 Hans Carl Hasselbalch1,2*
Hans Carl Hasselbalch1,2* Vibe Skov1
Vibe Skov1 Lasse Kjaer1
Lasse Kjaer1 Trine Alma Knudsen1Christina Schjellerup Eickhardt-Dalbøge1
Trine Alma Knudsen1Christina Schjellerup Eickhardt-Dalbøge1 Christina Ellervik2,3Sabrina Cordua1Anders Lindholm Sørensen1Sarah Friis Christensen1
Christina Ellervik2,3Sabrina Cordua1Anders Lindholm Sørensen1Sarah Friis Christensen1 Marie Hvelplund Kristiansen4
Marie Hvelplund Kristiansen4 Jes Sanddal Lindholt5
Jes Sanddal Lindholt5 Mads Thomassen6Torben A. Kruse6Niels Eske Bruun2,7Matias Greve Lindholm2,7
Mads Thomassen6Torben A. Kruse6Niels Eske Bruun2,7Matias Greve Lindholm2,7 Claus Henrik Nielsen2,8Miklos Egyed9
Claus Henrik Nielsen2,8Miklos Egyed9 Winfried März10Morten Kranker Larsen1,2
Winfried März10Morten Kranker Larsen1,2 Troels Wienecke2,4
Troels Wienecke2,4- 1Department of Hematology, Zealand University Hospital, Roskilde, Denmark
- 2Department of Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 3Department of Pathology, Harvard Medical School, Boston, MA, United States
- 4Department of Neurology, Zealand University Hospital, Roskilde, Denmark
- 5Department of Cardiothoracic and Vascular Surgery, Odense University Hospital, Odense, Denmark
- 6Department of Clinical Genetics, Odense University Hospital, Odense, Denmark
- 7Department of Cardiology, Zealand University Hospital, Roskilde, Denmark
- 8Institute for Inflammation Research, Center for Rheumatology and Spine Diseases, Rigshospitalet, Copenhagen University Hospital, Copenhagen, Denmark
- 9Department of Hematology, Somogy County Moritz Kaposi General Hospital, Kaposvar, Hungary
- 10Department of Laboratory Medicine, Heidelberg University, Heidelberg, Germany
Clonal Hematopoiesis of Indeterminate Potential (CHIP) is associated with an increased risk of cardiovascular diseases (CVD) and is a precursor stage to the BCR-ABL negative chronic myeloproliferative neoplasms (MPNs). These diseases are acquired stem cell neoplasms, arising due to mutations in the hematopoietic stem cell. The most prevalent is the JAK2V617F (JAK2) mutation, which potently generates reactive oxygen species (ROS), and accordingly contributes greatly to the chronic inflammatory state and the increased risk of thrombosis in MPNs. The MPNs are largely underdiagnosed blood cancers with a long pre-diagnostic phase of several years, when the elevated blood cell counts are considered reactive to smoking, blood clots, infections or chronic inflammatory diseases. Since the JAK2 mutation as CHIP-JAK2 associates with an increased risk of CVD and an increased risk of hematological and non-hematological cancers there is an urgent need to explore and validate the JAK2 mutation as a novel risk factor for CVD and to establish CHIP-clinics, which in an interdisciplinary collaboration between experts from several disciplines, and ensure timely diagnosis of the undiagnosed MPN patient and associated comorbidities. We envisage studies of the JAK2 mutation in large CVD cohorts to deliver the “Proof of Concept” for the JAK2 mutation to be implemented as a novel, highly important risk factor for CVD. These novel preventive strategies are considered to have the potential of reducing morbidity and mortality in a large population of citizens and patients, carrying the thrombosis- and CVD-promoting JAK2 mutation.
Highlights
● CHIP-JAK2V617F (JAK2) is associated with a chronic inflammatory state and an increased risk of cardiovascular diseases (CVD), including ischemic heart disease and stroke.
● The JAK2 mutation is envisaged as a novel risk factor for CVD and the CHIP-clinic as a novel interdisciplinary powerhouse for preventive medicine and much earlier diagnosis of MPNs and associated comorbidities.
1 Introduction
In 2014, seminal studies identified the presence of clonal hematopoiesis in normal individuals (1, 2). Clonal Hematopoiesis of Indeterminate Potential (CHIP) is defined by the presence of somatic mutations with an allele burden of more than 2%, but without the presence of a hematological abnormality or malignancy. This age-related entity is associated with an increased risk of cardiovascular diseases (CVD) (3, 4), hematological and non-hematological cancer and all-cause mortality (1), which is mainly driven by CVD. Taking into account that several of the CHIP mutations – JAK2V617F (JAK2), TET2, DNMT3A and ASXL1 – are “inflammatory” mutations, giving rise to a chronic inflammatory and thrombogenic state and the JAK2 mutation being associated with a 12-fold increase in coronary disease, there is an urgent need to explore and validate the JAK2 mutation as a novel risk factor for CVD in patients at high risk of having this mutation, such as patients with CVD. Screening of patients with CVD for the JAK2 mutation is foreseen to unravel a large number of JAK2-positive individuals at a much earlier stage in their development towards the Philadelphia-chromosome negative myeloproliferative neoplasms, essential thrombocythemia, polycythemia vera or myelofibrosis (MPNs) or already having overt but undiagnosed MPNs. Thus, Danish studies have for the first time demonstrated that MPNs are massively underdiagnosed chronic blood cancers (5). At least 10.000 citizens in Denmark have undiagnosed MPNs, and accordingly are at a constant risk of life-invalidating and potentially life-threatening thrombotic events (6, 7). By our current CHIP-project at the Department of Hematology, Zealand University Hospital, Roskilde, Denmark and Department of Hematology, Somogy County Moritz Kaposi General Hospital, Kaposvar, Hungary, and studies of the prevalence of the JAK2V617F mutation in the large German and Danish CVD cohorts (Figure 1) we envisage to deliver the “Proof of Concept” for the JAK2 mutation to be associated with incident events of CVD and as such to implement this mutation as a novel, highly important risk factor and predictive biomarker for CVD. Additionally, applying genomic, cytokine- and thrombophilia studies on our CHIP-cohort of 613 JAK2-positive citizens in the Danish General Suburban Population Study (GESUS) and in CVD patients (Figure 1), we aim to uncover novel mechanisms underlying the development of JAK2-associated CVD and the interplay between CVD and clonal expansion (8) from the earliest time point possible – the CHIP stage. By describing the association between the dynamics of clonal expansion and the evolution and development of CVD in the CHIP-stage and taking into account that CVD per se stimulates hematopoiesis and the production of inflammatory leukocytes (8), we have a unique platform for studying and deciphering mutations in blood cells as the common link between chronic inflammation, CVD and cancer development. Thereby, we hope to open the window for future preventive studies in the CHIP-stage with early stem-cell targeting therapy (pegylated interferon-alpha2 (IFN)) to eradicate the JAK2 mutated clone in addition to studies of the impact of old (statins and colchicine) and novel treatment strategies, e.g. IL1R-inhibitor or IL6-inhibitor, to target chronic inflammation, which is an important driver of both CVD and the malignant clone (9–21). Such novel strategies with preventive and personalized medicine are highly relevant and timely, considering their potential in reducing morbidity and mortality in a large population of citizens and patients, carrying the thrombosis- and CVD-promoting JAK2 mutation. In this paper, we wish to describe the rationales and perspectives of establishing CHIP-clinics at all departments of hematology worldwide, taking into account that such a CHIP-clinic is envisaged to be a powerhouse for much earlier diagnosis and treatment of the MPNs at an early stage, when they have not yet for years been suffering all the complications and inflammation-mediated comorbidities of unrecognized and undiagnosed MPN.
