 Binsah George*
Binsah George* Kok Hoe Chan
Kok Hoe Chan Adan Rios
Adan Rios- Division of Hematology/Oncology, Department of Internal Medicine, The University of Texas Health Science Center at Houston, Houston, TX, United States
Early precursor T-cell acute lymphoblastic leukemia (ETP-ALL) is a rare type of T-cell ALL (T-ALL) that was initially described in 2009. Since its initial description, it has been formally recognized as a distinct entity, according to the 2016 World Health Organization classification. ETP-ALL is characterized by unique immunophenotypic and genomic profiles. The diagnosis and management of ETP-ALL remain challenging. Poorer outcomes, high incidence of induction failure and relapsed/refractory disease demand novel therapies. This review emphasizes the challenges of initial diagnosis, the molecular mechanisms underlying leukemogenesis, and the available therapeutic options. Additionally, it discusses the importance of induction failure, the prognostic value of detecting minimal residual disease, and the implications of hematopoietic stem cell transplantation, along with emerging novel therapies.
1 Introduction
The bone marrow (BM) is an abode of progenitor cells of myeloid and lymphoid precursors. After entering the thymus, lymphoid precursor cells differentiate and proliferate into thymocytes, which are recognized as T-lineage cells. T-cell acute lymphoblastic leukemia (T-ALL) is the proliferation of immature thymocytes and infiltration of immature T cells in the peripheral blood and BM, with or without involvement of the mediastinum, cerebral spinal fluid, and other lymphoid organs. T-ALL represents 25% of adult ALL cases with a long-term overall survival (OS) of 50-60% compared to the OS of 75-80% in the general pediatric ALL population (1, 2). Immature forms of T-ALL are further subclassified into early precursor T-cell (ETP)-ALL, near-ETP-ALL, and pro-T-ALL (3).
ETP-ALL is a specific entity defined by the world health organization (WHO) in 2016 for its unique immunophenotype and genetic constitution compared to other subtypes of T-ALL (4). ETP-ALL consists a subset of immature T-ALL cells with the capability of multilineage differentiation into T cells and myeloid cells, but no B-cell lineage (5–7). ETP-ALL was initially described in pediatric patients based on the characteristic gene expression profile (GEP) and immunophenotype (5). ETP-ALL represents approximately 7% of adult ALL cases, and approximately 22% of adult T-ALL cases are immature (8, 9). Unfortunately, ETP-ALL has poor outcomes due to its genomic instability and refractoriness to standard chemotherapy (8, 10). The 5-year OS rate of adult patients with ETP-ALL treated with standard chemotherapy is less than 40%. Currently, an allogeneic hematopoietic stem cell transplant (Allo-HSCT) is recommended after a patient achieved first complete remission (CR1) (5, 8, 11–14). In contrast to adult ETP-ALL, pediatric or young adolescent patients with ETP and near-ETP phenotypes have similar EFS and OS as compared to the non-ETP group as reported in the COG AALL0434 study, despite experienced higher rates of induction failure (15).
This review summarizes the diagnostic features, current evaluations of genetic aberrations, therapeutic options, and insights into the biology and molecular pathogenesis of ETP.
2 Diagnosis of ETP
2.1 Immunophenotypic signature
T-ALL is generally classified into four major subtypes: cortical (CD1a+), mature (sCD3+), pre-T (cCD3+ plus CD2+, CD5+, and/or CD8+), and pro-T ALL (cCD3+ and CD7+). In 2009, the immature pro-T-ALL subtype was redefined as ETP-ALL. The near-ETP subtype is positioned between the ETP and early (or pre-T) ALL (5, 16).
The European Group for the Immunological Characterization of Leukemias classified T-ALL from pro-T-ALL, pre-T-ALL, and cortical T-ALL to mature T-ALL based on the expression of immunological markers at different stages of T-cell development in the thymus (Table 1). T-ALL was defined by the expression of TdT and CD3.
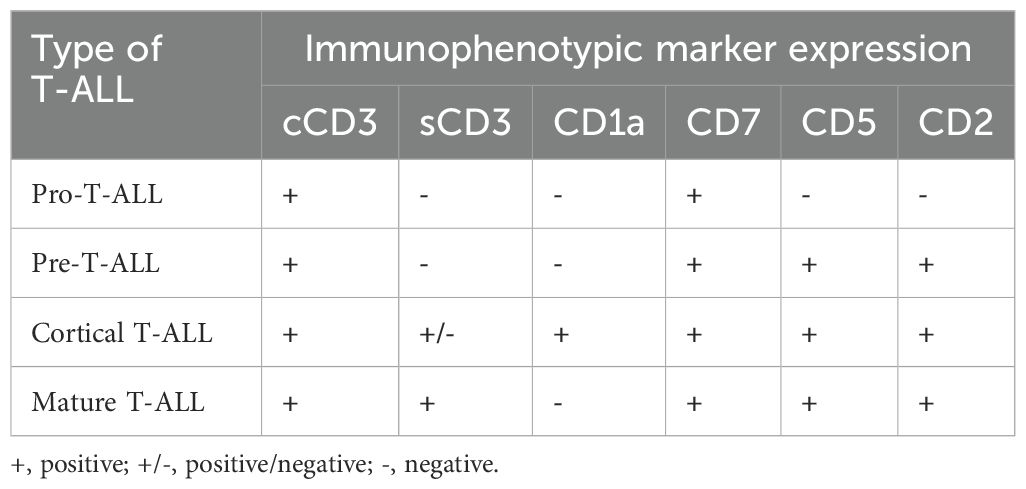
Table 1. EGIL classificaton of T-ALL (91).
Immunophenotyping is the cornerstone of ETP-ALL diagnosis; however, it is often challenging. The immunophenotype of ETP leukemic cells is distinct and was initially described by Coustan-Smith et al. in 2009; it typically lacks the expression of T lineage cell surface markers such as CD1a and CD8, shows absence or weak expression of CD5 (< 75% of the blast population), and the presence of at least one stem cell (CD34, CD117) marker and/or myeloid marker (CD11b, CD13, CD33, HLA-DR, CD65) in at least 25% of leukemic cells. This paved the path for the inclusion of ETP in WHO Classification 2016 and was defined as positive cCD3 and CD7 expression, negative CD1a and CD8, negative or dim expression of CD5 (< 75% blast population) and positive for at least one stem cell or myeloid antigen (≥ 25% positive) including CD34, HLA-DR, CD117, CD11b, CD13, CD33, CD65. According to the 2017 WHO classification, CD2 and CD4 may be positive for ETP-ALL (Table 2) (5). Although EGIL did not classify ETP-ALL as a separate entity, T-I ALL (pro-T-ALL) and T-II ALL (pre-T-ALL) cases were predominantly cCD3 and CD7 positive, surface CD3 negative (sCD3), CD1a negative and could represent ETP-ALL.

