
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Hematol. , 15 February 2024
Sec. Blood Cancer
Volume 3 - 2024 | https://doi.org/10.3389/frhem.2024.1339870
 Guillermo Garcia-Manero1*
Guillermo Garcia-Manero1* Uwe Platzbecker2
Uwe Platzbecker2 Kian-Huat Lim3
Kian-Huat Lim3 Grzegorz Nowakowski4Omar Abdel-Wahab5Hagop Kantarjian1Amit Verma6
Grzegorz Nowakowski4Omar Abdel-Wahab5Hagop Kantarjian1Amit Verma6 Daniel T. Starczynowski7*
Daniel T. Starczynowski7*The intracellular serine/threonine interleukin 1 receptor-associated kinase 4 (IRAK4) is necessary for most signaling by activated Toll-like receptors (TLRs). Activation of IRAK4 drives activation of nuclear factor kappa B (NF-κB) and so promotes cell survival, inflammation, and other aspects of the adaptive immune response. However, the IRAK4 pathway can be coopted by cancers and lead to the survival and proliferation of malignant cells. Inappropriate IRAK4 activity has been linked with the progression of myelodysplastic syndrome (MDS), other hematologic malignancies, and some solid tumors, and preclinical cancer models indicate that IRAK4 inhibition has anti-tumor effects. As such, inhibition of IRAK4 is an emerging and attractive target for tumor suppression. The growing interest in IRAK4 motivated the 1st Symposium on IRAK4 in Cancer held in October 2022 to bring together IRAK4 researchers and clinicians to discuss new insights into the biology of IRAK4 and development of IRAK4 inhibitors. Presentations and discussions at the meeting provided updates on the biology of IRAK4 and its links with mutations in the spliceosome, new outcomes from preclinical models that indicate synergy between inhibitors of IRAK4 and FLT3 and BTK inhibitors, and an update on the clinical development of the investigational IRAK4 inhibitor emavusertib, currently being assessed in ongoing phase 1/2 clinical studies in hematologic cancers and several solid tumors.
Innate immune responses mediated through toll-like receptors (TLRs) or interleukin-1 receptors (IL-1R) on sentinel cells such as macrophages and dendritic cells are an important first-line defense against pathogens (1–3). After recognizing generic molecular patterns on these pathogens, TLR/IL-1R activation triggers intracellular inflammatory signaling cascades promoting cell survival, cytokine production, inflammation, and priming of adaptive immune responses (1, 4).
Most innate immune responses downstream of activated TLRs, including response to lipopolysaccharide, require the intracellular serine/threonine interleukin 1 receptor-associated kinase 4 (IRAK4), which signals to the nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways (1, 5, 6). Cooption of the IRAK4 pathway plays a central role in the pathogenesis of specific hematologic malignancies and may also be implicated in certain solid tumors (1, 5, 7). As such, IRAK4 is considered an attractive therapeutic target for multiple cancers.
Recognizing the growing interest in this area of research, the 1st Symposium on IRAK4 in Cancer held virtually on 7 October 2022 brought together IRAK4 researchers and clinicians to discuss new insights into the biology of IRAK4 and development of anti-cancer agents focused on this target. The meeting was co-chaired by Drs Amit Verma (Montefiore Albert Einstein College of Medicine) and Guillermo Garcia-Manero and Hagop Kantarjian (MD Anderson Cancer Center). This article summarizes the topics that were discussed. Key meeting themes were the connections between IRAK4 and multiple processes important in immunity and cancer biology, including dysregulation of inflammation signaling as a key mechanism activating IRAK4 and driving proliferation in MDS and acute myeloid leukemia (AML); the links between spliceosome mutations, dysregulated ribonucleic acid (RNA) splicing, and IRAK4 activity; the clinical potential of IRAK4 inhibition in multiple cancers and its synergy with established treatments such as chemotherapy and targeted inhibitors of Bruton’s tyrosine kinase (BTK) and FMS-like tyrosine kinase 3 (FLT3); and early clinical trial results for the IRAK4 inhibitor emavusertib (CA-4948; Curis, Inc) in patients with AML, high-risk MDS, or non-Hodgkin lymphoma.
Dr Daniel Starczynowski (Cincinnati Children’s Hospital) opened the meeting with a review of IRAK4 biology. Figure 1 illustrates current understanding of the IRAK4 pathway, although much remains to be learned about the specific roles and functions of IRAK4 in different tissues and physiological contexts. TLRs on hematopoietic cells such as macrophages, lymphocytes, and dendritic cells recognize generic molecular patterns on bacteria, fungi, viruses, and cellular debris (1, 7). Activation of TLR or IL-1R cell surface receptors leads to assembly of the myddosome, a large multimeric signaling complex composed of IRAK4, IRAK1/IRAK2, and the cytoplasmic adaptor myeloid differentiation primary response (MyD88) (2, 15, 16). IRAK4 is a core component of the myddosome and associates with MyD88 and IRAK1/2 in a stoichiometric manner to form a helical macromolecular complex (2, 16). Once activated in the context of the myddosome, IRAK1 disassociates and activates E3 ligase tumor necrosis factor receptor-associated factor-6 (TRAF6) (2). This induces downstream NF-κB and MAPK pathways leading to expression of various pro-inflammatory genes, including those encoding cytokines and chemokines, to mediate an inflammatory response (2, 5, 17). IRAK1 activated within the myddosome also leads to rapid activation of the nucleotide binding and oligomerization leucine-rich repeat pyrin domain-containing 3 (NLRP3) inflammasome (18, 19).