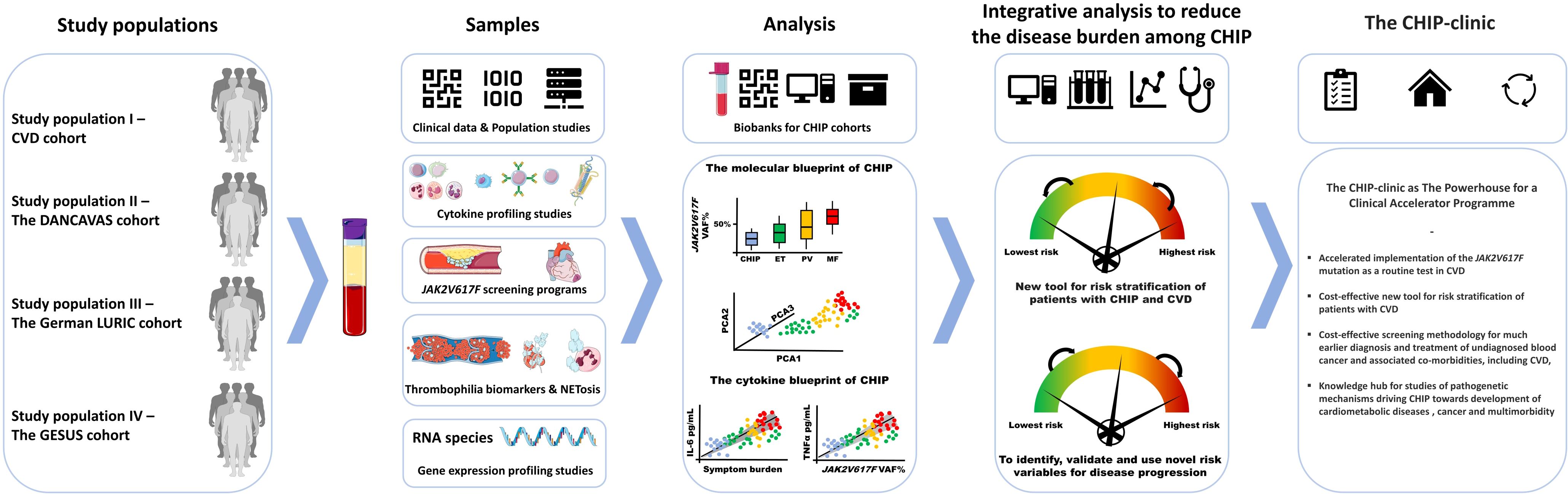
Figure 1. Workflow for analyses of biosamples from different large study cohorts and their integration in the CHIP-Clinic as the Powerhouse and Catalyst of Preventive Medicine.The figure contains elements from Medical Servier Art.
2 The rationales for a CHIP-clinic
2.1 The link between chronic inflammation, cardiovascular diseases, cancer and “inflammatory” mutations in the soil of clonal hematopoiesis of indeterminate potential
Somatic mutations in blood cells give rise to a chronic inflammatory state. These “inflammatory” mutations include among others the JAK2, which is prevalent in 70% of patients with MPNs (see below)) (Figure 2) (7). The JAK2 mutation per se is a generator of reactive oxygen radicals (ROS) (22–24) and thereby an inflammatory state, impacting not only the bone marrow but also the vascular system in general, taking into account that the blood cells with this mutation are constantly circulating in an activated state and thereby negatively impacting the microcirculation in several organs, implying an increased risk of blood clots in e.g. the heart, brain, and lungs (Figure 2). By ROS production the JAK2 mutation gives rise to DNA-damage and accordingly genomic instability via increased degradation of p53 (22–24). Importantly, through ROS production and secretion of lipocalin-2 the JAK2 mutated cells also evoke paracrine DNA damage to neighboring normal cells (25). The JAK2 mutation is present in the background population as CHIP (5), and as such associated with ischemic heart disease (26), a 12-fold increased risk of coronary artery disease (3, 6), and an increased risk of other cancers (26), which has also been reported in MPNs, both prior (27) and after the diagnosis of MPNs (28) with a particularly increased risk of esophagus, liver, lung, urogenital (kidney) and skin cancers, both malignant melanoma and non-melanoma (28). The JAK2 mutation is a thrombogenic factor per se and elicits ischemic heart lesions in murine models (“MPN-Cardiomyopathy” )? (29, 30), accelerates heart failure (29) and induces Neutrophil Extracellular Trap Formation (NETosis), which is closely linked to thrombosis (6, 30). Considering the vast amount of evidence for a link between CHIP and vascular diseases and other cancers (1, 3, 4, 26, 31, 32), and CHIP even predicting adverse outcomes of CVD (32) the time is ripe to investigate, if the JAK2 mutation per se as CHIP and when being assessed by a highly sensitive droplet digital PCR (ddPCR) is a unique new cardiovascular risk factor to be evaluated in large CVD-cohorts and in well-designed prospective studies of the frequencies of this mutation in “High-Risk-MPN-Profile” patients, such as patients with CVD, and accordingly also the frequencies of undiagnosed MPNs. The findings of JAK2 as a promotor of NETosis (30), the major role of NETosis in atherosclerosis and atherothrombosis development (33) and NETosis facilitating cancer invasiveness (34, 35) are highly intriguing in the context of the increased risk of cancers in patients with CHIP (26) and MPNs (27, 28). These associations may be causally linked to the JAK2 mutation, inducing a huge chronic inflammatory load and thereby predisposing not only to CVD (6, 7, 29, 30) but also to increasing genomic instability (22–25) and development of other cancers (26–28). Importantly, arterial thrombosis in MPNs