 Luce Perie1†
Luce Perie1† Selina Stippa1†
Selina Stippa1† Christoph Saffer2,3†
Christoph Saffer2,3† Andrea Hartmann1
Andrea Hartmann1 Mathias Mörgelin4Svante L. H. Zipfel5
Mathias Mörgelin4Svante L. H. Zipfel5 Marc Thilo Figge2,3
Marc Thilo Figge2,3 Thorsten Wiech6Christine Skerka1
Thorsten Wiech6Christine Skerka1 Peter F. Zipfel1,3*†
Peter F. Zipfel1,3*†- 1Department of Infection Biology, Leibniz Institute for Natural Product Research and Infection Biology, Jena, Germany
- 2Applied Systems Biology, Leibniz Institute for Natural Product Research and Infection Biology, Jena, Germany
- 3Institute of Microbiology, Friedrich-Schiller-University, Jena, Germany
- 4Colzyx AB, Sweden Medicon Village, Lund, Sweden
- 5Department of Cardiovascular Surgery, University Heart & Vascular Center, University Hospital Eppendorf Hamburg, Hamburg, Germany
- 6Section Nephropathology, University Hospital Eppendorf, Hamburg, Germany
Introduction: FHR1 is a multifunctional human plasma protein with three C-terminal domains, namely short consensus repeats (SCR) 3–5, showing 98% sequence-identity with the complement inhibitor Factor H. We show that FHR1 uses all three C-terminal SCR to make surface contact. The conserved C-terminal regions of FHR1 and Factor H are altered in patients with atypical-hemolytic-uremic-syndrome. Therefore, we compared FHR1 isoforms with sequence-variations in SCR3, and pathogenic mutants with sequence variations in SCR5.
Methods: FHR1 binding to apoptotic cells was evaluated EM and fluorescent microscopy and in kidney biopsies. FHR1 and Factor H variants and mutants were generated and expressed. The variants and mutant proteins were tested in binding studies to C3b , C3d and heparin, in hemolytic assays and for the induction of inflammatory cytokines. The action profiles of FHR1 and Factor H were calculated and compared.
Results: Functional data revealed that residues YVQ vs HLE in SCR3 and LA vs SV in SCR5 altered ligand binding and surface interaction, influenced target recognition and complement control. Amino-acid-sequence variations in SCR3 influenced FHR1 contact with surface constituents, such as glycosaminoglycans. By contrast, SCR5, the most C-terminal domain, was more relevant for C3b/C3d contact. Notably, wild-type FHR1LA selected C3d, while pathogenic aHUS-associated alterations FHR1SV selected C3b. In consequence mutant FHR1SV altered fined-tuned FHR1-directed effector functions while pathogenic Factor HLA modified C3-convertase control.
Discussion: This influences timing of complement control and inflammatory effector actions at modified self-surfaces. Pathogenic FHR1SV, directed to C3b-decorated targets, adds inflammatory activity at a time when C3-convertase control is appropriate and conversely, mutant Factor HLA adds C3-convertase control at C3d-coated surfaces when inflammatory effector functions are favorable. Further, our computational modeling approach confirms such distinct effects of FHR1 monomers and dimers as compared to flexible Factor H. These effects may explain inappropriate timing of complement regulation and inflammation of the aHUS-derived mutant proteins FHR1SV and Factor HLA.
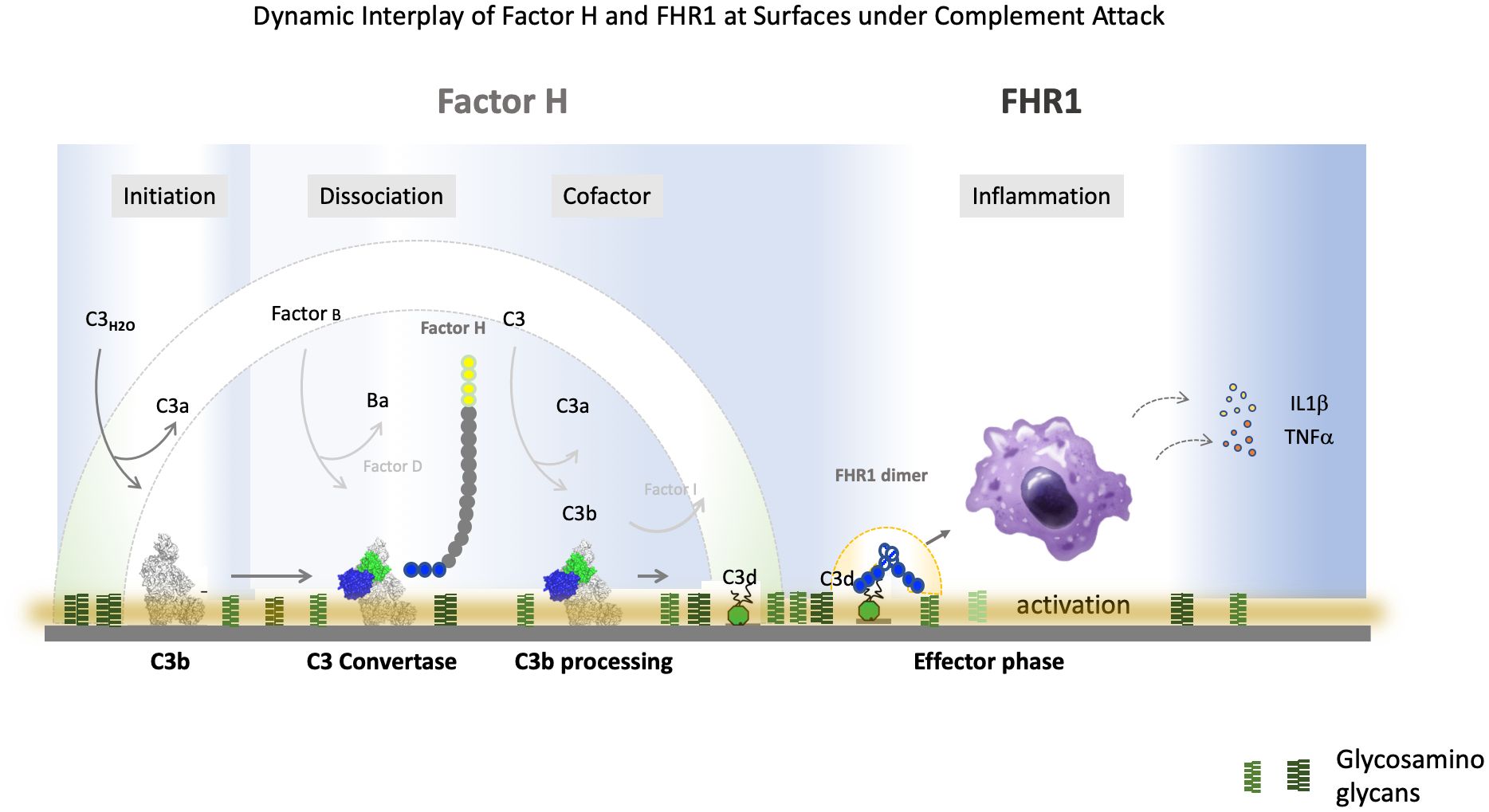
Graphical Abstract.
Introduction
The multiple functional human plasma protein FHR1 modulates complement activation, drives inflammatory and innate immune responses (1–3). FHR1 inhibits complement C5-convertase activity and terminal complement complex formation in vitro, and as inflammatory mediator, induces secretion of proinflammatory cytokines (4–8). Further FHR1-gene variants and mutant FHR1-proteins are associated with several human disorders including aHUS, C3 glomerulopathy, and age-related-macular-degeneration (2, 9–12).
FHR1 is composed of five SCRs and forms dimers via the N-terminal SCRs1-2, which additionally mediate inflammation. FHR13-5 show 98% sequence-identity to the three C-terminal SCRs of Factor H. This region includes binding sites for heparin, GAGs, sialic-acids, modified cell surfaces, monomeric C-reactive protein, malondialdehyde, CRP and C3-fragments (12–17). Pathogenic microbes including Borrelia burgdorferi, Candida albicans, Aspergillus fumigatus, Plasmodium falciparum, and Leptospira interogans utilize FHR1 for immune evasion (18–22).
We previously identified two separate cDNA clones termed FHR1-H36.1 and FHR1-H36.2 which show three amino acid variations in SCR3. Clone H36.1 encodes FHR1Y157V159Q175 and clone H36.2 encodes FHR1HLE (23). These isoforms have different separation profiles and pI-values: pI FHR1YVQ 7.10, pI FHR1HLE 7.53, and they bind differently to C3b (24, 25). The human Genome-Aggregation-Database (26) indicates for Europeans a higher allelic-frequency of FHR1YVQ.
FHR1 is a member of the FHR-Factor H protein-family, which also includes FHR2, FHR3, FHR4, FHR5, Factor H, and FHL1. All seven of these plasma proteins are exclusively composed of SCR-domains and their genes are located in the FHR-Factor H-cluster on human chromosome 1q32. This cluster is a hot spot for variations and chromosomal rearrangements reported for several human diseases (27).
FHR1 and Factor H are the only members of the FHR-Factor H family that share three almost identical C-terminal-domains. Except for the exchange of LA (FHR1) for SV (Factor H) in the most C-terminal SCR-domain, the three most C-terminal-domains of FHR1YVQ and Factor show complete sequence-identity. The other family members FHR2, FHR3, FHR4, and FHR5 share two C-terminal SCRs with Factor H but the sequence homology is lower (28, 29).
In aHUS, the two most C-terminal SCRs of Factor H, i.e., SCRs 19-20 are a hotspot for pathogenic mutations (7, 30). These C-terminal SCRs19-20 are characterized by their structure and binding to C3-fragments and pathogenic microbes. SCRs19-20 bind to sialic acids, heparin glucosamine-glycans, and C3b (26, 28, 31–39). At present, it is unclear whether the third most C-terminal domain, SCR3 of FHR1, corresponding to SCR18 in Factor H, is involved in surface contact and recognition (30, 31).
Homozygous deletion of a chromosomal-segment including FHR1 and FHR3 is strongly associated with a risk of developing aHUS, C3-Glomerulopathy (C3G), and systemic-lupus-erythematosus (SLE) (1, 9, 40–48). However, the same deletion protects against IgAN, AMD, and cardiovascular disease (CVD) (6, 49–51) and is also found in about 6–8% of the healthy Caucasian population (6, 52, 53). Furthermore, increased FHR1-levels are reported in ANCA (antineutrophil-cytoplasmic-antibodies) associated-vasculitis and IgA-nephropathy (5, 53); and the allele encoding FHR1YVQ is enriched in aHUS patients (24).
Chromosomal rearrangements in this FHR-Factor H-gene cluster are represented by intragenic and chromosomal deletions, as well as duplications, resulting in hybrid genes, deleted genes, or in novel hybrid genes. These FHR-variants, FHR::Factor H and Factor H::FHR hybrid-proteins, are expressed, cause pathology (26, 54).
Recently mutant FHR1-genes encoding FHR1S290V296, which contain the C-terminal region of Factor H and pathogenic Factor HL1191A1197 harboring the C-terminal domain of FHR1 have been reported in aHUS patients (9, 10, 54–56) (Figure 1). Thus, indicating that residues 290-FHR1/1191-Factor H and residues 296/1197 have a selective role in surface control. C3G-associated FHR1-variants have duplicated N-terminal interaction regions and have an altered FHR1 plasma repertoire (57, 58).