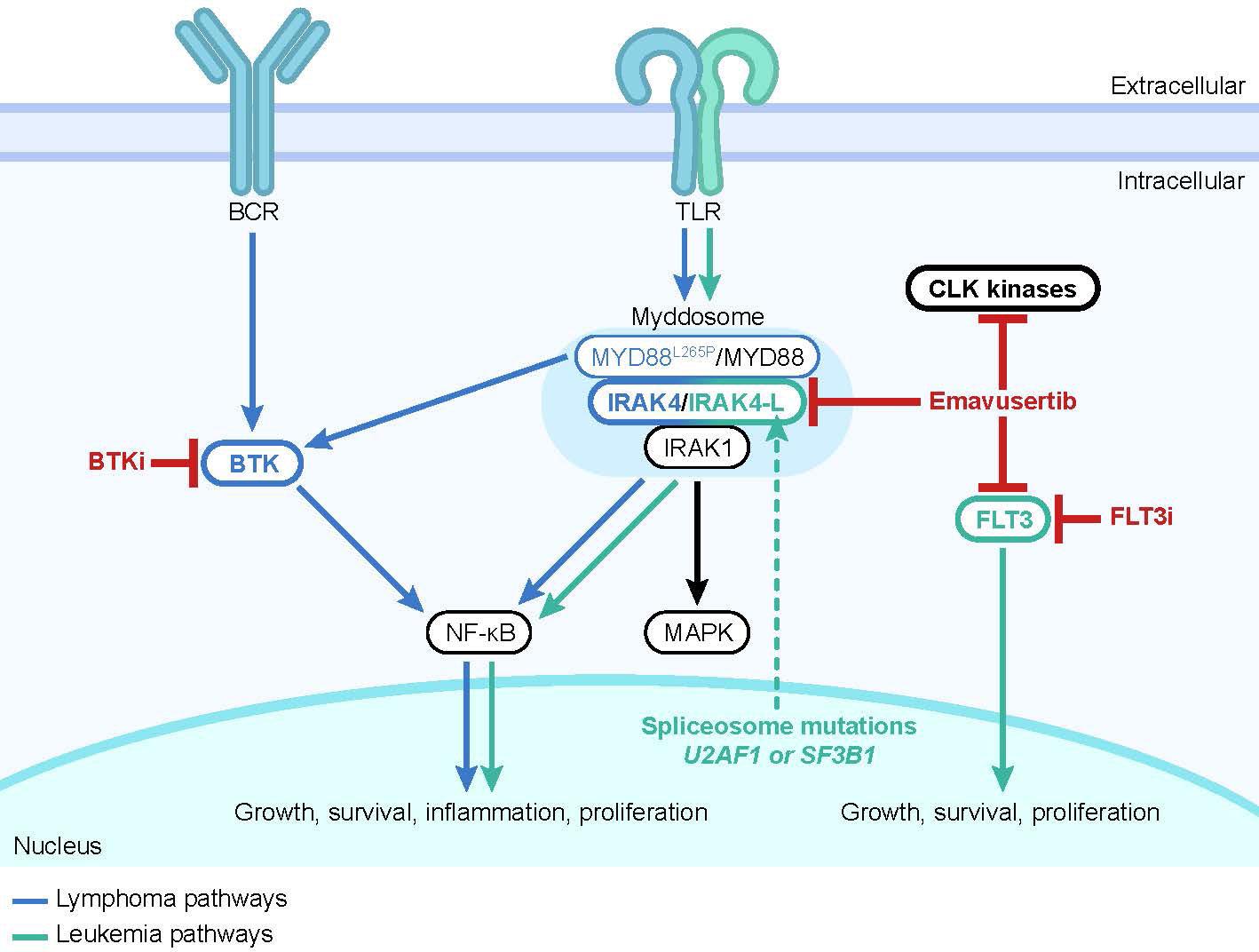
Figure 1 The IRAK4 pathway. Upstream signals from transmembrane receptors such as TLRs drive formation of the myddosome, activation of IRAK4, and activation of downstream intracellular pathways that result in activation of NF-κB and MAPK. Hypermorphic MYD88L265P is associated with BTK activation (8). Mutations in the spliceosome components U2AF1 or SF3B1 drive expression of the hypermorphic IRAK4-L isoform (9, 10). Small molecule inhibitors are shown in red. Multiple BTK and FLT3 inhibitors are approved for the treatment of hematological malignancies (11, 12). The investigational small molecule emavusertib inhibits CLK kinases in vitro and has shown inhibition of TLR/IRAK4 and FLT3 activity in preclinical leukemia models (9, 13, 14). Abbreviations: BCR, B-cell receptor; BTK, Bruton’s tyrosine kinase; CLK, CDC-like kinase; FLT3, FMS-like tyrosine kinase 3; i, inhibitor; IRAK1, interleukin 1 receptor-associated kinase 1; IRAK4, interleukin 1 receptor-associated kinase 4; MAPK, mitogen-activated protein kinase; MyD88, myeloid differentiation primary response; NF-κB, nuclear factor kappa B; TLR, toll-like receptor.
Dr Starczynowski explained further that in many hematologic and solid malignancies, innate immune signaling becomes dysregulated due to mutation and development of an aberrant inflammatory environment (1, 20). For example, MDS is characterized by defects in hematopoiesis and myelodysplasia due to the dominance of aberrant hematopoietic stem cell clones (1, 20, 21). The pathogenesis of MDS is often driven by a combination of genetic mutations and associated hyperactivation of innate immune signaling pathways (7, 20, 21). Cases of AML that develop from MDS generally maintain similar signaling dependencies to MDS itself (1).
The frequent and aberrant activation of the IRAK4 pathway seen in MDS and various leukemias is likely a function of its position linking signaling from most TLRs to activation of NF-κB and subsequent pro-survival and pro-proliferation consequences (1). MDS cells show increased expression of TLR and signaling of downstream components, especially in lower-risk disease (LR-MDS) (7, 20). In addition, deletion of chromosomal segment 5q (del5q), common in MDS, results in activation of TRAF6 and IRAK1 due to loss of negative regulation through multiple mechanisms (22, 23).
Other leukemias and lymphomas are driven by mutations or perturbations upstream of IRAK4, such as in TLR and MyD88, and these will also mediate their effects through IRAK4 (1, 24). L265P is a common hypermorphic driver mutation in MyD88 that is often found in Waldenström’s macroglobulinemia (WM), activated B-cell-like diffuse large B cell lymphoma (ABC-DLBCL), splenic marginal zone lymphoma, and central nervous system (CNS)/testicular-DLBCL (8, 25–32). L265P drives both activation of IRAK4 and BTK (8, 24).
Dr Garcia-Manero then summarized key findings regarding aberrant activation of the TLR/IRAK4 pathway in MDS and AML. He described how TLRs 1, 2, and 6 are upregulated in CD34+ cells from the bone marrow of patients with MDS, and some patients have an activating TLR2 mutation (33). TLR2 expression levels on CD34+ cells correlate with MDS risk status (33). TLR2 signaling through the IRAK4 pathway induces the histone demethylase JMJD3 that in turn epigenetically regulates expression of genes and cytokines involved in innate immunity (33, 34). Such TLR2 signaling may therefore comprise a positive feedback loop. Additionally, chronic TLR pathway activation impairs normal stem cell repopulation (7).
Dr Ulrich Steidl (Montefiore Einstein Cancer Center) described research exploring the functional role of IL-1 receptor accessory protein (IL1RAP) in MDS and AML. IL1RAP is a component of the IL-1 receptor complex directly upstream of IRAK4, and is frequently upregulated on (pre)leukemic stem cells from patients with MDS and AML (35–41). Preclinical models indicate that upregulation of IL1RAP plays multiple functionally important roles in AML cells (40, 42). Firstly, targeting IL1RAP inhibits the clonogenic capacity of leukemic stem cells and reduces tumor burden but does not impact healthy hematopoietic processes or viability; secondly, upregulation of IL1RAP and subsequent activation of downstream signaling is a cell-intrinsic process in MDS and AML leukemic stem cells; and thirdly, IL1RAP is often upregulated during disease progression (40, 42).
Dr Alan List (Precision BioSciences, Inc) outlined the multiple connections between three key multimeric protein complexes known as signalosomes or supramolecular organizing centers (SMOCs) that assemble within the cell cytoplasm in MDS (43). These comprise the myddosome, NLRP3 inflammasome, and the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)/stimulator of interferon genes (STING) cytosolic deoxyribonucleic acid (DNA) sensor complex (43). These complexes are involved in signaling in the innate immune response and induce inflammatory and lytic cell death responses (43). Assembly of each of these SMOCs is induced by specific stimuli. Formation of the myddosome is induced by the binding of TLRs to their cognate ligands. In LR-MDS, the key ligands are the inflammatory proteins S100A8 and S100A9, which are produced by cells of the myeloid lineage and are abundant in the inflammatory milieu and activate TLR4 (5, 7, 44).