may be an “early warning” of another cancer (36). In this context, this research innovatively links chronic inflammation, vascular diseases and cancer based upon MPNs as “Inflammatory and Vascular Diseases” (12, 37), “A Human Inflammation Model” and “A Human Inflammation Model for Cancer Development” (10, 11) as alluded to below. Most lately, the JAK2 mutation has also been demonstrated to be prevalent in patients with chronic kidney disease (CKD) (38) and we and others have previously shown MPNs to be associated with progressive impairment of kidney function (39) - indeed very similar to the development of progressive CKD in patients with dysregulated type II DM. The above findings make the JAK2 mutation even more intriguing to explore in regard to the association between pre-MPN in the CHIP-stage, overt but undiagnosed MPNs and the inflammatory co- and multimorbidity burden among patients at high-risk of housing this mutation, herein patients with CVD. Interestingly, recent studies have shown heart failure to be associated with cancer development (40) and a close link to exist between the myeloid cell lineage and CVD development (41). Indeed, increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis (42). The link between CVD (hypertension, atherosclerosis and myocardial infarction) and the hematopoietic system in terms of overproduction of inflammatory myeloid cells has most recently been shown to be consequent to bone marrow endothelial dysfunction with leakage of myeloid cells, angiogenesis and vascular fibrosis in the bone marrow (8). Accordingly, the association between CVD and hematopoiesis is likely a self-perpetuating vicious circle, in which CVD stimulates hematopoiesis and increases circulating levels of inflammatory leukocytes by remodeling the vascular bone marrow niche (8) and increased hematopoietic stem cell proliferation in CVD accelerates clonal hematopoiesis which further fuels the fire by the production of oxidative stress (43) and inflammatory cytokines, thereby accelerating atherosclerosis, atherothrombosis and clonal expansion and evolution towards MPNs and other cancers (Figure 2).
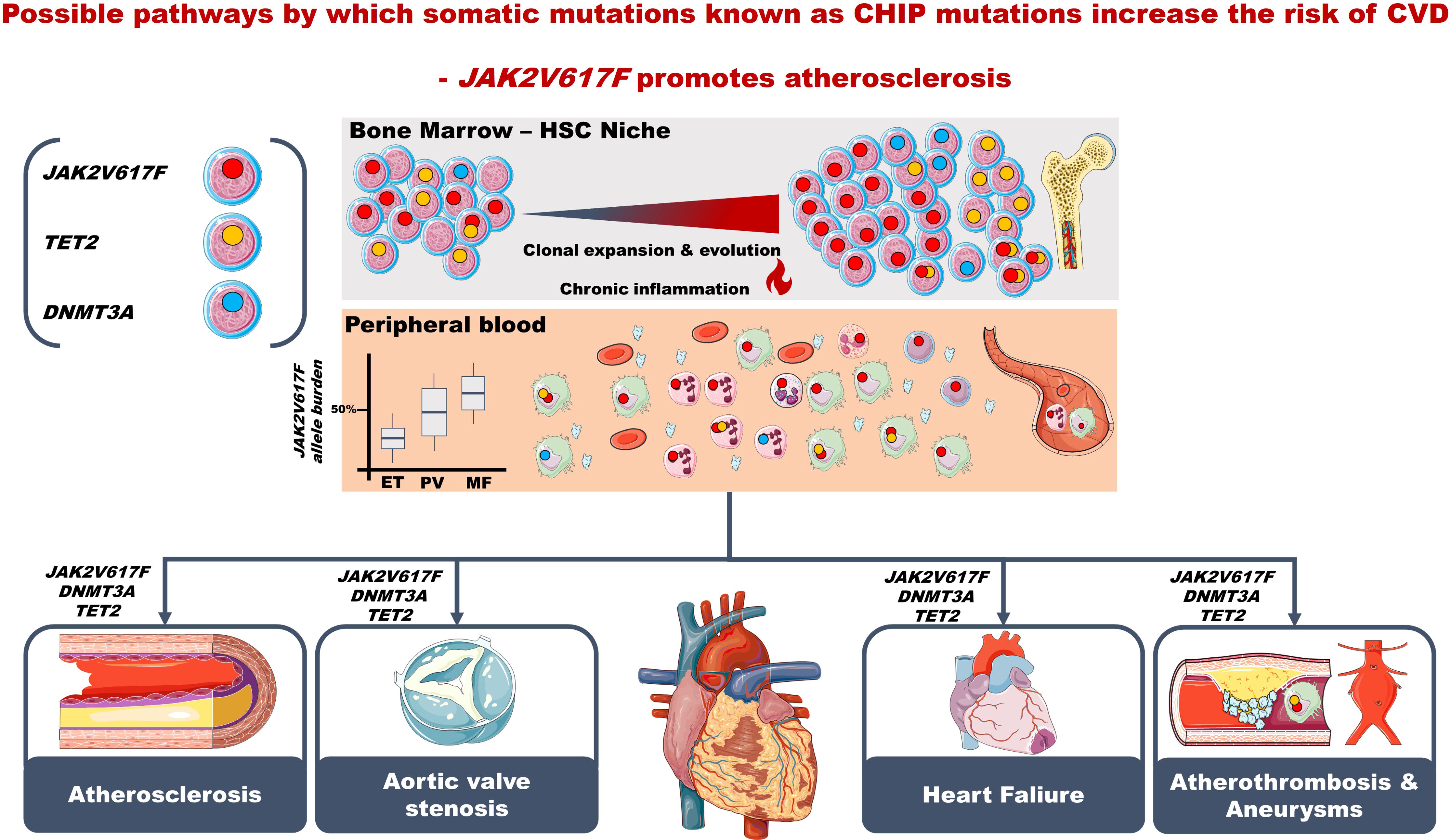
Figure 2. Suggested timeline of clonal hematopoiesis (CH). CH arises due to ongoing age-related somatic mutagenesis, which is likely attributed to a progressive decline of DNA repair mechanisms with ageing. Although factors affecting clonal expansion are currently unknown, the genetic background most likely has a significant role, both in promoting and inhibiting expansion. Secondary mutations are presumed to advance clonal expansion, and immune surveillance to keep mutant clones at bay. Modifying factors affecting CH likely include environmental factors and lifestyle factors (e.g., overweight, smoking), the common denominator being chronic inflammation, which both might elicit CH but also be generated by enhanced production by mutant cells of inflammatory cytokines such as IL-1beta and IL-6. Together with enhanced production of myeloid cells, either reactive to inflammation, infection or non-haematological or haematological cancer (e.g., MPNs) atherosclerosis is accelerated and consequently the risk of myocardial infarction and ischemic stroke increases. CH increases with ageing (inflammaging) and impaired immune surveillance (immuneaging). The figure contains elements from Medical Servier Art.