Table 2. Diagnostic criteria for ETP (5).
Since the initial description of ETP in 2009, multiple groups have attempted to define ETP to develop an immunotypic profile with high sensitivity and specificity. Khogeer et al. used 11 markers in the scoring system to diagnose ETP, a score of ≥ 8 suggested a diagnosis of ETP, expression of sCD3 (>20% blasts) and CD5 (>75% of blast) denoted -2 points in the scoring system (17). Zuurbier et al. devised an immunophenotypic profile of CD1a, CD4, and CD8 double negative, CD34 positive, and/or CD13/CD33 positive for the diagnosis of ETP (18), and The Tokyo Children’s Cancer Study Group proposed a 6- and 11-marker combination to validate the ETP-ALL cohort (12). The scoring system included CD2, sCD3, CD4, and CD10, and their study reported that these markers were expressed less frequently compared to the findings of Coustan-Smith et al. (5).
In the initial description of ETP, a subset of cases characterized by the expression of CD5 but not low enough to meet the criteria for ETP-ALL, was referred to as near ETP-ALL (5). The strong expression of CD5 is classified as near ETP and considered immunophenotypically and transcriptionally similar to ETP. A recent study by Morita et al., reported a 5-yr OS of near ETP-ALL and non-ETP-ALL to be identical while ETP-ALL showed a significantly worse OS (19).
The GEP of ETP cells shares similarities with that of hematopoietic stem cells and myeloid progenitor cells and is usually characterized by a definite molecular profile of frequent DNMT3A and FMS-like tyrosine kinase 3 (FLT3) mutations and a lower incidence of NOTCH1 mutations (11, 20, 21).
2.2 Genomic trademark
ETP-ALL derives its genetic profile from the earliest thymic progenitor cells (ETPs) rather than common lymphoid progenitors, which usually lose the B cell differentiation potential intra-thymically or pre-thymically but maintain their myeloid and T differentiation potential. Hence, their cellular origins and genetic profiles resemble those of myeloid progenitor cells (4, 22). Mutations in FLT3 are frequently observed in ETP-ALL. FLT3 activating mutation is an effective target in AML with multiple drugs available in the market and is being explored to alter the trajectory of the disease (23). A common mutation in FLT3, internal tandem duplication (ITD), or point mutation in the tyrosine kinase domain (FLT3-TKD), is observed in nearly 35% of adult ETP-ALL cases (11). However, gene mutations are observed in only 10% of the pediatric ETP-ALL (4). DNMT3A mutations, on the other hand, are usually seen in older patients and comprise approximately 12% of the cases (24).
Although ETP-ALL is derived from multipotent bone marrow progenitor cells, the genetic characteristics of pediatric and adult ETP vary. Even within the WHO classification of T-ALL, the ETP-ALL subset differs from T-ALL in terms of gene expression. Pediatric ETP-ALL frequently harbors mutations in RAS signaling pathways, such as FLT3, KRAS, BRAF, NRAS, JAK1, and JAK2, and hematopoietic regulators and developers, such as RUNX1, IKZF1, and GATA3 (4). However, adult ETP-ALL commonly exhibits mutated DNMT3A and FLT3 gene expression (24). Rearrangement and gene fusion in KMT2A, NUP214, NUP98, BCL11B, and MLLT10, and upregulation of HOX have recently been recognized in the genetic expression of ETP-ALL (25, 26). GATA3, which plays a significant role in the maturation of ETPs, has been observed in 30% of adult ETP-ALL cases. In addition, BCL11B expression, which leads to the maturation of the T cell lineage to double positive CD4 and CD8, was recently reported to be mutated in ETP-ALL (27, 28). IL7R mutations, which are required for early lymphoid maturation, are observed in nearly 45% of ETP-ALL cases. Mutations in these genes result in homodimerization, with subsequent hyperstimulation of the JAK/STAT and PI3K/AKT/mTOR pathways, leading to the development of leukemia (29). Mutation/deletion in transcription factor gene RUNX1 results in maturation arrest and loss of thymocyte differentiation (30, 31). Multiple genes have been implicated in the leukemogenesis of ETP-ALL, but the phenotype requires the gain of signaling driver mutations, such as NRAS and FLT3. Table 3 provides an overview of the genetic alterations in ETP-ALL. NOTCH1, PTEN, and FBXW7 oncogene mutations and deletions in CDKN2A/B are frequently observed in T-ALL; however, they are uncommon in ETP-ALL (32). Although near-ETP-ALL and ETP-ALL share some similarities, they exhibit different clinical and genetic profiles. Near ETP and non-ETP-ALL are enriched in TAL1 dysregulation, whereas ETP-ALL is enriched in LMO22/LYL1 aberrations (33).

3 Therapeutic options
3.1 Chemotherapeutic approach
At present, multiagent chemotherapy regimens represent the most effective therapeutic approach for ETP-ALL, despite their origin from pediatric protocols. These protocols typically involve induction, consolidation, dose intensification, intrathecal chemotherapy, and maintenance therapy. Studies involving chemotherapeutic agents such as HCVAD (hyperfractionated cyclophosphamide, vincristine, adriamycin, dexamethasone alternating with methotrexate, and high-dose cytarabine) with or without nelarabine (34), augmented Berlin-Frankfurt-Muenster (aBFM) protocol, and peg-asparaginase have shown that ETP-ALL has poorer survival rates than T-ALL, with higher induction failure rates and increased minimal residual disease (MRD) positivity at Day 33-35 post-induction (5, 8, 12, 35).
ETP-ALL, particularly in adults, has poorer outcome as compared to T-ALL (36). Therefore, the current consensus is to perform allo-HSCT in patients in CR1 to mitigate some of the adverse outcomes associated with ETP-ALL, especially in adults. Historically, both adult and pediatric outcomes of ETP-ALL have been poor. Allo-HSCT in CR1 has been shown to improve outcomes in multicenter studies and research by the GRAALL group (37). Recent reports indicate that achieving complete remission with FLAG-IDA (fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin) is promising, potentially overcoming initial molecular refractoriness and allowing progression to allo-HSCT (38).
Although cranial radiotherapy (CRT) can reduce CNS relapse, particularly in CNS3 disease, it does not translate into improved OS (39). Modern therapeutic approaches include intensive CNS-directed chemotherapy, which is incorporated into most regimens to significantly reduce long-term neurocognitive deficits and morbidity (40–42).
3.1.1 Addition of nelarabine
Nelarabine, a DNA synthesis inhibitor with selective toxicity to T lymphoblasts, received accelerated FDA approval in 2005 for treating patients with T-ALL who were unresponsive or relapsed after two chemotherapy regimens (43, 44). Its use and efficacy in pediatric and young adult populations have been studied extensively in a large randomized clinical trial (RCT) conducted by the Children’s Oncology Group (COG) (45). However, frontline use of nelarabine in adults remains to be explored in clinical trials (17, 34, 36).