The NLRP3 inflammasome is formed in response to danger signals (45), drives production of IL-1β and IL-18, and promotes caspase activation and a specific form of programmed cell death termed pyroptosis (5, 17, 43, 46). Activation of the inflammasome leads to assembly of apoptosis-associated speck-like protein containing a CARD (ASC) into a large protein complex known as a speck. Assembly of these potentially damaging ASC specks is driven by and serves as a marker of pyroptosis. Formation of and signaling by the NLRP3 inflammasome is induced by two signals: an initial priming signal that can be provided by activated TLR signaling and the myddosome, and a second activation signal that can be provided by cGAS/STING (5, 17, 43).
The third SMOC, cGAS/STING, forms in response to autologous cytosolic DNA. This complex drives production of type I interferon, and serves as an NLRP3 inflammasome activation signal (43). In vitro, inhibition of cGAS suppresses pyroptosis and induces terminal differentiation in primary MDS samples (43).
Dr List described the mechanism by which activation of the myddosome, NLRP3 inflammasome, and cGAS/STING creates an interdependent feed-forward cycle that underlies ineffective hematopoiesis and continued inflammation in LR-MDS. The inflammatory proteins S100A8 and S100A9 drive activation of TLR4, myddosome formation and activation, and then NLRP3 inflammasome priming and activation, which in turn drives further inflammation and production of cell debris that can further activate the innate immune system (5, 7, 17). Pyroptotic cell lysis driven by inflammasome activation releases ASC specks that are catalytically active and propagate inflammation. Various somatic gene mutations further drive activation of the NLRP3 inflammasome, including by exposing genomic DNA for detection by cGAS/STING (43). Together, this research suggests that that effective treatment of LR-MDS is likely to require concurrent suppression of myddosome and inflammasome signals and dissolution of catalytically active ASC specks.
Mutations in genes controlling the spliceosome – the machinery that creates mature mRNA molecules – are also common in MDS and AML (47). Spliceosome mutations can alter splicing by including or excluding specific exons from the mRNA leading to the creation of alternative protein isoforms (48). Mutations in the splicing factors U2AF1 (leading to retention of exon 4) and SF3B1 (leading to retention of exon 6) are now known to drive the expression of IRAK4-L, an elongated, hypermorphic isoform of IRAK4 (1, 9, 10, 49). This IRAK4-L isoform mediates maximal activation of the downstream NF-κB pathway, supporting leukemic cell survival, and is associated with clinical aggressiveness and a worse prognosis (Figure 2) (9, 10). More than 50% of AML patients preferentially express the IRAK4-L isoform (10). By contrast, the shorter IRAK4-S isoform is preferentially expressed in healthy hematopoietic tissues (10).
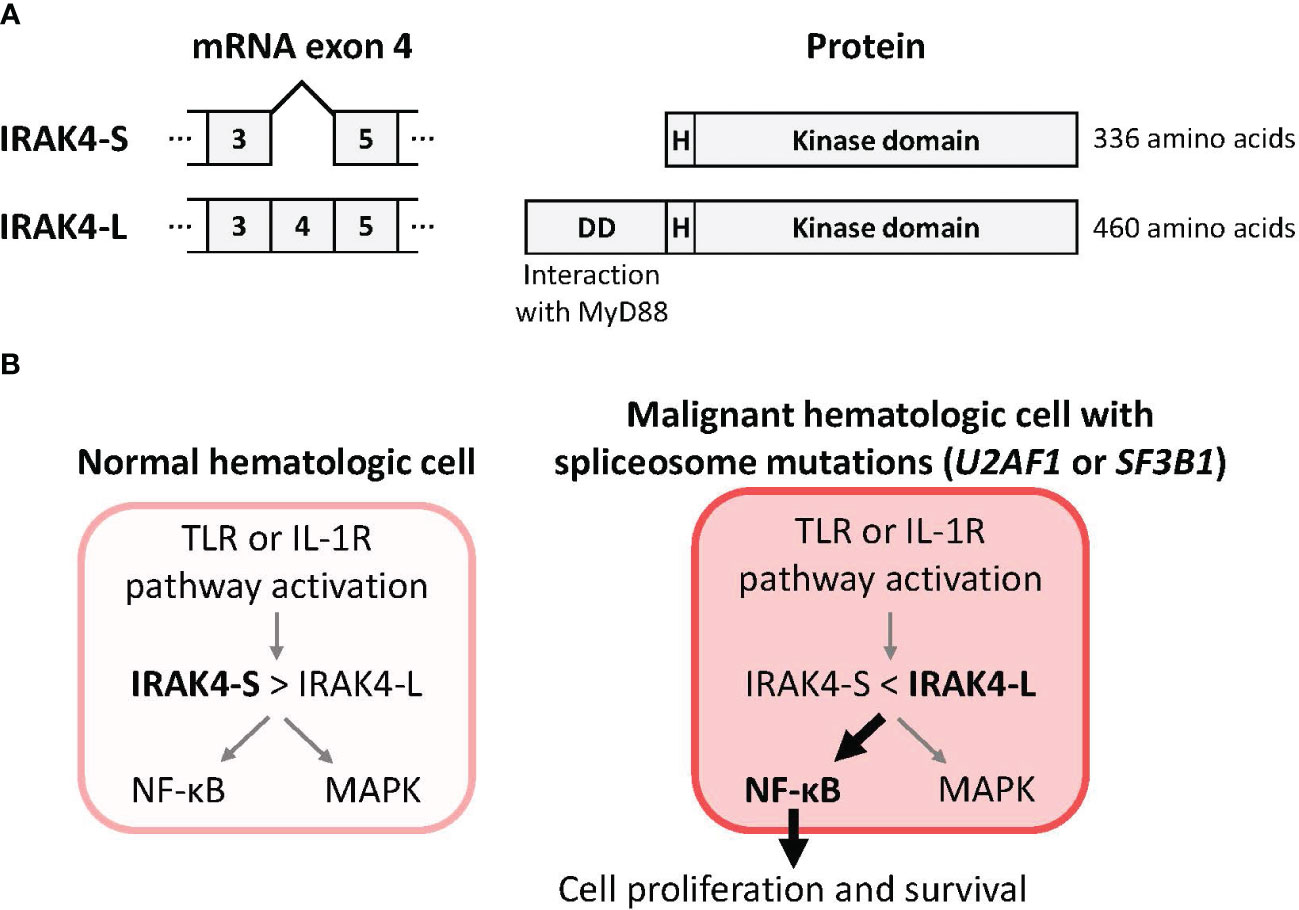
Figure 2 Mutations in the spliceosome components U2AF1 or SF3B1 drive production of IRAK4-L, a long isoform of IRAK4, and maximal activation of NF-κB. (A) Removal of IRAK4 exon 4 from the pre-mRNA by the spliceosome is predicted to result in use of an alternative translational start site and production of a 336 amino acid IRAK4 termed IRAK4-S that lacks the N-terminal death domain. Inclusion of exon 4 in the pre-mRNA is predicted to result in a 460 amino acid IRAK4 termed IRAK4-L that includes the death domain that mediates interaction with MyD88 (10). (B) A model for IRAK4 isoforms and their impacts (9, 10). The pool of IRAK4 isoforms in normal hematologic cells is skewed towards the IRAK4-S isoform that results in limited downstream activation of NF-κB. In malignant hematologic cells with mutations in the spliceosome components U2AF1 (10) or SF3B1 (9) the pool of IRAK4 isoforms is skewed towards the IRAK4-L isoform that results in maximal downstream activation of NF-κB. Abbreviations: DD, death domain; H, hinge domain; IRAK4, interleukin 1 receptor-associated kinase 4; IL-1R, interleukin 1 receptor; IRAK4-L, IRAK4 long isoform; IRAK4-S, IRAK4 short isoform; MAPK, mitogen-activated protein kinase; MyD88, myeloid differentiation primary response 88; NF-κB, nuclear factor kappa B; TLR, toll-like receptor.