2.2 The myeloproliferative neoplasms, chronic inflammation and association with co- and multimorbidities
The MPNs include essential thrombocythemia (ET), polycythemia vera (PV) and myelofibrosis (MF) (44). A long prediagnostic phase often precedes these chronic blood cancers with 10-20 years, in which patients experience repeated thrombotic episodes (45) and are prone to suffer several inflammation-mediated comorbidities, including CVD (e.g. ischemic heart disease, coronary calcifications, abdominal aortic aneurysms and pulmonary hypertension) (6, 7, 10–12, 26, 37, 39, 46–49) and second cancers (26–28). In 2012, Hasselbalch published a perspective paper entitled “Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer” (10). A few years later, it was suggested that MPNs might actually be “A Human Inflammation Model” and “A Human Inflammation Model for Cancer Development” (11). Today, the concept of chronic inflammation as the driving force for clonal evolution in MPNs is widely accepted (50) (Figure 2). In this context, it is crucial to improve our understanding of the huge co- and multimorbidity burden associated with MPNs and how it relates to the “inflammatory” mutations - JAK2, TET2, DNMT3A and ASXL1 - in the MPN-mutational landscape at the earliest time point possible. This time point is the very early stage of MPNs, pre-MPNs as CHIP, which is prevalent in the elderly population due to “inflammaging” and “immunoaging” (51) being tightly associated with “inflammatory” mutations, CVD and cancer in the background population. As such they are likely prevalent in “High-Risk MPN-Profile” patients as well – the topic of this paper, in which we focus upon the prevalence of the JAK2 mutation among patients with CVD and its implementation as a novel risk factor for CVD development and accordingly as a novel tool in risk stratification, assessment of prognosis and overall survival in patients with CVD.
2.3 The CHIP-clinic
As alluded to above, screening of CVD patients for the JAK2-mutation will unravel some patients to have undiagnosed MPNs and the remaining will be categorized as CHIP. Based upon preliminary follow-up data from the Danish GESUS-cohort, an increasing number of CHIP citizens/patients will suffer CVD and/or transform into overt MPNs (5), which without early diagnosis and treatment will have a high risk of major thrombosis and worsening of CVD burden. Therefore, we wish to develop and implement the first CHIP-clinics in Denmark and Hungary, the aim being to prohibit inflammation-mediated multimorbidity due to undiagnosed CVD and MPNs. The CHIP-clinics will be boarded by experts in hematology, cardiology, neurology and molecular biology and accordingly have several objectives. First, they will be knowledge hubs where experts share interdisciplinary expertise for the optimal care, guidance, and treatment of citizens who harbor somatic mutations in blood cells. In the future, such clinics are envisaged to improve the prevention of common diseases, such as CVD, through timely and interdisciplinary treatment of patients. Secondly, they will be knowledge hubs for research in the pathogenic factors that are determinants for CVD and MPN development from the earliest time point possible.
In Denmark, we take advantage that this first CHIP clinic will be built upon an already existing platform “Clinical Academic Group (CAG) within Greater Copenhagen Health Science Partners (GCHSPs)” (CAG ZIRI)”, which was established in 2021 as part of the Danish MPN-Consortium. CAG-ZIRI has pioneered the concept of chronic inflammation as the driving force for the development and progression of MPN cancers and associated comorbidities, such as CVD. During the last 3 years experts in hematology, cardiology, neurology, and molecular biology at Zealand University Hospital (ZUH), Roskilde, Denmark have had monthly meetings on ongoing projects between the departments as part of Ph.d-projects focusing on the impact of the JAK2 mutation in patients with CVD (screening study of 550 stroke patients) showing a prevalence of 11.3% (52) and a study on the CVD burden in MPN-patients, showing valve calcifications and coronary artery sclerosis to be prevalent amongst MPN-patients (48, 49). In this collaboration, the experience and expertise in the MPN-Consortium at ZUH and CAG-ZIRI will greatly support the implementation of a CHIP-clinic and - based upon the experience obtained - its implementation at other departments in Denmark and abroad in the coming years. This novel interdisciplinary workflow will provide timely care of citizens/patients, in whom a JAK2 mutation or other somatic mutations have been found, thereby providing novel standards – a paradigm shift – for future patient care with timely follow-up of citizens/patients with these CVD-and cancer promoting mutations. Within the interdisciplinary collaboration in the CHIP-Clinic we envisage to raise evidence for the JAK2V617F mutation as a novel risk factor for CVD and a predictive biomarker of prognosis and overall survival using several large patient and population cohorts (Figure 1).