In a significant phase 3 RCT for frontline pediatric and young adult populations (COG trial AALL0434) received the augmented BFM protocol (aBFM), participants were randomized to receive either high-dose MTX or Capizzi protocols, with further randomization to receive six 5-day courses of nelarabine (650 mg/m2/dose) during consolidation, delayed intensification, and maintenance. This study focused on disease-free survival (DFS). The 5-year DFS was 88% with high dose MTX + nelarabine compared to 82% without nelarabine, and the best outcome (91%) was observed with Capizzi protocols and nelarabine. Patients receiving nelarabine had a significantly lower cumulative incidence of CNS diseases (1.3% vs. 6.9% without nelarabine) (45).
3.1.2 HyperCVAD and nelarabine in adult T-ALL
Frontline use of nelarabine was tested in a phase 2 trial at the University of Texas MD Anderson Cancer Center for Adult T-ALL. In this trial, nelarabine was administered at 650 mg/m2/day IV for 5 days over 2 cycles following the standard 8 courses of HCVAD, alternating with high-dose MTX/cytarabine, and again for 2 cycles during maintenance. Initial analysis revealed no significant benefit (34). The protocol was then amended to include nelarabine in cycles 5 and 7 of induction and consolidation. Updated data revealed a 5-year OS of 83% with nelarabine compared to 38% without nelarabine in patients with non-ETP-ALL. Ongoing trial amendments (NCT00501826) include venetoclax and pegasparaginase (17).
3.1.3 Clinical trials using nelarabine frontline
Another major clinical trial was GRAALL-2014 (NCT02619630), a phase 2 multi-center study led by The Group for Research on Adult Acute Lymphoblastic Leukemia. This trial aimed to evaluate the efficacy of nelarabine in high-risk patients using a 4-gene classifier (NOTCH/FBXW7 and/or RAS/PTEN) and/or detectable MRD following induction and first consolidation. Nelarabine was administered at 1.5 g/m2 on days 1, 3, and 5 in up to 5 blocks during consolidation and maintenance. The primary endpoint of the 4-year DFS results is expected in 2025 (46).
The UK National Cancer Research Institute’s UK ALL14 (NCT 01085617) is a phase 3 RCT in adult patients, comparing the standard of care (SOC) with and without a single cycle of nelarabine (1.5 g/m2 IV, days 1, 3, and 5) after the second induction phase. Data presented at the 2021 American Society of Hematology (ASH) meeting showed a 3-year event-free survival (EFS) of 57% in the SOC arm versus 61.7% in the SOC-nelarabine arm. The relapse rates were similar in both arms (29.1% in the SOC arm vs. 28% in the SOC-nelarabine arm) (47). Differences in DFS outcomes between UKALL14 and AALL0434 may be due to the total number of doses (3 in UKALL14 vs. 30 in AALL0434) and the timing of nelarabine administration (following the second induction in UKALL14 vs. following the first induction in AALL0434) (46, 47).
Based on cumulative data from the phase 3 COG ALL0434 trial and adult trials by MD Anderson with HCVAD and nelarabine, many leukemia specialists in the USA are incorporating nelarabine into the initial treatment regimen for adult T-ALL.
3.2 Newer oral agents
Poor outcomes in ETP-ALL highlight the urgent need for novel therapeutic agents, particularly for relapsed and refractory diseases. By understanding the underlying molecular and biological mechanisms, we can potentially improve survival rates, similar to the success of inotuzumab and blinatumomab in B-ALL (48). Preclinical studies have indicated that ETP-ALL exhibits innate resistance to glucocorticoids compared to more differentiated T-ALL. Nevertheless, interventions targeting pathways such as JAK/STAT and BCL-2 have shown the potential to restore steroid sensitivity in ETP-ALL (29). These novel therapies are summarized in Table 4.
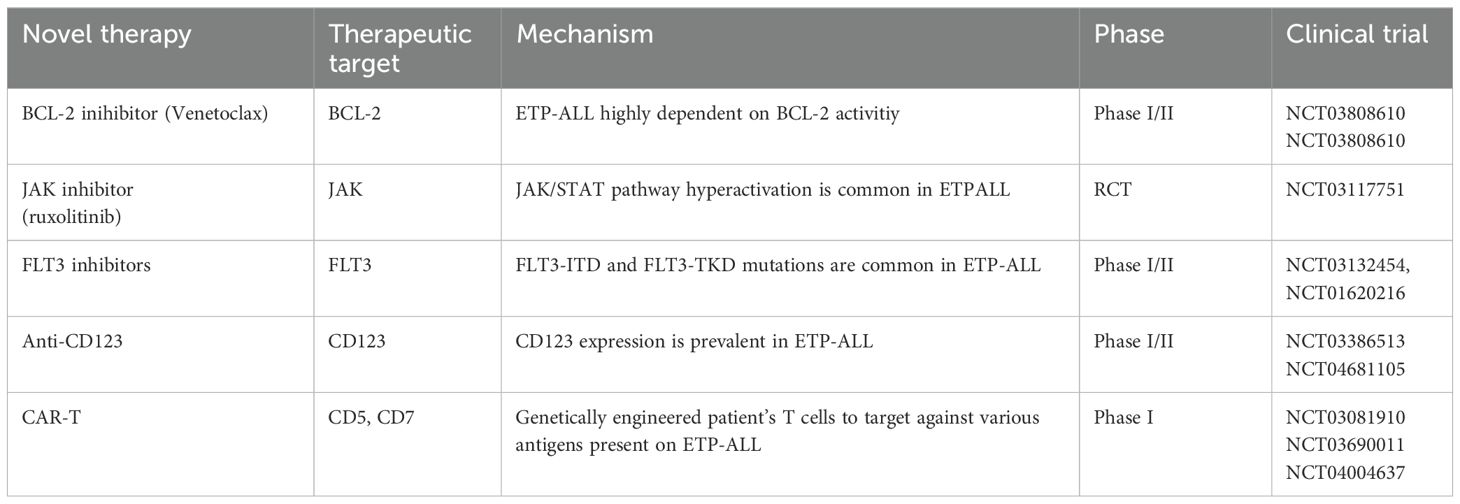
Table 4. Clinical trials with novel therapies.
3.2.1 Venetoclax
VEN is a BCL-2 inhibitor that has shown promising preclinical results in immature T-ALL and xenograft mouse models. Venetoclax is widely used to treat chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML) (49). In CLL, single-agent activity of VEN has been established; however, in AML, single-agent activity is poor due to the acquisition of resistance. ETP-ALL upregulates BCL-2 levels through its anti-apoptotic activity. The use of VEN in combination with chemotherapy or hypomethylating agents for the treatment (R/R) T-ALL is promising. Combination with other BH3 mimetic drugs, such as navitoclax and obatoclax (BCL-W and BCL2L inhibitors) or bortezomib (MCL-1 inhibitors), with or without chemotherapy/HMA, would be an attractive approach (50, 51). VEN and bortezomib have shown clinical activity in pilot studies on R/R T-ALL and may pave the way for future chemotherapy-free strategies (52).