Dr Amit Verma then added to this topic by reviewing the latest research into the distinct biology of the hypermorphic and hypomorphic IRAK4 isoforms. In healthy hematopoiesis, the IRAK4-S isoform is ubiquitinated and degraded, limiting the downstream effects of signaling through the IRAK4 pathway (9). However, in MDS and AML with spliceosome mutations such as SF3B1 and U2AF1, the stable larger active IRAK4-L isoform that is produced is able to associate with MyD88 and supports maximal downstream signaling to NF-κB, but also confers sensitivity to IRAK4 inhibitors (9, 10). IRAK4 knockdown in primary samples from a patient with SF3B1-mutated MDS led to an increase in myeloid differentiation (9). Treatment with the IRAK4 inhibitor emavusertib blocked TLR signaling in vitro and increased myeloid differentiation in primary SF3B1-mutated MDS samples. Furthermore, in preclinical patient-derived xenograft (PDX) models, emavusertib treatment decreased leukemic engraftment (9).
Dr Omar Abdel-Wahab (Memorial Sloan Kettering Cancer Center) then introduced a separate line of investigation into splicing dysregulation in AML that may help overcome treatment resistance. Studies have identified certain genes regulating RNA processing and metabolism whose loss confers increased sensitivity to AML-directed treatments including the B cell lymphoma 2 (BCL2) inhibitor venetoclax (50). These genes included RNA splicing factors that are substrates for the CDC-like kinase (CLK) and dual-specificity tyrosine-regulated kinase (DYRK) families (50). Inhibition of CLK/DYRK with the small molecule inhibitor SM09419 alongside venetoclax promoted intron retention and synergized with venetoclax treatment in vitro. The specific mechanism involved inclusion of a poison exon that contains a premature stop codon that impacts the FLT3 gene. SM09419 also overcame venetoclax resistance in AML cells and in preclinical PDX models. Together, these findings support administering CLK/DYRK inhibitors alongside BCL2 inhibitors in AML (50). Interestingly, in addition to its action on IRAK4, emavusertib also binds with high affinity to kinases in the CLK and DYRK families, suggesting additional mechanisms and therapeutic possibilities for emavusertib in AML.
The therapeutic potential of IRAK4 inhibition has led to the development of small molecule IRAK4 inhibitors. Key functional residues linked with IRAK4 kinase activity include the Tyr262 gatekeeper, the catalytic Lys213 residue, and the hinge residue Met265 (13, 51). IRAK4 differs structurally from other serine/threonine kinases (reviewed by Wang Z, et al), including a Schellman loop in the N-terminal extension and a longer loop between helices αD and αE, resulting in an extended ATP-binding pocket (51). However, the current investigational small molecule inhibitors of IRAK4 of which we are aware also inhibit additional targets; for example, in addition to inhibition of IRAK4, emavusertib in vitro also inhibits the activity of FLT3 and CLK-family kinases (9, 13, 14), (Figure 1).
Dr Grzegorz Nowakowski (Mayo Clinic Comprehensive Cancer Center) then discussed the growing interest in IRAK4 inhibition in multiple hematologic cancers (1, 52). Pro-oncogenic NF-κB signaling is driven by the activity of both the TLR/IRAK4 and the B-cell receptor (BCR)/BTK pathways (11, 27, 53). These parallel pathways are primary independent activators of NF-κB, and dysregulation of either pathway can drive excessive B-cell proliferation (Figure 1) (1, 11, 27, 52, 53). Inhibition of the BTK pathway has proven effective, leading to the approval of BTK inhibitors for the treatment of multiple B-cell malignancies (11). Indeed, in preclinical studies, IRAK4 inhibition has been shown to have anti-tumor activity in a variety of myeloid and lymphoid cell types (1, 14). Interestingly, inhibition of both IRAK4 and BTK pathways is synergistic (53, 54).
In cancers where inflammatory signaling is believed to play a role assessing the combinatorial effects of IRAK4 inhibition and additional agents that inhibit inflammatory signaling is an exciting area of future investigation.
Research is assessing whether there is a role for the IRAK4 pathway in driving adaptive resistance to targeted therapies. For example, Dr Starczynowski outlined how adaptive resistance in vitro to a FLT3 inhibitor by AML cells with a FLT3 internal tandem duplication (ITD) mutation demonstrates an evolution towards reduced sensitivity without FLT3 or NRAS mutations (55). This adaptive resistance to the FLT3 inhibitor is due to activation of the TLR/IRAK4 pathway and specifically TLR9 activation (55), suggesting that extracellular DNA is driving TLR9 and IRAK4 pathway activation. Subsequent knockdown or inhibition of IRAK4 restored sensitivity to FLT3 inhibitors (55). In addition to inhibiting IRAK4, emavusertib also binds with high affinity to FLT3-ITD proteins (56), suggesting that emavusertib may interact with both a common oncogenic driver and an important potential mechanism of adaptive resistance to FLT3 treatment.