3 Discussion and perspectives for JAK2 mutation screening of patients with CVD and future research directions
By molecular screening for mutations in blood cells from “high-risk MPN-profile” patients, herein patients with CVD, who often have been or are heavy smokers [smoking is a risk factor for MPN-development (53, 54)], we envisage to deliver the proof of concept for preventive, rational and personalized medicine by much earlier diagnosis and treatment of MPNs among patients with CVD. This change in clinical practice is most likely also highly “cost-effective”, since we have just recently published that the earlier IFN-treatment is instituted the shorter the time needed to achieve major molecular remissions with low-burden JAK2 (55). Since a JAK2 mutation burden above 50% in MPN-patients significantly associates with the development of coronary artery calcification (48, 49) and both CHIP, CVD and MPNs also associate with impaired kidney function (38, 39, 56–61), studies on the prevalence of the JAK2 mutation – both as CHIP-JAK2 and undiagnosed MPNs - amongst patients with CVD are urgently needed as underlined by the high prevalence in patients with ischemic stroke (52). Early screening for MPNs in high-risk MPN-profile patients is also dictated by common knowledge on cancer biology, implying increasing genomic instability, subclone formation, resistance to treatment and ultimately metastasis, if not being treated. In this perspective, the time is ripe for early screening to institute treatment with IFN at the earliest time point possible, since IFN is the only disease-modifying agent in terms of inducing MRD in a subset of MPN patients (62–68). Based upon the above associations between the JAK2 mutation and CVD development, early reduction of the JAK2 allelic burden is mandatory not only for prevention of thrombotic events but also likely for the development and progression of CVD, which share deregulation of several oxidative stress and atherosclerosis genes with the MPNs (69, 70) - genes which in MPNs are favorably influenced by treatment with IFN (71, 72). Accordingly, the ultimate outcome of early treatment with IFN may be the resolution of CVD manifestations and halting of CVD progression due to the normalization of elevated atherosclerosis promoting cell counts (e.g. leukocytes, monocytes, platelets) in concert with reduction of the atherosclerosis-and atherothrombosis promoting JAK2 mutation (6, 7, 26, 30, 48, 49), which also associates with increased NET formation (6, 30, 33). Importantly, in the Danish DALIAH-trial (73) treatment with IFN was associated with a reduction in NETosis activity (74), which may be beneficial, considering the role of NETosis in atherosclerosis and atherothrombosis development (30, 33) as alluded to above. Highly intriguing, most recent reports have shown IFN to induce resolution of angina pectoris (AP) in a CHIP-patient (75)) and in five MPN-patients with severe treatment refractory AP (76). The LURIC and the DANCAVAS studies (77, 78) have generated a large number of high-impact papers on several aspects of CVD, including genomics in the LURIC cohort (77). As such, both cohorts will allow us to perform a multitude of JAK2 association studies to already published biomarkers. In addition, we will have the possibility on baseline DNA-samples from all cohorts to perform additional molecular studies by panel NGS, including analyses for other inflammation-mediating mutations (e.g. TET2,DNMT3A,ASXL1) which are currently being performed on 300 citizens from the GESUS cohort. Taking into account that 80% of cardiac events and strokes are preventable, half through early detection and intervention, and this project is foreseen to unravel the JAK2-mutation in about 10-12% of the patients [prevalence of the JAK2-mutation in stroke patients 11,3% (52)] and thereby promoting the JAK2-mutation as a novel risk factor for CVD development the project will have the potential to reinforce the rationales for implementing this simple and inexpensive methodology as a new routine test to be performed in all patients admitted with CVD. Therefore, the JAK2-mutation is a candidate biomarker to be included in a multivariate precision tool, the “Know Your Risk” test, which is elaborated in the Danish PREPARE (Personalized Risk Estimation and Prevention of Cardiovascular Disease) Programme (https://www.sdu.dk/en/forskning/prepare). This innovative tool is being developed by machine learning to combine self-reported risks, advanced objective findings [including CT-scans (78)], registry data, genomic and proteomic biomarkers. The implementation of the JAK2-mutation as a new screening test in patients with CVD and potentially adding great value to the multivariate precision tool above will also introduce a paradigm shift by much earlier diagnosis and treatment of CVD-patients with undiagnosed MPNs, thereby prohibiting repeated hospitalizations of the undiagnosed MPN-patient due to JAK2-mediated thrombotic events and JAK2-mediated worsening of multi-organ atherosclerosis and organ failure (e.g. heart failure, chronic nephropathy, abdominal aneurysms, peripheral arterial insufficiency, dementia). In this context, the CHIP-clinic will be the optimal platform for an interdisciplinary collaboration for timely risk stratification, treatment and follow-up programs, based upon machine learning algorithms, which are being generated in the continuation study of DANCAVAS – the PREPARE program as alluded to above.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