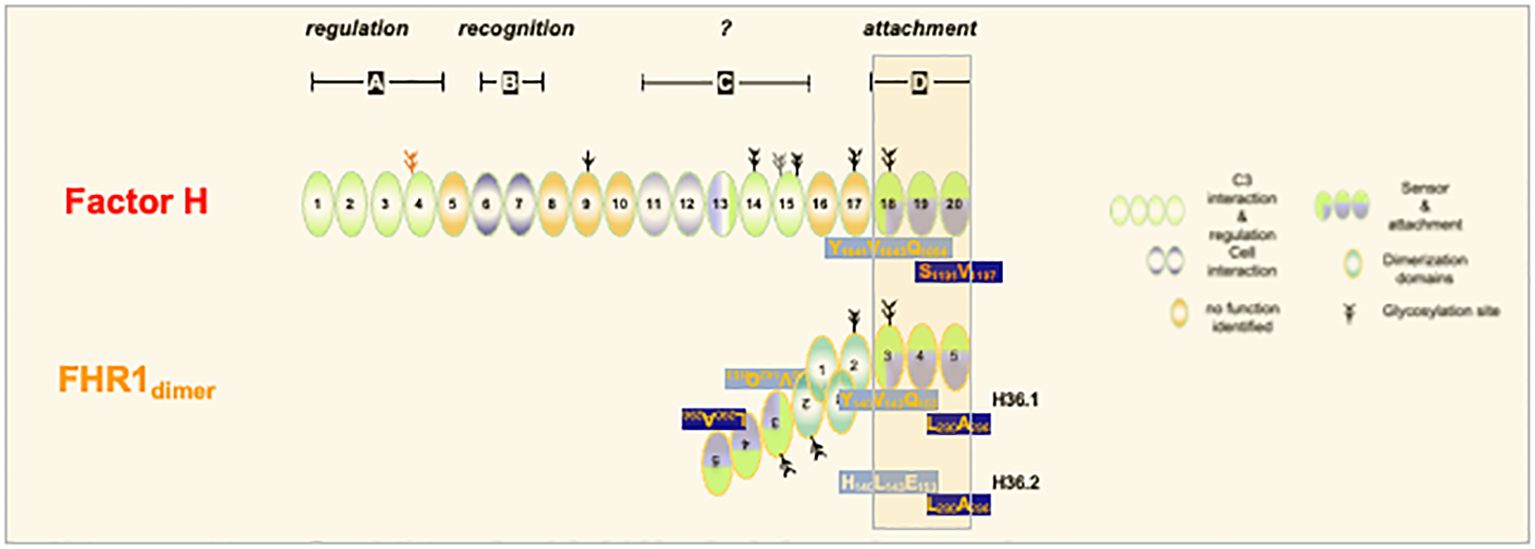
Figure 1. FHR1 and Factor H share C-terminal recognition regions. The three C-terminal SCR-domains from the recognition regions of FHR1 (SCR3-5) and Factor H (SCR18-20) (darked background). The domains show sequence identify except for the two residues in most C-terminal domain. Factor H uses S1191V1197 and FHR1 uses L290,A296. In addition two variants of FHR1 do exist which in domain 3 differ by three amino acids. FHR1 - H36.1 uses Y140V142Q153 in SCR3. This motif matches to the motif used in SCR 18 of Factor H. Variant H36.2 uses H140L142E153 in SCR3. The elongated form of Factor H is shown and FHR1 is presented in its homodimeric from.
We were interested in determining whether (i) SCR3, the first domain of the conserved C-terminal segment, is part of the recognition region; (ii) the two FHR1 isoforms differ in function, and (iii) LA vs SV-exchange in the most C-terminal domain of FHR1 influences protein function. To this end, we expressed and evaluated both natural FHR1 isoforms, with amino acid variations in SCR3, and generated FHR1 mutants, which in SCR5 have the FHR1-characteristic LA-residues exchanged for Factor H-typic SV-residues, and examined their interactions with the ligands heparin and GAGs, necrotic cell surfaces, and C3b and C3d. Further, we evaluated if the exchange of LA for SV in the most C-terminal SCR of FHR1 influences complement regulation. Last, we developed a three-dimensional to-scale model and in combination with Monte-Carlo simulations show vastly different control regions for FHR1 and Factor H.
Materials and methods
Human serum and antibodies
Normal human serum (NHS) with two copies of FHR1, serum with one chromosomal copy of FHR1 (HS+/Δ) and FHR1-FHR-3 deficient serum (HSΔ/Δ) were collected from healthy individuals, upon informed consent. FHR1 copy numbers were determined by multiplex ligation dependent probe amplification (MLPA); FHR1 and Factor H levels were evaluated by Western Blotting and ELISA. FHR1 was detected either with a monoclonal antibody JHD-7.10.1 (4, 59) or with polyclonal rabbit FHR1 antiserum, which reacts with FHR1 and Factor H. Factor H was detected with M16 antibody (11, 60) polyclonal goat anti-C3 antiserum and polyclonal rabbit anti-C3a antiserum were from CompTech (Tyler, TX).
Cell culture and treatments
HUVECs (CRL-1730; ATCC) were cultivated as described (53). Necrosis and apoptosis were induced as described in DMEM lacking FBS. After washing with PBS, necrotic, apoptotic, or intact cells were incubated with FHR1-variants (10 µg/ml), Factor H or BSA for 30 min at 37°C and evaluated by to flow cytometry. CHO-K1 cells (ATCC CCL-61) were cultivated in Ham’s F-12 medium (61).
Site-directed mutagenesis and protein expression
Recombinant FHR1YVQ was generated using FHR1 H36.2 cDNA (22). Mutations were introduced H36.1 and H36.2 cDNAs by site-directed mutagenesis according to the manufacturer’s protocol (Stratagene, LaJolla, CA). The following primers were used: FHR1YVQ_Fwd: 5’-CCGCCCACAGTACAAAATGCTTATATAGTGTCGAGACAGAT-3’ and FHR1YVQ_Rev: 5’-GGTGAGAGAGTACGTTATCAATGTAGGAGCCCTTAT-3’. Purified Factor H, C3b, C3d, and Factor I were from Complement Technology, Inc (Tyler, TX). In addition, the mutants FHR1YVQ-SV and FHR1HLE-SV were generated using FHR1LA_Fwd: 5’-GCCAAACAGAAGCTTTATTCGAGAACAGGTGAATCAGTTGAATTTGTGTGTAAACGG-3’. After sequencing, the four proteins were expressed in Pichia pastoris and purified as described (62).
Binding assays
Heparin (Fluka), heparan-sulfate, chondroitin-sulfate-A and chondroitinsulfate-4 (Sigma-Aldrich, Taufkirchen) (50 µg/ml in DPBS), were immobilized onto heparin binding plates (EpranEx, BD). C3b, C3d, C3 or iC3b (5 µg/ml) were coated to Maxisorp ELISA plates (Nunc) or heparin binding plates (6). After washing with PBS and blocking (2%; v/v) BSA-PBS) human serum (NHS, HS+Δ, HSΔΔ), FHR1 isotypes or mutants, Factor H or BSA were added at the indicated concentrations. The mixtures were incubated at 37°C for 1h. Bound FHR1 or Factor H were detected using JHD 7.10.1, diluted (1:1000) followed by the corresponding anti-mouse secondary antiserum (1:1000; Dako). OD values were measured at 450nm.
Western blot
Protein extracts from cell lysates or human serum were lysed with RIPA-buffer containing 20 mM Tris, 150 mM NaCl, 1% Triton X-100, supplemented with a cocktail of protease inhibitors (Thermofisher, #PIA32953) separated by 10% SDS-PAGE and proteins were transferred onto nitrocellulose membranes (GE Healthcare) as described (5).
RNA isolation and RT-PCR analysis
Total RNA was extracted from cells with RNeasy (Qiagen, #75144) and from tissues with TRIzol (Thermofisher, #15596018). 1 μg of total RNA was reverse transcribed into cDNA using the iScript™ cDNA Synthesis Kit (BioRad, #1708890). RT-PCR analysis was performed as previously described (5).
Flow cytometry
Binding of FHR1 variants, Factor H, or FHR1 and Factor H C-terminals fragmentsto intact and necrotic HUVECs was determined as described (61). Briefly, cells were incubated with 10 μg FHR1 variants or BSA for 30 min at 37 °C in PBS-BSA 1%. Cells were washed with PBS and bound FHR1 was assessed by staining with JHD-7.10.1 in combination with Alexa Fluor 488 labeled donkey anti-mouse IgG (1:500, Invitrogen). Fluorescence was measured by flow cytometry (LSRII according to FSC/SSC and VD/Annexin).
Complement activation
CHO cells were challenged with complement active HSΔΔ (20%; diluted in activation buffer (20 mM Hepes, 144 mM NaCl, 7 mM MgCl2, 10 mM EGTA, pH 7.4) in presence of the FHR1 variants (75 µg/ml), Factor H (50 µg/ml) or BSA (50 µg/ml) for 30 min at 37°C. After incubation, and washing and deposited C3b was analyzed by flow cytometry using a FITC-conjugated goat F(ab’)2 anti−human C3 (1:200 (Protos Immunoresearch, CA). C5b9 deposition was determined with mAb C5b (1:200) in combination with appropriate secondary antiserum.
Hemolytic assays
Chicken erythrocytes (CRBC) or sheep erythrocytes (both at 5x106/ml) were treated with FHR1 variants (75 µg/ml) added to complement active HSΔ/Δ (20%) (40). After 30 min incubated at 37°C hemoglobin release was recorded at 414 nm.
Electron microscopy
Apoptotic HUVECs were incubated with FHR1 and Factor H. The JHD10 antibody and Factor H antiserum were added followed by gold labeled specific secondary antiserum. Then the cells were fixed with 2.5% glutaraldehyde, dehydrated and embedded in Epon as described. Samples were then processed as previously reported (63).
Immunohistochemistry
Standard immunohistochemistry was performed in paraffin fixed biopsies obtained from patients with Lupus nephritis and ANCA nephritis (61). Recombinant FHR1 was added to the biopsies and following washing specific FHR1-antiserum was added and detected with appropriate immunolabeled secondary antiserum.
Modeling the action regions of FHR1 and Factor H
A computational to-scale model of FHR1 and Factor H was developed to analyze their respective action regions. This involved an analytical approach to compute the action regions approximated by geometric shapes and millions of Monte-Carlo simulations to explore probable locations of N-terminal SCRs and their distance to C-terminal SCRs for both proteins. Detailed information can be found in the Supplementary Information.
Statistical analysis
Data are represented as mean ± SD. The groups were compared by Student’s t-test or one-way analysis of variance (ANOVA). P-values <0.05 were considered statistically significant. All statistical analyses were performed using Prism 9-software (GraphPad-Software).
Results
FHR1 levels influence Factor H binding
We first assayed binding of FHR1 and Factor H to heparin. For NHS, i.e., serum from individuals with two alleles of FHR1 (HS++), heparin binding was set at 100% (Table 1). For serum from an individual carrying one FHR1 allele, i.e., HS+Δ, FHR1 binding was reduced by 50% (Table 1) and with serum lacking FHR1 (HSΔΔ), binding was reduced to background-levels. Factor H binding increased by 35% with HS+Δ, and by more than 5-fold with HSΔΔ. Thus, FHR1 levels strongly influenced Factor H binding to heparin (Table 1).
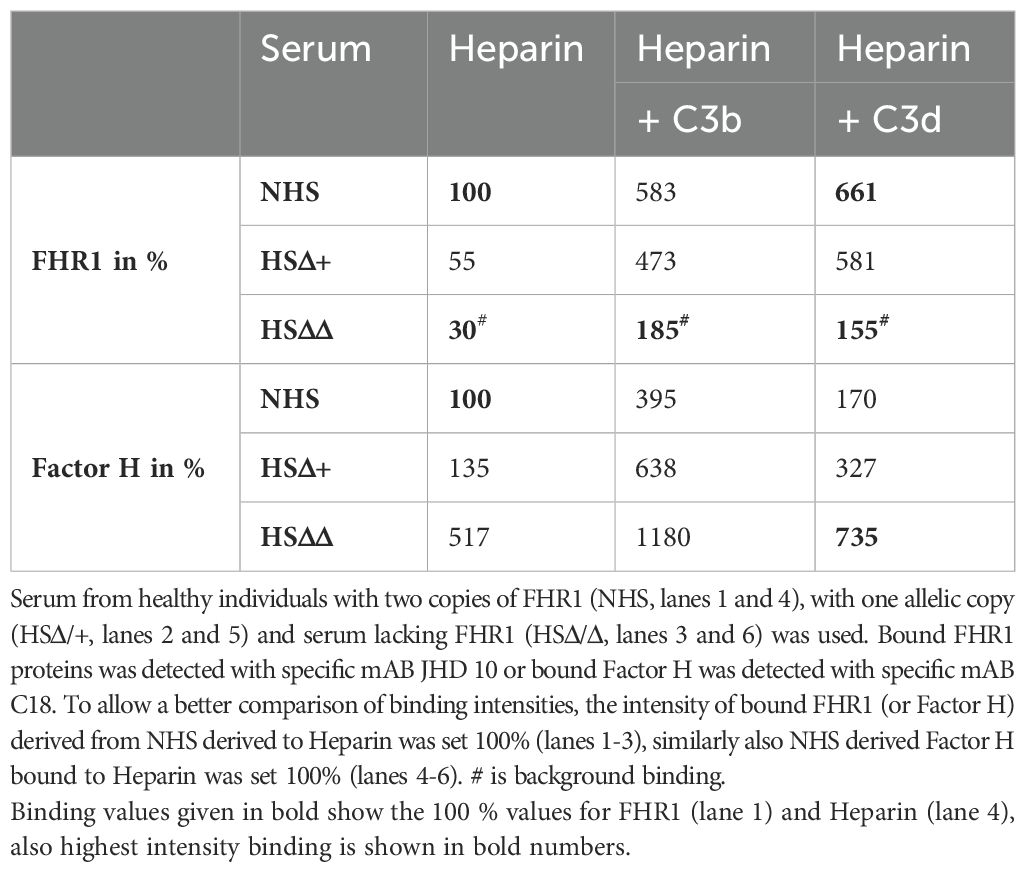
Table 1. Binding of FHR1 and Factor H to Heparin and Heparin::C3b, as well as Heparin::C3d combinations immobilized to the surface of an ELISA plate.