To date, our understanding of the functions of IRAK4 in cancer has largely come from research into hematological malignancies. However, as Dr Kian-Huat Lim (Washington University School of Medicine in St. Louis) explained, more is being learned about the roles of IRAK4 in solid tumors. In particular, research is revealing links between IRAK4 and chemoresistance and poorer outcomes in pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (1, 3, 57). PDAC is treated primarily with chemotherapy, and immunotherapy has proved largely ineffective (58). Activation of NF-κB driven by KRAS mutations is a common feature in PDAC and is associated with poor prognosis and treatment resistance (58). IRAK4 is constitutively activated in at least two-thirds of PDAC cases, and this has been positively correlated with activation of NF-κB (57) and inversely correlated with CD8+ T-cell abundance (59). Preclinical studies have shown that IRAK4 inhibition reduces NF-κB activation (57). Notably, Dr. Lim and colleagues created the first conditional IRAK knockout PDAC mouse model and showed that loss of tumor-intrinsic IRAK4 leads to adenocarcinomas that are more inflamed and infiltrated with activated CD4 and CD8 T cells, and are sensitive to checkpoint immunotherapy (59). Similarly, emavusertib treatment has been shown to synergize with chemotherapy and immunotherapy (59). These results led to a clinical trial that will be conducted through the NCI Experimental Therapeutics Clinical Trials Network mechanism combining standard chemotherapy and emavusertib for PDAC patients who have progressed through first line chemotherapy (Table 1). Similarly, in colorectal cancer, activation of IRAK4 and thereby NF-κB indicates poor prognosis (3). Chemotherapy further enhances TLR9 expression and subsequent IRAK4 and NF-κB activation (3). In preclinical models, IRAK4 inhibition potentiated the effects of chemotherapy in this setting as well (3) and these outcomes establish the rationale for a future clinical trial combining emavusertib with chemotherapy. Additional preclinical studies are currently underway to better understand the mechanisms of TLR and BCR signaling in cancers and the potential of IRAK4 inhibition with emavusertib.
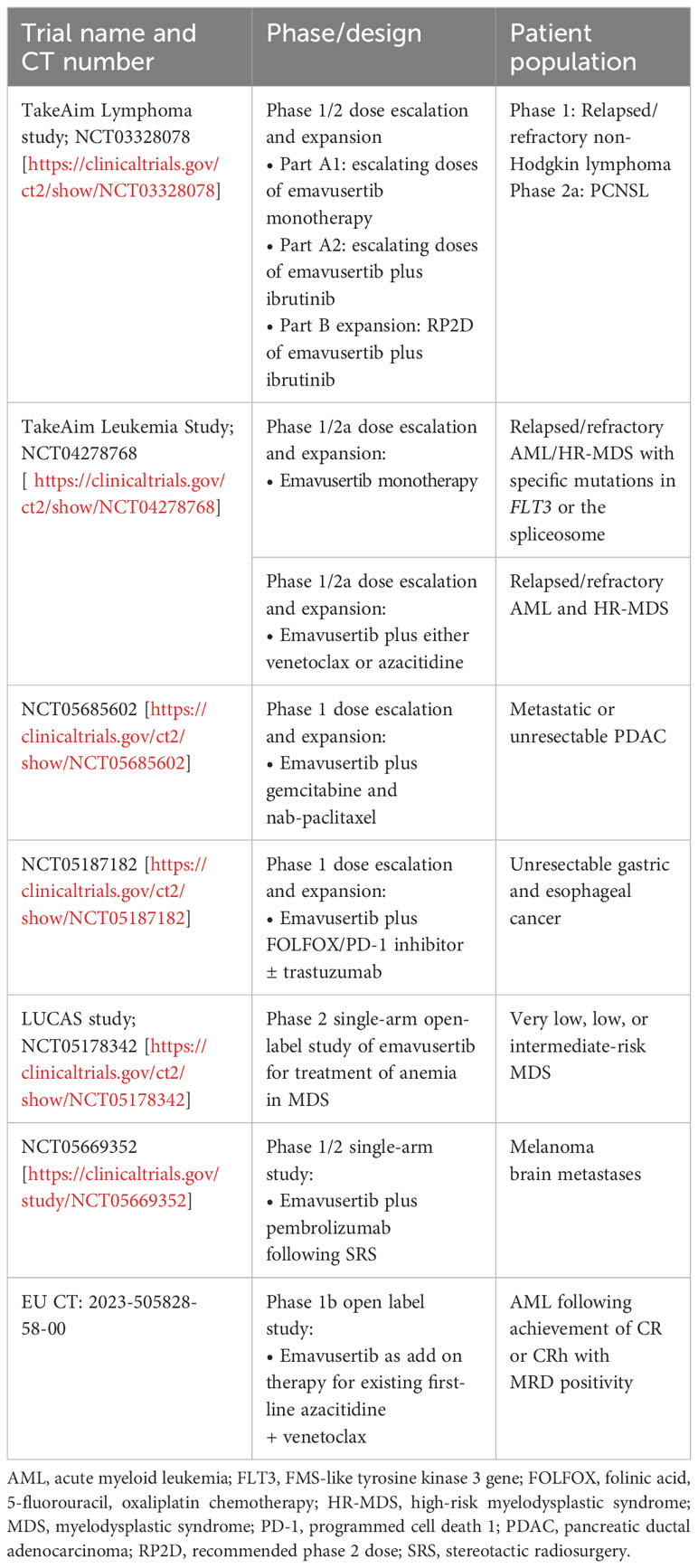
Table 1 Key ongoing clinical trials of emavusertib.
Dr Robert Martell (Curis, Inc) concluded the meeting by summarizing early findings from two key ongoing phase 1/2 clinical studies assessing emavusertib in hematologic cancers involving activation of the IRAK4 pathways (Table 1). These studies are exploring both monotherapy and combination regimens, given the preclinical evidence showing synergy between emavusertib or IRAK4 inhibition and treatments targeting FLT3 (55) and BTK (54, 60).
Early results from these studies are promising. In the dose-escalation portion of the phase 1/2a TakeAim Lymphoma trial evaluating the safety, efficacy, and recommended phase 2 dose of emavusertib in patients with relapsed or refractory non-Hodgkin lymphomas (NCT03328078), emavusertib monotherapy decreased tumor burden across several non-Hodgkin lymphoma subtypes, especially at higher doses (61). Anti-tumor responses were seen in both BTK inhibitor-naïve and -experienced patients. Of note, a heavily pretreated patient with WM showed a clear dose-dependent decrease in M protein levels in response to treatment with emavusertib (61). As of an October 12, 2023, data-cutoff, five of 16 evaluable, heavily pretreated patients showed a complete response (CR, 31%) (62). Three of five evaluable patients (60%) with primary CNS lymphoma (PCNSL), an area of high unmet need, achieved a CR. Accordingly, the ongoing phase 2a portion of the TakeAim Lymphoma trial is evaluating the safety and efficacy of emavusertib in combination with ibrutinib in patients with PCNSL.
There are also early signs of efficacy for single-agent emavusertib in AML and high-risk MDS (HR-MDS) in the ongoing phase 1/2a TakeAim Leukemia trial assessing the safety, efficacy, and recommended phase 2 dose of emavusertib (NCT04278768) (56, 61, 63). During the meeting, Dr. Garcia-Manero presented data from the TakeAim Leukemia trial, which includes patients with AML harboring FLT3 mutations and patients with AML or MDS with spliceosome mutations. In a more recent data cutoff of October 12, 2022, 71 patients with a median of 2 prior lines of therapy had been treated with emavusertib monotherapy. Of the 7 evaluable patients with AML with FLT3 mutations who received emavusertib, 2 achieved a CR (29%), 1 was in morphologic-free leukemia state, and 1 with gilteritinib-refractory disease achieved near-normalization of blast count and complete loss of detectable FLT3 clones. Evaluable patients with AML with a spliceosome mutation (n=9) receiving emavusertib achieved a CR/hematological CR rate of 22%. Finally, evaluable patients with MDS with a spliceosome mutation (n=11) achieved an objective response rate of 45%, with all responses achieving a marrow CR (63). Anti-tumor activity was also observed in patients without specific targeted mutations (56, 63).