HH: Visualization, Writing – original draft, Writing – review & editing, Conceptualization. VS: Writing – review & editing. LK: Writing – review & editing. TAK: Writing – review & editing. CE-D: Writing – review & editing. CE: Writing – review & editing. SC: Writing – review & editing. AS: Writing – review & editing. SFC: Writing – review & editing. MK: Writing – review & editing. JL: Writing – review & editing. MT: Writing – review & editing. TK: Writing – review & editing. NB: Writing – review & editing. ML: Writing – review & editing. CN: Writing – review & editing. ME: Writing – review & editing. WM: Writing – review & editing. ML: Visualization, Writing – review & editing. TW: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age- related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
2. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
3. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
4. Min KD, Kour A, Sano S, Walsh K. The role of clonal haematopoiesis in cardiovascular diseases: epidemiology and experimental studies. J Intern Med. (2020) 288:507–17. doi: 10.1111/joim.v288.5
5. Cordua S, Kjær L, Skov V, Pallisgaard N, Hasselbalch HC, Ellervik C, et al. Prevalence and phenotypes of jak2 v617f and calreticulin mutations in a Danish general population. Blood. (2019) 134:469–79. doi: 10.1182/blood.2019001113
6. Hasselbalch HC, Elvers M, Schafer AL. The pathobiology of thrombosis, microvascular disease, and hemorrhage in the myeloproliferative neoplasms. Blood. (2021) 137:2152–60. doi: 10.1182/blood.2020008109
7. Moliterno AR, Kaizer H, Reeves BN. JAK2 V617F allele burden in polycythemia vera: burden of proof. Blood. (2023) 141:1934–42. doi: 10.1182/blood.2022017697
8. Rohde D, Vandoorne K, Lee IH, Grune J, Zhang S, McAlpine CS, et al. Bone marrow endothelial dysfunction promotes myeloid cell expansion in cardiovascular disease. Nat Cardiovasc Res. (2022) 1:28–44. doi: 10.1038/s44161-021-00002-8
9. Fleischman AG, Aichberger KJ, Luty SB, Bumm TG, Petersen CL, Doratotaj S, et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. (2011) 118:6392–8. doi: 10.1182/blood-2011-04-348144
10. Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. (2012) 119:3219–25. doi: 10.1182/blood-2011-11-394775
11. Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res. (2013) 37(2):214–20. doi: 10.1016/j.leukres.2012.10.020doi: 10.1016/j.leukres.2012.10.020
12. Hasselbalch HC, Bjørn ME. MPNs as inflammatory diseases: The evidence, consequences, and perspectives. Mediators Inflamm. (2015) 2015:102476. doi: 10.1155/2015/102476
13. Hermouet S, Bigot-Corbel E, Gardie B. Pathogenesis of myeloproliferative neoplasms: Role and mechanisms of chronic inflammation. Mediators Inflamm. (2015) 2015:145293. doi: 10.1155/2015/145293
14. Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ, et al. Myeloproliferative neoplasms and inflammation: whether to target the Malignant clone or the inflammatory process or both. Leukemia. (2016) 30:1018–24. doi: 10.1038/leu.2016.12
15. Koschmieder S, Chatain N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. (2020) 42:100711. doi: 10.1016/j.blre.2020.100711
16. Rai S, Grockowiak E, Hansen N, Paz DL, Stoll CB, Hao-Shen Hv, et al. Inhibition of interleukin-1β reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat Commun. (2022) 13(1):5346. doi: 10.1038/s41467-022-32927-4
17. Rahman MF, Yang Y, Le BT, Dutta A, Posyniak J, Faughnan P, et al. Interleukin-1 contributes to clonal expansion and progression of bone marrow fibrosis in JAK2V617F-induced myeloproliferative neoplasm. Nat Commun. (2022) 13:5347. doi: 10.1038/s41467-022-32928-3
18. Hermouet S. Mutations, inflammation and phenotype of myeloproliferative neoplasms. Front Oncol. (2023) 13:1196817. doi: 10.3389/fonc.2023.1196817
19. Hasselbalch HC, Kristiansen MH, Kjær L, Skov V, Larsen MK, Ellervik C, et al. CHIP-JAK2V617F, chronic inflammation, abnormal megakaryocyte morphology, organ failure, and multimorbidties. Blood Adv. (2024) 8:681–2. doi: 10.1182/bloodadvances.2023012190
20. Rai S, Zhang Y, Grockowiak E, Kimmerlin Q, Hansen N, Stoll CB, et al. IL-1β promotes MPN disease initiation by favoring early clonal expansion of JAK2-mutant hematopoietic stem cells. Blood Adv. (2024) 8(5):1234–49. doi: 10.1182/bloodadvances.2023011338
21. Hermouet S, Hasselbalch HC. Interleukin-1b, JAK2V617F mutation and inflammation in myeloproliferative neoplasms. Blood Adv. (2024). doi: 10.1182/bloodadvances.2024013528
22. Marty C, Lacout C, Droin N, Le Couédic JP, Ribrag V, Solary E, et al. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia. (2013) 27:2187–95. doi: 10.1038/leu.2013.102
23. Bjørn ME, Hasselbalch HC. The role of reactive oxygen species in myelofibrosis and related neoplasms. Mediators Inflamm. (2015) 2015:648090. doi: 10.1155/2015/648090
24. Karantanos T, Moliterno AR. The roles of JAK2 in DNA damage and repair in the myeloproliferative neoplasms: Opportunities for targeted therapy. Blood Rev. (2018) 32:426–32. doi: 10.1016/j.blre.2018.03.007
25. Kagoya Y, Yoshimi A, Tsuruta-Kishino T, Arai S, Satoh T, Akira S, et al. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood. (2014) 124:2996–3006. doi: 10.1182/blood-2014-04-570572
26. Nielsen C, Birgens HS, Nordestgaard BG, Bojesen SE. Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. Br J Haematol. (2013) 160:70–9. doi: 10.1111/bjh.2012.160.issue-1
27. Pettersson H, Knutsen H, Holmberg E, Andréasson B. Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur J Haematol. (2015) 94:152–6. doi: 10.1111/ejh.2015.94.issue-2
28. Frederiksen H, Farkas DK, Christiansen CF, Hasselbalch HC, Sørensen HT. Chronic myeloproliferative neoplasms and subsequent cancer risk: a Danish population-based cohort study. Blood. (2011) 118:6515–20. doi: 10.1182/blood-2011-04-348755
29. Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, et al. JAK2V617F-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci. (2019) 4:684–97. doi: 10.1016/j.jacbts.2019.05.013
30. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. (2018) 10:eaan8292. doi: 10.1126/scitranslmed.aan8292
31. Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. (2019) 4:32–40. doi: 10.1001/jamacardio.2018.3965