The binding of FHR1 to C3b::Heparin and C3d::Heparin combinations was further tested. Using NHS, FHR1 bound 6-fold more to C3b::Heparin and almost 7-fold more to the C3d::Heparin surface (Table 1) than to heparin alone. Factor H binding to the C3b::Heparin combination increased almost 4-fold, but its binding to C3d::Heparin was lower (1.7-fold) (Table 1). FHR1 clearly preferred C3d::Heparin while Factor H preferred C3b::Heparin, suggesting a selective role for surface-attached C3-fragments.
Binding of FHR1YVQ and Factor H to late apoptotic HUVECs. Apoptotic HUVECs showed morphologic alterations and bleb formation (Figure 2A). FHR1YVQ bound to apoptotic cells, and adjacent dots revealed that FHR1 bound as a dimer (Figure 2A). Factor H also bound to apoptotic cells, but single and adjacent dots were observed. The double dots could potentially indicate that Factor H binds as a dimer to apoptotic surfaces (Figure 2A). When FHR1YVQ and Factor H were combined, and proteins were visualized using different sized gold particles, double-dots of different sizes were observed, indicating that FHR1 and Factor H colocalize at surfaces. Altogether, the results suggest that FHR1YVQ bind to apoptotic surfaces as a homodimer, and that it could potentially form heterodimers with Factor H (Figure 2A).
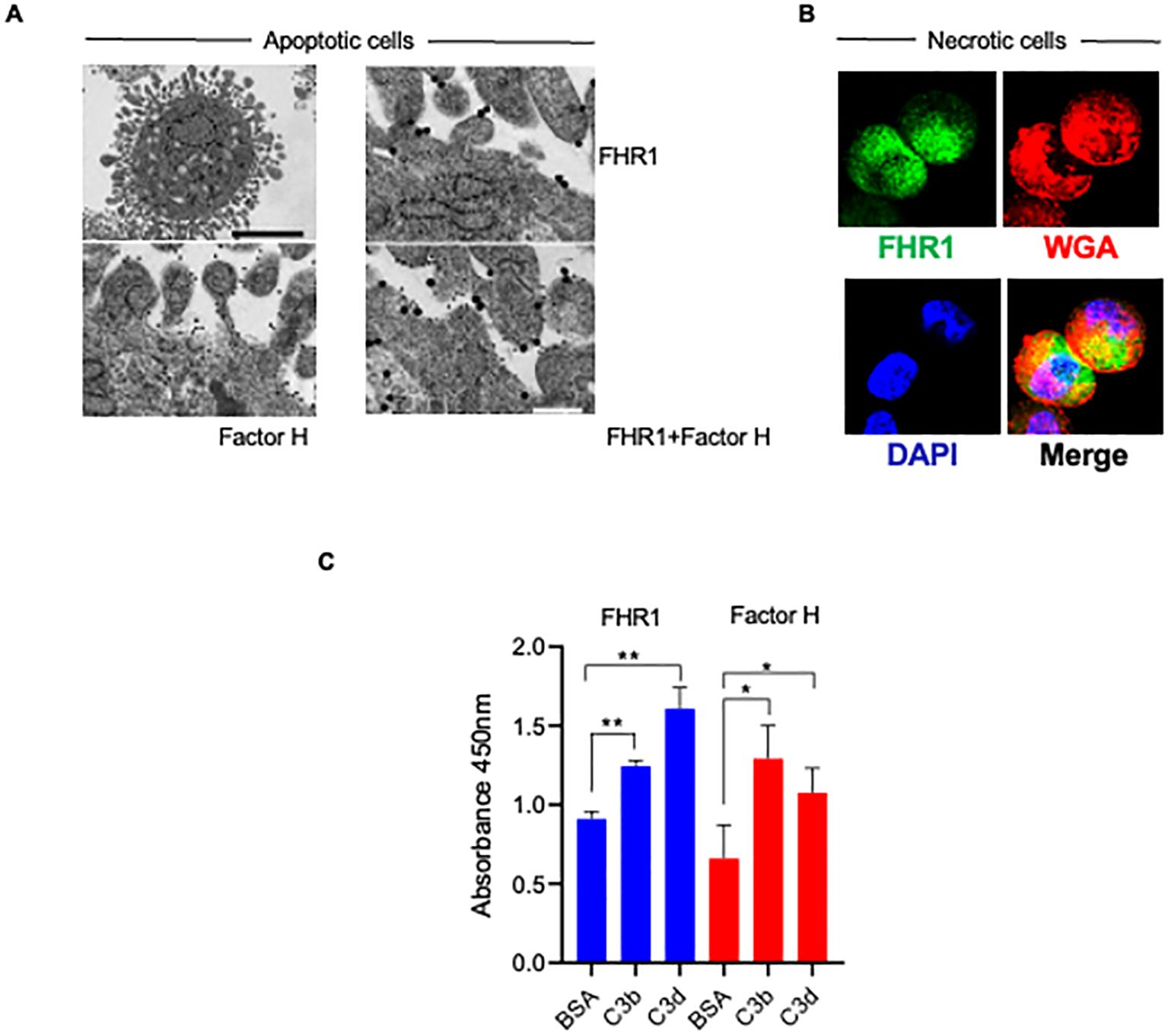
Figure 2. FHR1 binds as dimers to apoptotic surfaces, and binds to C3b and to C3d. (A) Apoptotic HUVEC-cells form blebs at their surface. Binding of FHR1, Factor H and FHR1::Factor H combinations to the surfaces of apoptotic cells evaluated by electron microscopy. FHR1 and Factor H were detected with specific antiserum in combination with gold labeled secondary antisera, either FHR1 5 um and Factor H 10 um. Representative images are shown. (B) FHR1 binding to necrotic HUVECs. FHR1 was detected with specific JHD antiserum in combination with fluorescently labeled secondary antiserum (green fluorescence) on the surface of necrotic HUVEC-cells. WGA (wheat germ agglutinin; red fluorescence) was used to show membrane integrity and disruption. DNA staining by DAPI (blue immunofluorescence). Representative images are shown. (C) FHR1, Factor H (each at 10 μg/ml) binding to immobilized C3b and C3d to was evaluated by ELISA; BSA was used as control. Results represent mean ± SD of three independent experiments. **p < 0.01; ***p < 0.001.
Confocal microscopy revealed that FHR1YVQ bound at low levels to intact HUVECs and more to necrotic HUVECs. FHR1 binding was most prominent at sites where membrane integrity had been lost, i.e., sites lacking WGA-staining (Figure 2B, Supplementary Figure S1).
Given their different binding strengths to C3d/C3d::Heparin combinations, we directly compared FHR1 and Factor H binding to C3b and C3d. FHR1YVQ bound to immobilized C3b, but it bound more strongly to C3d (Figure 2C) and Factor H showed the opposite profile bound more strongly to C3b and less to C3d (Figure 2C).
FHR1 deposition was evaluated in diseased glomeruli. FHR1 revealed granular positivity in close proximity to necrotic segments in ANCA-associated necrotizing glomerular areas (Figure 3A). In Lupus nephritis FHR1 bound strongly to tissue areas within the immune complexes (Figure 3B). FHR1 deposition at specific sites in glomeruli of the two kidney diseases agrees with a local action of this effector protein.
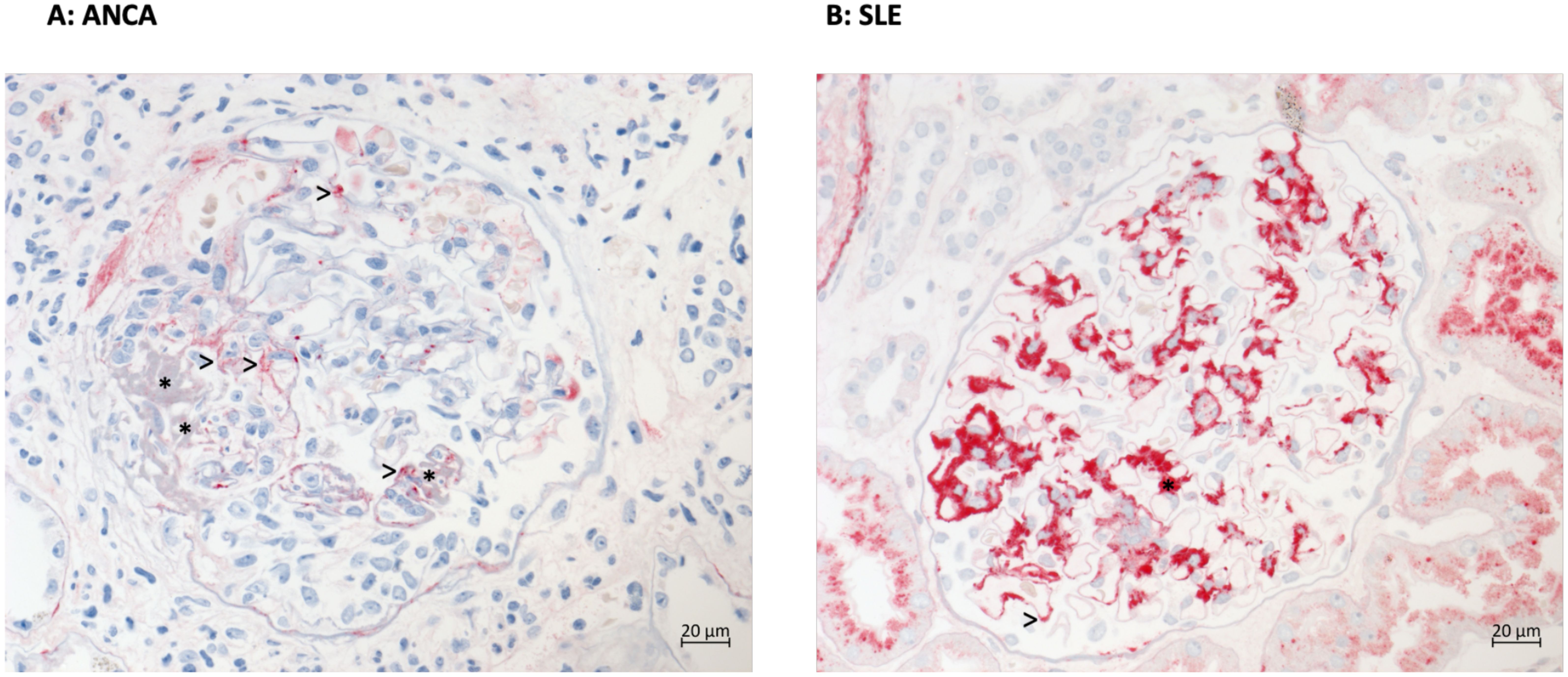
Figure 3. FHR1 shows distinct deposition in kidney of patients with ANCA vasculitis or Lupus nephritis. (A) Immunohistochemical staining for FHR1 reveals mild granular red positivity (>) in the mesangium and at the peripheral basement membranes especially in the vicinity of necrotic areas (* fibrin precipitates) but negativity in preserved capillaries of the glomerular tuft in a case of pauci-immune ANCA-associated necrotizing glomerulonephritis. (B) SLE Immunohistochemical staining for FHR1 protein in a case of lupus nephritis reveals abundant granular red positivity in all mesangial fields (e.g. *) and at some peripheral basement membranes (e.g. >) corresponding to all areas of immune complex deposition.
FHR1 isoforms regulate complement differently
FHR1YVQ and FHR1HLE differ by three amino acids in SCR3. Both variants are common in the European population but whether they are functionally different is unclear (Figure 4A, Supplementary Figure 1) (25). To determine whether the amino acid differences alter ligand binding and complement action, we first assessed their binding to heparin and GAGs. Both isoforms bound to heparin, heparan-sulfate, chondroitin-sulfate-A, and chondroitin-sulfate-4 (Figure 4B, Supplementary Table 1). The FHR1YVQ variant bound slightly better than the FHR1HLE variant to all four ligands, and the higher binding of FHR1YVQ to heparin, heparan-sulfate, and chondroitin-sulfate-A was significant. In addition, both variants bound to necrotic HUVECs. Again, FHR1YVQ bound better to necrotic HUVECs than FHR1HLE (~25%) (Figure 4C). Binding to C3 and C3-activation fragments revealed that both isoforms bound better to C3 and to C3d than to C3b and iC3b (Figure 4D). FHR1HLE bound less to C3d than FHR1YVQ. The differences for heparin and C3d binding demonstrate that SCR3 also contributes to ligand binding.