3.2.2 FLT-3 inhibitors
Tyrosine kinase inhibitors (TKI) are well-established therapy for FLT-3 mutation AML and have improved overall survival since the use of TKI in FLT-3 mutated AML (53). Although there is limited preclinical evidence to evaluate this hypothesis, the high occurrence of FLT3-TKD and FLT3-ITD (35%) in ETP-ALL makes it an innovative option for using TKI to improve CR and MRD-negative induction responses (11). FLT3-ITD may be a potential marker for MRD monitoring in ETP-ALL when immunoglobulin/T-cell receptor (IG/TR) markers are not available (54).
3.2.3 Ruxolinitib
Patients with ETP-ALL and PRC2 mutations exhibit increased protein expression of the JAK/STAT pathway. Similarly, IL7R mutations stimulate the hyperactivation of the IL7 pathway, which in turn upregulates JAK/STAT signaling (29). RUXO inhibits JAK1/2. The anti-leukemic effects of RUXO in ETP-ALL xenografts have been demonstrated; however, surprisingly, these effects were independent of mutations in JAK/STAT signaling (55). Clinical trial NCT03613428 is investigating the effects of RUXO in R/R ETP-ALL patients, and NCT03117751 is examining the effects of a combination of conventional chemotherapy and RUXO in newly diagnosed pediatric ALL, including T-ALL.
3.2.4 Monoclonal antibody
3.2.4.1 Anti-CD123
CD123 is expressed in multiple leukemia types, including blastic plasmacytoid dendritic cell neoplasms (AML), and was recently found to be expressed in 43% of T-ALL cases. Surprisingly, CD123 is robustly expressed (93%) in ETP and early non ETP-ALL (56–58). Phase 1 clinical trial (NCT04681105) with single-agent flotetuzumab (a monoclonal antibody against CD123) is currently recruiting patients over the age of 12 years with hematological malignancies, including T-ALL. In addition, a single-agent monoclonal antibody against CD123 with IMGGN 632 is currently used to recruit patients with hematological malignancies above 18 years of age, including those with T-ALL (NCT03386513).
3.2.4.2 Anti-CD33
Gemtuzumab ozogamicin (GO), a monoclonal antibody against CD33, is commonly used in AML to improve survival in patients with high-risk cytogenetics (59). ETP-ALL cells express CD33 (67%). Therefore, the use of GO appears to be a promising therapeutic option. However, further clinical trials are required to evaluate the role of GO in ETP-ALL (18).
3.2.5 Chimeric antigen receptor engineered T-cell therapy
CAR-T cell therapy has led to significant advances in B acute lymphoblastic leukemia (B-ALL). Nevertheless, there are limitations to applying CAR-T therapy to T-ALL, as normal and malignant T cells have similar antigen profiles. CAR-T cells targeting CD5 and CD7 expression in T-ALL, including ETP, are currently being recruited (NCT03081910) and (NCT03690011).
3.3 Roles of allo-HSCT in ETP-ALL
For patients with T-ALL exhibiting high-risk features, contemporary pediatric-inspired regimens recommend allo-HSCT for CR1 and relapsed/refractory diseases. High-risk criteria include ETP-ALL, detectable MRD by multiparameter flow cytometry (≥ 0.1% at the end of induction (EOI) or end of consolidation (EOC)), or adverse genetic factors such as PTEN gene alterations, N/K RAS mutations, or the presence of NOTCH1/FBXW7 mutations (10, 60, 61).
In the AALL0434 study, patients diagnosed with ETP-ALL and near-ETP were significantly more likely to have EOI MRD levels ≥ 0.1% (75.5% and 57.1%, respectively) than those diagnosed with Non-ETP (23.5%). They also had a five-fold higher risk of induction failure (6.2% vs. 1.2%). Moreover, nearly 60% of ETP-ALL patients had EOI MRD levels ≥ 1%, whereas this was observed in 37.8% of near-ETP patients and only 13.2% of non-ETP patients. Despite these disparities, the EFS and OS rates for patients with ETP and near-ETP were comparable to those in patients without ETP, suggesting that induction failure or high EOI MRD levels alone do not justify altering the therapy for ETP-ALL within contemporary treatment protocols (45).
In a recent multicenter review involving children and young adults treated using modern protocols, 10-year EFS rates were comparable between patients with ETP-ALL (51.3%) and those without ETP features (58.6%). However, notably, those who underwent HSCT in CR1 achieved a significantly better 10-year DFS rate of 63.8% compared to 45.5% for those who did not undergo HSCT. Although the analysis did not specifically assess outcomes based on ETP status, an exploratory analysis suggested a potential benefit of allo-HSCT irrespective of end-consolidation MRD status (62).
Because of the unfavorable outcomes observed in ETP-ALL patients who retain detectable MRD at the end of consolidation therapy, our recommendation is to consider allo-HSCT using the most suitable donor available. It is crucial that patients attain a state of MRD-negative remission before proceeding with allo-HSCT because the presence of detectable MRD prior to transplantation is linked to elevated rates of relapse (63).
4 Clinical utilities of MRD monitoring
Monitoring of MRD is universally acknowledged as the most essential prognostic factor in ALL management. The clinical impact and timing of MRD monitoring are significant. Multiple studies on adult ALL have established effective key points for early MRD monitoring while receiving therapy in combination with chemotherapy and/or immunotherapy at early MRD monitoring during end of induction or post-early consolidation. Early MRD achievement is correlated with a good overall response and survival (64). Furthermore, a few studies have noted that quick MRD negativity after only 2 weeks of therapy is associated with a favorable prognosis (65).
Recently, the MRD has been refined by the addition of genomic marker assessments. A pivotal study on molecular MRD assessment was led by the German Multicenter Study Group for Adult ALL (GMALL) in 580 Ph-ALL patients in CR with high- or standard-risk features. The only prognostic significance correlating survival with standard induction/consolidation therapy was the achievement of molecular MRD negative (66). This was further validated by other groups, including the PETHEMA, GIMEMA LAL 1913 and UKALL XII/ECOG2993 (67, 68).
4.1 Fusion transcript molecular MRD assessment and novel markers
Another significant method for MRD monitoring is based on fusion transcripts, which are considered the main drivers of leukemogenesis and are expressed in leukemia cells. It is observed in nearly 40% of the patients with ALL and is fairly stable during therapy, making it a reliable and ideal tool for MRD assessment (69, 70). The relevance of molecular MRD and chromosomal abnormalities in prognosis makes their detection at the time of diagnosis critical. In T-ALL, approximately 20% of patients have TAL1 deletions (SIL/TAL1), and other gene alterations involving EML1/ABL, NUP/ABL, ETV6/ABL, and KMT2A are also relevant (71, 72).