32. Gumuser ED, Schuermans A, Cho SMJ, Sporn ZA, Uddin MM, Paruchuri K, et al. Clonal hematopoiesis of indeterminate potential predicts adverse outcomes in patients with atherosclerotic cardiovascular disease. J Am Coll Cardiol. (2023) 81:1996–2009. doi: 10.1016/j.jacc.2023.03.401
33. Dong Y, Zhang Y, Yang X, Yan C, Feng Y. Recent insights into neutrophil extracellular traps in cardiovascular diseases. J Clin Med. (2022) 11:6662. doi: 10.3390/jcm11226662
34. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of neutrophil extracellular traps (NETs) in tumor progression and metastasis. Front Immunol. (2020) 11:1749. doi: 10.3389/fimmu.2020.01749
35. Mahmud Z, Rahman A, Mishu ID, Kabir Y. Mechanistic insights into the interplays between neutrophils and other immune cells in cancer development and progression. Cancer Metastasis Rev. (2022) 41:405–32. doi: 10.1007/s10555-022-10024-8
36. De Stefano V, Ghirardi A, Masciulli A, Carobbio A, Palandri F, Vianelli N, et al. Arterial thrombosis in Philadelphia-negative myeloproliferative neoplasms predicts second cancer: a case-control study. Blood. (2020) 135:381–6. doi: 10.1182/blood.2019002614
37. Frederiksen H, Szépligeti S, Bak M, Ghanima W, Hasselbalch HC, Christiansen CF. Vascular diseases in patients with chronic myeloproliferative neoplasms - impact of comorbidity. Clin Epidemiol. (2019) 11:955–67. doi: 10.2147/CLEP.S216787
38. Shi M, Sun X, Bick A, He J. The association between clonal hematopoiesis of indeterminate potential and inflammatory biomarkers among chronic kidney disease patients circulation. Circulation. (2020) 141:AP456. doi: 10.1161/circ.141.suppl_1.P456
39. Christensen AS, Møller JB, Hasselbalch HC. Chronic kidney disease in patients with the Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Res. (2014) 38:490–5. doi: 10.1016/j.leukres.2014.01.014
40. de Boer RA, Meijers WC, van der Meer P, van Veldhuisen DJ. Cancer and heart disease: associations and relations. Eur J Heart Fail. (2019) 21:1515–25. doi: 10.1002/ejhf.v21.12
41. Nahrendorf M. Myeloid cell contributions to cardiovascular health and disease. Nat Med. (2018) 24:711–20. doi: 10.1038/s41591-018-0064-0
42. Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. (2021) 184:1348–1361.e22. doi: 10.1016/j.cell.2021.01.049
43. Sørensen AL, Hasselbalch HC, Bjørn ME, Nielsen CH, Cordua S, Skov V, et al. Elevated levels of oxidized nucleosides in individuals with the JAK2V617F mutation from a general population study. Redox Biol. (2021) 41:101895. doi: 10.1016/j.redox.2021.101895
44. Spivak JL. Myeloproliferative neoplasms. N Engl J Med. (2017) 376:2168–81. doi: 10.1056/NEJMra1406186
45. Enblom A, Lindskog E, Hasselbalch H, Hersby D, Bak M, Tetu J, et al. High rate of abnormal blood values and vascular complications before diagnosis of myeloproliferative neoplasms. Eur J Intern Med. (2015) 26:344–7. doi: 10.1016/j.ejim.2015.03.009
46. Brabrand M, Hansen KN, Laursen CB, Larsen TS, Vestergaard H, Abildgaard N. Frequency and etiology of pulmonary hypertension in patients with myeloproliferative neoplasms. Eur J Haematol. (2019) 102:227–34. doi: 10.1111/ejh.2019.102.issue-3
47. Yokokawa T, Misaka T, Kimishima Y, Wada K, Minakawa K, Sugimoto K, et al. Crucial role of hematopoietic JAK2 V617F in the development of aortic aneurysms. Haematologica. (2021) 106:1910. doi: 10.3324/haematol.2020.264085
48. Solli CN, Chamat-Hedemand S, Elming H, Ngo A, Kjaer L, Skov V, et al. Coronary artery-and aortic valve calcifications in patients with Philadelphia-negative myeloproliferative neoplasms. Int J Cardiol. (2022) 364:112–8. doi: 10.1016/j.ijcard.2022.06.029
49. Solli CN, Chamat-Hedemand S, Elming H, Ngo A, Kjaer L, Skov V, et al. High JAK2V617F variant allele frequency is associated with coronary artery but not aortic valve calcifications in patients with Philadelphia-negative myeloproliferative neoplasms. Eur J Haematol. (2023) 111:400–6. doi: 10.1111/ejh.v111.3
50. Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ. m.fl. Myeloproliferative neoplasms and inflammation: whether to target the Malignant clone or the inflammatory process or both. Leukemia. (2016) 30:1018–24. doi: 10.1038/leu.2016.12
51. Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. (2020) 17:137–44. doi: 10.1038/s41569-019-0247-5
52. Kristiansen MH, Kjær L, Skov V, Larsen MK, Ellervik C, Hasselbalch HC, et al. JAK2V617F mutation is highly prevalent in patients with ischemic stroke: a case-control study. Blood Adv. (2023) 7:5825–34. doi: 10.1182/bloodadvances.2023010588
53. Hasselbalch HC. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk Res. (2015) S0145-2126:30373–8. doi: 10.1016/j.leukres.2015.09.002
54. Jayasuriya NA, Kjaergaard AD, Pedersen KM, Sørensen AL, Bak M, Larsen MK. Smoking, blood cells and myeloproliferative neoplasms: meta-analysis and Mendelian randomization of 2·3 million people. Br J Haematol. (2020) 189:323–34. doi: 10.1111/bjh.v189.2
55. Pedersen RK, Andersen M, Knudsen TA, Sajid Z, Gudmand-Hoeyer J, Dam MJB, et al. Data-driven analysis of JAK2V617F kinetics during interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms. Cancer Med. (2020) 9:2039–51. doi: 10.1002/cam4.2741
56. Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation. (2021) 143:1157–72. doi: 10.1161/CIRCULATIONAHA.120.050686
57. Gecht J, Tsoukakis I, Kricheldorf K, Stegelmann F, Klausmann M, Griesshammer M, et al. Kidney dysfunction is associated with thrombosis and disease severity in myeloproliferative neoplasms: implications from the german study group for MPN bioregistry. Cancers. (2021) 13:4086. doi: 10.3390/cancers13164086
58. Larsen MK, Skov V, Kjaer L, Møller-Palacino NA, Pedersen RK, Andersen M, et al. Clonal haematopoiesis of indeterminate potential and impaired kidney function-A Danish general population study with 11 years follow-up. Eur J Haematol. (2022) 109:576–85. doi: 10.1111/ejh.13845