At the time of the symposium, 49 patients with AML or MDS had been treated with emavusertib monotherapy and no dose-limiting myelosuppression had been reported, supporting its suitability to combine with other treatments such as chemotherapy (56). The pharmacokinetic profile of emavusertib supports twice-daily dosing and high target suppression at clinically relevant exposures (14, 61). Investigation of the safety and tolerability, pharmacokinetics, and change in measurable residual disease (MRD) status of emavusertib as an add-on therapy with existing first-line azacitidine and venetoclax therapy in patients with AML who achieved CR or CRh with MRD positivity is currently underway in a phase 1b open label study (EU CT Number 2023-505828-58-00).
Emavusertib treatment has also shown efficacy in combination with ibrutinib, including in patients with B-cell malignancies previously exposed to BTK inhibitors (60). Objective responses have been recorded, and preliminary efficacy data suggest that combination treatment with emavusertib may overcome ibrutinib resistance (60).
Further ongoing clinical trials are evaluating treatment outcomes with emavusertib in other therapeutic areas. Ongoing phase 1 clinical trials led by Dr Lim are also exploring the synergistic potential of emavusertib in gastric and esophageal cancer (NCT05187182). Furthermore, the ongoing phase 2 LUCAS trial (NCT05178342) is assessing the impacts of emavusertib treatment on anemia in patients with very low, low, and intermediate risk MDS. A phase 1/2 trial is evaluating the use of emavusertib plus pembrolizumab following stereotactic radiosurgery in patients with melanoma brain metastases (NCT05669352) (Table 1).
The 1st Symposium on IRAK4 in Cancer provided valuable insights into the biology and therapeutic potential of IRAK4 inhibition in cancers. IRAK4 is a key kinase necessary for intracellular signaling from most TLRs, and the TLR-IRAK4 pathway culminates in activation of NF-κB. Within an innate immune response, this activation is advantageous as it promotes cell survival, cytokine production, inflammation, and priming of adaptive immune responses. But in cancers that coopt the IRAK4 immune signaling pathway, dysregulation of inflammatory signaling leads to survival and proliferation of malignant cells.
IRAK4 activity has been implicated in the progression of multiple cancer types, including both hematologic malignancies and solid tumors. Advances are being made in our understanding of the role of IRAK4 signaling within these cancers and the potential applications of IRAK4 inhibitors. For example, recent research has discovered that U2AF1 and SF3B1 spliceosome mutations induce the expression of therapeutically targetable, hypermorphic IRAK4-L and provide a genetic link to the activation of chronic innate immune signaling in AML.
Preclinical studies have shown IRAK4 inhibition to have anti-cancer effects, synergize with several current treatments including FLT3 inhibitors and BTK inhibitors, overcome adaptive treatment resistance, and potentially unleash anti-tumor T-cell immunity. The IRAK4 inhibitor emavusertib has shown promise in multiple preclinical cancer models with several tumor responses reported in ongoing phase 1/2 clinical trials, as monotherapy and in combination with ibrutinib. Ongoing research and clinical trials into IRAK4 signaling will help further elucidate its role and consequences in cancer and define the opportunities for therapeutic IRAK4 inhibition.
GG: Writing – review & editing. UP: Writing – review & editing. K-HL: Writing – review & editing. GN: Writing – review & editing. OA-W: Writing – review & editing. HK: Writing – review & editing. AV: Writing – review & editing. DS: Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
The authors of this article would like to thank their co-presenters at the symposium for their contributions, insights and discussion: Dr. Ulrich Steidl, of the Montefiore Einstein Cancer Center; Dr. Alan List, of Precision BioSciences Inc; and Dr. Robert Martell, of Curis Inc.
GG-M received research funding from AbbVie, Amphivena Therapeutics, Astex Pharmaceuticals, Bristol Myers Squibb, H3 Biomedicine, Helsinn Therapeutics, Merck, Novartis, and Onconova Therapeutics, served in a consulting or advisory role for Acceleron Pharma, Astex Pharmaceuticals, Bristol Myers Squibb, Helsinn Therapeutics, and Jazz Pharmaceuticals, and received honoraria from AbbVie, Acceleron Pharma, Astex Pharmaceuticals, Bristol Myers Squibb, and Helsinn Therapeutics. UP received grant support from Bristol Myers Squibb, Celgene, Janssen, Novartis, Amgen, and Merck, served in a consulting or advisory role for Bristol Myers Squibb and Celgene, and received honoraria from Bristol Myers Squibb, Celgene, Janssen, Novartis, AbbVie, Geron, and Takeda. K-HL received research materials from Curis. GN received research funding from Celgene, NanoString Technologies, and MorphoSys and served in a consulting or advisory role or as a member of an entity’s Board of Directors for Celgene, MorphoSys, Genentech, Selvita, Debiopharm Group, and Kite/Gilead. OA-W received grant support from Nurix Therapeutics, Loxo/Lilly, and Minovina Therapeutics as well as served in a consulting or advisory role from H3B Biomedicine, Foundation Medicine Inc., Merck, Prelude Therapeutics, and Janssen. HK received grant support from AbbVie, Amgen, Ascentage Pharma, Bristol Myers Squibb, Daiichi-Sankyo, Immunogen, Jazz Pharmaceuticals, and Novartis and honoraria from AbbVie, Amgen, Amphista, Ascentage, Astellas, Biologix, Curis, Ipsen Biopharmaceuticals, KAHR Medical, Labcorp, Novartis, Pfizer, Shenzen Target Rx, Stemline, and Takeda. AV received research funding from Prelude, BMS, GSK, Incyte, Medpacto, Curis, and Eli Lilly, is a scientific advisor for Stelexis, Bakx, Novartis, Acceleron and Celgene, and has received honoraria from Stelexis, Curis and Janssen, and holds equity in Stelexis, Bakx, and Throws Exception. DS served in a consulting or advisory role and received research funding from Kymera Therapeutics, Kurome Therapeutics, Captor Therapeutics, and Tolero Therapeutics and has equity in Kurome Therapeutics.