The persistence of MRD is associated with higher relapse and shorter survival, which was validated in a study by the GRAALL group of 423 young patients with ALL (B and T), which showed that persistence of PTEN gene alteration, N/K RAS mutation, or the absence of NOTCH1/FBXW7 was associated with higher relapses (45).
4.2 Antigen receptor rearrangement molecular MRD assessment
During B and T lymphocyte development, immunoglobulin (IG) and T cell receptor (TR) genes undergo somatic rearrangement. This process involves combining the V, D, and J regions to form a single exon that encodes a variable region specific to either B or T lymphocytes. These rearrangements are essential for the normal physiological function of these cells and have not been implicated in the pathogenesis of leukemia. However, in leukemia, the malignant transformation of lymphoid cells results in clonal IG and TR gene rearrangements. These clonal signatures can be leveraged for the detection of MRD, even at low levels, in ALL cells. Leukemic cells express a distinct sequence of gene rearrangements compared with normal lymphoid cells.
Typically, TR rearrangements are associated with T lymphocytes, whereas IG rearrangements are associated with B lymphoid cells. However, in leukemia cases, instances of cross-lineage involvement are observed, where both B and T leukemia cells exhibit mixed-lineage features. This characteristic can be advantageous for evaluating the MRD.
Therefore, it is crucial that the DNA extracted from leukemia cells undergo PCR amplification and subsequent gene scanning to establish clonality. This approach aids in identifying unique IG and TR gene rearrangements specific to leukemia cells, facilitating accurate detection and monitoring of MRD (73–75).
4.3 Minimal residual disease monitoring and methodologies of MRD detection
Advances in technology have significantly enhanced the ability to detect and assess residual leukemia cells beyond traditional cytomorphology. Various approaches including flow cytometry (FCM), cytogenetics, high-throughput sequencing, and PCR-related tools have been implemented globally. Standard methods for assessing MRD in ALL involve amplifying IG/TR gene rearrangements using PCR or multiparametric flow cytometry to identify leukemia-associated immunophenotypes (LAIP). Current MRD assessments can detect one leukemic blast in 10,000 normal cells (76–79).
Technological advancements have expanded the applicability of conventional methods to achieve greater sensitivity (> 95%). These include digital droplet PCR (ddPCR), which represents the third generation of conventional PCR methods and provides faster and more reliable results. ddPCR enables nucleic acid quantification without the need for calibration curves (80, 81). Next-generation flow cytometry (NGF) for MRD is a novel technique that evaluates the degree of immunophenotypic variability between normal B cell precursors and ALL blast cells. It can also distinguish therapy-induced immunophenotypic shifts in the ALL population, achieving a sensitivity of 10^-6 compared to the 10^-4 -10^-5 sensitivity of conventional FCM-MRD (82–84).
Another promising method for MRD detection is next-generation sequencing (NGS), which detects clonal sequences and clone-specific IG/TR gene rearrangements before and after treatment. NGS offers a sensitivity of 10^-6, an improvement over the 10^-5 sensitivity achieved with real-time quantitative PCR (RQ-PCR) (85, 86). Comparative studies from the Berlin-Frankfurt-Munster trials have shown that NGS is more reliable than qPCR for detecting relapse post-induction and post-allogeneic HSCT in ALL (85, 87). The integration of innovative technologies, especially ddPCR and NGS, represents an attractive and feasible alternative to conventional MRD assessment for more precise evaluation of post-therapy responses in patients with ALL.
The choice between using peripheral blood (PB) versus bone marrow (BM) for MRD assessment depends on the lineage (B-cell or T-cell). In B-cell precursor ALL (BCP-ALL), BM samples generally provide MRD levels that–1-3 logs better than those of PB samples, making BM the preferred source for reliable MRD assessment. In contrast, for T-ALL, both the BM and PB samples are considered reliable sources for MRD assessment. However, most institutions continue to rely on BM samples to assess MRD in T-lineage ALL, although further validation is needed to exclusively transition to PB samples (88, 89).
Despite the potential of newer NGS-based MRD assessment techniques, their widespread adoption is limited in academic centers owing to their high costs and technical complexity. As these assays become more cost-effective, broader and more robust applications of these technologies are expected to become available (90).
5 Conclusion
The diagnosis and management of ETP-ALL are challenging. Close monitoring of MRD during induction/consolidation and post-transplantation can offer timely opportunities to add or alter treatment with early detection of MRD. Novel therapies based on molecular pathogenesis have demonstrated promising outcomes in preclinical models of VEN and RUXO; however, larger clinical trials are needed to validate the outcomes of ETP-ALL. FLT-3 mutations and epigenetic aberrations in ETP-ALL may justify the use of FLT-3 inhibitors and hypomethylating agents in this group of diseases. Another innovative option is the use of monoclonal antibodies against CD123, CD33, and CAR-T cells. Advances in CRISPR/Cas9 technology and CD5, CD7 targeted CAR-T cells may pave the way for better outcomes and prognoses of ETP-ALL.
Author contributions
BG: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Validation, Writing – original draft, Writing – review & editing. KC: Data curation, Investigation, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. AR: Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rafei H, Kantarjian HM, Jabbour EJ. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk Lymphoma. (2019) 60:2606–21. doi: 10.1080/10428194.2019.1605071
2. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. (2006) 354:166–78. doi: 10.1056/nejmra052603