To evaluate complement regulation, each variant was added to active HSΔ/Δ serum and following incubation, C3b and C5b-9 deposition were recorded. Neither isoform affected C3b deposition (Figure 4A). Factor H, used as a control, reduced C3b deposition (data not shown). However, both isoforms reduced C5b-9 deposition and chicken erythrocytes lysis (Figures 4B, C). FHR1HLE showed ~20% higher inhibitory effect than FHR1YVQ. Thus, SCR3 contributes to ligand binding, surface recognition, and complement control.
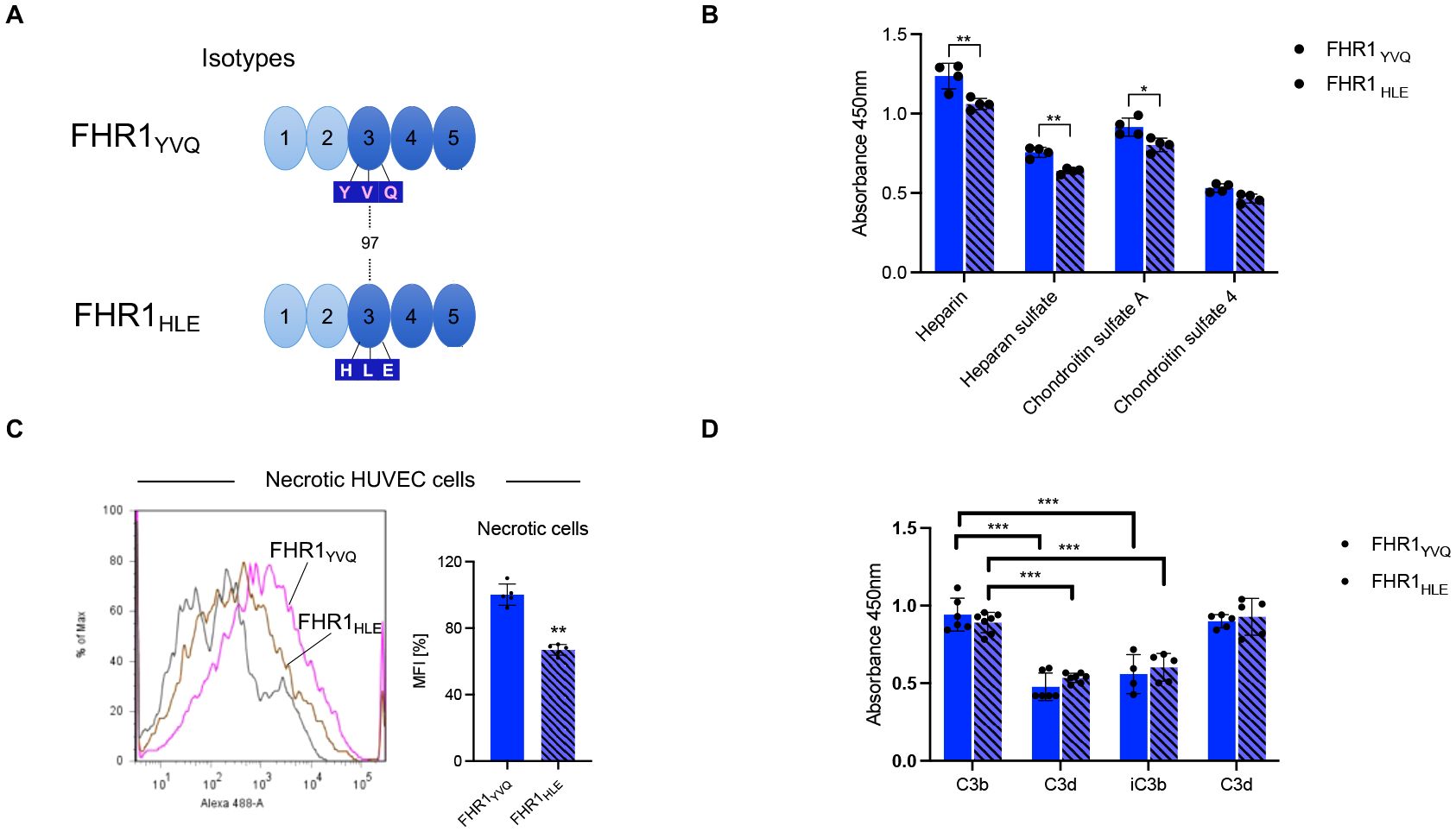
Figure 4. FHR1YVQ and FHR1HLE bind to Heparin, GAGs, necrotic surfaces and to C3 fragments. (A) Domain structure of FHR1 and sequence variation in SCR3 representing the isotypes FHR1YVQ and FHR1HLE. (B) FHR1YVQ or FHR1HLE (each at 10 μg/ml) were added to immobilized Heparin, Heparan-sulfate, Chondroitin sulfate-A or Chondroitin sulfate-4 coated on ELISA plates After incubation and washing bound proteins were detected by JHD. (C) FHR1 binding to necrotic HUVEC-cells. The FHR1 isoforms (at 10 μg/ml) were added to necrotic HUVEC-cells. Following incubation and washing bound proteins were detected and analyzed by flow cytometry. The panel on the right shows the MFI in %, with FHR1YVQ set to 100%. (D) Binding of FHR1 isotypes variants (each at 10 μg/ml) to immobilized C3, C3b, iC3b and C3d was evaluated. After incubation and washing bound proteins were detected by FHR1 JHD antibody. Results represent mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
FHR1 induces inflammatory cytokines
To compare their proinflammatory effects FHR1-variants were added to HSΔ/Δ and the supplemented serum was added to THP1-macrophages. Both isoforms induced IL-1β and TNFα secretion and FHR1HLE was around 25% more effective than FHR1YVQ (Figure 4D). Similarly, when added together with LPS, both isoforms enhanced cytokine releases and again, FHR1HLE was more effective (Figure 4D). Thus, SCR3 influences FHR1 mediated C5b-9 deposition, hemolysis, and secretion of proinflammatory cytokines.
C-terminal FHR1-Factor H-fragments affect complement control
The biological effects of the C-terminal fragments, which lack the N-terminal interaction and regulatory-regions, i.e., FHR13-5 with LA and Factor H18-20 with SV-motifs, were compared (Figure 5A, Supplementary Figure 1B).
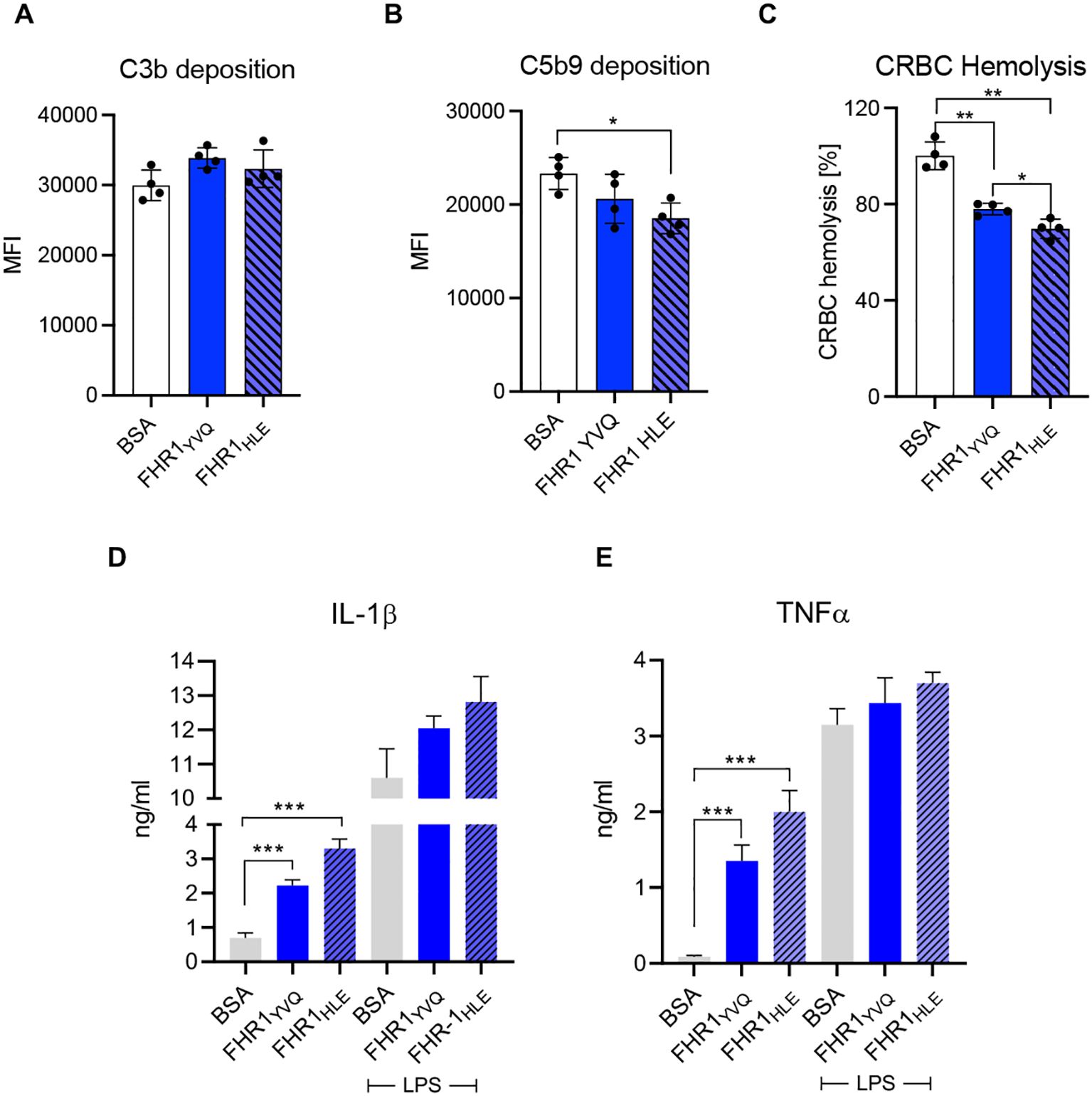
Figure 5. FHR1YVQ and FHR1HLE isotypes do not promote C3b deposition, but reduce downstream complement action and induce flammation. (A, B) CHO cells were incubated with complement active HSΔΔ (20%) deficient for FHR1/FHR3 supplemented with one FHR1 isoform (50ug) or BSA. (A) C3b and (B) C5b-9 deposition were analyzed by flow cytometry. (C) Chicken erythrocytes were challenged in complement active HSΔΔ (20%) in presence of FHR1YVQ, FHR1HLE (each at 75 μg/ml) or BSA. Lysis was recorded after 1 h. (D) Both FHR1 variants (each at 10 μg/ml) or BSA induced IL-1β and (E) TNFα release. The variants had the similar stimulatory effects when they were added together with LPS also THP1. Results represent mean ± SD of three independent experiments. * p < 0.05 **; p < 0.01; *** p < 0.001.
FHR13-5 or Factor H18-20 were added to HSΔ/Δ and following incubation, C3b-deposition was examined. FHR13-5 increased C3b-deposition, but Factor H18-20 was more effective (Figure 5B). However, FHR13-5 reduced and Factor H18-20 increased C5b-9 deposition (Figure 5C). These opposite effects highlight the relevance of the LA and SV-residues for target access and complement control. FHR13-5 and Factor H18-20 at 3.0 ug/ml had similar effects on erythrocyte-lysis. At higher doses, lysis by FHR13-5 reached a plateau, but Factor H18-20 continued to increase lysis dose dependently (Figure 5D). Furthermore, both fragments induced CCL2, CXC10, IL6 transcription in human monocytes (Figure 5E). However, Factor H18-20 induced a 2-fold higher CCL2 expression levels than FHR13-5.
FHR1YVQ-SV mutants induce complement activation and in inflammation
The effects of the C-terminal fragments suggest that residues 290/296 of FHR1 influence complement functions. To show this more clearly, we generated FHR1YVQ and FHR1HLE mutants containing the Factor HSV motive in SCR5 (FHR1YVQ-SV and FHR1HLE-SV) (Figure 6A, Supplementary Figures S1A, B). FHR1YVQ-SV and FHR1YVQ bound more strongly to heparin, heparan-sulfate, chondroitin-sulfate-A, and chondroitin sulfate-4 than did FHR1HLE and FHR1HLE-SV (Figures 5B, 6B, Supplementary Table 2), confirming the importance of SCR3 and SCR5 for heparin and GAG binding.
