/ Bristol Myers Squibb; EP: Research Funding- Bristol Myers Squibb, Syntrix Pharmaceuticals, Kura, Incyte; Honoraria- Taiho, Stemline, Blueprint; RK: Abbvie- speaker bureau, advisory board, BMS- research grant, advisory board, DSI- advisory board, Geronconsultancy, Janssen- consultancy, Jazz- speaker bureau, advisory board, Pharma Essentiaspeaker bureau, advisory board, Rigel- speaker bureau, advisory board, Servio- speaker bureau, advisory board, Sobi- speaker bureau, advisory board, Sumitomo Pharma- consultancy, advisory board; AK has received consulting fees/honoraria from Karyopharm, Incyte, Abbvie, MorphoSys, GSK, BMS, CTI Biopharma and research support from Novartis, GSK, MorphoSys, Protagonist, Janssen, Geron, and Blueprint.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol Jan. (2021) 96:145–62. doi: 10.1002/ajh.26050
2. Tefferi A, Guglielmelli P, Lasho TL, Gangat N, Ketterling RP, Pardanani A, et al. MIPSS70+ Version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J Clin Oncol. (2018) 36:1769–70. doi: 10.1200/JCO.2018.78.9867
3. Barraco D, Elala YC, Lasho TL, Begna KH, Gangat N, Finke C, et al. Molecular correlates of anemia in primary myelofibrosis: a significant and independent association with U2AF1 mutations. Blood Cancer J May 6. (2016) 6:e416. doi: 10.1038/bcj.2016.24
4. Verstovsek S, Manshouri T, Pilling D, Bueso-Ramos CE, Newberry KJ, Prijic S, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp Med. (2016) 213:1723–40. doi: 10.1084/jem.20160283
5. Pardanani A, Finke C, Abdelrahman RA, Lasho TL, Tefferi A. Associations and prognostic interactions between circulating levels of hepcidin, ferritin and inflammatory cytokines in primary myelofibrosis. Am J Hematol. (2013) 88:312–6. doi: 10.1002/ajh.23406
6. Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. (2011) 29:392–7. doi: 10.1200/JCO.2010.32.2446
7. Elena C, Passamonti F, Rumi E, Malcovati L, Arcaini L, Boveri E, et al. Red blood cell transfusion-dependency implies a poor survival in primary myelofibrosis irrespective of IPSS and DIPSS. Haematologica. Jan. (2011) 96:167–70. doi: 10.3324/haematol.2010.031831
8. Kuykendall AT, Shah S, Talati C, Al Ali N, Sweet K, Padron E, et al. Between a rux and a hard place: evaluating salvage treatment and outcomes in myelofibrosis after ruxolitinib discontinuation. Ann Hematol Mar. (2018) 97:435–41. doi: 10.1007/s00277-017-3194-4
9. Verstovsek S, Mesa RA, Livingston RA, Hu W, Mascarenhas J. Ten years of treatment with ruxolitinib for myelofibrosis: a review of safety. J Hematol Oncol. (2023) 16:82. doi: 10.1186/s13045-023-01471-z
10. Gupta V, Harrison C, Hexner EO, Al-Ali HK, Foltz L, Montgomery M, et al. The impact of anemia on overall survival in patients with myelofibrosis treated with ruxolitinib in the COMFORT studies. Haematologica. Dec. (2016) 101:e482–4. doi: 10.3324/haematol.2016.151449
11. Maffioli M, Mora B, Ball S, Lurlo A, Elli EM, Finazzi MC, et al. A prognostic model to predict survival after 6 months of ruxolitinib in patients with myelofibrosis. Blood Adv. (2022) 6:1855–64. doi: 10.1182/bloodadvances.2021006889
12. Oh ST, Talpaz M, Gerds AT, Gupta V, Verstovsek S, Mesa R, et al. ACVR1/JAK1/JAK2 inhibitor momelotinib reverses transfusion dependency and suppresses hepcidin in myelofibrosis phase 2 trial. Blood Adv. (2020) 4:4282–91. doi: 10.1182/bloodadvances.2020002662
13. Oh ST, Mesa RA, Harrison CN, Bose P, Gerds AT, Gupta V, et al. Pacritinib is a potent ACVR1 inhibitor with significant anemia benefit in patients with myelofibrosis. Blood Adv. (2023) 7:5835–42. doi: 10.1182/bloodadvances.2023010151
14. Verstovsek S, Gerds AT, Vannucchi AM, Al-Ali HK, Lavie D, Kuykendall AT, et al. Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis (MOMENTUM): results from an international, double-blind, randomised, controlled, phase 3 study. Lancet. (2023) 401:269–80. doi: 10.1016/S0140-6736(22)02036-0
15. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
16. Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. (2008) 22:437–8. doi: 10.1038/sj.leu.2404914
17. Tefferi A, Nicolosi M, Mudireddy M, Lasho TL, Gangat N, Begna KH, et al. Revised cytogenetic risk stratification in primary myelofibrosis: analysis based on 1002 informative patients. Leukemia. (2018) 32:1189–99. doi: 10.1038/s41375-018-0018-z
18. Artz AS, Logan B, Zhu X, Akpek G, Bufarull RM, Gupta V, et al. The prognostic value of serum C-reactive protein, ferritin, and albumin prior to allogeneic transplantation for acute myeloid leukemia and myelodysplastic syndromes. Haematologica. (2016) 101:1426–33. doi: 10.3324/haematol.2016.145847
19. Tefferi A, Nicolosi M, Penna D, Mudireddy M, Szuber N, Lasho TL, et al. Development of a prognostically relevant cachexia index in primary myelofibrosis using serum albumin and cholesterol levels. Blood Adv. (2018) 2:1980–4. doi: 10.1182/bloodadvances.2018018051
20. Kuykendall AT, Mo Q, Sallman DA, Al Ali N, Chan O, Yun S, et al. Disease-related thrombocytopenia in myelofibrosis is defined by distinct genetic etiologies and is associated with unique prognostic correlates. Cancer. (2022) 128:3495–501. doi: 10.1002/cncr.34414
21. Kuykendall AT, Ball S, Mora B, Mo Q, Al Ali N, Maffioli M, et al. Investigation of serum albumin as a dynamic treatment-specific surrogate for outcomes in patients with myelofibrosis treated with ruxolitinib. JCO Precis Oncol. (2024) 8:e2300593. doi: 10.1200/PO.23.00593
Keywords: cytopenic myelofibrosis, prognostic model, overall survival, albumin, thrombocytopenia, SRSF2, TP53
Citation: Ball S, Al Ali NH, Jain AG, Aguirre LE, Yun S, Chan O, Xie Z, Sallman DA, Lancet J, Padron E, Komrokji RS and Kuykendall AT (2024) Distinct clinico-genomic factors drive outcomes in patients with myelofibrosis and disease-related anemia. Front. Hematol. 3:1492680. doi: 10.3389/frhem.2024.1492680
Received: 07 September 2024; Accepted: 04 November 2024;
Published: 22 November 2024.
Edited by:
Hans Carl Hasselbalch, Zealand University Hospital, DenmarkReviewed by:
Ioanna Triviai, Max Planck Institute for Molecular Genetics, GermanyGhaith Abu-Zeinah, NewYork-Presbyterian, United States
Copyright © 2024 Ball, Al Ali, Jain, Aguirre, Yun, Chan, Xie, Sallman, Lancet, Padron, Komrokji and Kuykendall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew T. Kuykendall, YW5kcmV3Lmt1eWtlbmRhbGxAbW9mZml0dC5vcmc=