3. Paietta E. Proposals for the immunological classification of acute leukemias. Leukemia. (1995) 9:2147–8.
4. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. (2012) 481:157–63. doi: 10.1038/nature10725
5. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. (2009) 10:147–56. doi: 10.1016/s1470-2045(08)70314-0
6. Wada H, Masuda K, Satoh R, Kakugawa K, Ikawa T, Katsura Y, et al. Adult T-cell progenitors retain myeloid potential. Nature. (2008) 452:768–72. doi: 10.1038/nature06839
7. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
8. Jain N, Lamb AV, O'Brien S, Ravandi F, Konopleva M, Jabbour E, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. (2016) 127:1863–9. doi: 10.1182/blood-2015-08-661702
9. Mao J, Xue L, Wang H, Zhu Y, Wang J, Zhao L. A new treatment strategy for early T-cell precursor acute lymphoblastic leukemia: A case report and literature review. Onco Targets Ther. (2021) 14:3795–802. doi: 10.2147/OTT.S312494
10. Bond J, Graux C, Lhermitte L, Lara D, Cluzeau T, Leguay T, et al. Early response-based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: A group for research on adult acute lymphoblastic leukemia study. J Clin Oncol. (2017) 35:2683–91. doi: 10.1200/jco.2016.71.8585
11. Neumann M, Coskun E, Fransecky L, Mochmann LH, Bartram I, Sartangi NF, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PloS One. (2013) 8:e53190. doi: 10.1371/journal.pone.0053190
12. Inukai T, Kiyokawa N, Campana D, Coustan-Smith E, Kikuchi A, Kobayashi M, et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children's Cancer Study Group Study L99-15. Br J Haematol. (2012) 156:358–65. doi: 10.1111/j.1365-2141.2011.08955.x
13. Ma M, Wang X, Tang J, Xue H, Chen J, Pan C, et al. Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med. (2012) 6:416–20. doi: 10.1007/s11684-012-0224-4
14. Patrick K, Wade R, Goulden N, Mitchell C, Moorman AV, Rowntree C, et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol. (2014) 166:421–4. doi: 10.1111/bjh.12882
15. Wood BL, Devidas M, Summers RJ, Chen Z, Asselin B, Rabin KR, et al. Prognostic significance of ETP phenotype and minimal residual disease in T-ALL: a Children's Oncology Group study. Blood. (2023) 142:2069–78. doi: 10.1182/blood.2023020678
16. Genescà E, la Starza R. Early T-cell precursor ALL and beyond: immature and ambiguous lineage T-ALL subsets. Cancers (Basel). (2022) 14(8):1873. doi: 10.3390/cancers14081873
17. Morita K, Jain N, Kantarjian H, Takahashi K, Fang H, Konopleva M, et al. Outcome of T-cell acute lymphoblastic leukemia/lymphoma: Focus on near-ETP phenotype and differential impact of nelarabine. Am J Hematol. (2021) 96:589–98. doi: 10.1002/ajh.26144
18. Khogeer H, Rahman H, Jain N, Angelova EA, Yang H, Quesada A, et al. Early T precursor acute lymphoblastic leukaemia/lymphoma shows differential immunophenotypic characteristics including frequent CD33 expression and in vitro response to targeted CD33 therapy. Br J Haematol. (2019) 186:538–48. doi: 10.1111/bjh.15960
19. Zuurbier L, Gutierrez A, Mullighan CG, Canté-Barrett K, Gevaert AO, de Rooi J, et al. Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica. (2014) 99:94–102. doi: 10.3324/haematol.2013.090233
20. Neumann M, Heesch S, Schlee C, Schwartz S, Gökbuget N, Hoelzer D, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. (2013) 121:4749–52. doi: 10.1182/blood-2012-11-465138
21. Neumann M, Heesch S, Gökbuget N, Schwartz S, Schlee C, Benlasfer O, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. (2012) 2:e55. doi: 10.1038/bcj.2011.49
22. Bell JJ, Bhandoola A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature. (2008) 452:764–7. doi: 10.1038/nature06840
23. Zhao JC, Agarwal S, Ahmad H, Amin K, Bewersdorf JP, Zeidan AM. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev. (2022) 52:100905. doi: 10.1016/j.blre.2021.100905
24. González-Gil C, Morgades M, Fuster-Tormo F, García-Chica J, Montesinos P, Torrent A, et al. Genomic data improves prognostic stratification in adult T-cell acute lymphoblastic leukemia patients enrolled in measurable residual disease-oriented trials. Blood. (2021) 138:3486–6. doi: 10.1182/blood-2021-148263
25. La Starza R, Pierini V, Pierini T, Nofrini V, Matteucci C, Arniani S, et al. Design of a comprehensive fluorescence in situ hybridization assay for genetic classification of T-cell acute lymphoblastic leukemia. J Mol Diagn. (2020) 22:629–39. doi: 10.1016/j.jmoldx.2020.02.004
26. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. (2016) 16:494–507. doi: 10.1038/nrc.2016.63
27. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49:1211–8. doi: 10.1038/ng.3909
28. Di Giacomo D, La Starza R, Gorello P, Pellanera F, Kalender Atak Z, De Keersmaecker K, et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood. (2021) 138:773–84. doi: 10.1182/blood.2020010510
29. Delgado-Martin C, Meyer LK, Huang BJ, Shimano KA, Zinter MS, Nguyen JV, et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia. (2017) 31:2568–76. doi: 10.1038/leu.2017.136
30. Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. (2007) 204:1945–57. doi: 10.1084/jem.20070133
31. Sin CF, Man PM. Early T-cell precursor acute lymphoblastic leukemia: diagnosis, updates in molecular pathogenesis, management, and novel therapies. Front Oncol. (2021) 11:750789. doi: 10.3389/fonc.2021.750789
32. Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood. (2013) 122:74–82. doi: 10.1182/blood-2013-03-491092
33. Tavakoli Shirazi P, Eadie LN, Heatley SL, Hughes TP, Yeung DT, White DL. The effect of co-occurring lesions on leukaemogenesis and drug response in T-ALL and ETP-ALL. Br J Cancer. (2020) 122:455–64. doi: 10.1038/s41416-019-0647-7
34. Abaza Y, M Kantarjian H, Faderl S, Jabbour E, Jain N, Thomas D, et al. Hyper-CVAD plus nelarabine in newly diagnosed adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma. Am J Hematol. (2018) 93:91–9. doi: 10.1002/ajh.24947
35. Fuhrmann S, Schabath R, Möricke A, Zimmermann M, Kunz JB, Kulozik AE, et al. Expression of CD56 defines a distinct subgroup in childhood T-ALL with inferior outcome. Results of the ALL-BFM 2000 trial. Br J Haematol. (2018) 183:96–103. doi: 10.1111/bjh.15503
36. Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. (2009) 114:5136–45. doi: 10.1182/blood-2009-08-231217
37. Bond J, Graux C, Lhermitte L, Lara D, Cluzeau T, Leguay T, et al. Early response–based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: A group for research on adult acute lymphoblastic leukemia study. J Clin Oncol. (2017) 35:2683–91. doi: 10.1200/jco.2016.71.8585
38. Bataller A, Garrote M, Oliver-Caldés A, López-Guerra M, Colomer D, Aymerich M, et al. Early T-cell precursor lymphoblastic leukaemia: response to FLAG-IDA and high-dose cytarabine with sorafenib after initial refractoriness. Br J Haematol. (2019) 185:755–7. doi: 10.1111/bjh.15601
39. Vora A, Andreano A, Pui CH, Hunger SP, Schrappe M, Moericke A, et al. Influence of cranial radiotherapy on outcome in children with acute lymphoblastic leukemia treated with contemporary therapy. J Clin Oncol. (2016) 34:919–26. doi: 10.1200/jco.2015.64.2850
40. Stark B, Avrahami G, Nirel R, Abramov A, Attias D, Ballin A, et al. Extended triple intrathecal therapy in children with T-cell acute lymphoblastic leukaemia: a report from the Israeli National ALL-Studies. Br J Haematol. (2009) 147:113–24. doi: 10.1111/j.1365-2141.2009.07853.x
41. Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. (2009) 360:2730–41. doi: 10.1056/nejmoa0900386
42. Spiegler BJ, Kennedy K, Maze R, Greenberg ML, Weitzman S, Hitzler JK, et al. Comparison of long-term neurocognitive outcomes in young children with acute lymphoblastic leukemia treated with cranial radiation or high-dose or very high-dose intravenous methotrexate. J Clin Oncol. (2006) 24:3858–64. doi: 10.1200/jco.2006.05.9055
43. Rodriguez CO Jr, Mitchell BS, Ayres M, Eriksson S, Gandhi V. Arabinosylguanine is phosphorylated by both cytoplasmic deoxycytidine kinase and mitochondrial deoxyguanosine kinase. Cancer Res. (2002) 62:3100–5.