The authors declare that this study received funding from Curis, Inc. The funder had the following involvement: support for the IRAK4 Symposium discussed within, and support for manuscript preparation. Editorial support was provided by BOLDSCIENCE and was supported by Curis, Inc.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bennett J, Starczynowski DT. IRAK1 and IRAK4 as emerging therapeutic targets in hematologic Malignancies. Curr Opin Hematol (2022) 29:8–19. doi: 10.1097/MOH.0000000000000693
2. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem (2009) 284:25404–11. doi: 10.1074/jbc.M109.022392
3. Li Q, Chen Y, Zhang D, Grossman J, Li L, Khurana N, et al. IRAK4 mediates colitis-induced tumorigenesis and chemoresistance in colorectal cancer. JCI Insight (2019) 4:130867. doi: 10.1172/jci.insight.130867
4. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med (2021) 218:e20201544. doi: 10.1084/jem.20201544
5. Sallman DA, List A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood (2019) 133:1039–48. doi: 10.1182/blood-2018-10-844654
6. Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh K-H, et al. A critical role for IRAK4 kinase activity in Toll-like receptor–mediated innate immunity. J Exp Med (2007) 204:1025–36. doi: 10.1084/jem.20061825
7. Paracatu LC, Schuettpelz LG. Contribution of aberrant toll like receptor signaling to the pathogenesis of myelodysplastic syndromes. Front Immunol (2020) 11:1236. doi: 10.3389/fimmu.2020.01236
8. Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood (2013) 122:1222–32. doi: 10.1182/blood-2012-12-475111
9. Choudhary GS, Pellagatti A, Agianian B, Smith MA, Bhagat TD, Gordon-Mitchell S, et al. Activation of targetable inflammatory immune signaling is seen in myelodysplastic syndromes with SF3B1 mutations. Elife (2022) 11:e78136. doi: 10.7554/eLife.78136
10. Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid Malignancies. Nat Cell Biol (2019) 21:640–50. doi: 10.1038/s41556-019-0314-5
11. Ahn IE, Brown JR. Targeting Bruton’s tyrosine kinase in CLL. Front Immunol (2021) 12:687458. doi: 10.3389/fimmu.2021.687458
12. Zhao JC, Agarwal S, Ahmad H, Amin K, Bewersdorf JP, Zeidan AM. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev (2022) 52:100905. doi: 10.1016/j.blre.2021.100905
13. Gummadi VR, Boruah A, Ainan BR, Vare BR, Manda S, Gondle HP, et al. Discovery of CA-4948, an orally bioavailable IRAK4 inhibitor for treatment of hematologic Malignancies. ACS Med Chem Lett (2020) 11:2374–81. doi: 10.1021/acsmedchemlett.0c00255
14. Booher RN, Nowakowski GS, Patel K, Lunning MA, Samson MES, Atoyan R, et al. Preclinical activity of IRAK4 kinase inhibitor CA-4948 alone or in combination with targeted therapies and preliminary phase 1 clinical results in non-Hodgkin lymphoma. Blood (2018) 132:4168. doi: 10.1182/blood-2018-99-117383
15. O’Carroll A, Chauvin B, Brown JWP, Meagher A, Coyle J, Schill J, et al. Pathological mutations differentially affect the self-assembly and polymerisation of the innate immune system signalling adaptor molecule MyD88. BMC Biol (2018) 16:149. doi: 10.1186/s12915-018-0611-7
16. Lin S-C, Lo Y-C, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature (2010) 465:885–90. doi: 10.1038/nature09121
17. Sallman DA, Cluzeau T, Basiorka AA, List A. Unraveling the pathogenesis of MDS: the NLRP3 inflammasome and pyroptosis drive the MDS phenotype. Front Oncol (2016) 6:151. doi: 10.3389/fonc.2016.00151
18. Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Toll-like receptor signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol (2013) 191:3995–9. doi: 10.4049/jimmunol.1301681
19. Lin K-M, Hu W, Troutman TD, Jennings M, Brewer T, Li X, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A (2014) 111:775–80. doi: 10.1073/pnas.1320294111
20. Winter S, Shoaie S, Kordasti S, Platzbecker U. Integrating the “Immunome” in the stratification of myelodysplastic syndromes and future clinical trial design. J Clin Oncol (2020) 38:1723–35. doi: 10.1200/JCO.19.01823
21. Cazzola M. Myelodysplastic syndromes. N Engl J Med (2020) 383:1358–74. doi: 10.1056/NEJMra1904794
22. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med (2010) 16:49–58. doi: 10.1038/nm.2054
23. Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
24. Bennett JR, Ishikawa C, Agarwal P, Yeung J, Sampson A, Uible E, et al. Paralog-specific signaling by IRAK1/4 maintains MyD88-independent functions in MDS/AML. Blood (2023) 142:989–1007. doi: 10.1182/blood.2022018718
25. Poulain S, Roumier C, Decambron A, Renneville A, Herbaux C, Bertrand E, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood (2013) 121:4504–11. doi: 10.1182/blood-2012-06-436329
26. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl. J. Med. (2012) 367:826–33. doi: 10.1056/NEJMoa1200710
27. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470:115–9. doi: 10.1038/nature09671
28. Martinez-Lopez A, Curiel-Olmo S, Mollejo M, Cereceda L, Martinez N, Montes-Moreno S, et al. MYD88 (L265P) somatic mutation in marginal zone B-cell lymphoma. Am. J. Surg. Pathol. (2015) 39:644–51. doi: 10.1097/PAS.0000000000000411
29. Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood (2014) 123:1637–46. doi: 10.1182/blood-2013-09-525808
30. Kraan W, Horlings HM, van Keimpema M, Schilder-Tol EJM, Oud MECM, Scheepstra C, et al. High prevalence of oncogenic MYD88 and CD79B mutations in diffuse large B-cell lymphomas presenting at immune-privileged sites. Blood Cancer J (2013) 3:e139. doi: 10.1038/bcj.2013.28
31. Kraan W, van Keimpema M, Horlings HM, Schilder-Tol EJM, Oud MECM, Noorduyn LA, et al. High prevalence of oncogenic MYD88 and CD79B mutations in primary testicular diffuse large B-cell lymphoma. Leukemia (2014) 28:719–20. doi: 10.1038/leu.2013.348
32. Lee J-H, Jeong H, Choi J-W, Oh H, Kim Y-S. Clinicopathologic significance of MYD88 L265P mutation in diffuse large B-cell lymphoma: a meta-analysis. Sci Rep (2017) 7:1785. doi: 10.1038/s41598-017-01998-5
33. Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia (2013) 27:1832–40. doi: 10.1038/leu.2013.180
34. Wei Y, Chen R, Dimicoli S, Bueso-Ramos C, Neuberg D, Pierce S, et al. Global H3K4me3 genome mapping reveals alterations of innate immunity signaling and overexpression of JMJD3 in human myelodysplastic syndrome CD34+ cells. Leukemia (2013) 27:2177–86. doi: 10.1038/leu.2013.91
35. Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood (2012) 120:1290–8. doi: 10.1182/blood-2012-01-404699
36. Askmyr M, Ågerstam H, Hansen N, Gordon S, Arvanitakis A, Rissler M, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood (2013) 121:3709–13. doi: 10.1182/blood-2012-09-458935
37. Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, et al. (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood (2014) 123:3951–62. doi: 10.1182/blood-2013-10-536078
38. Herrmann H, Sadovnik I, Eisenwort G, Rülicke T, Blatt K, Herndlhofer S, et al. Delineation of target expression profiles in CD34+/CD38– and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. (2020) 4:5118–32. doi: 10.1182/bloodadvances.2020001742
39. Ho T-C, LaMere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood (2016) 128:1671–8. doi: 10.1182/blood-2016-02-695312
40. Mitchell K, Barreyro L, Todorova TI, Taylor SJ, Antony-Debré I, Narayanagari S-R, et al. IL1RAP potentiates multiple oncogenic signaling pathways in AML. J Exp Med (2018) 215:1709–27. doi: 10.1084/jem.20180147
41. de Boer B, Sheveleva S, Apelt K, Vellenga E, AB M, Huls G, et al. The IL1-IL1RAP axis plays an important role in the inflammatory leukemic niche that favors acute myeloid leukemia proliferation over normal hematopoiesis. Haematologica (2021) 106:3067–78. doi: 10.3324/haematol.2020.254987
42. Ågerstam H, Karlsson C, Hansen N, Sandén C, Askmyr M, von Palffy S, et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc Natl Acad Sci USA (2015) 112:10786–91. doi: 10.1073/pnas.1422749112
43. McLemore AF, Hou H-A, Meyer BS, Lam NB, Ward GA, Aldrich AL, et al. Somatic gene mutations expose cytoplasmic DNA to co-opt the cGAS/STING/NLRP3 axis in myelodysplastic syndromes. JCI Insight (2022) 7:e159430. doi: 10.1172/jci.insight.159430
44. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MAD, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13:1042–9. doi: 10.1038/nm1638
45. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
46. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
47. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature (2011) 478:64–9. doi: 10.1038/nature10496
48. Niño CA, Scotto di Perrotolo R, Polo S. Recurrent spliceosome mutations in cancer: mechanisms and consequences of aberrant splice site selection. Cancers (Basel) (2022) 14:281. doi: 10.3390/cancers14020281
49. Pollyea DA, Kim HM, Stevens BM, Lee FF-Y, Harris C, Hedin BR, et al. MDS-associated SF3B1 mutations enhance proinflammatory gene expression in patient blast cells. J Leukoc Biol (2021) 110:197–205. doi: 10.1002/JLB.6AB0520-318RR
50. Wang E, Bello Pineda JM, Bourcier J, Stahl M, Penson AV, Wakiro I, et al. Modulation of RNA splicing enhances response to BCL2 inhibition in acute myeloid leukemia. Blood (2021) 138:507. doi: 10.1182/blood-2021-146373
51. Wang Z, Wesche H, Stevens T, Walker N, Yeh W-C. IRAK-4 inhibitors for inflammation. Curr Top Med Chem (2009) 9:724–37. doi: 10.2174/156802609789044407
52. Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood (2007) 109:2700–7. doi: 10.1182/blood-2006-07-025809
53. Kelly PN, Romero DL, Yang Y, Shaffer AL III, Chaudhary D, Robinson S, et al. Selective interleukin-1 receptor–associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid Malignancy. J Exp Med (2015) 212:2189–201. doi: 10.1084/jem.20151074
54. Guidetti F, Arribas AJ, Sartori G, Spriano F, Barnabei L, Tarantelli C, et al. Targeting IRAK4 with emavusertib in lymphoma models with secondary resistance to PI3K and BTK inhibitors. J Clin Med (2023) 12:399. doi: 10.3390/jcm12020399
55. Melgar K, Walker MM, Jones LM, Bolanos LC, Hueneman K, Wunderlich M, et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci Transl Med (2019) 11:eaaw8828. doi: 10.1126/scitranslmed.aaw8828
56. Garcia-Manero G, Winer ES, DeAngelo DJ, Tarantolo S, Sallman DA, Dugan J, et al. S129: TakeAim Leukemia - a phase 1/2a study of the IRAK4 inhibitor emavusertib (CA-4948) as monotherapy or in combination with azacitidine or venetoclax in relapsed/refractory AML or MDS. HemaSphere (2022) 6:30–1. doi: 10.1097/01.HS9.0000843408.31385.3f
57. Zhang D, Li L, Jiang H, Knolhoff BL, Lockhart AC, Wang-Gillam A, et al. Constitutive IRAK4 activation underlies poor prognosis and chemoresistance in pancreatic ductal adenocarcinoma. Clin. Cancer Res. (2017) 23:1748–59. doi: 10.1158/1078-0432.CCR-16-1121
58. Dodhiawala PB, Khurana N, Zhang D, Cheng Y, Li L, Wei Q, et al. TPL2 enforces RAS-induced inflammatory signaling and is activated by point mutations. J. Clin. Invest. (2020) 130:4771–90. doi: 10.1172/JCI137660
59. Somani VK, Zhang D, Dodhiawala PB, Lander VE, Liu X, Kang L-I, et al. IRAK4 signaling drives resistance to checkpoint immunotherapy in pancreatic ductal adenocarcinoma. Gastroenterology (2022) 162:2047–62. doi: 10.1053/j.gastro.2022.02.035
60. Joffe E, Nowakowski GS, Tun HW, Rosenthal AC, Lunning MA, Ramchandren R, et al. Open-label, dose-escalation, and expansion trial of CA-4948 in combination with ibrutinib in patients with relapsed or refractory hematologic Malignancies. J Clin Oncol (2022) 40:7575–5. doi: 10.1200/JCO.2022.40.16_suppl.7575
61. Nowakowski G. Safety, Pharmacokinetics and Activity of CA-4948, an IRAK4 Inhibitor, for Treatment of Patients with Relapsed or Refractory Hematologic Malignancies: Results from the Phase 1 Study (2020). ASH. Available at: https://ash.confex.com/ash/2020/webprogram/Paper140857.html (Accessed November 4, 2022).
62. Grommes C, Tun H, Rosenthal AC, Lunning MA, Ramchandren R, Regales L, et al. Takeaim lymphoma: an open-label, dose escalation and expansion trial of emavusertib (CA-4948) in combination with ibrutinib in patients with relapsed or refractory hematologic Malignancies. Blood (2023) 142:4497. doi: 10.1182/blood-2023-189746
63. Metzeler KH, Winer ES, Platzbecker U, Verma A, DeAngelo DJ, Tarantolo SR, et al. Molecular characterization of clinical response in relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndrome patients treated with single agent emavusertib. Blood (2022) 140:9048–9. doi: 10.1182/blood-2022-167477
Keywords: IRAK4, inflammasome, myddosome, TLR, AML, MDS, BTK, FLT3
Citation: Garcia-Manero G, Platzbecker U, Lim K-H, Nowakowski G, Abdel-Wahab O, Kantarjian H, Verma A and Starczynowski DT (2024) Research and clinical updates on IRAK4 and its roles in inflammation and malignancy: themes and highlights from the 1st symposium on IRAK4 in cancer. Front. Hematol. 3:1339870. doi: 10.3389/frhem.2024.1339870
Received: 16 November 2023; Accepted: 29 January 2024;
Published: 15 February 2024.
Edited by:
Matteo Molica, S. Eugenio, ItalyReviewed by:
Elspeth Payne, University College London, United KingdomCopyright © 2024 Garcia-Manero, Platzbecker, Lim, Nowakowski, Abdel-Wahab, Kantarjian, Verma and Starczynowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermo Garcia-Manero, Z2dhcmNpYW1AbWRhbmRlcnNvbi5vcmc=; Daniel T. Starczynowski, RGFuaWVsLnN0YXJjenlub3dza2lAY2NobWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.