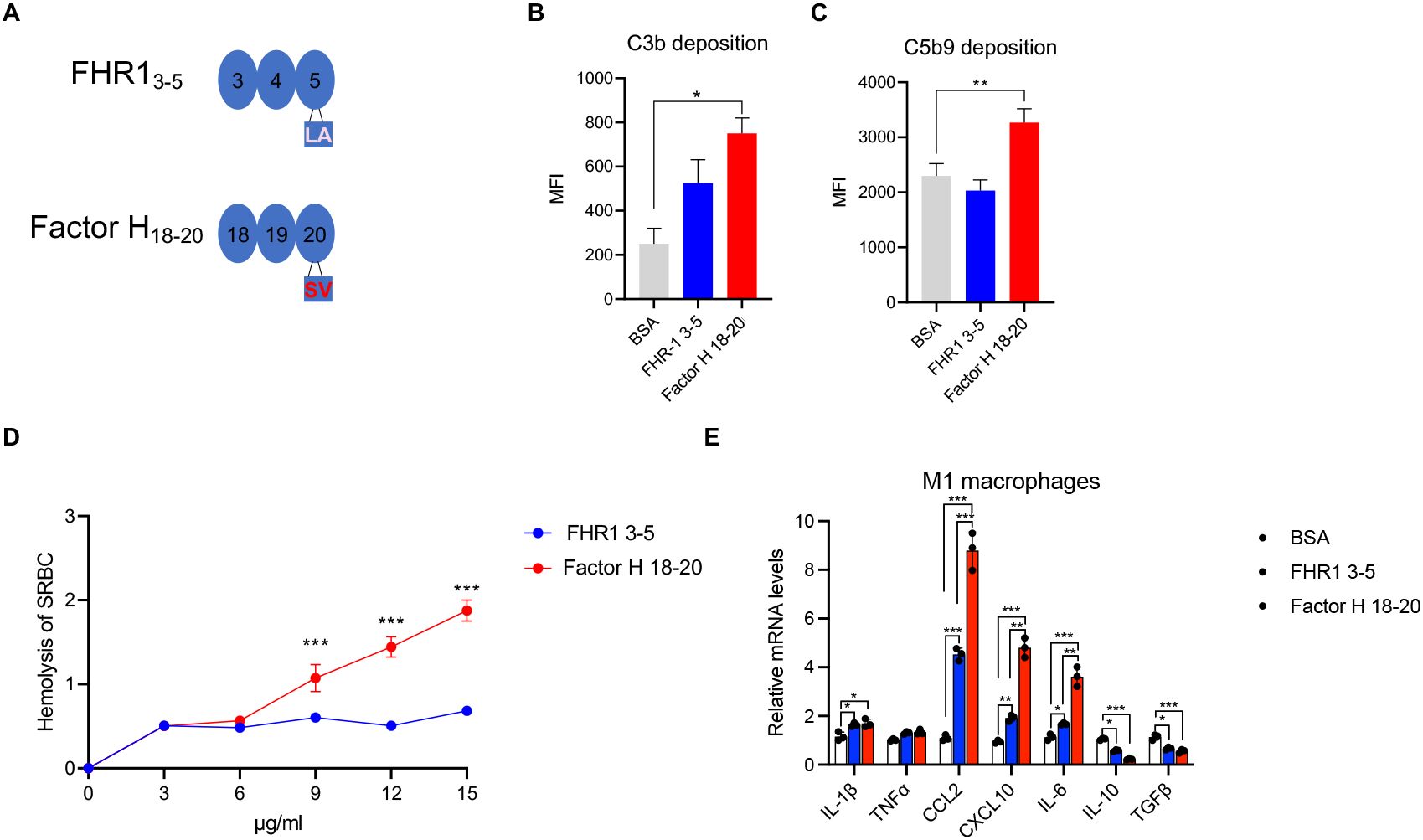
Figure 6. C-terminal fragments: FHR13-5 and Factor H18-20 bind to C3b and C3d, but do not induce complement activation. (A) Position of LA and SV residues in a structural model of the three C-terminal domains i.e. SCRs3-5 of FHR1 or in SCRs18-20 of Factor H. (B, C) FHR13-5, Factor H18-20 or BSA were added to complement active HSΔΔ (20%) deficient for FHR1 and FHR3 and this mixture was added to CHO cells. Following incubation surface deposited C3b (B) and C5b-9 (C) were detected by flow cytometry using specific antibodies. (D) Sheep erythrocytes were challenged with complement active HSΔΔ (30%) complemented with FHR13-5 or Factor H18-20 used at concentrations ranging from 3–15 μg/ml. (E) Cytokine release by FHR1 3-5, Factor H18-20 (10 μg/ml). the C terminal fragments were added to THP1 macrophages and following incubation mRNA levels were determined by RT-PCR. Results represent mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
FHR1YVQ-SV and FHRHLE-SV mutants bound more strongly to necrotic HUVECs than did wild-type equivalents (Figures 5C, 6C, D). The FHR1-mutants bound less (~20%) to C3b and C3d than did their wild-type counterparts (Figures 5D, 6C, D), confirming that the LA-SV residues in SCR5 influence FHR1 binding to necrotic surfaces and C3b/C3d.
FHR1YVQ-SV mutant promotes complement activation and inflammation. Both proteins with the YVQ-motif did affect C3a-generation to a larger extent than the YVQ forms (Figures 7A, B). Full-length Factor H reduced C3a-generation. By contrast, both FHR1SV mutants increased C3b-deposition (by 35% and 30%), while the FHR1-isoforms showed minor effects (Figure 7C).
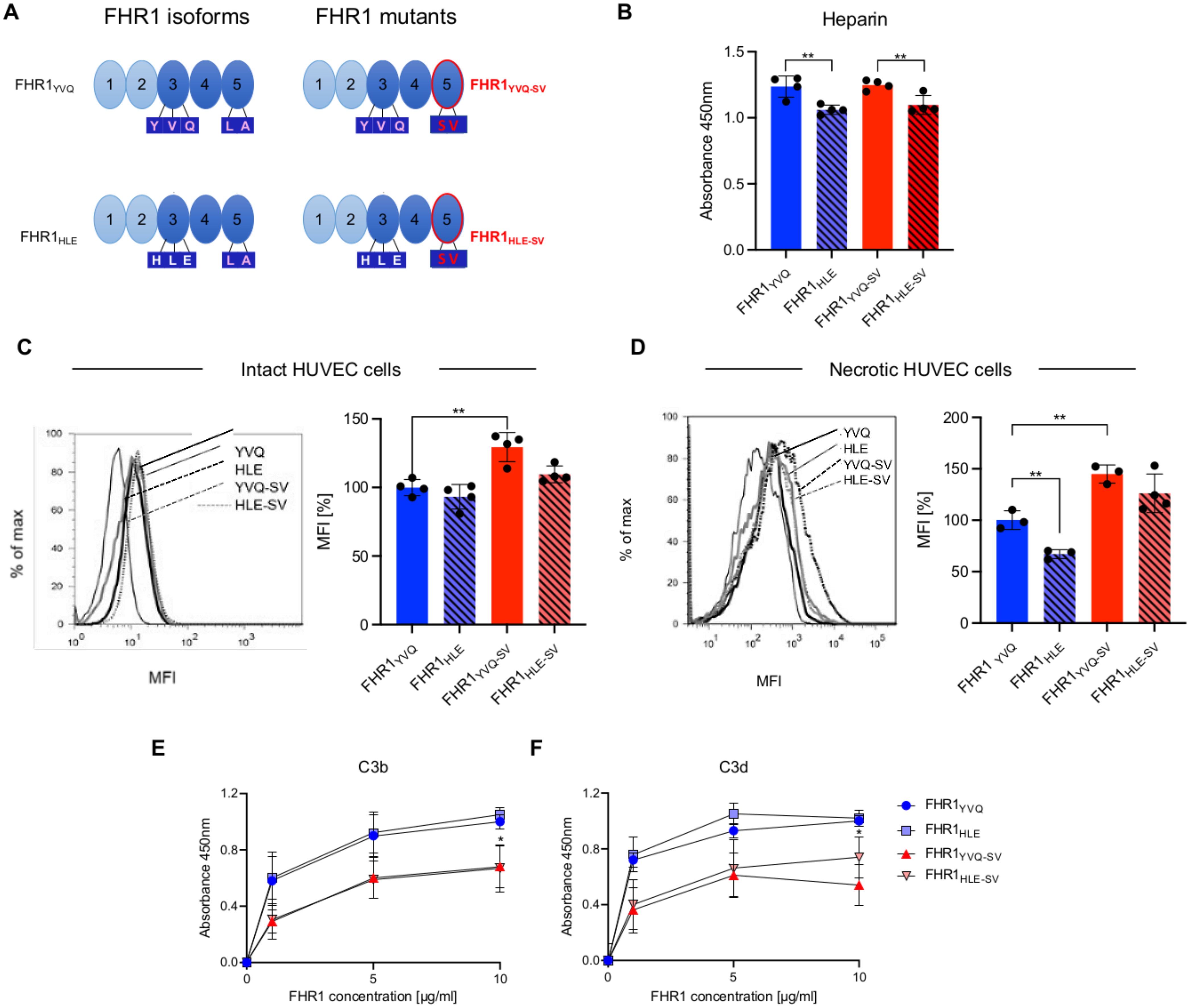
Figure 7. FHR1 mutants - ligand and cell surfaces binding and interaction with C3 fragments. (A) Schematic representation of the two FHR1 isoforms and FHR1 mutants harboring in the most C-terminal domain the SV motive of Factor H. (B) Binding of FHR1YVQ, FHR1HLE, FHR1 YVQ-SV and FHR1HLE-SV each at 10 μg/m l to Heparin coated wells. After washing bound FHR1 was detected with JHD10 (C, D) FHR1 mutants and isoforms were added to (C) intact or to (D) necrotic HUVECs. After incubation and extensive washing bound FHR1s were detected and recorded by flow cytometry. The panels on the left show the graphs and the MFIs are presented in the panel of the right. (E, F) FHR1YVQ, FHR1HLE, FHR1YVQ-SV and FHR1HLE-SV used at the indicated concentrations were added to immobilized C3b (E) or to C3d (F). After washing bound proteins were detected with specific JHD antibody and appropriate secondary antiserum. Results represent mean ± SD of three independent experiments. *p < 0.05; **p < 0.01.
Both FHR1SV-mutants also increased C5b-9 deposition (35% and 30%), whereas the FHR1 isoforms reduced C5b-9 deposition (Figure 7D). Factor H reduced C5b-9 deposition by 80% (Figure 7D). Similar effects were observed for erythrocyte lysis. Both FHR1-isoforms reduced chicken erythrocyte lysis by 25% and 30%, whereas both FHR1SV-mutants increased chicken erythrocyte lysis by 25% and 18% (Figure 7E). The increase in lysis by the FHR1SV-mutants was dose-dependent (Figure 7F). These opposite effects of the FHR1 variants and FHR1SV-mutants show that the LA-SV residues play an important role in complement control at surfaces.
FHR1 mutants - effect on complement activation
The effect of the FHR1 variants and FHR1 mutants were tested on complement activation with FHR1 deficient complement active human serum. C3a generation upon addition of FHR1YVQ, FHR1HLE and also of FHR1YVQ-SV, FHR1HLE-SV to HSΔΔ resulted in C3a (Figures 8A, B). Factor H showed reduction. Similarly, the FHR1 variants and mutants influence C3b deposition and the effects were different to Factor H. (Figure 8C). The FHR1 variants were less efficient in C3b generation as compared to the FHR1 mutants, with the SV motive in the most C-terminal SCR. This effect can be interpreted by a more efficient competition of Factor H. Highly related effects were observed for C5b-9 deposition (Figure 8D) and lysis of both chicken and sheep erythrocytes (Figures 8E–G).