44. Cohen MH, Johnson JR, Massie T, Sridhara R, McGuinn WD Jr, Abraham S, et al. Approval summary: nelarabine for the treatment of T-cell lymphoblastic leukemia/lymphoma. Clin Cancer Res. (2006) 12:5329–35. doi: 10.1158/1078-0432.ccr-06-0606
45. Dunsmore KP, Winter SS, Devidas M, Wood BL, Esiashvili N, Chen Z, et al. Children's oncology group AALL0434: A phase III randomized clinical trial testing nelarabine in newly diagnosed T-cell acute lymphoblastic leukemia. J Clin Oncol. (2020) 38:3282–93. doi: 10.1200/jco.20.00256
46. Clare J Rowntree AAK, Clifton-Hadley L, Farah N, Mansour MR, Hussain J, Marks DI, et al. Fielding, first analysis of the UKALL14 randomized trial to determine whether the addition of nelarabine to standard chemotherapy improves event free survival in adults with T-cell acute lymphoblastic leukaemia (CRUK/09/006). Blood. (2021) 138:366.
47. Rowntree CJ, Kirkwood AA, Clifton-Hadley L, Farah N, Mansour MR, Hussain J, et al. First analysis of the UKALL14 randomized trial to determine whether the addition of nelarabine to standard chemotherapy improves event free survival in adults with T-cell acute lymphoblastic leukaemia (CRUK/09/006). Blood. (2021) 138:366–6. doi: 10.1182/blood-2021-152355
48. George B, Kantarjian H, Jabbour E, Jain N. Role of inotuzumab ozogamicin in the treatment of relapsed/refractory acute lymphoblastic leukemia. Immunotherapy. (2016) 8:135–43. doi: 10.2217/imt.15.108
49. George B, Kantarjian H, Baran N, Krocker JD, Rios A. TP53 in acute myeloid leukemia: molecular aspects and patterns of mutation. Int J Mol Sci. (2021) 22(19):10782. doi: 10.3390/ijms221910782
50. Moujalled DM, Pomilio G, Ghiurau C, Ivey A, Salmon J, Rijal S, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia. (2019) 33:905–17. doi: 10.1038/s41375-018-0261-3
51. Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. (2018) 8:1566–81. doi: 10.1158/2159-8290.cd-18-0140
52. La Starza R, Cambò B, Pierini A, Bornhauser B, Montanaro A, Bourquin JP, et al. Venetoclax and bortezomib in relapsed/refractory early T-cell precursor acute lymphoblastic leukemia. JCO Precis Oncol. (2019) 3:PO.19.00172. doi: 10.1200/PO.19.00172
53. Lo Nigro L, Andriano N, Buldini B, Silvestri D, Villa T, Locatelli F, et al. FLT3-ITD in children with early T-cell precursor (ETP) acute lymphoblastic leukemia: incidence and potential target for monitoring minimal residual disease (MRD). Cancers (Basel). (2022) 14(10):2475. doi: 10.3390/cancers14102475
54. Daver N, Venugopal S, Ravandi F. FLT3 mutated acute myeloid leukemia: 2021 treatment algorithm. Blood Cancer J. (2021) 11:104. doi: 10.1038/s41408-021-00495-3
55. Maude SL, Dolai S, Delgado-Martin C, Vincent T, Robbins A, Selvanathan A, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. (2015) 125:1759–67. doi: 10.1182/blood-2014-06-580480
56. Angelova E, Audette C, Kovtun Y, Daver N, Wang SA, Pierce S, et al. CD123 expression patterns and selective targeting with a CD123-targeted antibody-drug conjugate (IMGN632) in acute lymphoblastic leukemia. Haematologica. (2019) 104:749–55. doi: 10.3324/haematol.2018.205252
57. Lane AA. Targeting CD123 in AML. Clin Lymphoma Myeloma Leuk. (2020) 20 Suppl 1:S67–s68. doi: 10.1016/S2152-2650(20)30466-3
58. Pemmaraju N, Lane AA, Sweet KL, Stein AS, Vasu S, Blum W, et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N Engl J Med. (2019) 380:1628–37. doi: 10.1056/nejmoa1815105
59. Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. (2014) 15:986–96. doi: 10.1016/s1470-2045(14)70281-5
60. Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. (2011) 118:2077–84. doi: 10.1182/blood-2011-03-338707
61. O'Dwyer KM. Optimal approach to T-cell ALL. Hematol Am Soc Hematol Educ Program. (2022) 2022:197–205. doi: 10.1182/hematology.2022000337C
62. Raetz EA, Rebora P, Conter V, Schrappe M, Devidas M, Escherich G, et al. Outcome for children and young adults with T-cell ALL and induction failure in contemporary trials. J Clin Oncol. (2023) 41:5025–34. doi: 10.1200/jco.23.00088
63. Summers RJ, Teachey DT, Hunger SP. How I treat early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) in children. Blood. (2024). doi: 10.1182/blood.2023023155
64. Gökbuget N, Dombret H, Giebel S, Bruggemann M, Doubek M, Foà R, et al. Minimal residual disease level predicts outcome in adults with Ph-negative B-precursor acute lymphoblastic leukemia. Hematology. (2019) 24:337–48. doi: 10.1080/16078454.2019.1567654
65. Šálek C, Folber F, Froňková E, Procházka B, Marinov I, Cetkovský P, et al. Early MRD response as a prognostic factor in adult patients with acute lymphoblastic leukemia. Eur J Haematol. (2016) 96:276–84. doi: 10.1111/ejh.12587
66. Gökbuget N, Kneba M, Raff T, Trautmann H, Bartram CR, Arnold R, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. (2012) 120:1868–76. doi: 10.1182/blood-2011-09-377713
67. Ribera JM, Oriol A, Morgades M, Montesinos P, Sarrà J, González-Campos J, et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol. (2014) 32:1595–604. doi: 10.1200/jco.2013.52.2425
68. Patel B, Rai L, Buck G, Richards SM, Mortuza Y, Mitchell W, et al. Minimal residual disease is a significant predictor of treatment failure in non T-lineage adult acute lymphoblastic leukaemia: final results of the international trial UKALL XII/ECOG2993. Br J Haematol. (2010) 148:80–9. doi: 10.1111/j.1365-2141.2009.07941.x
69. Chen X, Wood BL. Monitoring minimal residual disease in acute leukemia: Technical challenges and interpretive complexities. Blood Rev. (2017) 31:63–75. doi: 10.1016/j.blre.2016.09.006
70. Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, et al. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. (2003) 17:2318–57. doi: 10.1038/sj.leu.2403135
71. Elia L, Mancini M, Moleti L, Meloni G, Buffolino S, Krampera M, et al. A multiplex reverse transcriptase-polymerase chain reaction strategy for the diagnostic molecular screening of chimeric genes: a clinical evaluation on 170 patients with acute lymphoblastic leukemia. Haematologica. (2003) 88:275–9.