59. Dawoud AAZ, Gilbert RD, Tapper WJ, Cross NCP. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia. (2022) 36:507–15. doi: 10.1038/s41375-021-01382-3
60. Kestenbaum B, Bick AG, Vlasschaert C, Rauh MJ, Lanktree MB, Franceschini N, et al. Clonal hematopoiesis of indeterminate potential and kidney function decline in the general population. Am J Kidney Dis. (2023) 81:329–35. doi: 10.1053/j.ajkd.2022.08.014
61. Vlasschaert C, Robinson-Cohen C, Chen J, Akwo E, Parker AC, Silver SA, et al. Clonal hematopoiesis of indeterminate potential is associated with acute kidney injury. Nat Med. (2024) 30:810–7. doi: 10.1038/s41591-024-02854-6
62. Hasselbalch HC. Myelofibrosis with myeloid metaplasia: the advanced phase of an untreated disseminated hematological cancer. Time to change our therapeutic attitude with early upfront treatment? Leuk Res. (2009) 33:11–8. doi: 10.1016/j.leukres.2008.06.002
63. Larsen TS, Møller MB, de Stricker K, Nørgaard P, Samuelsson J, Marcher C, et al. Minimal residual disease and normalization of the bone marrow after long-term treatment with alpha-interferon2b in polycythemia vera. A report on molecular response patterns in seven patients in sustained complete hematological remission. Hematology. (2009) 14:331–4. doi: 10.1179/102453309X12473408860587
64. Hasselbalch HC. A new era for IFN-α in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev Hematol. (2011) 4:637–55. doi: 10.1586/ehm.11.63
65. Utke Rank C, Weis Bjerrum O, Larsen TS, Kjær L, de Stricker K, Riley CH, et al. Minimal residual disease after long-term interferon-alpha2 treatment: a report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk Lymphoma. (2016) 57:348–54. doi: 10.3109/10428194.2015.1049171
66. Bjørn ME, Hasselbalch HC. Minimal residual disease or cure in MPNs? Rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert Rev Hematol. (2017) 10:393–404. doi: 10.1080/17474086.2017.1284583
67. Hasselbalch HC, Holmström MO. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure? Semin Immunopathol. (2019) 41:5–19. doi: 10.1007/s00281-018-0700-2
68. Hasselbalch HC, Silver RT. New perspectives of interferon-alpha2 and inflammation in treating philadelphia-negative chronic myeloproliferative neoplasms. Hemasphere. (2021) 5:e645. doi: 10.1097/HS9.0000000000000645
69. Hasselbalch HC, Thomassen M, Hasselbalch Riley C, Kjær L, Stauffer Larsen T, Jensen MK, et al. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of nrf2 for genomic instability and disease progression. PloS One. (2014) 9(11):e112786. doi: 10.1371/journal.pone.0112786
70. Skov V, Thomassen M, Kjaer L, MK L, TA K, Ellervik C, et al. Whole blood transcriptional profiling reveals highly deregulated atherosclerosis genes in Philadelphia-chromosome negative myeloproliferative neoplasms. Eur J Haematol. (2023) 111:805–14. doi: 10.1111/ejh.v111.5
71. Skov V, Thomassen M, Kjær L, Ellervik C, Larsen MK, Knudsen TA, et al. Interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms favourably impacts deregulation of oxidative stress genes and antioxidative defence mechanisms. PloS One. (2022) 17(6):e0270669. doi: 10.1371/journal.pone.0270669
72. Skov V, Thomassen M, Kjaer L, Kruse TA, Hasselbalch HC. Interferon-alpha2 treatment of patients with polycythemia vera favorably impacts deregulation of atherosclerosis genes. Abstract ASH New Orleans December. (2022) 140(Supplement 1):9604–5. doi: 10.1371/journal.pone.0270669
73. Knudsen TA, Skov V, Stevenson K, Werner L, Duke W, Laurore C, et al. Genomic profiling of a randomized trial of interferon-α vs hydroxyurea in MPN reveals mutation-specific responses. Blood Adv. (2022) 6:2107–19. doi: 10.1182/bloodadvances.2021004856
74. Massarenti L, Knudsen TA, Enevold C, Skov V, Kjaer L, Larsen MK, et al. Interferon alpha-2 treatment reduces circulating neutrophil extracellular trap levels in myeloproliferative neoplasms. Br J Haematol. (2023) 202:318–27. doi: 10.1111/bjh.v202.2
75. Egyed M, Kajtar B, Foldesi C, Skov V, Kjær L, Hasselbalch HC. Ropeginterferon-alfa2b resolves angina pectoris and reduces JAK2V617F in a patient with clonal hematopoiesis of indeterminate potential: A case report. Front Hematol. (2022) 1:1005666. doi: 10.3389/frhem.2022.1005666
76. Egyed M, Kovacs E, Karadi E, Herczeg J, Katjar B, Kjaer L, et al. Resolution of refractory angina pectoris in five MPN-patients treated with interferon. J Leukemia. (2023) 11:347. doi: 10.35248/2329-6917.23.11.347
77. Winkelmann BR, März W, Boehm BO, Zotz R, Hager J, Hellstern P, et al. LURIC Study Group (LUdwigshafen RIsk and Cardiovascular Health). Rationale and design of the LURIC study–a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics. (2001) 1(Suppl 1):S1–73. doi: 10.1517/14622416.2.1.S1
Keywords: JAK2V617F mutation, cardiovascular disease, stroke, myeloproliferative neoplasms (MPNs), CHIP-clinic, preventative medicine
Citation: Hasselbalch HC, Skov V, Kjaer L, Knudsen TA, Eickhardt-Dalbøge CS, Ellervik C, Cordua S, Sørensen AL, Christensen SF, Kristiansen MH, Lindholt JS, Thomassen M, Kruse TA, Bruun NE, Lindholm MG, Nielsen CH, Egyed M, März W, Larsen MK and Wienecke T (2024) The CHIP-clinic as the catalyst of preventive medicine. Front. Hematol. 3:1459154. doi: 10.3389/frhem.2024.1459154
Received: 03 July 2024; Accepted: 05 September 2024;
Published: 18 October 2024.
Edited by:
Hugh Young Rienhoff Jr., Institute for Further Study, United StatesReviewed by:
Francesco Tarantini, University of Bari Aldo Moro, ItalyCopyright © 2024 Hasselbalch, Skov, Kjaer, Knudsen, Eickhardt-Dalbøge, Ellervik, Cordua, Sørensen, Christensen, Kristiansen, Lindholt, Thomassen, Kruse, Bruun, Lindholm, Nielsen, Egyed, März, Larsen and Wienecke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans Carl Hasselbalch, aGFucy5oYXNzZWxiYWxjaEBnbWFpbC5jb20=