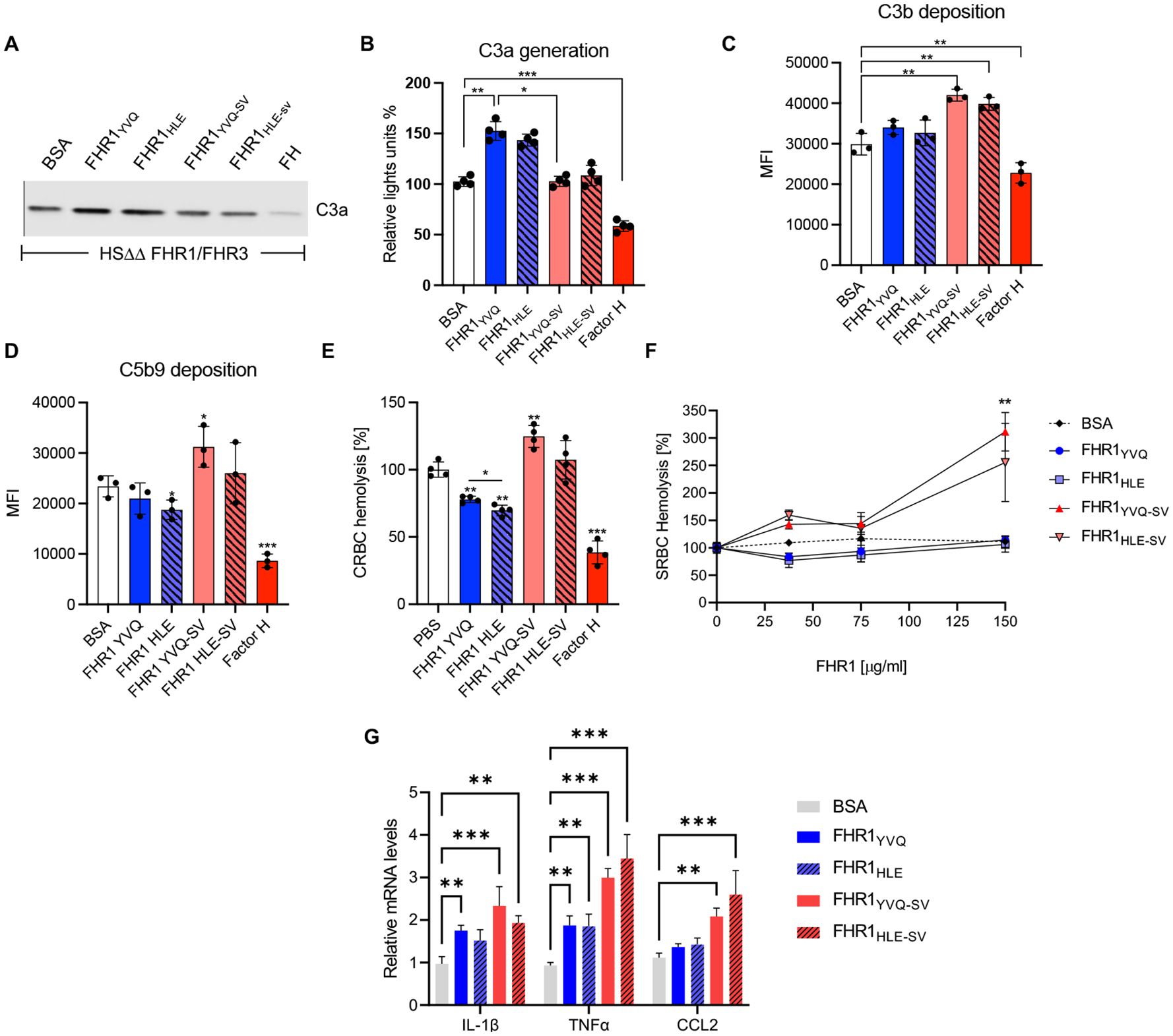
Figure 8. FHR1 mutants - effect on complement activation. (A, B) C3a generation upon treatment with HSΔΔ (i.e. FHR1/FHR3 deficient serum supplemented with FHR1YVQ, FHR1HLE, FHR1YVQ-SV, FHR1HLE-SV each at 50 μg/ml or BSA. (C, D) CHO cells were incubated with HSΔΔ serum (20%) and the indicated FHR1 constructs, Factor H or BSA were added (75ug/ml) (C) C3b and (D) C5b9 deposition were detected with C3b/d and C5b-9 antibodies by flow cytometry. (E) Chicken erythrocytes were challenged in HSΔΔ serum (20%) in combination with the indicated FHR1 isoforms or mutants (each at 75 μg/ml), Factor H (50 ug/ml) or BSA. (F) Sheep erythrocytes were challenged with HSΔΔ (30%) and the FHR1 constructs were added at the indicated concentrations. Results represent mean ± SD of three independent experiments. (G) shows induction of pro-inflammatory cytkeins by human monocytes challenged with FHR1/FHR3 deficient serum supplemented with the indicated FHR1 variants and FHR1 mutants. *p < 0.05; **p < 0.01.
Surface FHR1 and Factor H control different regions
When bound via their C-termini FHR1 and Factor H control different surface areas or three-dimensional hemispheres. Our modeling approach revealed that FHR1-dimers attached via its C-termini control a rectangular area of 72 nm2 and a hemicylinder with a volume of 509 nm³. Its N-terminal region occupy about two-fifth (39.3%) and one-tenth (11.1%) of its corresponding surface area and volume respectively (Table 2, Figure 9A). The FHR1*-dimer, an FHR1-dimer with only one C-terminal domain attached, can bend in two dimensions and controls twice the surface area and volume of the FHR1-dimer, yet occupies only half of its corresponding surface area and volume (Table 2, Figure 9B). By contrast, Factor H which exposes a flexible 17SCRs-long segment, controls a large circular surface area of 8,638 nm2 and a hemisphere with a volume of 303,023 nm3. The N-terminal SCRs of one Factor H occupy 0.33% of its surface area and only 0.02% of its volume (Table 2, Figure 9C). However, Factor H is able to reach over 100 (50) times the surface area and over 500 (250) times the volume of FHR1 (or FHR1*) (Table 2). Interestingly, probable locations of N-terminal SCRs of FHR1 and FHR1* are uniformly distributed regarding their distance to the C-terminal SCRs (Figure 9D, Supplementary Videos 1, 2), while for Factor H, distances from C-terminal SCRs to probable locations of N-terminal SCRs form a normal distribution (Figure 9D, Supplementary Video 3). This reveals compelling differences in the action radius of both proteins.
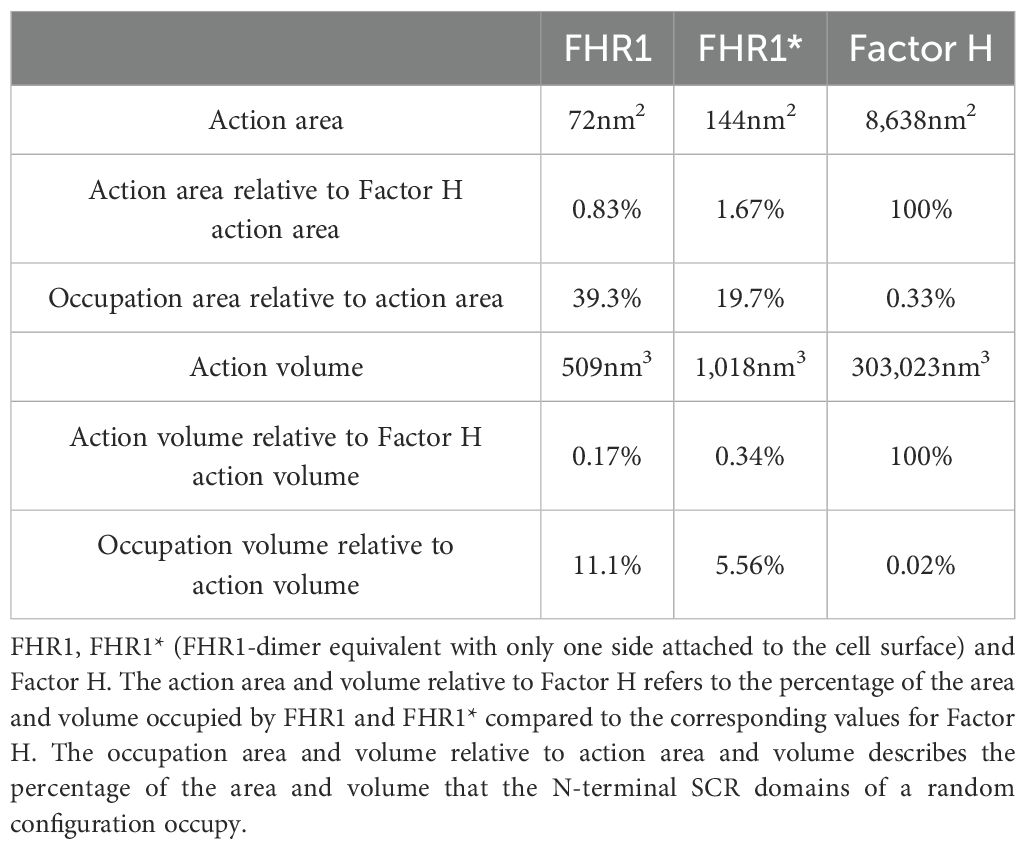
Table 2. Analytically computed action areas and volumes for FHR1 and Factor H.
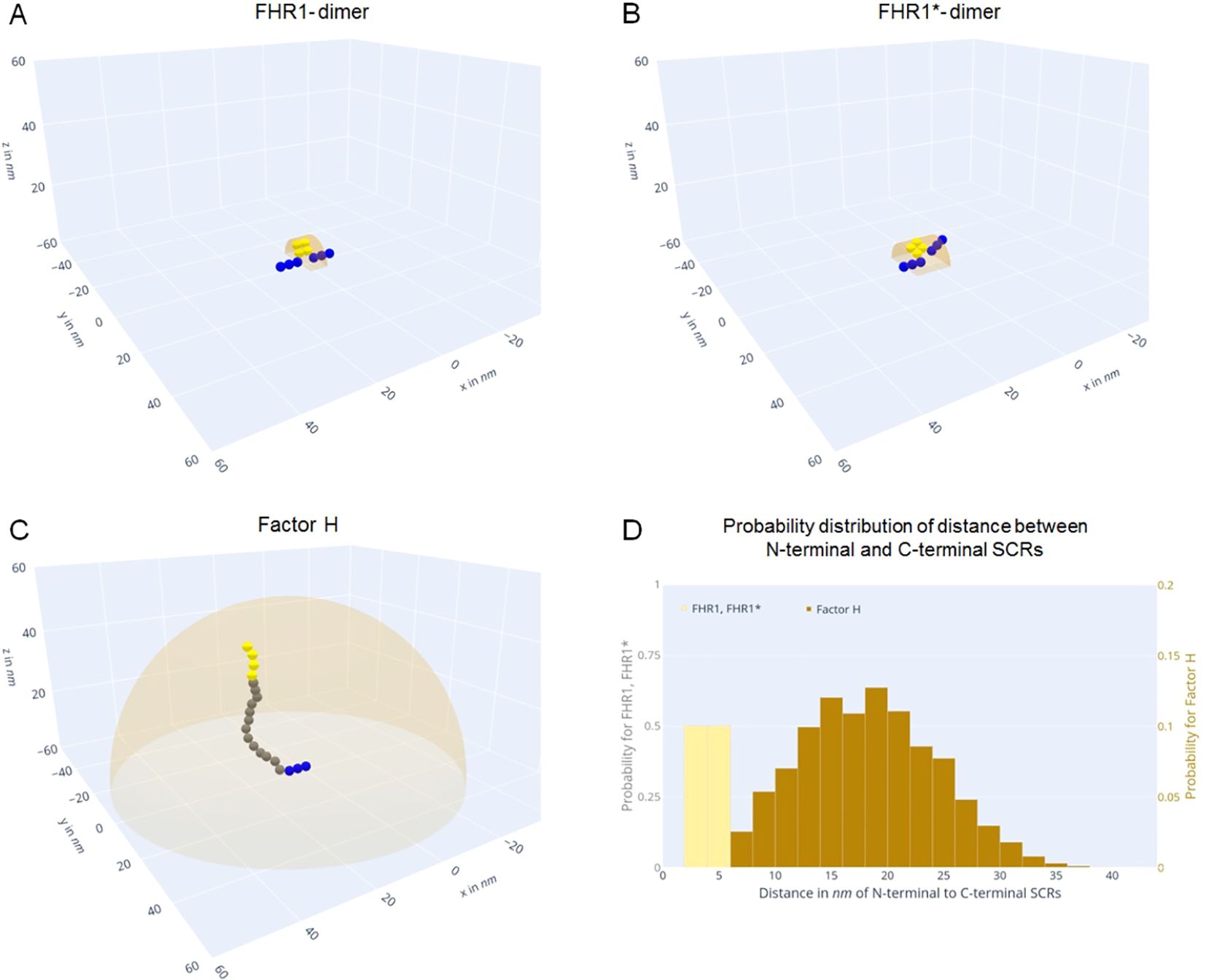
Figure 9. FHR1 and Factor H control different action areas. (A-C) Blue spheres represent C-terminal SCR domains. Green spheres represent freely movable SCRs5-17 in Factor (H) Yellow spheres signify N-terminal SCR domains bound to the cell surface (z = 0), while orange hemicylinders or hemispheres denote the analytically computed action volumes. (A) Exemplary FHR1-dimer. It can reach a volume of 509nm3, with one configuration occupying more than one-tenth (11.1%) of its action volume. (B) Exemplary FHR1*-dimer. It can move in two dimensions and reaches twice the region of FHR1. (C) Exemplary Factor H configuration. It can reach 303,023nm3, with one configuration occupying only 0.02% of its hemispherical action volume. (D) Histograms of distances from the N-terminal SCRs to the C-terminal SCRs over 106 Monte-Carlo simulations for each FHR1, FHR1* (yellow) and Factor H (brown) for maximum flexibility. FHR1 and FHR1* are uniformly distributed in the interval 2nm to 6nm. Factor H resembles a normal distribution with a mean of 18.2nm and standard deviation of 6.1nm (compare spatial distributions see Supplementary Videos 1–3).