72. D'Angiò M, Valsecchi MG, Testi AM, Conter V, Nunes V, Parasole R, et al. Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: a 10-year experience of the AIEOP group. Haematologica. (2015) 100:e10–3. doi: 10.3324/haematol.2014.112151
73. van Dongen JJ, Langerak AW, Brüggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. (2003) 17:2257–317. doi: 10.1038/sj.leu.2403202
74. Szczepański T, Pongers-Willemse MJ, Langerak AW, Harts WA, Wijkhuijs AJ, et al. Ig heavy chain gene rearrangements in T-cell acute lymphoblastic leukemia exhibit predominant DH6-19 and DH7-27 gene usage, can result in complete V-D-J rearrangements, and are rare in T-cell receptor alpha beta lineage. Blood. (1999) 93:4079–85.
75. Verhagen OJ, Willemse MJ, Breunis WB, Wijkhuijs AJ, Jacobs DC, Joosten SA, et al. Application of germline IGH probes in real-time quantitative PCR for the detection of minimal residual disease in acute lymphoblastic leukemia. Leukemia. (2000) 14:1426–35. doi: 10.1038/sj.leu.2401801
76. Flohr T, Schrauder A, Cazzaniga G, Panzer-Grümayer R, van der Velden V, Fischer S, et al. Minimal residual disease-directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T-cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia. (2008) 22:771–82. doi: 10.1038/leu.2008.5
77. George BS, Yohannan B, Gonzalez A, Rios A. Mixed-phenotype acute leukemia: clinical diagnosis and therapeutic strategies. Biomedicines. (2022) 10(8):1974. doi: 10.3390/biomedicines10081974
78. Cazzaniga G, Valsecchi MG, Gaipa G, Conter V, Biondi A. Defining the correct role of minimal residual disease tests in the management of acute lymphoblastic leukaemia. Br J Haematol. (2011) 155:45–52. doi: 10.1111/j.1365-2141.2011.08795.x
79. Dworzak MN, Gaipa G, Ratei R, Veltroni M, Schumich A, Maglia O, et al. Standardization of flow cytometric minimal residual disease evaluation in acute lymphoblastic leukemia: Multicentric assessment is feasible. Cytomet B Clin Cytom. (2008) 74:331–40. doi: 10.1002/cyto.b.20430
80. Whale AS, Huggett JF, Cowen S, Speirs V, Shaw J, Ellison S, et al. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. (2012) 40:e82. doi: 10.1093/nar/gks203
81. Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. (2013) 10:1003–5. doi: 10.1038/nmeth.2633
82. Gaipa G, Basso G, Maglia O, Leoni V, Faini A, Cazzaniga G, et al. Drug-induced immunophenotypic modulation in childhood ALL: implications for minimal residual disease detection. Leukemia. (2005) 19:49–56. doi: 10.1038/sj.leu.2403559
83. van Dongen JJ, van der Velden VH, Brüggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. (2015) 125:3996–4009. doi: 10.1182/blood-2015-03-580027
84. Pedreira CE, Costa ES, Lecrevisse Q, van Dongen JJ, Orfao A, EuroFlow Consortium. Overview of clinical flow cytometry data analysis: recent advances and future challenges. Trends Biotechnol. (2013) 31:415–25. doi: 10.1016/j.tibtech.2013.04.008
85. Kotrova M, Muzikova K, Mejstrikova E, Novakova M, Bakardjieva-Mihaylova V, Fiser K, et al. The predictive strength of next-generation sequencing MRD detection for relapse compared with current methods in childhood ALL. Blood. (2015) 126:1045–7. doi: 10.1182/blood-2015-07-655159
86. Eckert C, Flohr T, Koehler R, Hagedorn N, Moericke A, Stanulla M, et al. Very early/early relapses of acute lymphoblastic leukemia show unexpected changes of clonal markers and high heterogeneity in response to initial and relapse treatment. Leukemia. (2011) 25:1305–13. doi: 10.1038/leu.2011.89
87. Kotrova M, van der Velden VHJ, van Dongen JJM, Formankova R, Sedlacek P, Brüggemann M, et al. Next-generation sequencing indicates false-positive MRD results and better predicts prognosis after SCT in patients with childhood ALL. Bone Marrow Transplant. (2017) 52:962–8. doi: 10.1038/bmt.2017.16
88. Coustan-Smith E, Sancho J, Hancock ML, Razzouk BI, Ribeiro RC, Rivera GK, et al. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood. (2002) 100:2399–402. doi: 10.1182/blood-2002-04-1130
89. Brüggemann M, Kotrova M. Minimal residual disease in adult ALL: technical aspects and implications for correct clinical interpretation. Blood Adv. (2017) 1:2456–66. doi: 10.1182/bloodadvances.2017009845
90. Della Starza I, Nunes V, Cavalli M, De Novi LA, Ilari C, Apicella V, et al. Comparative analysis between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell receptor gene rearrangements to monitor minimal residual disease in acute lymphoblastic leukaemia. Br J Haematol. (2016) 174:541–9. doi: 10.1111/bjh.14082
91. You MJ, Medeiros LJ, Hsi ED. T-lymphoblastic leukemia/lymphoma. Am J Clin Pathol. (2015) 144:411–22. doi: 10.1309/AJCPMF03LVSBLHPJ
Keywords: early precursor T cell-acute lymphoblastic leukemia, T-acute lymphoblastic leukemia, minimal residual monitoring, CAR-T therapy, near-ETP, immunophenotype
Citation: George B, Chan KH and Rios A (2024) Early T-cell precursor acute lymphoblastic leukemia: diagnostic pitfalls, genomic alteration, novel therapeutics, and minimal residual disease monitoring. Front. Hematol. 3:1463410. doi: 10.3389/frhem.2024.1463410
Received: 11 July 2024; Accepted: 03 October 2024;
Published: 29 October 2024.
Edited by:
Valentina Giudice, University of Salerno, ItalyCopyright © 2024 George, Chan and Rios. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Binsah George, Qmluc2FoLnMuZ2VvcmdlQHV0aC50bWMuZWR1
†Present address: Kok Hoe Chan, CAMC Cancer Center, Charleston, WV, United States