Discussion
To define the role of FHR1 and the FHR1::Factor H interplay at target surfaces, and delineate the pathogenic role of FHR1SV mutants, we characterized how sequence variations in SCR3 and SCR5 affect ligand recognition and complement action. SCR3 of FHR1 (corresponding to Factor H SCR18) contributed to target recognition. YVQ-HLE variations influenced heparin, GAG-binding, attachment to necrotic surfaces, and assisted in C3b and C3d binding. Variations in SCR5 (SCR20 of Factor H) altered binding profiles and complement regulation at target surfaces. Wild-type FHR1LA interacted selectively with C3d, and Factor HSV selectively with C3b, but pathogenic FHR1SV switched its preferred interaction partner to C3b. Such altered surface contact by the aHUS-mutant modified C5b-9 deposition and hemolysis.
FHR1 inhibits the terminal pathway, blocks hemolysis, and induces cytokine and chemokine synthesis (3, 5). Here we showed that FHR1 prefers C3d and uses its C-terminal recognition-region to contact C3d, GAGs and surface ligands, simultaneously. Furthermore FHR1 plasma levels influenced Factor H access to C3b/C3d::Heparin combinations and balanced local Factor H-mediated C3 convertase control vs FHR1-mediated C5b-9 control and inflammation.
SCR3 of FHR1 mediates target recognition, binding to C3d, GAG-binding, and contact to necrotic surfaces and is part of the recognition- region. The FHR1YVQ and FHR1HLE variants bound with different strengths to heparin, heparan-sulfate, chondroitin-sulfate-A, and chondroitin-sulfate-4. FHR1HLE bound ca 15% more weakly to necrotic surfaces, but binding to C3d and C3b was affected to a minor degree. However, an additional heparin-binding element is also localized in the most C-terminal domain of Factor H (53, 57, 64), suggesting that the C-terminal region can bind to heparin, GAGs and/or sialic acids via two separated cooperating sites; one located in SCR3 (Factor HSCR18) and the other in SCR5 (Factor HSCR20) (15, 53, 57). The FHR1HLE variant was functionally more efficient for controlling C5b-9 deposition, hemolysis, and cytokine synthesis. Thus SCR3 is relevant for heparin and GAG binding, and sequence variations in SCR3 influence surface contact and local complement control.
FHR1SV-mutants displaying the Factor HSV-motif in SCR5 had different binding profiles to wild-type FHR1, and resulted in higher C5b-9 deposition and hemolysis, demonstrating the importance of residues 290 and 296 for C3b/C3d contact, protein function and explaining a pathophysiological role of the LA-vs-SV exchange. The preference of FHR1 for C3d and Factor H for C3b selects functionally different proteins and adjusts complement control at surfaces. Thereby adjusting C3-convertase inhibition vs C5-b9 inhibition and inflammatory effector functions.
Cell surfaces displaying carbohydrates, together with deposited C3b or C3d, provide different binding affinities for FHR1 and Factor H. Thus, the surface composition and C3-fragments can select for regulator vs effectors. Modified surfaces with specific GAG-profiles together with C3 fragments can select functionally distinct proteins and thereby adjust regulator vs modulator attachment. Such surface-characteristics apparently influence spatiotemporal complement dynamics, directing C3-convertase control, C3b-processing, and inflammatory opsonophagocytosis (2, 53). Using immunoelectron microscopy, we found that FHR1-homodimers attach to apoptotic surfaces and interestingly, also FHR1::Factor H heterodimers were identified.
Modeling the action regions of FHR1 and Factor H shows that Factor H controls more than 100 (500) times the surface (volume) of FHR1. However, this can be associated with limitations of different types; while FHR1-dimer configurations occupy a large proportion of their corresponding action region, the N-terminal of Factor H configuration only occupies 0.33% (0.02%) of its action area (volume) (Supplementary Videos 1–3). This indicates a rapid binding proclivity for FHR1, because potential binding partners in their controlled region must already be close (6nm) to the corresponding N-terminal SCRs, which is not the case for Factor H (Figure 9D). These binding profiles and diverse dimensions of controlled regions, show for FHR1 a rather restricted action region than for Factor H, highlighting unique local functions.
The altered binding profiles of wt FHR1LA, vs FHR1SV and Factor HLA-mutants cause different complement regulation at target-sites, which could explain aspects of aHUS-pathogenesis. The Genome-Aggregation-Data-base (GenomAD) describes the FHR1-variations at 290 and 296 as missense exchanges of likely pathogenic relevance. We show however that the FHR1SV, Factor HLA mutants have significantly altered ligand and target binding properties and opposite functions in C5b-9-deposition and hemolysis compared to wide type FHR1LA fragments. This is in line with the different binding characteristics of full-length FHR1 and Factor H to heparin and glycosaminoglycans, as shown here and as reported (23, 43, 53, 63). Furthermore, structural data on aHUS-mutants in Factor HSCRs19-20 suggested that the pathogenic LA-residues are ‘buried mutations’ that might alter the domain-fold (23, 43, 53, 63, 65). The LA or SV exchange appears to induce structural alterations that alter ligand interaction and affect complement regulation. In consequence, pathogenic FHR1 and Factor H aHUS-mutants show disturbances in their recognition profiles and interactions at endothelial surfaces under complement attack (66, 67).
The opposite effects of FHR1SV and Factor HLA-mutants show that LA and SV residues drive selectivity and influence surface targeting. The aHUS-associated, pathogenic FHR1SV and Factor HLA mutants increase the number of already known recognition and regulatory regions of FHR1 proteins combinations of recognition and regulatory regions. Both mutant proteins provide new combinations of the N-terminal dimer forming region (FHR1) or the regulatory region (Factor H) with alternate recognition segments. The Factor HLA mutant attached to C3d coated surfaces adds C3-convertase control and cofactor activity at the stage when C5b-9 inhibition and inflammatory effector action is required. Similarly, FHR1SV attached to C3-convertases or to C3b-coated surfaces displays C5b-9 inhibitory and inflammatory functions at a stage when C3 convertase regulation or cofactor activity is required. Thus, altered surface targeting of mutant FHR1SV and Factor HLA cause complement deregulation, which could explain aHUS pathology.
This study reveals a dynamic interplay of FHR1 and Factor H at complement activating surfaces. Despite their overall similarity, the recognition regions of the two proteins are functionally distinct and have differential effects on temporal complement control. Factor H controls immediate complement action at the C3 convertase level. FHR1 acts when deposited C3b is processed to C3d and then drives a secondary inflammatory response. Thus altered surface actions of the aHUS-associated mutants, FHR1SV and Factor HLA, adds a new chapter to our current understanding of misdirected complement activation at surfaces in aHUS patients.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Medical Faculty of the Friedrich Schiller University, Jena, Germany. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
LP: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. SS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. CSa: Formal analysis, Investigation, Methodology, Software, Visualization, Writing – review & editing. AH: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. MM: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. SZ: Data curation, Investigation, Methodology, Validation, Visualization, Writing – review & editing. MF: Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing – review & editing. TW: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – review & editing. CSk: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. PZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Deutsche Forschungsgemeinschaft SFB 1192, Immune mediated Glomerular diseases. project (B6 PZ & TW). PZ acknowledges support by Kidneeds Iowa, Iowa USA. This work was funded by the Deutsche Forschungsgemeinschaft SFB/TR 124, FungiNet with project number 210879364 (Project B4 to MTF) and the excellence graduate school Jena School for Microbial Communication (JSMC).õ
Conflict of interest
PZ received honoraria and speaker fees from Alexion, Alnylam, Bayer, CSL Vifor, Eleva Biologics, Novartis and Samsung Bioepis. MM is Chief Scientific Officer at Colzyx AB, Sweden.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2024.1469176/full#supplementary-material
References
1. Zipfel PF, Wiech T, Grone HG, Skerka Cl . Complement catalyzing glomerular diseases. Cell Tissue Res. (2021) 385:355–70. doi: 10.1007/s00441-021-03485-w
2. Skerka C, Pradel G, Halder LD, Zipfel PF, Zipfel SLH, Strauß O. Factor H-related protein 1: a complement regulatory protein and guardian of necrotic-type surfaces. Br J Pharmacol. (2021) 178:2823–31. doi: 10.1111/bph.v178.14
3. Lucientes-Continente L, Márquez-Tirado B, Goicoechea de Jorge E. The Factor H protein family: The switchers of the complement alternative pathway. Immunol Rev. (2023) 313:25–45. doi: 10.1111/imr.v313.1
4. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood. (2009) 114:2439–47. doi: 10.1182/blood-2009-02-205641
5. Irmscher S, Brix SR, Zipfel SLH, Halder LD, Mutlutürk S, Wulf S, et al. Serum FHR1 binding to necrotic-type cells activates monocytic inflammasome and marks necrotic sites in vasculopathies. Nat Commun. (2019) 10:2961. doi: 10.1038/s41467-019-10766-0
6. Irmscher S, Zipfel SLH, Halder LD, Ivanov L, Gonzalez-Delgado A, Waldeyer C, et al. Factor H-related protein 1 (FHR-1) is associated with atherosclerotic cardiovascular disease. Sci Rep. (2021) 11:22511. doi: 10.1038/s41598-021-02011-w
7. Jozsi M, Oppermann M, Lambris JD, Zipfel PF. The C-terminus of complement factor H is essential for host cell protection. Mol Immunol. (2007) 44:2697–706. doi: 10.1016/j.molimm.2006.12.001
8. Zipfel PF, Jokiranta TS, Hellwage J, Koistinen V, Meri S. The factor H protein family. Immunopharmacology. (1999) 42:53–60. doi: 10.1016/S0162-3109(99)00015-6
9. Piras R, Valoti E, Alberti M, Bresin E, Mele C, Breno M, et al. CFH and CFHR structural variants in atypical Hemolytic Uremic Syndrome: Prevalence, genomic characterization and impact on outcome. Front Immunol. (2023) 30:1011580. doi: 10.3389/fimmu.2022.1011580
10. Bernabéu-Herrero ME, Jiménez-Alcázar M, Anter J, Pinto S, Sánchez Chinchilla D, Garrido S, et al. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. (2015) 67:276–86. doi: 10.1016/j.molimm.2015.06.021
11. Oppermann M, Manuelian T, Józsi M, Brandt E, Jokiranta TS, Heinen S, et al. The C-terminus of complement regulator Factor H mediates target recognition: evidence for a compact conformation of the native protein. Clin Exp Immunol. (2006) 144:342–52. doi: 10.1111/j.1365-2249.2006.03071.x
12. Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, et al. A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol. (2008) 181:2610–9. doi: 10.4049/jimmunol.181.4.2610
13. Mihlan M, Stippa S, Jozsi M, Zipfel PF. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. (2009) 16:1630–40. doi: 10.1038/cdd.2009.103
14. Marquez-Tirado B, Gutiérrez-Tenorio J, Tortajada A, Lucientes Continente L, Caravaca-Fontán F, Malik TH, et al. Factor H-related protein 1 drives disease susceptibility and prognosis in C3 glomerulopathy. J Am Soc Nephrol. (2022) 33:1137–53. doi: 10.1681/ASN.2021101318
15. Csincsi AI, Szabó Z, Bánlaki Z, Uzonyi B, Cserhalmi M, Kárpáti É, et al. FHR-1 binds to C-reactive protein and enhances rather than inhibits complement activation. J Immunol. (2017) 199:292–303. doi: 10.4049/jimmunol.1600483
16. Alic L, Papac-Milicevic N, Czamara D, Rudnick RB, Ozsvar-Kozma M, Hartmann A, et al. A genome-wide association study identifies key modulators of complement factor H binding to malondialdehyde-epitopes. Proc Natl Acad Sci U S A. (2020) 117:9942–51. doi: 10.1073/pnas.1913970117
17. Simon N, Lasonder E, Scheuermayer M, Kuehn A, Tews S, Fischer R, et al. Malaria parasites co-opt human factor H to prevent complement-mediated lysis in the mosquito midgut. Cell Host Microbe. (2013) 13:29–41. doi: 10.1016/j.chom.2012.11.013
18. Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF. Binding of human factor H-related protein 1 to serum-resistant Borrelia burgdorferi is mediated by borrelial complement regulator-acquiring surface proteins. J Infect Dis. (2007) 196:124–33. doi: 10.1086/518509
19. Dasari P, Shopova IA, Stroe M, Wartenberg D, Martin-Dahse H, Beyersdorf N. Aspf2 from aspergillus fumigatus recruits human immune regulators for immune evasion and cell damage. Front Immunol. (2018) 9:1635. doi: 10.3389/fimmu.2018.01635
20. Röttgerding F, Kraiczy P. Immune evasion strategies of relapsing fever spirochetes. Front Immunol. (2020) 23:11:1560. doi: 10.3389/fimmu.2020.01560
21. Castiblanco-Valencia MM, Fraga TR, Silva LB, Monaris D, Abreu PA, Strobel S. Leptospiral immunoglobulin-like proteins interact with human complement regulators factor H, FHL-1, FHR-1, and C4BP. J Infect Dis. (2012) 205:995–1004. doi: 10.1093/infdis/jir875
22. Skerka C, Horstmann RD, Zipfel PF. Molecular cloning of a human serum protein structurally related to complement factor H. J Biol Chem. (1991) 266:12015–20. doi: 10.1016/S0021-9258(18)99058-7
23. Abarrategui-Garrido C, Martínez-Barricarte R, López-Trascasa M, de Córdoba SR, Sánchez-Corral P, et al. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. (2009) 114:4261–71. doi: 10.1182/blood-2009-05-223834
24. Xu B, Kang Y, Du Y, Guo W, Zhu L, Zhang H. Atypical hemolytic uremic syndrome-associated FHR1 isoform FHR1*B enhances complement activation and inflammation. Front Immunol. (2022) 13:755694. doi: 10.3389/fimmu.2022.755694
25. gnomAD . Available online at: https://gnomad.broadinstitute.org (Accessed August 3, 2024).
26. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy. J Am Soc Nephrol. (2020) 31:241–56. doi: 10.1681/ASN.2019050515
27. Renner B, Laskowski J, Poppelaars F, Ferreira VP, Blaine J, Antonioli AH, et al. Factor H related proteins modulate complement activation on kidney cells. Kidney Int. (2022) 102:1331–44. doi: 10.1016/j.kint.2022.07.035
28. Ruiz-Molina N, Parsons J, Decker EL, Reski R. Structural modelling of human complement FHR1 and two of its synthetic derivatives provides insight into their in-vivo functions. Comput Struct Biotechnol J. (2023) 21:1473–86. doi: 10.1016/j.csbj.2023.02.002
29. Kolodziejczyk R, Mikula KM, Kotila T, Postis VLG, Jokiranta TS, Goldman A, et al. Crystal structure of a tripartite complex between C3dg, C-terminal domains of factor H and OspE of Borrelia burgdorferi. PloS One. (2017) 12:e0188127. doi: 10.1371/journal.pone.0188127
30. Meri T, Amdahl H, Lehtinen MJ, Hyvärinen S, McDowell JV, Bhattacharjee A, et al. Microbes bind complement inhibitor factor H via a common site. PloS Pathog. (2013) 9:e1003308. doi: 10.1371/journal.ppat.1003308
31. Jozsi M, Oppermann M, Lambris JD, Zipfel PF. The C-terminus of complement factor H is essential for host cell protection. Mol Immunol. (2007) 44:2697–706. doi: 10.1016/j.molimm.2006.12.001
32. Ruiz-Molina N, Parsons J, Decker EL, et al. Structural modelling of human complement FHR1 and two of its synthetic derivatives provides insight into their in-vivo functions. Comput Struct Biotechnol J. (2023) 21:1473–86. doi: 10.1016/j.csbj.2023.02.002
33. Sánchez-Corral P, Pouw RB, López-Trascasa M, Józsi M. Self-damage caused by dysregulation of the complement alternative pathway: relevance of the factor H protein family. Front Immunol. (2018) 9:1607. doi: 10.3389/fimmu.2018.01607
34. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrín D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol. (2015) 11:77–82. doi: 10.1038/nchembio.1696
35. Blaum BS, Deakin JA, Johansson CM, Herbert AP, Barlow PN, Lyon M. Lysine and arginine side chains in glycosaminoglycan-protein complexes investigated by NMR, cross-linking, and mass spectrometry: a case study of the factor H-heparin interaction. J Am Chem Soc. (2010) 132:6374–81. doi: 10.1021/ja1000517
36. Schmidt CQ, Herbert AP, Hocking HG, Uhrín D, Barlow PN. Translational mini- review series on complement factor H: structural and functional correlations for factor H. Clin Exp Immunol. (2008) 151:14–24. doi: 10.1111/j.1365-2249.2007.03553.x
37. van Beek AE, Pouw RB, Brouwer MC, van Mierlo G, Geissler J, Ooijevaar-de Heer P, et al. Factor H-related (FHR)-1 and FHR-2 form homo- and heterodimers, while FHR-5 circulates only as homodimer in human plasma. Front Immunol. (2007) 8:1328. doi: 10.3389/fimmu.2017.01328
38. Zipfel PF, Mihlan M, Skerka C. The alternative pathway of complement: a pattern recognition system. Adv Exp Med Biol. (2007) 598:80–92.
39. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy J Am Soc Nephrol. (2020) 31:241–56. doi: 10.1681/ASN.2019050515
40. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. (2007) 38:1173–7.
41. Gharavi AG, Kiryluk K, Choi Y, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. (2011) 43:321–7. doi: 10.1038/ng.787
42. Dopler A, Stibitzky S, Hevey R, Mannes M, Guariento M, Höchsmann B, et al. Deregulation of factor H by factor H-related protein 1 depends on sialylation of host surfaces. Front Immunol. (2021) 12:615748. doi: 10.3389/fimmu.2021.615748
43. Józsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. (2008) 111:1512–4. doi: 10.1182/blood-2007-09-109876
44. Xie J, Kiryluk K, Li Y, Mladkova N, Zhu L, Hou P, et al. Fine mapping implicates a deletion of CFHR1 and CFHR3 in protection from igA nephropathy in Han Chinese. J Am Soc Nephrol. (2016) 27:3187–94. doi: 10.1681/ASN.2015111210
45. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD) Hum. Mol Genet. (2010) 19:4694–704. doi: 10.1093/hmg/ddq399
46. Medjeral-Thomas NR, Lomax-Browne HJ, Beckwith H, Willicombe M, McLean AG, Brookes P, et al. Circulating complement factor H-related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. (2018) 92:942–952. doi: 10.1016/j.kint.2017.03.043
47. Tortajada A, Gutiérrez E, Goicoechea de Jorge E, Anter J, Segarra A, Espinosa M, et al. Elevated factor H-related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int. (2017) 92:953–963. doi: 10.1016/j.kint.2017.03.041
48. Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PloS One. (2013) 8:e60352. doi: 10.1371/journal.pone.0060352
49. Martin Merinero H, Subías M, Pereda A, Gómez-Rubio E, Juana Lopez L, Fernandez C, et al. Molecular bases for the association of FHR-1 with atypical hemolytic uremic syndrome and other diseases. Blood. (2021) 137:3484–94. doi: 10.1182/blood.2020010069
50. Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, et al. Atypical hemolytic uremic syndrome associated with a hybrid complement gene. PloS Med. (2006) 3:e431. doi: 10.1371/journal.pmed.0030431
51. Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Garcia-Fernandez J, Gastoldi S, Besso L, et al. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol. (2015) 26:209–19. doi: 10.1681/ASN.2013121339
52. Li X, Hao Z, Liu X, Li W. Deficiency of mouse FHR-1 homolog, FHR-E, accelerates sepsis, and acute kidney injury through enhancing the LPS-induced alternative complement pathway. Front Immunol. (2020) 11:1123. doi: 10.3389/fimmu.2020.01123
53. Márquez-Tirado B, Gutiérrez-Tenorio J, Tortajada A, Lucientes Continente L, Caravaca-Fontán F, Malik TH, et al. Factor H–related protein 1 drives disease susceptibility and prognosis in C3 Glomerulopathy. J Am Soc Nephrol. (2022) 33:1137–1153. doi: 10.1681/ASN.2021101318
54. Chen Q, Manzke M, Hartmann A, Büttner M, Amann K, Pauly D, et al. Complement factor H-related 5-hybrid proteins anchor properdin and activate complement at self-surfaces. J Am Soc Nephrol. (2016) 27:1413–25. doi: 10.1681/ASN.2015020212
55. Rudnick RB, Chen Q, Stea DE, Hartmann A, Papac-Milicevic N, Person F, et al. FHR5 binds to laminins, uses separate C3b and surface-binding sites, and activates complement on malondialdehyde-acetaldehyde surfaces. J Immunol. (2018) 200:2280–90. doi: 10.4049/jimmunol.1701641
56. Lauer N, Mihlan M, Hartmann A, Schlötzer-Schrehardt U, Keilhauer C, Scholl HP, et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J Immunol. (2011) 187:4374–83. doi: 10.4049/jimmunol.1002488
57. Micklisch S, Lin Y, Jacob S, Karlstetter M, Dannhausen K, Dasari P, et al. Age-related macular degeneration associated polymorphism rs10490924 in ARMS2 results in deficiency of a complement activator. J Neuroinflammation. (2017) 14:4. doi: 10.1186/s12974-016-0776-3
58. Challis RC, Araujo GS, Wong EK, Anderson HE, Awan A, Dorman AM, et al. A De Novo Deletion in the Regulators of Complement Activation Cluster Producing a Hybrid Complement Factor H/Complement Factor H-Related 3 Gene in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol. (2016) 27(6):1617–24. doi: 10.1681/ASN.2015010100
59. Mihlan M, Blom AM, Kupreishvili K, Lauer N, Stelzner K, Bergström F, et al. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. (2011) 25:4198–210. doi: 10.1096/fj.11-186460
60. Irmscher S, Döring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
61. Michelfelder S, Fischer F, Wäldin A, Hörle KV, Pohl M, Parsons J, et al. The MFHR1 fusion protein is a novel synthetic multitarget complement inhibitor with therapeutic potential. J Am Soc Nephrol. (2018) 29:1141–53. doi: 10.1681/ASN.2017070738
62. Herwald H, Mörgelin M, Svensson HG, Sjöbring U. Zinc-dependent conformational changes in domain D5 of high molecular mass kininogen modulate contact activation. Eur J Biochem. (2001) 268:396–404. doi: 10.1046/j.1432-1033.2001.01888.x
63. Schmidt T, Afonso S, Perie L, Heidenreich K, Wulf S, Krebs CF, et al. An interdisciplinary diagnostic approach to guide therapy in C3 glomerulopathy. Front Immunol. (2022) 13:826513. doi: 10.3389/fimmu.2022.826513
64. Loeven MA, Maciej-Hulme ML, Yanginlar C, Hubers MC, Kellenbach E, de Graaf M, et al. Selective binding of heparin/heparan sulfate oligosaccharides to factor H and factor H-related proteins: therapeutic potential for C3 glomerulopathies. Front Immunol. (2021) 12:676662. doi: 10.3389/fimmu.2021.676662
65. Hyvärinen S, Uchida K, Varjosalo M, Jokela R, Jokiranta TS. Recognition of malondialdehyde-modified proteins by the C terminus of complement factor H is mediated via the polyanion binding site and impaired by mutations found in atypical hemolytic uremic syndrome. J Biol Chem. (2014) 289:4295–306. doi: 10.1074/jbc.M113.527416
66. Loeven MA, Rops AL, Lehtinen MJ, van Kuppevelt TH, Daha MR, Smith RJ, et al. Mutations in complement factor H impair alternative pathway regulation on mouse glomerular endothelial cells in vitro. J Biol Chem. (2016) 291:4974–81. doi: 10.1074/jbc.M115.702506
67. Meinel C, Spartà G, Dahse HM, Hörhold F, König R, Westermann M, et al. Streptococcus pneumoniae From Patients With Hemolytic Uremic Syndrome Binds Human Plasminogen via the Surface Protein PspC and Uses Plasmin to Damage Human Endothelial Cells. J Infect Dis. (2018) 217:358–70. doi: 10.1093/infdis/jix305
Keywords: complement, FHR1, complement hemolysis, hemolytic uremic syndrome, FHR1 Factor H balance at surfaces
Citation: Perie L, Stippa S, Saffer C, Hartmann A, Mörgelin M, Zipfel SLH, Figge MT, Wiech T, Skerka C and Zipfel PF (2024) Unique target binding by the C-terminal region of FHR1 provides a new perception of aHUS pathology. Front. Hematol. 3:1469176. doi: 10.3389/frhem.2024.1469176
Received: 23 July 2024; Accepted: 07 November 2024;
Published: 26 December 2024.
Edited by:
Ralph Kettritz, Charité University Medicine Berlin, GermanyReviewed by:
Francesco La Rocca, Madonna delle Grazie Hospital, ItalyNirmal Banda, University of Colorado Hospital, United States
Copyright © 2024 Perie, Stippa, Saffer, Hartmann, Mörgelin, Zipfel, Figge, Wiech, Skerka and Zipfel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter F. Zipfel, cGV0ZXIuemlwZmVsQGxpdmUuY29t
†These authors have contributed equally to this work